
Abstract
Adsorption energy is a fundamental thermodynamic quantity in the description of gas–surface interactions. In general, it depends not only on the ad-species and on the chemical nature of the substrate but also on the surface coverage and the density and nature of the surface defects. It can also be significantly affected by the presence of coadsorbates and by temperature when the latter determines a different arrangement of species on the substrate or even the formation of different moieties at the surface. On the one hand, the measure of the heat of adsorption by the isosteric method can be used only in selected cases, where adsorption occurs reversibly and a reliable control of the coverage over a large enough range of temperatures is experimentally accessible. On the other hand, temperature-programmed desorption, which until now has been the most widely used technique, is not applicable when adsorption occurs irreversibly, and care is needed to properly analyze the experimental data, especially when the heat of adsorption is strongly coverage dependent. Single-crystal calorimetry (SCAC) provides a means of overcoming most such limitations. Since it is impossible to cover all the results obtained by this technique in a single review, a rich bibliography is provided and only selected results (in the opinion of the authors) are summarized here. The first section addresses the results obtained by the infrared calorimeter of the Cambridge group, which mostly deals with the coverage dependence of the heat of adsorption of different gases on single-crystal (both low and high Miller index) surfaces. The second section deals with the results obtained by the pyroelectric calorimeter by the Washington group, which cover both the adsorption of large organic molecules and of nongaseous species. The impressive results obtained to date have drawn other research groups into the field, and new designs have appeared that enable, for example, the study of the adsorption of gas molecules at the surface of nanoparticles and at oxide surfaces. The last section of this chapter is devoted to such results.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


- Calorimetry
- Heat of Adsorption
- Heat of Reaction
- Defects at Surfaces
- Hydrocarbons
- CO
- Propylene Oxide
- Water
- Oxide Surface
- Lateral interaction
- Nanoparticle
- Sticking Probability
- Supersonic Molecular Beam
- Single Crystal Microcalorimetry
The heat of adsorption of molecules at surfaces is a key thermodynamic parameter for the understanding of adsorption and reaction at surfaces and, consequently, also for heterogeneous catalysis. It is also a key parameter in providing a test for the first principles quantum theory calculations now widely used to study processes at surfaces.
1 Methods for Calorimetry
The isosteric method, which measures the pressure required to keep a constant coverage of the adsorbate at the surface as a function of temperature, can be effectively used only when it is possible to check the equilibrium coverage in a reliable way over a large enough range of temperatures. It cannot, thus, be employed when irreversible adsorption occurs, and it can be affected by the changes in the arrangements of adsorbates at the surface while the temperature and the pressure are being modified [30.1]. Thermal desorption spectroscopy (TDS or TPD) is more routinely used, since it is compatible with conventional UHV experimental setups; it cannot, however, be used when desorption does not occur. Moreover, the extraction of thermodynamic data from TPD curves is affected significantly by the method employed for the analysis. The basic idea of calorimetry is to measure the temperature change occurring at the surface when adsorption occurs. While measurements for non monocrystalline surfaces have been conducted over a long period of time [30.2], measurements for single-crystal surfaces became systematically available only in the 1990s. After a few previous attempts by Kyser and Masel [30.3], the first single-crystal calorimeter was developed by the group of King in Cambridge [30.4, 30.5], and a systematic investigation of adsorption of different gases at single-crystal surfaces was undertaken. These results have already been reviewed in detail [30.1]. Here, we shall present only some of them: the effect of promoters on the heat of adsorption, the measurement of the energy difference between surface phases, the heat of reaction, lateral interactions among adsorbates, and the role of defects in adsorption. The main limitation of the first Cambridge calorimeter, which essentially monitored the change in the infrared emission caused by the temperature increase upon adsorption, was the restriction to room temperature adsorption and to nonactivated systems. A different calorimeter, based on pyroelectric detection, was then developed by the same group; this calorimeter allowed the measurement of heats of adsorption at different temperatures [30.6]. This design was extensively improved by the group of Campbell [30.7, 30.8] at Washington University in Seattle, who extended the method to the measurement of the heat of adsorption of metals and of nanoparticles at surfaces. Some of their latest achievements will also be reviewed in this chapter. Recently, similar experimental setups have been developed also by other research groups [30.10, 30.9] improving the sensitivity and extending the set of systems for which the heat of adsorption can be measured.
2 Definition of the Heat of Adsorption
Adsorption at surfaces is an exothermic process, and the differential molar enthalpy change is, thus, negative. According to the usual convention we define the differential molar adsorption heat q as its opposite. The isosteric adsorption heat qst is defined according to the Clausius–Clapeyron equation
where P is the pressure, T the absolute temperature, R the gas constant, and θ the fractional surface coverage. In practice it is necessary to measure the change in the gas pressure needed to keep the fractional surface coverage constant while changing the temperature. This approach suffers from two drawbacks: firstly it can be applied only when adsorption takes place reversibly, and secondly, a reliable indicator of the actual surface coverage is needed; θ can be checked in different ways, i. e., monitoring the work function, the diffraction pattern, or some specific spectroscopic signal (vibrational or electronic) related to the adsorbate, but care is needed since these indicators may be affected in some cases also by change in the temperature, especially because a relatively wide range of temperatures is required for an accurate measurement. Moreover, when the temperature is increased, the pressure required to maintain a constant coverage may increase beyond the capabilities of traditional UHV techniques, thus further limiting the practical feasibility of this approach. An example is reported in Fig. 30.1, which shows the pioneering work of Tracy and Palmberg [30.11]. They first measured thechange in the work function ΔΦ of Pd(100) as a function of temperature for different CO pressures. From these curves of pressure versus inverse temperature were obtained (Fig. 30.1b). The coverage of CO on Pd(100) is monitored by monitoring the work function. The coverage for each ΔΦ was obtained as follows. In a series of experiments CO was adsorbed at room temperature until the work function changed by ΔΦ.


(a) Heat of adsorption as a function of coverage for CO on Pd(100). (b) Pressure as a function of the inverse temperature required to keep a constant CO coverage on Pd(100). Coverage is monitored by the work function of the surface. Curves corresponding to different work functions and, thus, to different coverages are reported (Adapted from [30.11], with permission from Elsevier)
The sample was then rapidly flashed to 500∘C; the integral of the pressure rise is proportional to the amount of CO that has desorbed. The absolute calibration of the coverage was possible using the c(4 × 2)-45∘ low-energy electron diffraction (LEED) pattern which corresponds to a coverage of 0.5 ML. Using the Clapeyron equation, the heat of adsorption as a function of θ is obtained.
3 Experimental Setups
As outlined above, the basic principle behind the development of a single-crystal calorimeter (SCAC in the following) is the measurement of the change in the temperature of a sample upon adsorption of a measured dose of adsorbate. In order to obtain a quantitative result for the heat of adsorption it is necessary to measure accurately and reliably the heat released to the sample upon adsorption and the amount of adsorbing gas. The result was attained by using sample of very low thermal capacity (i. e., single crystals with a radius of some mm, and a thickness of some micrometers) and by dosing the desired gas by use of a supersonic molecular beam (SMB). The flux of the SMB is measured prior to each experiment by a spinning rotor gauge. The surface of the sample is then exposed at normal incidence using a pulsed SMB and the sticking probability and the increase in the temperature are measured simultaneously. The coverage is obtained by integrating the amount of gas adsorbed at each pulse. The different SCAC setups can be classified on the basis of the method used to measure the heat released and the temperature change.
The calorimeter initially developed by the group of King (Fig. 30.2a) measures the change in the infrared intensity emitted by the back of the sample upon adsorption of gas. The intensity is measured by a suitable IR detector and is calibrated by comparing the signal measured upon adsorption with the one recorded when the clean surface is exposed to a laser of known power impinging at normal incidence and correcting for the reflectivity of the sample at the laser wavelength. The reflectivity is measured in a separate experiment by optical methods. The change in radiated power from the back of the crystal is proportional to the third power of the temperature times the change in T due to adsorption. The sensitivity, thus, drops as T3 and for this reason the infrared detection technique is inadequate for low-temperature measurements.


In order to overcome this limitation another detector was developed that employs the pyrolectric effect; the increase in the temperature produced by adsorption is measured by monitoring the pyroelectric signal in place of the IR emission. In the first design [30.6], the detector element was a LiTaO3 wafer 0.3 mm thick onto which AuCr electrodes had been deposited. The single crystal (which had a thickness of ≈ 2 μm) was bonded to the wafer by cold welding onto an intermediate layer of polycrystalline Ni, which, in turn, was evaporated onto one AuCr electrode. The voltage signal generated between the two electrodes was passed through a specially designed high-impedance amplifier and then acquired digitally. In subsequent designs (Fig. 30.2b) [30.7, 30.8], a pyroelectric ribbon is moved carefully until it touches the backside of the sample and the pyroelectric signal is recorded. A similar method is used in the latest experimental setups [30.10, 30.9].
4 Overview of Experimental Results by the Cambridge Group
A very large amount of data has been obtained by the Cambridge group starting from the 1990s. We shall briefly overview the most significant results in the opinion of the authors. We shall consider the following topics here:
-
a)
The dissociative adsorption of oxygen from the regime of chemisorption to the onset of oxidation on low Miller index Ni surfaces
-
b)
The effect of surface temperature and potassium precoverage on Ni oxidation
-
c)
The experimental determination of the energy difference between surface phases
-
d)
The effect of lateral interactions between adsorbates on the heat of adsorption and their role in surface chemistry
-
e)
The heat of adsorption in presence of defects
-
f)
Heat of adsorption of hydrocarbons and determination of the metal–C bond energy
-
g)
The heat of reaction for CO oxidation
-
h)
The determination of prefactors for desorption.
4.1 Dissociative Adsorption
Dissociative oxygen adsorption and substrate oxidation are among the most common phenomena occurring when reactive surfaces are exposed to O2. Figure 30.3a shows the sticking probability s measured at normal incidence and at room temperature (RT) by the King and Wells method using a pulsed supersonic molecular beam of O2 impinging onto a clean Ni(100) surface. By integrating s over the exposure time and multiplying it by the beam flux it is possible to obtain the total amount of oxygen that has adsorbed (apparent oxygen adatom coverage). In Fig. 30.3a, s is plotted versus such apparent coverage. Three regimes are present:
-
i)
Initially s is large (≈ 0.55) and decreases rapidly with increasing exposure to the beam.
-
ii)
After reaching a minimum s increases again and remains relatively large (> 0.1) until an apparent coverage of ≈ 5 ML of oxygen adatoms is reached.
-
iii)
Thereafter, s remains constant, indicating that a stable regime has been attained in which the amount of oxygen adsorbing during the length of the pulse is desorbed in the time between two subsequent pulses.


(a) Sticking probability versus apparent O adatom coverage on Ni(100) at RT, showing the chemisorption, oxidation, and reversible regimes. The smooth line running through the experimental data is the sum of the other lines used to fit the data, including two straight lines as an empirical fit to the chemisorption sticking probability, a peaked curve corresponding to oxidation by island growth, and a curve corresponding to reversible molecular oxygen adsorption. (b) Net sticking probability (obtained from the one in (a) by subtracting the reversible contribution) as a function of absolute coverage. (c) Oxygen differential adsorption heat on different Ni single-crystal surfaces at 300 K (Reprinted from [30.12], with the permission of AIP Publishing)
The contributions of these three regimes have been deconvoluted and are shown by the continuous lines in the figure. Figure 30.3b shows the net sticking probability (obtained by subtracting the reversible contribution to s) as a function of the net adatom coverage (obtained by subtracting the amount of oxygen that desorbs between subsequent pulses). The first regime corresponds to the rapid building up of an adsorbed oxygen layer, while the second one indicates the formation of an oxide layer at the surface. Ni oxidation is believed to start at nucleation points and to go on by island growth. Oxygen incorporation occurs only at the edges of the islands; after an initial increase s reaches a maximum and decreases as long as islands get larger until the oxide growth stops at ≈ 3.5 ML. Formation of a thicker oxide then becomes extremely slow or suppressed under the experimental conditions of this experiment.
30.3c shows the heat of adsorption as a function of coverage for Ni(100), as well as for the other low Miller index surfaces (Ni(110) and Ni(111)) at RT. The heat of adsorption is relatively large for all surfaces but exhibits different behaviors. On Ni(100) a rapid decrease with increasing coverage is present. This decrease is due to repulsive lateral interactions between second-nearest neighbors. The interaction between third-nearest neighbors must be, on the contrary, weakly attractive, since an ordered p(2 × 2) structure forms. For Ni(110), the initial heat (475 kJ ∕ mol) is lower than for Ni(100), and the drop with θ is less steep because of the formation of Ni–O chains, which shield the O atoms, thus decreasing the magnitude of repulsive interactions between them. Also, for Ni(111), q is initially (440 kJ ∕ mol) lower than for Ni(100), since adsorption occurs at threefold hollow sites; q initially increases due to the rearrangements of the adatoms at the surface to minimize the repulsive interactions. Such a reordering is made possible by the presence of an effective precursor-mediated adsorption path (evident from s(θ), which is, however, not reported here). Repulsive interactions set in thereafter, causing a decrease of q. The three low Miller index surfaces studied here behave similarly in the oxidation regime with heat of adsorption of the order of 200 kJ ∕ mol. Finally, when oxide growth stops, and reversible adsorption of oxygen starts to occur on the oxide film, the heat of adsorption decreases to ≈ 87 kJ ∕ mol.
4.2 Surface Temperature and Potassium Precoverage on Ni Oxidation
The heat of adsorption of O2 on Ni(100) was also measured as a function of the surface temperature using a pyroelectric detector [30.12]. The results are shown in Fig. 30.4. It is apparent that: i) for θ ≤ 0.25 ML, q is constant (and possibly slightly increasing with θ) at 410 K, while it is initially lower and decreases with increasing coverage for lower temperatures. ii) Above 0.25 ML, q decreases rapidly at 410 K than at lower T. A simple Monte Carlo simulation using a lattice-gas model provides a consistent explanation for all these observations [30.12]. The interaction between first neighbors is prohibitively high, since the saturation coverage does not exceed 0.5 ML in the chemisorption regime. The interaction between second-nearest neighbors must be strongly repulsive (by 30 kJ ∕ mol) in order to account for the sudden drop observed at 410 K above 0.25 ML when the sites of the c(2 × 2) structure starts to be occupied. The third-nearest neighbors' interaction must be weakly attractive (by 1.5 kJ ∕ mol) to explain the tendency to form the p(2 × 2) structure and in this way to account for the slight increase in q between 0 and 0.25 ML at 410 K. Since 0.25 ML is the coverage expected for an ordered p(2 × 2) overlayer, these findings indicate that at 410 K, the mobility of the O adatoms is high enough to allow the system to minimize the repulsive interactions by avoiding the occupation of second-nearest neighbor sites and possibly even to maximize the heat of adsorption, exploiting the fact that third-nearest neighbor interaction is weakly attractive. At RT, on the contrary, the mobility is not high enough to allow for the formation of a fully-ordered overlayer; repulsive interactions show up already < 0.25 ML, because of the occupation of second-nearest neighbor sites. This effect becomes even more evident at 90 K, where the limit of immobile adsorbate is reached and a nearly monotonic decrease in q is observed. The effect of K preadsorption [30.15] on q and s for oxygen adsorption on Ni(100) is apparent from Fig. 30.4b,c. For θK = 0.3 ML, the initial sticking coefficient is 0.85, i. e., more than two times larger than for the bare Ni(100) surface, and remains constant up to θO = 0.33. The heat of adsorption is initially higher (by 40 kJ ∕ mol) than for the bare surface and decreases similarly with increasing coverage. In the range of coverage corresponding to the formation of the oxide, the presence of K completely modifies the behavior of the system; s remains nearly constant up to an oxygen coverage of 2 ML, while q is constant until 1.3 ML of oxygen and decreases towards the reversible adsorption regime afterwards. The presence of K thus affects the kinetics much more effectively than the thermodynamics of oxygen adsorption at Ni(100). The increased charge density in the surface region due to K adsorption increases the probability of dissociation of O2 in the molecular precursor. For a K precoverage > 0.15 ML, the molecular state is stabilized and the rate of oxidation of the substrate is increased. The measurements of the effect of the K precoverage on the heat of adsorption of O2 on Ni(111) and Ni(100) are discussed in larger detail in [30.1].


(a) Differential heat of adsorption of O2 on Ni(100) versus apparent O adatom coverage on Ni(100) at different temperatures in the low-coverage regime (Adapted from [30.12], with the permission of AIP Publishing). (b) Sticking probability (lower graph) and differential heat (upper graph) for O2 on Ni(100) at RT without K. (c) Sticking probability (lower graph) and differential heat (upper graph) for O2 on Ni(100) at RT for a K precoverage of 0.3 ML (Reprinted from [30.13], with permission from Elsevier)
4.3 Energy Difference Between Surface Phases
We now show that it is possible to use microcalorimetry also to measure the energy difference between surface phases, information previously obtained only by theoretical methods [30.16]. It is known that the bare Pt(100) surface can exist at RT in two different phases: a metastable (1 × 1) and a hex phase. Upon annealing to 500 K the (1 × 1) phase turns into the hex one. It is, however, possible to adsorb molecules such as CO (or NO) at RT starting from both phases and ending with a (1 × 1) phase. The differential heat of adsorption of CO was then measured at RT starting from both phases. Since the final state is the same, i. e., a c(2 × 2) CO-covered (1 × 1) phase with a coverage of 0.5 ML, by comparing the integral heat of adsorption it was possible to obtain experimentally the energy difference between the hex and the (1 × 1) phase [30.16]. The integral heat of adsorption is defined as
The difference between the integral heats qint at a coverage of 0.5 ML multiplied by the coverage is, thus, equal to the difference in energy between the bare hex and (1 × 1) phases, which comes out to be 12 kJ ∕ mol.


(a) Sticking probability S (top) and differential heat of adsorption q (bottom) for CO adsorption on Pt(100)-hex and Pt(100)-(1 × 1) at RT. (b) The two initial states of clean Pt(100), the two different CO adsorption pathways (a and b), and the identical final state, Pt(100)-(1 × 1)–c(2 × 2)-CO. The difference in the integral adsorption heat qi (c) along paths a and b is the energy difference between the initial states (12 kJ ∕ mol). ([30.1], © ACS 1998; adapted from [30.14], with the permission of AIP Publishing)
4.4 The Role of Lateral Interactions in Surface Chemistry
The heat of adsorption of molecules is often strongly coverage dependent. Such dependence can arise for different reasons: direct and indirect interactions between adsorbed molecules, coexistence of molecular and dissociative adsorption, etc. Lateral interactions may arise from three contributions: dipole–dipole interaction, Pauli repulsion, and substrate-mediated interactions.
Dipole–dipole interactions are long-range forces but they are too small to explain the experimentally observed decrease in q. Pauli repulsion is definitely stronger, but since it arises from the overlap of electron wavefunctions it can play a role only between first neighbors. Thus, we can conclude that the major contribution to lateral interactions is provided by substrate-mediated interactions. They can be either repulsive or attractive, and their magnitude can be estimated by the change in the heat of adsorption with increasing coverage. One example is provided in Fig. 30.6a-c for CO adsorption on Pt(111). For θ = 0.33 ML, a \((\sqrt{3}\times\sqrt{3})\mathrm{R}30^{\circ}\) overlayer of CO molecules forms (Fig. 30.6a-c). In this structure, each CO admolecule is surrounded by six next-nearest neighbors. If we assume that adsorbate–adsorbate interactions can be described as sum of forces between couples of molecules (or atoms), i. e., if pairwise addictivity holds, then the repulsive interaction ω2 can be easily estimated


Differential heat of adsorption for CO on Pt(111) (a) and sticking probability (b) versus coverage and (c) model of the \((\sqrt{3}\times\sqrt{3})\mathrm{R}30^{\circ}\) (Adapted from [30.17], with the permission of AIP Publishing)
Lateral interactions play a key role in surface chemistry. A paradigmatic example is provided in Fig. 30.7a-c. The differential heat of adsorption of NO on the bare Ni(100) is initially quite high and decreases rapidly with increasing NO coverage, due to lateral interactions. The relatively high initial value indicates that dissociative adsorption takes place. The same figure shows also the differential heat for NO on the same surface precovered with different amounts of O adatoms; the initial heat decreases rapidly with increasing O precoverage. The data can be modeled by considering the sites occupied by the adatoms generated by the dissociation of NO into N and O adatoms or by the oxygen adatoms generated by the dissociation of O2 [30.18]. The integral heat is shown in Fig. 30.7a-cb. The model (MC) assumes an initial heat of adsorption equal to the experimentally measured initial value and fits the decrease in the heat of adsorption by assuming the same repulsive interaction between N–O, N–N, and O–O adatoms in second-nearest neighboring sites. Occupation of first-neighbor sites does not occur, as indicated by the value of the saturation coverage in the chemisorption regime, which is < 0.5 ML. The model properly describes the data up to a certain coverage of adatoms (≈ 0.12 ML for the bare surface and ≈ 0.09 ML for a surface precovered with O adatoms) and thereafter drops more rapidly than experimentally observed. Above such coverage, the experimentally measured differential heat exhibits a plateau, which is assigned to nondissociative chemisorption. We can, thus, draw the following picture. As long as lateral interactions are not too strong, the heat of adsorption is higher for dissociative than for molecular adsorption, and O and N adatoms form. Dissociation eventually ends (and molecular chemisorption starts) at the coverage for which the heat of dissociative adsorption falls below the heat of molecular chemisorption.


Differential heat q (a) and integral heat qi (b) of adsorption on NO on Ni(100) for different precoverages of O atoms (MC: Monte Carlo simulation). (c) Possible adsorption sites for the NO molecule (X) and for the N and O adatoms resulting from its dissociation (Y, Y′ and Z. See text.) (Adapted from [30.18], with the permission of AIP Publishing; [30.19], © Wiley 1996)
The schematic in Fig. 30.7a-cc clarifies what happens in the different geometrical arrangements of the O and N adatoms present on the surface. If a NO molecule lands at the X site, the adatoms resulting from its dissociation can try to occupy second and third-neighbor sites. If the preadsorbed adatoms occupy the sites shown in the left part of the scheme, dissociative adsorption cannot occur because occupation of first-neighbor sites is forbidden. If, on the contrary, the sites around the X site are free, dissociation can occur by occupying the Y sites. Occupation of the Z sites is not expected to occur, it being thermodynamically less favored. These results indicate that the array of empty sites that are required for dissociative adsorption to occur is quite demanding if the repulsive interactions are large, as in the present case. The model is able to describe consistently both the coverage dependence of q and of the sticking probability (not shown, [30.19]). We expect this scenario to occur for several different systems; a similar argument has, in fact, been invoked to explain the onset of molecular adsorption for NO on Ni(211) [30.20, 30.21].
4.5 Heat of Adsorption at Defects
It is well established that high Miller index surfaces can be model systems for the investigation of adsorption at well-defined defects; such surfaces indeed exhibit a high amount of majority defects arranged in a regular way [30.22]. Several stepped surfaces have been studied by microcalorimetry (i. e., adsorption of CO on Fe(211) [30.23], of NO on Ni(211) [30.20], O2 and NO on stepped Pt surfaces [30.24], and O2 on Ni(211) [30.21]). Here, we will summarize here the results obtained for two different cases: oxygen adsorption on stepped Ni(211) [30.21] and NO dissociation on stepped Pt surfaces [30.24]. In Fig. 30.8, the differential heat of adsorption and the sticking probability of O2 on Ni(211) are reported as a function of the apparent adatom coverage. The initial differential heat of adsorption q (620 kJ ∕ mol) is significantly higher than for low Miller index surfaces (Fig. 30.3) because of the presence of low-coordinated adsorption sites at the steps; q then decreases rapidly with coverage due to strong lateral repulsions between adatoms until a coverage of 1.8 ML of oxygen adatoms is reached. The drop in q from 620 to 460 kJ ∕ mol between 0 and 0.5 ML is due to repulsions between adatoms at a distance of about 4.98 Å (corresponding to ω2 = 40 kJ ∕ mol in Fig. 30.8). At 1 ML, q has dropped to 300 kJ ∕ mol due to an additional repulsive interaction between adatoms at a distance of 3.45 Å. The addition of further adatoms at the steps would cause further and stronger repulsive interactions between adatoms; at this coverage, the steps are, thus, saturated and adsorption at terrace sites must set in until a coverage of 1.8 ML is reached. At this coverage, q suddenly increases, reaching a maximum value of 310 kJ ∕ mol at 2.3 ML and remains constant at about 270 kJ ∕ mol up to 4 ML. The coverage dependence of the differential heat of adsorption is closely related to the sticking probability shown in the inset of Fig. 30.8. As for low Miller index surfaces, oxidation starts at nucleation points on the surface and proceeds via formation of islands of oxide. The number of sites at the boundaries of the islands increases until the islands starts to merge; at this point, the length of the island boundaries progressively diminishes, and the sticking probability approaches zero. Above this point, q decreases slowly to attain the steady-state value of 90 kJ ∕ mol around 9 ML. In Fig. 30.9a,b, the differential heat and the sticking probability for NO adsorption on Pt(111), Pt(211) and Pt(411) are compared. The fcc(211) surface consists of two-atom wide (111) terraces separated by a single-atom step of (100) orientation. The fcc(411) surface is more complex; it consists of two terraces of (100) character, one being two atoms wide and the other one atom wide, separated by a single-atom step of (111) orientation. The sticking probability behaves similarly on these surfaces; it decreases quite slowly with coverage, indicating that a precursor mechanism is active and is not affected significantly by the presence of an ordered array of steps or by the size of the terraces. The initial differential heat of adsorption is, on the contrary, significantly affected by the presence of the steps: q is higher on the (411) surface (217 kJ ∕ mol), intermediate on Pt(211) (192 kJ ∕ mol), and lower on Pt(111) (182 kJ ∕ mol). This result is consistent with the following trend: NO adsorbs molecularly on Pt(111), while it dissociates at steps on Pt(211) and Pt(411). The amount of dissociated NO is found to be greater for the latter surface because of the larger size of the (100) facets on Pt(411). For Pt(211), q indeed indicates dissociative adsorption up to 0.26 molecules ∕ cm2, while a similar argument suggests dissociative adsorption to occur up to 0.31 molecules ∕ cm2 for Pt(411). At higher coverage, when the steps are saturated, molecular adsorption sets in, thus explaining the similar heat of adsorption observed for Pt(111) and Pt(211). For Pt(411), q remains relatively high up to a larger coverage; probably incoming NO molecules will not be as close to the adsorbed N and O adatoms and lateral interactions are, thus, weaker, and a lower drop in q is observed [30.24].


(a) Differential and integral heat of adsorption for O2 on Ni(211) as a function of the O adatom coverage. The inset shows the coverage dependence of the sticking probability. (b) Model of the (211) surface with the sites occupied at 0.5 ML (top) and at 1 ML (bottom) oxygen coverage. The contributions to pairwise lateral repulsive interactions between adatoms at different distances are shown schematically (see text) (Adapted from [30.21], © 2004 American Chemical Society)


Differential heat (a) and sticking probability (b) for NO adsorption on low and high Miller index Pt surfaces (Adapted from [30.24], with permission from Elsevier)
4.6 Heat of Adsorption of Hydrocarbons
The measurement of the heat of adsorption of hydrocarbons is particularly relevant, since it also provides a way to determine the metal–C bond energy. This value cannot be obtained by thermal desorption experiments because adsorption of hydrocarbons at reactive metal surfaces in most cases occurs irreversibly, producing many different surface species, with different degrees of dehydrogenation and with different rehybridizations of the C–C bond inside the molecule. An essay of such complexity is provided in Fig. 30.10a-d, which shows the heat of adsorption of a simple hydrocarbon like ethene (C2H4) on different Pt surfaces. The different initial value of q and its different coverage dependences can only be understood by considering that different radicals form. On Pt(100)-hex [30.25] quad-σ acetylene forms on the bare surface, followed by ethylidyne between 0.06 and 0.18 ML. At higher coverage di-σ ethylene forms. Finally, reversible adsorption of π-bonded ethylene occurs. These assignments explain the decrease in q and the plateau present at the different coverages. On Pt(100)-(1 × 1), the situation is similar, except for the fact that ethylidyne does not form because of the absence of threefold sites on this surface; quad-σ acetylene forms on the bare surface, followed by di-σ-bonded ethylene, and then by reversibly adsorbed π-bonded ethylene. For Pt(110), the situation is quite different; it has been suggested that ethylylidine (C–CH2–) forms on the bare-surface bonding in the troughs of the (1 × 2) missing-row reconstruction. The next species is ethylidyne (C–CH3), which probably adsorbs on the (111) microfacets of the Pt(110) surface; above 0.5 ML, di-σ ethylene and then reversibly adsorbed π-bonded ethylene are observed [30.26]. On Pt(111) [30.27] an unusual, nonmonotonic, dependence of q on coverage is obtained. In the present case, several reactions can occur
-
Ia)
\(\mathrm{C}_{2}\mathrm{H}_{4}(\mathrm{g})+2^{*}\rightarrow\mathrm{CHCH}_{3}(\mathrm{a})\)
-
Ib)
\(\mathrm{CHCH}_{3}(\mathrm{a})+2^{*}\rightarrow\mathrm{CCH}_{3}(\mathrm{a})+\mathrm{H}(\mathrm{a})\)
-
II)
\(\mathrm{C}_{2}\mathrm{H}_{4}(\mathrm{g})+3^{*}+\mathrm{H}(\mathrm{a})\rightarrow\mathrm{CCH}_{3}(\mathrm{a})+\mathrm{H}_{2}(\mathrm{g})\)


At low coverage, no loss of hydrogen by recombination takes place, and reactions Ia) and Ib) occur. At 0.17 ML, reaction II) starts to occur, and a minimum in q is observed, since hydrogen desorption is endothermic. After consuming all the preadsorbed H, eventually formation of ethylidyne (reaction Ib) occurs. As the ethylidyne islands grow, fewer and fewer threefold sites are available and only bridge sites are vacant. They then favor the formation of CHCH3 and direct formation of ethylidyne stops. Since no H2 is lost by desorption, the measured heat of adsorption increases, giving rise to a maximum at ≈ 0.35 ML. Thereafter, q decreases again, declining towards reversible π-bonded ethylene adsorption. Similar experiments were performed also for Pd(100) and Ni(100). On Pd(100), adsorption of ethylene occurs reversibly at RT, and the measurement of q is difficult. The data for Ni(100) are shown in Fig. 30.11a-c. As for Pt surfaces, the interpretation of the results relies entirely on the identification of the species present on the surface. According to the literature (see the original papers and [30.1] for details), CH and CCH initially forms on this surface. The initial heat is associated with the formation of methylidyne (CH) followed by acetylide (CCH). The heat of adsorption was measured at RT also for acetylene on Ni(100) and Pd(100). The results are shown in Fig. 30.11a-c. On Ni(100), the formation of CH and then of CCH can account for the observed results. The lower value observed for Pd(100) is explained by the fact that on the latter surface, dehydrogenation does not occur and that di-σ acetylene is present at low coverage after exposure at RT. The calorimetric data have, thus, been used to estimate the metal–carbon bond energy for Pt, Ni, and Pd. The results are summarized in Table 30.1. It is apparent that the metal–C bond energy is strongly dependent on the nature of the substrate, being 20% higher for Pt than for Ni and 30% higher than for Pd. It is, however, nearly independent of the surface structure, at least for Pt surfaces. The database should be extended further in order to gain deeper insight into such general trends [30.29].
4.7 Heat of Reaction: CO Oxidation
The heat of adsorption of CO on an oxygen precovered surfaces have been measured calorimetrically for Pt(110) [30.30] and Pt(111) [30.17]. The results of the experiments are shown in Fig. 30.12a-d. The initial heat measured when CO is adsorbed onto an O-precovered surface is 177 kJ ∕ mol for Pt(110) and 157 kJ ∕ mol for Pt(111). The heat of reaction ΔHr can be easily estimated by considering the reaction


Differential heat of adsorption for C2H4 adsorption on Ni(100) (a) and for C2H2 adsorption on Ni(100) (b) and Pd(100) (c) (Adapted from [30.28], with permission from Elsevier)


Differential heat of adsorption (a) and sticking probability (b) for CO adsorption on Pt(111) precovered with a saturation coverage of oxygen. ([30.17], © AIP 1997). Differential heat of adsorption for: CO adsorption on O-saturated Pt(110) (c) and for O2 adsorption on CO-saturated Pt(110) (d) (Adapted from [30.1], © 1998 American Chemical Society)
and it reads
If the CO2 molecules would leave the surface without any excess energy, we would expect a differential heat of adsorption for CO on an oxygen-covered surface of 171 kJ ∕ mol. The difference between the calculated heat of reaction and the measured differential heat is \((-6\pm 17)\,\mathrm{kJ/mol}\) and is, thus, compatible with a vanishing excess energy. On the contrary, when O2 is dosed on a CO-precovered Pt(110) surface, the measured differential heat is 190 kJ ∕ mol. Indeed, the following relations hold
and this reads
An adsorption heat of 287 kJ ∕ mol would be measured if the reaction products would leave the surface without excess energy. The measured value of the differential heat is, however, significantly lower than expected! These results indicate that the desorbing CO2 molecules have an average kinetic energy of (49 ± 21) kJ ∕ mol [30.1]. Adsorption of CO on O-precovered Pt(111) leads to similar results [30.17].
4.8 Frequency Factor for Desorption
In all SCAC experiments, a steady state is reached characterized by a constant sticking probability and a constant differential heat of adsorption. In order to understand the meaning of this fact we must remember that in a SCAC experiment, a pulsed molecular beam is used. Typically, pulses last for 50 ms and are separated by 2.5 s. When a steady state is reached, the molecules adsorbed during each pulse desorb in the time between subsequent pulses. The number of molecules adsorbing during each pulse is, thus, just the number of molecules present in each pulse multiplied by the sticking probability. It is usual to express the rate of desorption using the following expression
where n is the desorption order and νdes the desorption frequency factor (also called prefactor for desorption). Since the number of molecules desorbing in the time between pulses is equal to the number of molecules adsorbing during the pulses in the steady-state region, it should be possible to determine νdes. Since under steady-state conditions the Gibbs free energy change for adsorption vanishes, it is possible to determine also the entropy change due to adsorption directly from the calorimetric data
and then
The standard entropy for a gas can be calculated using the law of perfect gases as
where S0 is the standard entropy of the gas (i. e., 198 J ∕ (K mol) for CO at 1 atm and 298 K), p the gas pressure, and p0 the standard pressure. By using the experimentally obtained ΔH, it is possible to extract the differential entropy of the adsorbed phase Sa
The results of this analysis for CO adsorption at RT on different single-crystal Ni surfaces are summarized in Table 30.2. The extraction of prefactors from calorimetric data, however, requires some care. The same analysis for CO Pt(110) [30.31] leads to a prefactor for desorption of 2.5 × 10(9±1.2) Hz. This is a value that is remarkably lower than usually assumed (≈ 1014 Hz)! The reason for this discrepancy is that the desorption rate is not exactly given by (30.8). This simple and widely-used expression is somewhat oversimplified, since it neglects several processes that may take place; for example, a mobile precursor may be involved also in desorption, thus providing a path back to the chemisorbed state. If these processes are considered, the desorption rate is reduced by a factor F, which can be written as [30.33]
where fd, fm, and fa are the probabilities of desorption, migration to a neighbor site, and chemisorption for a molecule in the precursor state; K is the Kisliuk constant. The smaller K is, the more mobile the precursor, while K = 1 represents an immobile precursor. Since F cannot be directly obtained by the experimental data, care must be taken in extracting the frequency factor directly from steady-state values using (30.8).
5 Overview of Experimental Results by the Washington Group
The main limitations of the Cambridge IR calorimeter arise from the need to operate it at room temperature and to use pulsed supersonic molecular beams of gases. In particular, the IR setup did not allow the study of activated adsorption systems, adsorption of gases with low vapor pressure around room temperature, and adsorption of nongaseous species (such a metals). Most of these limitations were overcome by the use of pyrolectric detectors, which enabled an extension of the range of temperatures at which the measurements can be performed, and by the development of modified sources enabling also the use of effusive beams of low vapor pressure molecules [30.7, 30.8] and adatoms [30.34, 30.35, 30.36, 30.37].
A large set of systems have been investigated in such ways obtaining calorimetric data regarding in particular:
-
a)
Heat of adsorption of both simple [30.38, 30.39, 30.40, 30.41] and more complex organic compounds [30.42, 30.43, 30.44, 30.45, 30.46, 30.47, 30.48, 30.49, 30.50] and subsequent determination of bond energies and heat of formation of chemically important radicals
-
b)
Heat of adsorption of adatoms at both metal [30.51, 30.52], oxides [30.53, 30.54, 30.55, 30.56, 30.57, 30.58, 30.59, 30.60], semiconductors [30.61], and insulating surfaces [30.62].
Dedicated reviews have appeared [30.37, 30.63, 30.64, 30.65, 30.66, 30.67] and just a few selected examples will be highlighted here.
5.1 Heat of Adsorption of “Large” Molecules at Surfaces
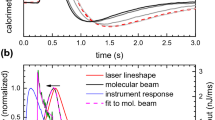
The supersonic molecular beam source successfully used for gases by the Cambridge group cannot be used to dose molecules having a vapor pressure < 100 Torr at room temperature. Under these conditions, an effusive beam with a source operating < 400 K can be used for molecules with a vapor pressure between 0.01 and 100 Torr at room temperature [30.7, 30.8]. The source is schematically shown in Fig. 30.13. Thanks to this improvement it was possible to measure the heat of adsorption and the sticking probability of species like benzene and other larger organic molecules with molar mass exceeding 70 g ∕ mol. The measurement of the heat of adsorption of such “large” organic molecules sometimes presents some additional complexity, which is schematically shown in Fig. 30.14 for the case of cyclohexene adsorption on Pt(111).


Schematic of the effusive molecular beam and its separate elements: gas handling system and beam line, with five orifices indicated by numbers: (1)–(5). The beam line consists of two differential pumping stages separated from the main chamber by a gate valve. The purpose of the gas handling system is to deliver the low vapor pressure molecules in the constant temperature bath, kept at 280 K, to the glass capillary array (GCA). The temperature of the GCA is monitored by a thermocouple and is resistively heated to maintain a temperature of 300 K. The line between the constant temperature bath and the GCA is maintained at 290 K, using heating tape, to prevent condensation of the low vapor pressure molecules along the way. The beam line consists of five orifices, only two of which are beam defining: (1) and (5). The inner diameter (ID) of (1) and (5) are 2.2 and 4.0 mm, respectively, and the distance between them is 403 mm. Orifices (2), (3), and (4), which are not beam defining, but only slightly larger, have IDs of 2.8, 3.8, and 4.0 mm, respectively. Also indicated are ports for the laser and prism for calibration with the laser beam, the shutter, used to stop the molecular beam, and the chopper, used to chop both the laser and molecular beams (Adapted from [30.7], with the permission of AIP Publishing)


(a) Differential heat of adsorption for cyclohexene adsorption on Pt(111). (b) Comparison of the pyroelectric and QMS signal versus time (see text). (c) Energetic of cyclohexane dehydrogenation on Pt(111), as obtained by calorimetric data. Only the heats of adsorption of H2 and cyclohexane were obtained by analysis of TPD data (see the original papers for details) ([30.63], © Roy. Soc. Chem. 2008; adapted from [30.44], © 2008 American Chemical Society)
The lower trace in Fig. 30.14a shows the QMS signal when the molecular beam hits the inert gold flag. The duration of the pulse is 50 ms. The above trace shows the QMS trace when the molecular beam hits the Pt(111) surface. It is apparent that the cyclohexene signal does not end when the molecular beam is interrupted but continues thereafter. This result indicates that, under the conditions of that experiment (263 K and coverage > 0.15 ML) transient desorption occurs off the surface.
The corresponding calorimetric signal exhibits a more complex behavior. Firstly, there is a delay between the instant at which adsorption and release of heat starts and the onset of the calorimetric signal, because of the limited thermal contact between the Pt(111) sample and the pyroelectric polymer. The heat deposited by the molecules is measured by the slope of the calorimetry pulse during the initial steep rise, typically 40 − 140 ms after the pulse hits the sample. Since the calorimetry signal after the initial steep rise is not used for calculating the amount of heat deposited, any desorption occurring after that point in time (140 ms) is neglected in the evaluation of q. However, desorption occurring during the first 140 ms will affect the measured heat. In order to account for this effect, a correction was introduced [30.44]. A short-time sticking probability was defined as the fraction of the impinging molecules, which is considered to determine the heat of adsorption. This fraction is higher than the fraction of molecules, which has not desorbed before the next cyclohexene pulse starts to hit the surface (the so-called “long-term sticking probability”) and which is used to determine the surface coverage. This somewhat rough correction, which is essentially a linear interpolation scheme neglecting desorption after the pulse has reached its maximum intensity, is sufficient when transient desorption is limited. Its validity is questionable when transient desorption is relevant, e. g., for cyclohexene adsorption Pt(111) at 263 K and > 0.15 ML; q for cyclohexene on Pt(111) was measured as a function of coverage for temperatures in the range from 100 to 300 K. The results are shown in Fig. 30.14. At 100 K, cyclohexene adsorbs as di-σ-bonded cyclohexene with an initial adsorption heat of 130 kJ ∕ mol. From this calorimetric result and using the standard enthalpy of formation of gas phase cyclohexene (−5 kJ ∕ mol), the standard enthalpy of formation of adsorbed di-σ-bonded cyclohexene was obtained (−135 kJ ∕ mol). A similar argument was used to analyze the experimental data recorded at 281 K; at this temperature, cyclohexene dehydrogenates upon adsorption, forming adsorbed 2-cyclohexenyl (π-allyl-c-C6H9) plus a hydrogen adatom. The initial heat of adsorption is 174 kJ ∕ mol, so adding the heat of formation of gas cyclohexene and one-half the heat of adsorption of H2 on Pt(111) (72 kJ ∕ mol), the heat of formation of adsorbed π-allyl-c-C6H9 was estimated (−143 kJ ∕ mol). Finally, from the measured heat of adsorption of benzene on Pt(111) at 300 K (197 kJ ∕ mol) and using the enthalpy of formation of gaseous benzene (83 kJ ∕ mol), the standard enthalpy of formation of adsorbed benzene was found (−114 kJ ∕ mol). By combining all these calorimetric data a partial energy landscape of the dehydrogenation of cyclohexane to benzene on Pt(111) was obtained. The reader is referred to the original papers for the details of this complex diagram, which nicely illustrates the extent of information attainable from calorimetry [30.42, 30.44, 30.63]. Inspection of the diagram provides deeper understanding of cyclohexane and cyclohexene dehydrogenation as well as of benzene hydrogenation in presence of a Pt catalyst. In particular, since the barrier for cyclohexene desorption is higher than the barrier for its further dehydrogenation it is difficult to stop the dehydrogenation of cyclohexane to cyclohexene. Similarly, the barrier for benzene desorption is too high at low coverage, thus explaining why no benzene is obtained from dehydrogenation of cyclohexane and cyclohexene. It is, however, possible to obtain benzene by increasing the coverage of co-adsorbed species; this way, the barrier to overcome for further dehydrogenation increases and desorption of benzene becomes competitive with its dissociation.
Under the high-pressure conditions at which real catalysts operate, the desorption of dehydrogenated products is, thus, kinetically competitive with complete dehydrogenation. Note that the latter should also be avoided to completely coke the catalyst itself.
Campbell and co-workers undertook a systematic investigation of the heat of adsorption of several species on Pt(111) surfaces: methanol on an oxygen precovered surface to obtain methoxy [30.45] (Fig. 30.15), formic acid on oxygen-saturated Pt(111) to obtain formate [30.49] (Fig. 30.16) and water on oxygen precovered Pt(111) [30.38]. The results are summarized in Table 30.3, reporting the adiabatic PtOR bond dissociation enthalpies for three oxygen-bound molecular fragments to the Pt(111) surface: deuterated hydroxyl (OD) [30.38], monodentate formate (O(O)CH) [30.49], and methoxy (OCH3) [30.45], where R is the part of the fragment bonded with oxygen. Quite interestingly, these ROPt(111) bond enthalpies vary linearly with the ROH bond enthalpies in the corresponding gas-phase molecules (water, methanol, and formic acid), and the slope is compatible with 1.00. This result resembles the known trend for organometallic complexes and is explained by the local character of chemical bonding, even on extended metal surfaces. It is, thus, possible to predict bond enthalpies for many other molecular fragments at metal surfaces and derive the energetics of relevant catalytic reactions [30.41].


(a) Differential heat of adsorption of methanol on clean Pt(111) at 100 K versus total methanol coverage. (b) Differential heat of adsorption of methanol versus total methanol coverage on (circles) clean and (triangles) oxygen-saturated Pt(111) at 150 K. (c) Thermodynamic cycle at 150 K used in calculating the bond enthalpy and standard heat of formation of adsorbed methoxy. The values are expressed in kJ ∕ mol (Adapted from [30.45], © 2012 American Chemical Society)


(a) Short-term (upper panel) and long-term sticking probabilities (lower panel) of formic acid on clean Pt(111) at 100 K and O-saturated Pt(111) at 150 and 190 K as a function of the total formic acid coverage. (b) Heats of adsorption measured at various temperatures on clean Pt(111) at 100 K (black circles) and O-saturated Pt(111) at 100 K (hollow and solid green circles), 150 K (hollow and solid blue triangles), and 190 K (red squares) as a function of total coverage of adsorbed HCOOH, irrespective of its final structure. Hollow symbols represent fast heat deposition steps, while solid symbols represent total heats (i. e., the sum of fast and slow steps). The multilayer heat of adsorption is indicated by a red line through the 100 K data in the lower right corner. (c) Thermodynamic cycle used to determine the enthalpy of formation of adsorbed monodentate formate on Pt(111) and its HCOOPt(111) bond enthalpy from the integral fast S1 heat of adsorption at 150 K and 3 ∕ 8 ML, shown as the bottom right-hand step. The values are expressed in kJ ∕ mol (Adapted from [30.49], © 2014 American Chemical Society)
5.2 Heat of Adsorption of Metal Adatoms at Surfaces
The measurement of the heat of adsorption of metal adatoms at surfaces [30.68] became feasible thanks to substantial upgrades of the Cambridge calorimeter. Such modifications involved both the source and the detector of the released heat. The heat of adsorption q for Ag on Si(100)-(2 × 1) is shown in Fig. 30.17. Chopped pulses of a silver atom beam from a hot effusive source are directed at the surface of a thin Si(100) sample, releasing energy into the crystal upon adsorption. This causes a small transient temperature rise, detected by a thin pyroelectric polymer ribbon pressed into contact with the backside of the crystal during the measurement. The signal provided by the pyroelectric detector is calibrated by comparison with the signal produced by light pulses of known energy provided by a He-Ne laser. The sticking probability is obtained by measuring the amount of metal in a pulse that is reflected off the sample with a line-of-sight quadrupole mass spectrometer. The incident flux is measured with a quartz crystal microbalance. For adsorption on the bare surface, q = 347 kJ ∕ mol. It decreases rapidly until at 0.55 ML it drops below the bulk heat of sublimation of Ag (285 k ∕ mol). At 1 ML, q rises again reaching a value compatible with the bulk heat of sublimation at 2.5 ML. The sticking probability is surprisingly monotonic; indeed, it increases with coverage despite the complicated behavior of q.


(a) Heat of adsorption of Ag atoms on Si(100)-(1 × 2) versus adatom coverage. The solid curve is formed by a linear fit to the data over the first ML and a double exponential fit to the data at coverages > 1.0 ML. (b) Sticking probability of Ag atoms on the same substrate (Adapted from [30.61], © 2001 by the American Physical Society)
The data can be rationalized as follows. The initial value of q is higher than the value expected for isolated adatoms due to the formation of chains of adatoms. Such elongated Ag islands, seen by STM at 300 K up to a coverage of 1 ML, would normally imply attractive Ag–Ag interactions. The observation of a globally decreasing q with coverage implies that they are, on the contrary, repulsive. The formation of Ag–Si bonds induces significant strain in the neighboring surface sites, thus explaining the overall repulsive interaction between islands. If the growth were controlled by thermodynamics alone, 3-D growth should start ≈ 0.55 ML i. e., when q becomes lower than the heat of sublimation. Due to kinetic constraints, this happens, however, only ≈ 1 ML; indeed, at such a coverage, q rises quickly towards the bulk sublimation value. Once large 3-D particles have formed on top of the wetting layer, they provide more stable sites for Ag than in the wetting layer. This example shows that the information provided by calorimetry can be successfully used to explain the different regimes in the growth of metallic films on different substrates.
The same kind of measurements are able to also highlight size effects in the growth of nanoparticles. The calorimeter measures sticking probability and heat of adsorption versus coverage. In order to obtain information on the effect of particle size it is necessary to correlate the coverage with the size of the nanoparticles. Quantitative analysis of spectral intensities in Auger electron spectroscopy and ion scattering spectroscopy for the different elements as a function of Ag coverage was employed to characterize the growth morphology of the Ag films on magnesia and ceria surfaces. The data were well fitted by assuming that the Ag grows as three-dimensional (3-D) Ag particles with an hemispherical shape and a fixed, coverage independent, number N of Ag particles for unit of area. The data are not sensitive to the exact shape of the nanoparticles but are very sensitive to their aspect ratio. The analysis of Auger data showed that, > 0.03 ML, N is independent of coverage. Dividing the Ag coverage (atoms ∕ cm2) by N (particles ∕ cm2), the average number of Ag atoms per particle is obtained at any given Ag coverage. Measurement of q for Ag adsorption on different substrates was performed and compared as a function of particle size and of the number of atoms in the nanoparticle. The results are summarized in Fig. 30.18. It is apparent that the initial value of q, i. e., the value proper to very small particles, is strongly dependent on the nature of the substrate; for nanoparticles containing < 1000 atoms, Ag atoms are 30 − 70 kJ ∕ mol more stable when they are deposited on ceria than on MgO surfaces.


(a) Measured heat of Ag atom adsorption qAg versus the Ag particle diameter DAg to which it adds, for Ag adsorption onto four different surfaces. (b) Measured energy of a Ag atom, relative to its energy in bulk Ag(solid), versus the number of metal atoms already in the particle, for Ag particles on different surfaces (From [30.58]. Reprinted with permission from AAAS)
This result nicely explains the pivotal role of the support in the sintering of nanoparticles. As long as a metal atom is less stable in a nanoparticle of a certain size than in the bulk, it will prefer to move and to form larger particles. The point is that this driving force for sintering drops below 10 kJ ∕ mol for Ag nanoparticles of 400 atoms (3 nm) on ceria and for nanoparticles of 3000 atoms (6 nm) on MgO(100). For nanoparticles consisting of ≈ 5000 atoms, the energy of the added atoms reaches the value of the bulk metal also on MgO. Data for Pb exhibits a similar behavior [30.56].
Very recently, a similar study was performed also for Cu deposited on ceria. By comparing the heat of adsorption at 300 and at 100 K, it was possible to obtain the chemical potential as a function of particle size [30.67]. Ion scattering spectroscopy (ISS) was employed to determine the number of nanoparticles for unit of area; the normalized ISS signals directly provide the fraction of the ceria surface, which is masked by Cu particles, and the fraction of the total maximum Cu signal (due to complete coverage by Cu) that is observed at any given Cu coverage. The heat of adsorption for Cu nanoparticles with a diameter from 0.45 to 0.8 nm is lower at 100 K than at room temperature by some 30 − 50 kJ ∕ mol on the same CeO1.95(111) surface; this difference was attributed to cluster nucleation on terrace sites at 100 K and at step-edge sites at 300 K. Hence, at 100 K, the number of nanoparticles for unit of area (5.3 × 1013 particles ∕ cm2) is about seven times higher than at 300 K (7.8 × 1012 particles ∕ cm2). Since the entropic contribution to the free energy changes very little with particle size compared with the enthalpic one, the chemical potential of a metal atom in a particle of diameter D, μ(D), is higher than that in the bulk metal, μ(∞), by an amount equal to the heat of sublimation of bulk Cu minus the differential heat of Cu adsorption at diameter D. It is, thus, possible to obtain μ(D) from the calorimetrically measured q as a function of particle size. The results are shown in Fig. 30.19. The chemical potential decreases with increasing particle size. The Cu chemical potential is 110 kJ ∕ mol higher for isolated Cu adatoms on stoichiometric terrace sites than for Cu in nanoparticles exceeding 2.5 nm diameter, where it reaches the bulk Cu(solid) limit. In Cu dimers, the Cu chemical potential is notably 57 kJ ∕ mol lower at step edges than on stoichiometric terrace sites.


(a) Cu atom heat of adsorption at 300 K (diamonds) and 100 K (squares) on CeO1.95(111) as a function of Cu coverage. (b) Chemical potential of Cu atoms in Cu nanoparticles on CeO1.95(111) relative to that in bulk Cu (solid) versus the effective diameter of the Cu particle down to the single atom limit (Adapted from [30.67], © 2015 American Chemical Society)
We finally mention the possibility to use metal adatoms to characterize the defectivity of oxide surfaces [30.57]. Figure 30.20a,b shows the heat of adsorption for Ca and Li on pristine and ion-bombarded MgO. While ion sputtering causes a strong increase in the initial adsorption energy for Li on MgO(100) at 300 K, the initial adsorption energy for Ca is independent of the extent of sputtering. In order to explain this intriguing result, density functional theory (DFT) calculations were also performed. DFT revealed that while Ca adatoms thermally diffuse to look for the remaining defect sites, Li adatoms remain blocked when they reach a terrace site nearby an occupied step or kink site. The measured dependence was simulated with a kinetic model taking the binding energies and the adatom migration barriers provided by DFT as inputs. The lines is the figure show the result of this model, which evidently captures the essential physics ruling the behavior of this system.


Heat of adsorption of Ca (a) and Li (b) versus coverage at 300 K for adsorption on (a) pristine MgO(100) and (b) and (c) MgO(100) irradiated with increasing Ar+ ion doses (from 1014 to 1016 ions ∕ cm2) to create more defects. Each curve is labeled with the percentage of total MgO sites that were defect sites (Adapted from [30.57], © 2009 American Chemical Society)
6 Results of Other Research Groups
The impressive achievements of single-crystal calorimetry obtained in Cambridge and in Washington stimulated further developments of the technique [30.10, 30.9]. In recent years, some papers have appeared focussing on the heat of adsorption of gases at nanoparticles of different size [30.69], on water adsorption at oxide surfaces [30.70], on the adsorption of chiral molecules [30.71], and on the effect of surface temperature on the coverage dependence of the heat of adsorption [30.72]. Such results are briefly reviewed in the final part of this chapter.
6.1 Heat of Adsorption of CO on Pd Nanoparticles
Although results obtained on single-crystal surfaces have provided valuable insight into the behavior of more complex systems, researchers are presently trying to investigate directly adsorption and reactions on nanoparticles supported on oxide substrates. Such efforts have also increased the range of operation of microcalorimetry. Figure 30.21 shows the dependence of the heat of adsorption of CO and of O2 on Pd nanoparticles of different size deposited on a thin Fe3O4 film grown on Pt(111). By depositing different amounts of Pd on the film, nanoparticles of different sizes were obtained. The particle size was determined by STM the smallest nanoparticles correspond to ≈ 120 atoms ∕ particle, while largest ones consist of ≈ 4900 atoms ∕ particle. The data are compared with the results obtained for a Pd(111) single crystal. The dependence of the initial adsorption energy for oxygen on the particle size exhibits a clear trend. It strongly rises from about 205 to 250 − 275 kJ ∕ mol when moving from Pd(111) to large Pd nanoclusters where it passes through a maximum and decreases thereafter, reaching a value of 205 kJ ∕ mol on the smallest Pd nanoparticles. The initial rise is accounted for by the fact that on Pd(111) oxygen adatoms adsorb at threefold-hollow sites, while on large Pd nanoclusters, oxygen first occupies undercoordinated sites at the edges of the particle, which usually have a higher heat of adsorption. For smaller nanoparticles, the first sites to be occupied are still undercoordinated sites at the edges. The decrease of q is, thus, due to a pure size effect; in smaller nanoparticles, the lattice spacing is lower, the d bands are downshifted [30.74], and this causes a significant decrease in the heat of adsorption [30.75]. The decrease of the particle size causes, moreover, a lower van der Waals contribution to the heat of adsorption, thus resulting in an overall decreasing value. The data for the initial heat of adsorption of CO on the same nanoparticles exhibits, on the contrary, a monotonic decrease with particle size. Since the heat of adsorption of CO on Pd(111) does not change so much from terrace to corner sites, the behavior of the heat of adsorption is dominated by size effects over the whole range considered in these studies.


(a) Initial adsorption energy for (upper panel) O2 and (lower panel) CO molecules plotted as a function of the nominal Pd coverage on the Fe3O4 ∕ Pt(111) samples with the nominal deposition thickness of 0.3 (only for CO), 0.6, 1.5, 4, and 7 Å and on the Pd(111) single-crystal surface. Next to the data points, the average diameters of the Pd particles are given. Error bars show the standard error of the mean. The dashed lines are given to guide the eye. (b) Adsorption heat of CO on Pd(111) and on a Pd ∕ Fe3O4 ∕ Pt(111) model catalyst plotted as a function of CO surface coverage. Each curve is an average of four to six experimental runs measured on freshly prepared samples. (c) A model describing two microscopic trends affecting the binding energy: the local configuration of the adsorption site, and the particle size. NPs = nanoparticles (Adapted from [30.73], © 2010 by the American Physical Society; [30.69], © Wiley VCH 2013)
6.2 Heat of Adsorption of Propylene Oxide
In Fig. 30.22a-d, the heat of adsorption of the R and S enantiomers of propylene oxide on bare and NEA-covered Pt(111) surfaces are reported. 1-(1-Naphthyl)ethylamine (NEA) is a model chiral modifier; it has an aromatic group for binding to a metal surface and an amino group close to the chiral centre capable of hydrogen bonding. Adsorption of S- and R-propylene oxide (PO) on the bare Pt(111) surface shows an initial heat of adsorption of 50 decreasing to 42 − 43 kJ ∕ mol at a coverage of \(\approx{\mathrm{1.5\times 10^{14}}}\,{\mathrm{molecules/cm^{2}}}\) corresponding to 1 PO molecule every 10 P atoms. Above this coverage, q either goes through a small local maximum (S-PO) or, rather, remains constant (R-PO) up to the coverage of 4.1 × 1014 molecules ∕ cm2. Saturation is then reached at a coverage of 5.5 × 1014 molecules ∕ cm2. When PO adsorbs on the pristine Pt(111) surface, its binding energy rapidly reaches a constant level; this plateau is most likely due to the formation of PO islands. After some PO nuclei consisting of a few PO molecules are formed on the surface, further PO molecules diffuse to the boundary of these nuclei and adsorb with a constant binding energy. It is only after the PO surface coverage increases to such an extent that neighboring PO islands start to coalesce that the binding energy of the last incoming PO molecules decreases and approaches the value characteristic for PO adsorption in the second layer. The coverage dependence of q changes completely if NEA is present on the surface; as evident from Fig. 30.22a-db–d a monotonically decreasing heat of adsorption is measured in this case. When the surface is covered with 50% of NEA, i. e., when half of the surface still exposes Pt atoms, the plateau in q has completely disappeared. Most likely, there is a preferred adsorption of PO in the vicinity of NEA, preventing PO island formation on the still available metal sites. All these results suggest a preferential interaction between PO and the adsorbed modifier possibly resulting in the formation of a complex between these molecules.


Adsorption energy of S-PO and R-PO as a function of the number of adsorbed molecules on (a) bare Pt(111), as well as R-NEA-covered Pt(111) with the R-NEA relative coverage of (b) 0.5; (c) 0.75; (d) 1. The data points are averages of 35 independent measurements; the error bars show the error of the mean value. ([30.71], published by the PCCP Owner Societies licensed under a Creative Commons Attribution 3.0 Unported Licence 2015)
6.3 Heat of Adsorption of Water at Metal and Oxide Surfaces
We conclude our review by presenting in Fig. 30.23 the microcalorimetric results for H2O adsorption on Pt(111) and on two thin iron-oxide films. On Pt(111) and on FeO ∕ Pt(111), the heat of adsorption is initially 47 kJ ∕ mol and remains nearly constant until ice is formed. On Fe3O4(111)∕Pt(111), q is initially much higher (87 kJ ∕ mol) and decreases thereafter, monotonically leveling at a coverage of 1.2 × 1015 molecules ∕ cm2. The high value of q is indicative of water dissociation. Measurements performed at different temperatures are shown in Fig. 30.23b. The initial adsorption energy at higher temperatures reaches 101 kJ ∕ mol and decreases, leveling off at a lower coverage without forming multilayer ice. An increase of the initial binding energy from 87 kJ ∕ mol at 120 K to 101 kJ ∕ mol at temperatures > 180 K is most likely related to a kinetic hindrance of water dissociation at 120 K. Since the initial adsorption energy remains constant for all temperatures > 180 K, this value can be considered as the true thermodynamic value that is not affected by the slow kinetics of water dissociation. The leveling of q corresponds, as is usual in calorimetric experiments using pulsed beams, to a steady-state equilibrium between molecules adsorbing during the water pulse and molecules desorbing in the time between subsequent pulses. The initial heat of adsorption at high temperature is higher than that obtained by thermal desorption methods (65 kJ ∕ mol) [30.76]. These calorimetric data shed light on a possible failure of the generally-accepted simple model of water dissociation leading to two individual OH groups. IRAS measurements using isotopically labeled water molecules and DFT calculations suggested, on the contrary, a more complex mechanism: a dimer form consisting of one H2O molecule dissociated into two OH groups and another nondissociated H2O molecule [30.70].


(a) Differential adsorption energy measured at 120 K plotted as a function of the number of adsorbed H2O molecules for Pt(111), FeO(111)∕Pt(111), and Fe3O4 (111)∕Pt(111). (b) Differential adsorption energy of H2O on Fe3O4(111)∕Pt(111) as a function of the number of adsorbed H2O molecules measured at different temperatures. ([30.70], © Wiley VCH 2015)
7 Conclusions
Although SCAC is only about 20 years old, it has already developed into a mature and powerful technique. The complexity of the experimental setups and the need for accurate calibrations are still probably limiting its further utilization despite the huge amount of information that could be provided.
In our opinion, we foresee and welcome two possible future directions of development:
-
a)
A systematic use of the existing SCAC setups with an extension of the sets of investigated systems, thus enriching the amount of data and the development of a more extended database covering more and more systems of catalytic interest. In this effort, the calorimetric information provided by SCAC will, however, need more and more precise identification of the moieties forming upon adsorption, which can be provided mainly by spectroscopic techniques, as has already happened for the analysis of calorimetric data obtained for simple organic molecules.
-
b)
Further developments of the technique will be necessary to allow the investigation of currently inaccessible adsorption systems, in particular of activated adsorption systems. This direction will most likely require the use of supersonic beams to overcome adsorption barriers and to attain high-enough adsorption probabilities and, thus, an acceptable signal-to-noise ratio in the calorimetric signal.
References
W.A. Brown, R. Kose, D.A. King: Femtomole adsorption calorimetry on single-crystal surfaces, Chem. Rev. 98, 797–832 (1998)
S. Cerny: Adsorption microcalorimetry in surface science studies sixty years of its development into a modern powerful method, Surf. Sci. Rep. 26, 1–59 (1996)
D.A. Kyser, R.J. Masel: Design of a calorimeter capable of measuring heats of adsorption on single crystal surfaces, Rev. Sci. Instrum. 58, 2141–2144 (1987)
C.E. Borroni-Bird, D.A. King: An ultrahigh vacuum single crystal adsorption microcalorimeter, Rev. Sci. Instrum. 62, 2177–2185 (1991)
A. Stuck, C.E. Wartnaby, Y.Y. Yeo, J.T. Stuckless, N. Al-Sarraf, D.A. King: An improved single crystal adsorption calorimeter, Surf. Sci. 349, 229–240 (1996)
S.J. Dixon-Warren, M. Kovar, C.E. Wartnaby, D.A. King: Pyroelectric single crystal adsorption microcalorimetry at low temperatures: Oxygen on Ni{100}, Surf. Sci. 307–309, 16–22 (1994)
H.M. Ajo, H. Ihm, D.E. Moilanen, C.T. Campbell: Calorimeter for adsorption energies of larger molecules on single crystal surfaces, Rev. Sci. Instrum. 75, 4471–4480 (2004)
W. Lew, O. Lytken, J.A. Farmer, M.C. Crowe, C.T. Campbell: Improved pyroelectric detectors for single crystal adsorption calorimetry from 100 to 350 K, Rev. Sci. Instrum. 81, 024102 (2010)
J.-H. Fischer-Wolfarth, J. Hartmann, J.A. Farmer, J.M. Flores-Camacho, C.T. Campbell, S. Schauermann, H.-J. Freund: An improved single crystal adsorption calorimeter for determining gas adsorption and reaction energies on complex model catalysts, Rev. Sci. Instrum. 82, 024102 (2011)
P. Hörtz, R. Schäfer: A compact low-temperature single crystal adsorption calorimetry setup for measuring coverage dependent heats of adsorption at cryogenic temperatures, Rev. Sci. Instrum. 85, 074101 (2014)
J.C. Tracy, P.W. Palmberg: Simple technique for binding energy determinations: CO on Pd(100), Surf. Sci. 14, 274–277 (1969)
J.T. Stuckless, C.E. Wartnaby, N. Al-Sarraf, St J.B. Dixon-Warren, M. Kovar, D.A. King: Oxygen chemisorption and oxide film growth on Ni{100}, {110}, and {111}: Sticking probabilities and microcalorimetric adsorption heat, J. Chem. Phys. 106, 2012–2030 (1997)
N. Al-Sarraf, J.T. Stuckless, C.E. Wartnaby, D.A. King: Adsorption microcalorimetry and sticking probabilities on metal single crystal surfaces, Surf. Sci. 283, 427–437 (1993)
Y.Y. Yeo, L. Vattuone, D.A. King: Energetics and kinetics of CO and NO adsorption on Pt{100}: Restructuring and lateral interactions, J. Chem. Phys. 104, 3810–3821 (1996)
N. Al-Sarraf, J.T. Stuckless, D.A. King: Direct measurement of potassium-promoted change in heat of adsorption of CO on Ni{100}, Nature 360, 243–245 (1992)
Y.Y. Yeo, C.E. Wartnaby, D.A. King: Calorimetric measurement of the energy difference between two solid surface phase, Science 268, 1731–1732 (1995)
Y.Y. Yeo, L. Vattuone, D.A. King: Calorimetric heats for CO and oxygen adsorption and for the catalytic CO oxidation reaction on Pt{111}, J. Chem. Phys. 106, 392–401 (1997)
L. Vattuone, Y.Y. Yeo, D.A. King: Adatom bond energies and lateral interaction energies from calorimetry: NO, O2, and N2 adsorption on Ni{100}, J. Chem. Phys. 104, 8096–8102 (1995)
L. Vattuone, Y.Y. Yeo, D.A. King: Lateral interactions as the determinant in the switch from molecular to dissociative chemisorption: NO on Ni{100}, Catal. Lett. 41, 119–123 (1996)
A.D. Karmazyn, V. Fiorin, D.A. King: Calorimetric studies of NO on Ni{2 1 1}: Criteria for switching from dissociative to molecular adsorption, Surf. Sci. 547, 184–192 (2003)
A.D. Karmazyn, V. Fiorin, D.A. King: Direct sticking and differential adsorption heats as probes of structural transitions: O2 on the stepped Ni{211} surface, J. Am. Chem. Soc. 126, 14273–14277 (2004)
L. Vattuone, L. Savio, M. Rocca: Bridging the structure gap: Chemistry of nanostructured surfaces at well-defined defects, Surf. Sci. Rep. 63, 101–168 (2008)
D. Borthwick, V. Fiorin, S.J. Jenkins, D.A. King: Facile dissociation of CO on Fe{2 1 1}: Evidence from microcalorimetry and first-principles theory, Surf. Sci. 602, 2325–2332 (2008)
V. Fiorin, D. Borthwick, D.A. King: Microcalorimetry of O2 and NO on flat and stepped platinum surfaces, Surf. Sci. 603, 1360–1364 (2009)
Y.Y. Yeo, A. Stuck, C.E. Wartnaby, R. Kose, D.A. King: Microcalorimetric study of ethylene adsorption at 300 K on Pt{100}-hex and Pt{100}-(1 × 1), J. Mol. Catal. 131, 31–38 (1998)
A. Stuck, C.E. Wartnaby, Y.Y. Yeo, D.A. King: Microcalorimetric study of ethylene on Pt{110}-(1 × 2), Phys. Rev. Lett. 74, 578–581 (1995)
Y.Y. Yeo, A. Stuck, C.E. Wartnaby, D.A. King: Microcalorimetric study of ethylene adsorption on the Pt{111} surface, Chem. Phys. Lett. 259, 28–36 (1996)
L. Vattuone, Y.Y. Yeo, R. Kose, D.A. King: Energetics and kinetics of the interaction of acetylene and ethylene with Pd{100} and Ni{100}, Surf. Sci. 447, 1–14 (2000)
H. Gross, C.T. Campbell, D.A. King: Metal–Carbon bond energies for adsorbed hydrocarbons from calorimetric data, Surf. Sci. 572, 179–190 (2004)
C.E. Wartnaby, A. Stuck, Y.Y. Yeo, D.A. King: Calorimetric measurement of catalytic surface reaction heat: CO oxidation on Pt{110}, J. Chem. Phys. 102, 1855–1858 (1995)
C.E. Wartnaby, A. Stuck, Y.Y. Yeo, D.A. King: Microcalorimetric heats of adsorption for CO, NO, and oxygen on Pt{110}, J. Phys. Chem. 100, 12483–12488 (1996)
J.T. Stuckless, N. Al-Sarraf, C. Wartnaby, D.A. King: Calorimetric heats of adsorption for CO on nickel single crystal surface, J. Chem. Phys. 99, 2202–2212 (1993)
D.A. King: The influence of weakly bound intermediate states on thermal desorption kinetics, Surf. Sci. 64, 43–51 (1977)
J.T. Stuckless, N.A. Frei, C.T. Campbell: A novel single-crystal adsorption calorimeter and additions for determining metal adsorption and adhesion energies, Rev. Sci. Instrum. 69, 2427–2438 (1998)
C.T. Campbell, J.C. Sharp, Y.X. Yao, E.M. Karp, T.L. Silbaugh: Insights into catalysis by gold nanoparticles and their support effects through surface science studies of model catalysts, Faraday Discuss. 152, 227–239 (2011)
J.R.V. Sellers, T.E. James, S.L. Hemmingson, J.A. Farmer, C.T. Campbell: Adsorption calorimetry during metal vapor deposition on single crystal surfaces: Increased flux, reduced optical radiation, and real-time flux and reflectivity measurements, Rev. Sci. Instrum. 84, 123901 (2013)
S. Schauermann, T.L. Silbaugh, C.T. Campbell: Single-crystal adsorption calorimetry on well-defined surfaces: From single crystals to supported nanoparticles, Chem. Rec. 14, 759–774 (2014)
W. Lew, M.C. Crowe, E.M. Karp, O. Lytken, J.A. Farmer, L. Árnadóttir, C. Schoenbaum, C.T. Campbell: The energy of adsorbed hydroxyl on Pt(111) by microcalorimetry, J. Phys. Chem. C 115, 11586–11594 (2011)
W. Lew, M.C. Crowe, C.T. Campbell, J. Carrasco, A. Michaelides: The energy of hydroxyl coadsorbed with water on Pt(111), J. Phys. Chem. C 115, 23008–23012 (2011)
E.M. Karp, C.T. Campbell, F. Studt, F. Abild-Pedersen, J.K. Nørskov: Energetics of oxygen adatoms, hydroxyl species and water dissociation on Pt(111), J. Phys. Chem. C 116, 25772–25776 (2012)
E.M. Karp, T.L. Silbaugh, C.T. Campbell: Bond energies of molecular fragments to metal surfaces track their bond energies to H atoms, J. Am. Chem. Soc. 136, 4137–4140 (2014)
H. Ihm, H.M. Ajo, J.M. Gottfried, P. Bera, C.T. Campbell: Calorimetric measurement of the heat of adsorption of benzene on Pt(111), J. Phys. Chem. B 108, 14627–14633 (2004)
J.M. Gottfried, E.K. Vestergaard, P. Bera, C.T. Campbell: Heat of adsorption of naphthalene on Pt(111) measured by adsorption calorimetry, J. Phys. Chem. B 110, 17539–17545 (2006)
O. Lytken, W. Lew, J.J.W. Harris, E.K. Vestergaard, J.M. Gottfried, C.T. Campbell: Energetics of cyclohexene adsorption and reaction on Pt(111) by low-temperature microcalorimetry, J. Am. Chem. Soc. 130, 10247–10257 (2008)
E.M. Karp, T.L. Silbaugh, M.C. Crowe, C.T. Campbell: Energetics of adsorbed methanol and methoxy on Pt(111) by microcalorimetry, J. Am. Chem. Soc. 134, 20388–20395 (2012)
T.L. Silbaugh, J.B. Giorgi, Y. Xu, A. Tillekaratne, F. Zaera, C.T. Campbell: Adsorption energy of tert-butyl on Pt(111) by dissociation of tert-butyl iodide: Calorimetry and DFT, J. Phys. Chem. C 118, 427–438 (2013)
E.M. Karp, T.L. Silbaugh, C.T. Campbell: Energetics of adsorbed CH3 and CH on Pt(111) by calorimetry: Dissociative adsorption of CH3I, J. Phys. Chem. C 117, 6325–6336 (2013)
E.M. Karp, T.L. Silbaugh, C.T. Campbell: Energetics of adsorbed CH3 on Pt(111) by calorimetry, J. Am. Chem. Soc. 135, 5208–5211 (2013)
T.L. Silbaugh, E.M. Karp, C.T. Campbell: Energetics of formic acid conversion to adsorbed formates on Pt(111) by transient calorimetry, J. Am. Chem. Soc. 136, 3964–3971 (2014)
C.A. Wolcott, I.X. Green, T.L. Silbaugh, Y. Xu, C.T. Campbell: Energetics of adsorbed CH2 and CH on Pt(111) by calorimetry: The dissociative adsorption of diiodomethane, J. Phys. Chem. C 118, 29310–29321 (2014)
H. Larsen, D.E. Starr, C.T. Campbell: Enthalpies of adsorption of metal atoms on single-crystalline surfaces by microcalorimetry, J. Chem. Thermodyn. 33, 333–345 (2001)
J.F. Zhu, S.F. Diaz, L.R. Heeb, C.T. Campbell: Adsorption of Pb on NiAl(1 1 0): Energetics and structure, Surf. Sci. 574, 34–42 (2005)
D.E. Starr, D.J. Bald, J.E. Musgrove, J.T. Ranney, C.T. Campbell: Microcalorimetric measurements of the heat of absorption of Pb on well-defined oxides: MgO(100) and p(2×1)-oxide on Mo(100), J. Chem. Phys. 114, 3752–3764 (2001)
D.E. Starr, S.F. Diaz, J.E. Musgrove, J.T. Ranney, D.J. Bald, L. Nelen, H. Ihm, C.T. Campbell: Heat of absorption of Cu and Pb on hydroxyl-covered MgO(100), Surf. Sci. 515, 13–20 (2002)
D.E. Starr, C.T. Campbell: Low-temperature adsorption microcalorimetry: Pb on MgO(100), J. Phys. Chem. B 105, 3776–3782 (2001)
C.T. Campbell, S.C. Parker, D.E. Starr: The effect of size-dependent nanoparticle energetics on catalyst sintering, Science 298, 811–814 (2002)
J.A. Farmer, C.T. Campbell, L. Xu, G. Henkelman: Defect sites and their distributions on MgO(100) by Li and Ca adsorption calorimetry, J. Am. Chem. Soc. 131, 3098–3103 (2009)
J.A. Farmer, C.T. Campbell: Ceria maintains smaller metal catalyst particles by strong metal-support bonding, Science 329, 933–936 (2010)
J.C. Sharp, Y.X. Yao, C.T. Campbell: Silver nanoparticles on Fe3O4(111): Energetics by Ag adsorption calorimetry and structure by surface spectroscopies, J. Phys. Chem. C 117, 24932–24936 (2013)
T.E. James, S.L. Hemmingson, T. Ito, C.T. Campbell: Energetics of Cu adsorption and adhesion onto reduced CeO2(111) surfaces by calorimetry, J. Phys. Chem. C 119, 17209–17217 (2015)
D.E. Starr, J.T. Ranney, J.H. Larsen, J.E. Musgrove, C.T. Campbell: Measurement of the energetics of metal film growth on a semiconductor: Ag/Si(100)-(2×1), Phys. Rev. Lett. 87, 106102 (2001)
M. Smedh, S.F. Diaz, C.T. Campbell: Adsorption and adhesion energies of Pb on (1×1)-Mo2C∕Mo(100) by calorimetry, Phys. Rev. B 67, 205401 (2003)
O. Lytken, W. Lew, C.T. Campbell: Catalytic reaction energetics by single crystal adsorption calorimetry: Hydrocarbons on Pt(111), Chem. Soc. Rev. 37, 2172–2179 (2008)
M.C. Crowe, C.T. Campbell: Adsorption microcalorimetry: Recent advances in instrumentation and application, Annu. Rev. Anal. Chem. 4, 41–58 (2011)
C.T. Campbell, L. Árnadóttir, J.R.V. Sellers: Kinetic prefactors of reactions on solid surfaces, Z. Phys. Chem. 227, 1435–1454 (2013)
C.T. Campbell: The energetics of supported metal nanoparticles: Relationships to sintering rates and catalytic activity, Acc. Chem. Res. 46, 1712–1719 (2013)
T.E. James, S.L. Hemmingson, C.T. Campbell: Energy of supported metal catalysts: From single atoms to large metal nanoparticles, ACS Catalysis 5, 5673–5678 (2015)
J.T. Stuckless, D.E. Starr, D.J. Bald, C.T. Campbell: Metal adsorption calorimetry and adhesion energies on clean single-crystal surfaces, J. Chem. Phys. 107, 5547–5553 (1997)
M. Peter, J.M. Flores-Camacho, S. Adamovski, L.K. Ono, K.-H. Dostert, C.P. O’Brien, B. Roldan Cuenya, S. Schauermann, H.-J. Freund: Trends in the binding strength of surface species on nanoparticles: How does the adsorption energy scale with the particle size?, Angew. Chem. Int. Ed. 52, 5175–5179 (2013)
P. Dementyev, K.-H. Dostert, F. Ivars-Barceló, C.P. O’Brien, F. Mirabella, S. Schauermann, X. Li, J. Paier, J. Sauer, H.-J. Freund: Water interaction with iron oxides, Angew. Chem. Int. Ed. 54, 13942–13946 (2015)
P. Dementyev, M. Peter, S. Adamovsky, S. Schauermann: Chirally-modified metal surfaces: Energetics of interaction with chiral molecules, Phys. Chem. Chem. Phys. 17, 22726–22735 (2015)
P. Hörtz, P. Ruff, R. Schäfer: A temperature dependent investigation of the adsorption of CO on Pt(111) using low-temperature single crystal adsorption calorimetry, Surf. Sci. 639, 66–69 (2015)
J.-H. Fischer-Wolfarth, J.A. Farmer, J.M. Flores-Camacho, A. Genest, I.V. Yudanov, N. Rösch, C.T. Campbell, S. Schauermann, H.-J. Freund: Particle-size dependent heats of adsorption of CO on supported Pd nanoparticles as measured with a single-crystal microcalorimeter, Phys. Rev. B 81, 241416(R) (2010)
B. Hammer, Y. Morikawa, J.K. Nørskov: CO chemisorption at metal surfaces and overlayers, Phys. Rev. Lett. 76, 2141–2144 (1996)
I.V. Yudanov, M. Metzner, A. Genest, N. Rösch: Size-dependence of adsorption properties of metal nanoparticles: A density functional study on palladium nanoclusters, J. Phys. Chem. C 112, 20269–20275 (2008)
W. Weiss, W. Ranke: Surface chemistry and catalysis on well-defined epitaxial iron-oxide layers, Prog. Surf. Sci. 70, 1–151 (2002)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter
Cite this chapter
Vattuone, L., King, D.A. (2020). Energetics of Adsorption: Single Crystal Calorimetry. In: Rocca, M., Rahman, T.S., Vattuone, L. (eds) Springer Handbook of Surface Science. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-030-46906-1_30
Download citation
DOI: https://doi.org/10.1007/978-3-030-46906-1_30
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-46904-7
Online ISBN: 978-3-030-46906-1
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)