
Abstract
Excessive fat accumulation in adipocytes leads to obesity, which is a major contributing risk factor for many metabolic diseases such as metabolic syndrome, type 2 diabetes, and cardiovascular diseases. A number of studies showed that overnutrition causes oxidative stress and chronic low-grade inflammation, which both play a crucial role both in obesity prevention and in the development of obesity-related complications. Adipose tissue, especially in the visceral compartment, is considered not only as an energy depository tissue, but also as an active endocrine organ releasing a variety of biologically active molecules known as adipokines, with many of them having pro-inflammatory properties. Here, we summarize current data on the relationship between oxidative stress and inflammation in obesity, with emphasis on metabolic switches and the involvement of redox-responsive signaling pathways such as NF-κB and Nfr2. Experimental data suggest the dual role of Nrf2 signaling in prevention and aggravation of obesity and obesity-related inflammation; the potential mechanisms of Nrf2 duality are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Obesity is a chronic metabolic condition that has become a global problem of twenty-first century, and medical conditions associated with obesity are grouped in the metabolic syndrome [1, 2]. Excessive fat accumulation, especially of visceral fat, increases the risk for various chronic metabolic complications, including hypertension, hyperglycemia, hyperlipidemia, nonalcoholic fatty liver disease, insulin resistance, atherosclerosis, type 2 diabetes, and obesity-related cancer [1, 3, 4].
Overconsumption of high caloric food is a main reason for world growth of obesity. When nutritional supplies exceed the energy needs, excess nutrients may be stored in the form fat reserves in adipose tissue [5]. Adipose tissue, in addition to its function of storing energy reserves, has important functions as an endocrine organ, which consists of adipocytes and many immune cells and produces a variety of biologically active compounds, including adipokines, such as adiponectin and leptin, chemokines, such as monocyte chemoattractant protein (MCP-1), and pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), plasminogen activator inhibitor (PAI-1), interleukins 1β (IL-1β), and 6 (IL-6) [6,7,8,9]. Upon the accumulation of excessive fat, the adipocyte size/number is increased and the profile of adipokine secretion is altered, leading to a low-grade chronic systemic inflammation in adipose tissue and other peripheral tissues [1, 8,9,10,11]. Adipocytes secrete various chemokines, leading to the recruitment of pro-inflammatory M1 macrophages and other types of immune cells into adipose tissue. Enlarged adipocytes and infiltrated immune cells increase the production of pro-inflammatory cytokines and chemokines, resulting in systemic inflammatory status [11]. Inflammation increases production of reactive oxygen species (ROS) by the immune cells as a part of the immune response [12]. In addition, overloading mitochondria with energy substrates and activation of adipose NADPH oxidase contribute to enhanced ROS production in enlarged adipocytes [3, 7, 10,11,12,13,14]. Moreover, pro-inflammatory and nutrient-induced oxidative stress may also promote further production of pro-inflammatory cytokines, which, in turn, aggravate ROS production in a “vicious cycle” [5, 10]. Thus, chronic oxidative stress and chronic inflammation should be considered as two main interconnecting players in obesity-related metabolic complications.
Increased ROS production is followed by activation of a number of redox-responsive transcription factors, including nuclear factor-κB (NF-κB) and nuclear factor erythroid 2-related factor 2 (Nrf2) [7, 10, 15,16,17]. Whereas NF-κB is considered as a major regulator of immune response [1], Nrf2 activates cellular defense systems against the cytotoxic effects of oxidative stress [18]. Several studies have demonstrated that Nrf2 also contributes to the anti-inflammatory processes [19, 20]. In addition, pharmacological activation of Nrf2 inhibits inflammation and impairs degenerative diseases providing an interface between redox and anti-inflammatory responses [21, 22]. Given that the inflammation together with oxidative stress act as interacting inductors of obesity-associated complications, antioxidant and anti-inflammatory activities of Nrf2 received much attention in studies on obesity and obesity-related complications. Experimental data found that Nrf2 pathway could have both anti-obesity and obesity-promoting effects, suggesting the complicated regulatory role of Nrf2 in energy metabolism.
2 Obesity and Metabolic Syndrome and as a Worldwide Health Problem
With the successful conquest of many old infectious diseases in the world, non-communicable diseases have become the major cause of morbidity and mortality [23]. One of the most common non-infectious diseases is obesity. Obesity is recognized as a chronic metabolic condition that has become a global problem twenty-first century. This is a medical, social, and economic issue. This pathology affects a significant portion of the human population in both developed and developing countries. According to the World Health Organization, at least 2.8 million people die each year from complications of obesity. The United States holds first place in the world in the number of people who have a high degree of obesity. In Central Europe, 20 to 24% of the adult population is obese. In particular, in Ukraine, 21.3% of people suffer from obesity, and 53.5% are overweight [2, 5, 24].
Overconsumption of high caloric food is thought to be a main reason for obesity. When nutritional supplies exceed the energy needs, they may be stored in the form of carbohydrate or fat reserves for use later under conditions as starvation, stress, or infection [5]. However, chronic overeating, combined with a sedentary lifestyle, leads to an increasing accumulation of storage fats in the body of both adults and children. Importantly, obese children and adolescents have a high probability of remaining obese as adults. In the development of obesity, especial attention is paid to the periods of pre-school and adolescence regarded as times of risk for the development and maintenance of obesity [24, 25]. There is, however, consistent experimental and epidemiological data evidencing that the risk for developing obesity may largely depend on conditions of early life. Accumulating research findings indicate that epigenetic regulation of gene expression also plays a role in linking prenatal malnutrition to the risk of later-life metabolic disorders [26]. Accordingly, obesity is a condition with genetic and acquired etiology.
There are many ways in which a person’s health can be classified in relation to the weight, but the most widely used method is a calculating body mass index (BMI). BMI is not used to definitively diagnose obesity because people who are very muscular sometimes have a high BMI without excess fat.
Overweight and obesity are caused by the increase in the size and the number of fat cells in the body. The latter leads to a number of metabolic complications collectively called as metabolic syndrome. The concept of the metabolic syndrome (MetS) has existed for at least 100 years [27]. This metabolic disturbance was first described in the 1920s by Kylin, a Swedish physician, as the clustering of signs such as hypertension, hyperglycemia, and gout [27, 28]. Later, in 1947, Vague drew attention to upper body adiposity as the obesity phenotype that was commonly associated with metabolic abnormalities related to type 2 diabetes and cardiovascular diseases [27, 29]. Today, MetS is recognized a multifaceted disorder, including dyslipidemia, hyperglycemia and hypertension, insulin resistance, and increased incidence of oxidative stress [4, 5, 30,31,32,33].
Nonetheless, there is debate surrounding the etiology and pathogenesis of MetS because a single unifying mechanism is still unknown [32, 33]. Obesity is not always synonymous with MetS. Not all obese people develop MetS and not all people with MetS are obese. For example, Asian Americans have greater prevalence of metabolic syndrome despite lower body mass index [34]. There are the so-called metabolically healthy obese individuals who have high level of insulin sensitivity and no hypertension and hyperlipidemia and other features of MetS [23]. Experimental data suggest that MetS itself has a multifactorial etiology, involving complex interactions between genetic background, hormones, and nutrition [33]. That is why some aspects that make it possible to find relationships between obesity and MetS should be further analyzed.
3 Obesity, Metabolic Complications, and Inflammation
3.1 Adipose Tissue: Functions and Distribution
In mammals, adipose tissue is heterogeneous and can be divided into brown and white adipose tissue (BAT and WAT, respectively) (Fig. 1). BAT is specialized in dissipating energy in the form of heat. This adipose tissue is localized in the interscapular, cervical, and paravertebral regions around large arteries, where it is able to take up glucose and trigacylglycerides from the blood. During lipogenesis, these nutrients are temporarily stored in intracellular lipid droplets of BAT [35]. WAT is not homogenous and includes subcutaneous and visceral compartments. Subcutaneous adipose tissue stores fats and is divided further into upper and lower body adipose tissue [4, 36]. Visceral adipose tissue supplies inner organs with energy and is divided into omental (subcutaneous) and mesenteric adipose [4, 37]. During long time, WAT was defined as an inactive organ, only capable of storing energy in the form of trigacylglycerides (TAGs). However, at present, its role as an endocrine organ playing important functions in whole-body metabolism has been well recognized [9, 35]. WAT was found to be responsible for the synthesis of various hormones, which are crucial in regulation of satiety and insulin sensitivity [6, 9, 11, 35]. WAT also participates in regulation of energy homeostasis because it is capable of releasing TAG-derived fatty acids into the bloodstream, which can subsequently be used by other organs as an energy substrate or be packaged in TAG-rich lipoproteins in the liver [9, 35]. In the case of the accumulation of excess fats in WAT, this may cause many metabolic dysfunctions (insulin resistance, diabetes mellitus, heart dysfunction, etc.) that are combined in the term “metabolic syndrome.”

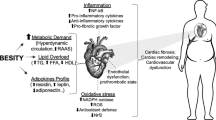
Potential mechanisms of obesity-related inflammation and metabolic disturbances. With a chronic excessive energy intake and low physical activity, a positive energy balance causes body weight gain and higher nutrient flux into visceral white adipose tissue (WAT). Visceral WAT adipocytes primarily respond to the higher demand for energy storage by increasing their size (adipocyte hypertrophy). Adipocyte hypertrophy is typically associated with increased cellular and tissue stress, disturbed adipokine secretion, increased production of pro-inflammatory cytokines, including interleukins (IL-6, IL-1β), TNF-α, PAI-1, and MCP-1, and anti-inflammatory molecules such as IL-10. These signal molecules can then act through a number of cell signaling pathways to induce dyslipidemia, insulin resistance, vascular dysfunction, and atherosclerosis
3.2 Adipose Tissue and Insulin Resistance
A major determinant of metabolic health is the ability of subcutaneous adipose tissue to store excess fat rather than allowing it to accumulate in ectopic depots including liver (i.e., in nonalcoholic fatty liver disease), muscle and heart, or in epicardial/pericardial and visceral fat depots, which cause the metabolic complications of obesity [38]. The ability to recruit and differentiate precursor cells into adipose cells (adipogenesis) in subcutaneous adipose tissue is under genetic regulation. Dysregulation of these signaling pathways is associated with impaired adipogenesis. This leads to hypertrophic, dysfunctional, and insulin-resistant adipose cells with a reduced content of GLUT4, the major insulin-regulated glucose transporter. This, in turn, reduces adipose tissue glucose uptake and de novo lipogenesis [38]. The described above events are the inductors of genetically induced insulin resistance. At the same time, under any circumstances, adipocyte cell is a critical mediator between insulin and liver glucose output. Insulin acts on adipose tissue (1) by stimulation of glucose uptake and TAG synthesis and (2) by suppression of TAG hydrolysis and release of free fatty acids (FFAs) and glycerol into the circulation [39]. It has been hypothesized that FFAs, released during lipolysis in visceral adipose tissue, are important inductors of acquired insulin resistance [4, 40]. Excessive abdominal fat mass is associated with increased concentration of FFAs in the blood plasma. Surplus of circulating FFAs may ectopically be accumulated in insulin-sensitive tissues and impair insulin action. Increased basal lipolysis may also modify the secretory profile of adipose tissue, influencing whole-body insulin sensitivity [4, 36, 40, 41]. Finally, excessive FFA release may also worsen adipose tissue inflammation, a well-known process contributing to insulin resistance [41].
It is possible that insulin resistance of adipocyte itself can be a major cause of the dysregulation of carbohydrate metabolism in the prediabetic state. The inability of insulin to perform its numerous roles leads to an impaired glucose metabolism and an increase in blood glucose levels [42, 43]. In the latter case, subjects undergo hyperglycemia and insulin deficiency. Approximately one quarter of insulin-resistant patients has a normal glucose tolerance test, but this condition increases significantly the risk of development of type 2 diabetes mellitus. If pancreatic β cells function normally, the compensatory hyperinsulinemia will be raised to maintain normal fasting and postprandial glucose concentrations [4, 42].
3.3 Adipose Tissue and Cardiovascular Diseases
People with a central deposition of adipose tissue can experience elevated cardiovascular diseases and mortality, including stroke, congestive heart failure, myocardial infarction, and cardiovascular death [44, 45]. It has long been recognized that an extensive capillary network surrounds adipose tissue. Adipocytes are located close to vessels with the highest permeability, the lowest hydrostatic pressure, and the shortest distance for transport of molecules to and from the adipocytes [27, 45]. Classical cardiovascular risk factors include high low-density lipoprotein (LDL), high cholesterol, hypertension, and dysfunctions in glucose metabolism [27, 44]. The adipose tissue is not simply a passive storehouse for fat but an endocrine organ that synthesizes and releases into the bloodstream a variety of important peptides and non-peptide compounds involved in cardiovascular homeostasis and in its disturbance [4, 27, 45]. Existing data allowing assume that excess fat provoking cardiovascular diseases via the modulation of pro-inflammatory processes.
3.4 Bioactive Substances of Adipose Tissue
Adipose tissue comprises ~50% adipocytes and ~50% other cells including pre-adipocytes, vascular, neural, and immune cells [46]. Under normal conditions, adipose tissue is involved not only in lipid synthesis, but also in storage and secretion of anti-inflammatory molecules. Provoking factors such as increased fat content can also induce adipose tissue to secrete a number of pro-inflammatory factors [4, 36, 46]. By acting as transmitters of endocrine or paracrine signals, the secreted pro-/anti-inflammatory factors can induce either inflammation or altered insulin sensitivity of the adipocyte [36]. Generally, the adipose tissue derivatives known as adipokines include, besides inflammatory cytokines, other molecules acting as appetite regulators (leptin), insulin sensitizers, and atheroprotectors (adiponectin) [4, 9, 36, 47]. Adipocytes, pre-adipocytes, and macrophages within adipose tissue secrete pro-inflammatory cytokines, including interleukins (IL-6, IL-1β), TNF-α, PAI-1, and MCP-1, and anti-inflammatory molecules such as IL-10 [4, 9, 47] (Fig. 1).
3.4.1 Leptin
Leptin is a small peptide (16 kDa), belonging to a group of anti-inflammatory cytokines [48, 49]. It is an anorexigenic peptide, which increases energy expenditure [9]. Leptin is expressed mainly by adipose tissue although low levels have been detected in the placenta, skeletal muscle, gastric and mammary epithelium, and the brain [9, 49]. Levels of leptin are increased by glucocorticoids, acute infection, and pro-inflammatory cytokines. In addition, its levels are higher in females than males, partly because of inhibition by androgens and stimulation by estrogen [9].
The functional leptin receptor is in the hypothalamus where it functions to increase energy expenditure and to reduce appetite. The leptin receptor is also found in other organs such as the heart, liver, kidneys, and pancreas; it is also present in the smooth muscle and endothelium of heart, brain vasculature, and myometrium [47, 49]. Adipose tissue and plasma leptin concentrations depend on the amount of stored fats as well as the status of energy balance. Therefore, leptin levels are higher in obese individuals and increase with overeating. Conversely, lean individuals have lower leptin levels, and fasting reduces circulating leptin levels. Many studies have shown a positive correlation between leptin levels and metabolic syndrome [47, 50, 51]. Nutritional regulation of leptin is mediated at least partially by insulin, as leptin decreases in response to low insulin levels [9, 52].
3.4.2 Adiponectin
Structurally, adiponectin is related to the complement 1q family [9] and circulates in three isoforms: a trimer of low-molecular mass, a hexamer of medium molecular mass, and a multimeric high molecular mass isoform [9, 47, 53]. Adiponectin, like leptin, is an adipose-derived plasma protein with broad range of effects. However, unlike leptin, it is secreted exclusively by adipocytes. The receptors for adiponectin were found in adipocytes, brain, muscle, liver, artery, etc. [54].
Adiponectin levels display no great fluctuations in the blood; thus, its release is not acute but regulated by long-term metabolic changes [9]. Adiponectin plays an important role in protection against insulin resistance/diabetes and atherosclerosis. It has insulin-sensitizing activity, lipid oxidation enhancement, and vasodilatation activities [9, 47, 53]. Levels of adiponectin are low in subjects with essential hypertension and in the obese, but can increase with a loss of weight [9, 47]. Adiponectin suppresses almost all processes involved in atherosclerotic vascular changes: the expression of adhesion molecules in vascular endothelial cells, adhesion of monocytes to endothelial cells (via TNF-α inhibition), vascular smooth muscle cell proliferation and migration [9, 47]. Levels of adiponectin, unlike that of leptin, are inversely correlated with metabolic syndrome [39, 41, 47].
3.4.3 Interleukin 6
Interleukin 6 (IL-6) is a multifunctional cytokine produced by various cells. Its effects are usually aimed at the development of inflammation, immune response, and hematopoiesis [55]. At the same time, IL-6 is an interleukin, which acts as both a pro-inflammatory cytokine and an anti-inflammatory myokine [56]. IL-6 is often secreted by M1 macrophages as part of the normal inflammatory response against infection and injury [9, 47, 55]. Role of IL-6 as an anti-inflammatory myokine is mediated through inhibition of TNF-alpha and IL-10 [56].
In metabolic syndrome, adipocyte dysfunction is frequently present and is associated with an increase in M1 macrophage population within adipose tissue. This can result in the increased secretion of IL-6 and other pro-inflammatory cytokines from adipose tissue. These pro-inflammatory cytokines can then act through a number of cellular signaling pathways, including mTOR and protein kinase C (PKC), to induce insulin resistance. Due to its inflammatory properties, IL-6 has been implicated in the endothelial cell damages within blood vessels leading to vascular dysfunction and atherosclerosis [47].
3.4.4 Tumor Necrosis Factor Alpha
Tumor necrosis factor alpha (TNF-α) is synthesized as a 26 kDa transmembrane protein that undergoes cleavage by a metalloproteinase to be released into the circulation as a 17 kDa soluble TNF-α molecule [9, 52]. TNF-α is a cell signaling cytokine involved in systemic inflammation and is one of the cytokines of acute phase inflammation. It is produced preferentially by activated macrophages, and less by other cell types such as CD4+ lymphocytes, neutral killers, neutrophils, mast cells, eosinophils, and neurons. Adipocytes are also able to produce TNF-α [9, 47,48,49].
For some years, it was assumed that adipocytes are the principal source of elevated TNF-α level in obesity. Nevertheless, it has been recognized more recently that not adipocytes but macrophages from the stromal vascular fraction are the primary source of adipose-derived TNF-α [9, 52]. Macrophages, which constitute about 10% of the stromal vascular fraction, are present in visceral adipose tissue in larger quantities than in the subcutaneous one [9]. Recent studies also postulate that the increased infiltration of adipose tissue with M1 macrophages contributes to the increased TNF-α in obesity [9, 47,48,49]. Since metabolic syndrome is often characterized by adipocyte dysregulation, and these dysregulated adipocytes tend to secrete TNF-α, IL-6, and other pro-inflammatory adipokines at higher levels, the central obesity often encountered in metabolic syndrome could be a risk factor for elevated TNF-α levels [9, 47].
3.4.5 Plasminogen Activator Inhibitor 1
Plasminogen activator inhibitor 1 (PAI-1) is a single chain 45-kDa glycoprotein involved in microvascular events. Endothelial and vascular smooth muscle cells are presumably the main sources of PAI-1 but other cells, such as platelets, hepatocytes, mesangial cells, fibroblasts, monocytes, macrophages, adipocytes, and stromal cells, have also been shown to secrete PAI-1 [9, 57]. Increased size of adipocytes and adipose tissue mass contribute to higher adipose production to circulating PAI-1. Experimental data show that visceral adipose tissue has a higher capacity to produce PAI-1 than subcutaneous adipose tissue. Studies in human adipocytes indicate that PAI-1 synthesis is up-regulated by insulin, glucocorticoids, angiotensin II, some fatty acids, and potently by cytokines such as TNF-α and transforming growth factor-β, whereas catecholamines reduce PAI-1 production [9, 57].
PAI-1 is involved in fibrinolysis and its levels are altered in obesity [57]. Plasma PAI-1 levels are increased proportionally to increase in visceral adiposity, indicating the possible role of PAI-1 as the link between abdominal/central obesity and cardiovascular diseases. PAI-1 protein can change the balance between fibrinolysis and fibrinogenesis, contributing to the remodeling of vascular architecture and the atherosclerotic process [9]. An altered function of the endocrine system and an impaired auto-/paracrine function at adipocyte levels may mediate this disturbance in fibrinolytic system and thereby increase the risk for cardiovascular diseases [9, 47, 57].
3.4.6 Monocyte Chemoattractant Protein-1
Monocyte chemoattractant protein-1 (MCP-1) is produced predominantly by macrophages and endothelial cells and is a potent chemotactic factor for monocytes [58, 59]. Expression of this pro-inflammatory chemokine is increased in atherosclerotic lesions, and inhibition of its expression reduces the extent of atheroma. These observations indicate that MCP-1 plays an important role in atherogenesis. In mammal models, the increased MCP-1 expression in adipose tissue was shown to contribute to the macrophage infiltration into this tissue, insulin resistance, and hepatic steatosis associated with obesity [58, 59].
3.4.7 Interleukin 10
Interleukin 10 (IL-10) is a predominantly anti-inflammatory cytokine, which is secreted by monocytes or M2 macrophages and functions as a modulation of systemic inflammation; in particular, it helps to promote normal tissue remodeling following an inflammatory response. One of the ways by which IL-10 moderates the inflammatory response is the inhibition of NADPH oxidase, and therefore the decrease in oxidative stress resulting from this enzyme [47]. Anti-inflammatory cytokine IL-10 is secreted by human WAT [60]. Furthermore, one study found that IL-10 levels inversely correlated with levels of total cholesterol, LDL, TAGs, blood glucose, and positively correlated with HDL levels [47, 61]. IL-10 antagonizes pro-inflammatory actions of IL-6, TNF-α, and IL-10 and appears to confer a protective effect against increase in these cytokines, which both are associated with metabolic syndrome and its comorbidities [47].
3.5 Inflammation and Macrophage Infiltration in Adipose Tissue
Macrophage count is increased in adipose tissue during obesity. Increased levels of FFAs, cholesterol, and bacterial lipopolysaccharide (LPS) are well-known inductors of macrophage recruitment. The inductors bind to and activate toll-like receptor 4 (TLR4) and its downstream signaling pathways in adipose resident cells. The activated macrophages secrete cytokines and chemokines, such as MCP-1, and express C–C motif chemokine receptor-2 (CCR2) and CCR5, which, in turn, augment the recruitment of more monocytes and other leukocytes into adipose tissue [62, 63]. Macrophages share the same differentiation and recruitment molecules with other myeloid cells in many inflammatory conditions [63, 64].
M1 macrophages are associated with a pro-inflammatory profile. These macrophages are generally stimulated by T-helper 1 (Th1) type of cytokines, such as interferon γ (IFN-γ), or by pathogen-associated molecular patterns (PAMPs), such as LPS [63]. In turn, M1 macrophages secrete cytokines, including IL-6, TNF-α, IL-1β, IL-12, and IL-23 [9, 47, 48, 49, 63]. M1 macrophages can also induce Th1 responses [63, 65, 66]. On the whole, these cells express high levels of major histocompatibility complex class II (MHC-II), CD80 and CD86 co-stimulatory molecules and CD68 [63]. Moreover, M1 macrophages express Th1 cell-attracting chemokines, including CXCL9 and CXCL10 [63, 67].
M2 macrophages are associated with tissue remodeling and inflammation resolution [63, 68]. M2 macrophages have immunosuppressive properties, have high phagocytic capacity, and secrete extracellular matrix components, angiogenic and chemotactic factors, anti-inflammatory cytokines, and growth factors, such as IL-10 and transforming growth factor-β (TGF-β) [63, 68, 69].
Macrophages are central mediators of obesity-induced inflammation and insulin resistance. They also are key cells for maintenance of adipose tissue homeostasis. Recently, several reports described the importance of these cells as regulators of insulin sensitivity, which requires the activation of innate immune receptors, transcription factors, and intracellular metabolism to support either pro- or anti-inflammatory adipose tissue phenotype. Thus, macrophages have a dual role, changing their status in obesity to reinforce immune responses, obesity progression, and development of related diseases [63].
3.6 Adipocyte Dysfunction and Inflammation
Inflammation is a process of activation of innate immune system in response to exogenous and endogenous factors, such as infection by microorganisms, tissue stress, and injury. The inflammation associated with obesity is triggered by the excessive intake of high caloric food rich in carbohydrates and fats (Fig. 1). Obesity causes the enlargement of adipose tissue that is accompanied by the release of a number of adipokines [47]. The release of chemokines, which induce recruitment of macrophages from the bloodstream, increases macrophage infiltration and inflammation with enhanced production of pro-inflammatory cytokines such as TNF-α, IL-6, and MCP-1 [70]. The macrophage and adipose tissue-derived adipokines act in a paracrine or autocrine way that exacerbates adipose tissue inflammation [47, 70]. This is accomplished by increased release of FFAs and dysregulated secretion of leptin and adiponectin. At the systemic level, altered adipokine secretion, can lead to decreased muscle and liver insulin sensitivity through enhanced ectopic lipid deposition [47]. The overproduction of adipokines leads to pathological conditions, such as obesity and adipose tissue inflammation that can develop insulin resistance and favor the pathogenesis of type 2 diabetes [70].
4 Role of Oxidative Stress in Obesity
4.1 Oxidative Stress as a Mechanism of Obesity-Associated Metabolic Complications
Excessive fat accumulation, especially of visceral fat, increases risk of metabolic syndrome and various chronic diseases, including type 2 diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer [12]. A number of mechanisms linking obesity to these associated diseases have been proposed, and oxidative stress together with inflammation response was supposed to have a crucial role among them. Oxidative stress has been implicated in vascular complications of diabetes and in pancreatic β cell destruction. Meanwhile, obese people without diabetes also display elevated intensity of oxidative stress. In addition, levels of oxidative stress markers were found to be increased in the adipose tissue of obese mouse models [3, 7, 12, 71].
The typical redox alterations in obese people and models of animal obesity include: (1) activation of cellular systems involved in the production of reactive oxygen species (ROS) and reactive carbonyl species (RCS), (2) increased levels of oxidatively damaged biomolecules (lipid peroxides, malondialdehyde, oxidized LDL, protein carbonyls, 3,5-dinitrotyrosine, advanced glycation end products (AGEs), 8-hydroxy-2′-deoxyguanosine, etc.), (3) an increase or a decrease in antioxidant defense capacity (superoxide dismutase, catalase, glutathione and thioredoxin, and associated enzymes [glutathione S-transferase, glutathione reductase, thioredoxin reductase], peroxiredoxins, NAD(P)H:ubiquinone oxidoreductase (NQO1), paraoxonase, etc.) [3, 5, 12, 14, 71,72,73,74,75,76,77,78,79]. It was shown that ROS/RCS may contribute to the development of obesity-associated insulin resistance and type 2 diabetes [75, 76, 77, 79], cause unfavorable changes in in the brain and arterial walls [80,81,82], as well as they are implicated in the pathogenesis associated with hypertension, atherosclerosis, and cancer [12, 82].
A clear relationship between obesity and oxidative stress is not yet fully defined. Many studies report that oxidative stress can be as a result, but also a trigger of obesity. Furthermore, oxidative stress can have both obesity-promoting and anti-obesity effects [14, 17, 76, 83]. It seems that fat content and its distribution in adipose tissue can affect the intensity of oxidative stress determining its downstream effects in obesity [5]. To support it, levels of oxidative damages were found to be higher in obese individuals and correlate directly with BMI and the percentage of body fat and TAG levels [73, 84]; in contrast, antioxidant defense capacity was lower if the amount of body fat and central obesity were higher [85]. However, there are no clear correlations between body fat accumulation and the protective capacity of antioxidant systems. An increase, decrease, or no change in the activity of antioxidant enzymes was reported in different tissues of obese subjects [12, 72, 73, 74, 79, 86,87,88]. These controversial data may indicate both tissue-specific responses and time-dependent effects [89].
4.2 Sources of ROS and RCS in Adipose Tissue
The generation of reactive oxygen/carbonyl species (ROS/RCS) is an inevitable aspect of aerobic life [3, 90,91,92,93]. Energy resources, mainly glucose and fatty acids, are oxidized via the Krebs cycle with generation of reducing equivalents in the form NADH and FADH2 (Fig. 2). These equivalents are subsequently re-oxidized in the electron transport chain (ETC) of mitochondria and their energy released is used for synthesis of ATP. Transport of protons from NADH and FADH2 across the inner mitochondrial membrane into the intermembrane space is coupled with transfer of electrons through various complexes of ETC. The end acceptor of electrons is oxygen, which undergoes four-electron reduction to water. A consecutive one-electron reduction of oxygen is also possible due to electron escaping from mitochondrial ETC. As a result, reactive oxygen species (ROS) such as superoxide anion radical (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•) are formed. Mitochondria are supposed to be responsible for over 90% of ROS production [91]. Under physiological conditions, ROS are maintained at low steady-state levels by functioning of complex antioxidant defense systems [91, 92].

Relationship between metabolic processes, ROS/RCS production, and inflammation at obesity development. Overnutrition causes intensification mitochondrial respiration followed by ROS increase. Increased ROS levels stimulate adipocyte proliferation and differentiation and promote activation of adipose NF-kB transcriptional factor, which triggers acute inflammation response followed by synthesis and releasing pro-inflammatory mediators. These mediators aggravate macrophage M2 recruitment and infiltration in adipose tissue. Executing their protective function, macrophage produces high levels of ROS resulting in chronic oxidative stress. In turn, chronic oxidative stress causes further increase in pro-inflammatory cytokine synthesis and macrophage infiltration leading inflammation to become chronic. ROS-activated NADPH oxidase (NOX4) and reactions of non-enzymatic glycosylation and monosaccharide oxidation also contribute to an increase in ROS levels under overnutrition. Low increase in ROS levels promotes activation of Nrf2 that triggers adaptive response to combat with raised oxidative stress and inflammation and to inhibit excessive adipocyte proliferation. At the same time, under chronic oxidative stress, Nrf2-mediated induction of protective mechanisms seems to be not able to cope with increased oxidative and inflammatory challenges. In addition, constantly enhanced Nrf2 activity may stimulate adipogenesis
Overconsumption of food rich in fats and carbohydrates leads to excessive supply of energy substrates in adipose and non-adipose cells (Fig. 2). Oxidation of free fatty acids and monosaccharides (glucose or fructose) increases levels of acetyl-CoA, which enter the Krebs cycle and may intensify the latter. In turn, the Krebs cycle intensification enhances generation of NADH and FADH2, which overload mitochondrial ETC and promote an increase in ROS production by ETC [90, 91]. Increased ROS levels are first dangerous for mitochondria since many Krebs cycle and ETC proteins and mitochondrial DNA are very sensitive to ROS attack. Mitochondrial ETC dysfunction together with the inhibition of the Krebs cycle, are involved in TAG accumulation via the redirection of acetyl-KoA for synthesis of fatty acids. The latter are further converted to storage lipids in adipose tissue or in the case of overloading of adipose tissue can be appear in the bloodstream and other organs provoking lipotoxicity [94].
Oxidation of free fatty acids in peroxisomes is another source of superoxide anion radial and hydrogen peroxide in adipose tissue [7]. Furthermore, the plasma membrane NADPH oxidase (NOX), especially adipose-specific NOX4 isoform, which converts molecular oxygen to its superoxide anion radical, can be involved in ROS formation in adipocytes under excessive supply of energy resources [12, 76]. This is confirmed by increased ROS levels in cultured adipocytes loaded with free fatty acids or glucose [14, 95, 96]. Excess nutrients activate NADPH oxidase and the pentose phosphate pathway (PPP), which is a major source of cellular NADPH using by NOX [96]. In obese mice, mRNA NOX4 levels were increased in adipose tissue but not in the liver or muscles [14]. Furthermore, ROS production can be inhibited by treatment with apocynin, an NADPH oxidase inhibitor [14] or the PPP inhibitor dehydroepiandrosterone [97] or via deletion of NOX4 gene [95]. These data suggest the involvement of plasma membrane NOX4 in ROS generation by excess of both glucose and free fatty acids. High glucose and FFAs, especially palmitate, was shown to stimulate ROS generation through protein kinase C-dependent activation of NOX [98].
Non-enzymatic glycosylation of proteins and nucleic acids, polyol (for glucose) and hexosamine (for fructose) pathways, glyceraldehyde, and monosaccharide autoxidation can be also important sources of ROS on high carbohydrate diets [77, 78, 90, 93, 99,100,101,102]. In addition to ROS, reactive carbonyl species (RCS), which include various compounds with one or more carbonyl groups, can be formed in many enzymatic and non-enzymatic processes such as lipid peroxidation, amino acid oxidation, monosaccharide autoxidation, and glycation [78, 100,101,102]. For example, methylglyoxal, the most common RCS found in biological systems, is produced by the triosephosphate isomerase reaction of glycolysis [78]. Both ROS and RCS as well as the so-called advanced glycation end products (AGEs) are thought to be involved in obesity progression and development of associated metabolic disorders [12, 99].
4.3 ROS as a Regulators Adipocyte Proliferation and Differentiation
Obesity is characterized by adipocyte hypertrophy which resulting from an increase in size and/or number of adipocytes. Adipocyte differentiation and function have been shown to be affected by cellular redox status. Enhanced ROS levels may alter food intake [103] and stimulate proliferation of pre-adipocytes and enlargement of differentiated adipocytes (Fig. 2) [13, 104, 105]. Exposure to low doses of H2O2 inhibited adipose differentiation of adipocyte 3T3L1 cells, but higher concentration of this oxidant markedly induced differentiation [106]. Moreover, ROS generated by both NOX4 and mitochondrial ETC are involved in adipocyte differentiation in adipogenic stem cells [13, 105]. Moreover, down-regulation of NOX4 gene inhibits production of ROS and differentiation of pre-adipocytes, while NOX4 overexpression was found to have the opposite effect [107]. In obese mice, treatment with NOX inhibitors reduces ROS levels in adipose tissue and improves symptoms of diabetes, hyperlipidemia, and hepatic steatosis [14]. In contrast, Li et al. [108] reported that NOX4 deficiency accelerated obesity in mice fed high-fat diet. It appears that the role of NOXs in obesity is complicated and should be elucidated further. Adipocyte differentiation is also characterized by an increase in mitochondrial metabolism [109], but whether this is essential for differentiation or a by-product of the differentiation process is not clearly understood [13].
4.4 Relationship Between Inflammatory and Oxidative Processes in Obesity
The main function of adipose tissue is to synthesize and to store fats in the form of TAGs and to release free fatty acids during fasting. Under conditions of chronic overnutrition, the excess energy nutrients lead to an increase in adipose tissue stimulating adipocyte proliferation and enlargement. Together with adipocyte size increment, free radical and inflammatory processes are intensified in adipose tissue (Fig. 2). These processes seem to be developed in parallel but reinforce each other over obesity progression: inflammation leads to the intensification of ROS production, and enhanced ROS levels stimulate inflammatory signaling cascades, thus forming a vicious circle [5].
In addition to metabolic functions, adipose tissue is an important endocrine organ. As described above, adipose tissue consists of adipocytes and various immune cells that produce a variety of biologically active molecules, including adipokines, chemokines, and pro-inflammatory cytokines such as TNF-α and interleukins IL-1β and IL-6 [6, 7, 8, 49, 55, 110]. Production of these proteins by adipose tissue, especially by that forming visceral fat, is increased in obesity; the obese people are, therefore, characterized by a state of chronic low-grade inflammation, which has a functional link with insulin resistance and metabolic syndrome [36, 53, 61, 66, 111]. However, the primary events triggering this inflammation are still unclear since a complex of endocrine and immune factors act to regulate this adipose tissue microenvironment [110].
In visceral adipose tissue, the induction of inflammatory responses can be connected with the involvement of toll-like receptors (TLRs), which were found to be present in plasma membrane of adipocytes [112]. Normally, these TLRs (TLR2 and TLR4) are activated by bacterial lipoproteins and lipopolysaccharides [113]. The activation of TLRs triggers a signaling cascade leading to translocation of nuclear factor-κB (NF-κB) to the nucleus. NF-κB protein is a main transcriptional regulator of adaptive immune response and its activation triggers a synthesis and release by adipose tissue of several pro-inflammatory cytokines, chemokines, and adhesion molecules [7, 10, 15]. Recent studies have showed that non-esterified fatty acids can be modulators of adipokine secretion by adipocytes. It has been shown that TLRs receptors may be activated by specific types of lipids, in particular saturated fatty acids was fond to activate both TLR2 and TLR4, whereas unsaturated fatty acids inhibit TLR-mediated signaling and gene expression [114]. Enlarged adipocytes are characterized by releasing free fatty acids with increasing their levels in the blood and adipose tissue microenvironment. Therefore, FFAs can contribute to a low-grade inflammation in adipose tissue. Besides hyperlipidemia, high glucose levels can also activate inflammatory pathways. Hyperglycemia may lead to the non-enzymatic glycation of proteins and lipids forming advanced glycation end products (AGEs), stimulating activation of the pattern recognition receptor RAGE, and eliciting an immune response through the activation of NF-kB [1, 90]. In obesity-related pro-inflammatory states, the increased size of adipocytes plays a decisive role because, to some extent, it increases adipose tissue and production of adipocytokines, and this triggers a number of inflammation-related pathophysiological processes [11].
Nutrient-induced oxidative stress can also contribute to the activation of inflammation responses in adipose tissue. For example, several pro-inflammatory transcription factors, including NF-κB and activator protein-1 (AP-1), are redox-sensitive and undergo the activation when ROS levels are increased [7, 10, 15]. Thus, mitochondria-derived or/and NOX4-stimulated ROS can mediate the production pro-inflammatory proteins during overnutrition. The production of adipokines by enlarged adipocytes increases migration of macrophages in adipose tissue, which worsens inflammation. Among chemokines produced by adipocytes, MCP-1, which attracts and triggers monocyte and macrophage migration, plays essential roles in this process [12, 96]. In turn, macrophages are known to produce ROS as a part of the immune response; therefore, macrophages may promote overproduction of ROS in adipose tissue [7, 12, 14]. Enhanced ROS levels, in turn, lead to the development of chronic oxidative stress with a gradual increase in oxidative damages of macromolecules and depletion of protective mechanisms [71]. Moreover, ROS have been shown to increase expression of MCP-1 and NADPH oxidase subunits in adipocytes [14], thus leading to more permanent state of inflammation. Further, enhanced secretion of pro-inflammatory factors by enlarged white adipose tissue contributes to constantly increasing ROS production. Thus, in obesity, oxidative stress may contribute to the establishment of a vicious cycle that promotes increased inflammation in the adipose tissue [10, 12]. The products of the adipocytes, in turn, may modify the metabolic and inflammatory processes in surrounding and other tissues (kidney, liver, brain, pancreatic gland, heart, etc.) causing various obesity-related complications [3, 12].
In addition, to the activation of inflammation signaling cascades, the increased intensity of oxidative stress in adipose tissue results in the induction of protective mechanisms against oxidative damages. Current data suggest that the induction of adaptive and inflammatory responses can have both anti-obesity and obesity-promoting effects [17, 83], that, probably, is a result of time- and intensity-dependent effects of oxidative stress. On the short-term scale, excess nutrients may induce low/moderate increase in ROS production followed by activation of a number of redox-responsive transcription factors such as NF-κB and AP-1, which were mentioned above, as well as nuclear factor erythroid 2 = related factor 2 (Nrf2) and FoxO1, a member of the forkhead box O family of proteins [7, 10, 15, 16, 17, 115]. The activation of NF-κB and AP-1 pathways triggers acute inflammation response, which includes a synthesis and release by adipose tissue of pro-inflammatory cytokines, e.g., TFN-α and IL-6. The increased level of IL-6 stimulates the liver to synthesize and secrete acute phase proteins (C-reactive protein) [1, 7, 10, 15]. The normal acute inflammatory response involves the delivery of plasma components and leucocytes to the site of insult and is initiated by tissue-resident macrophages leading to a production of different types of inflammatory mediators. If successful, the injurious agent is eliminated followed by inflammation cease and tissue repair. This is achieved by switching the lipid mediators from pro-inflammatory to anti-inflammatory and by the action of tissue-resident and newly recruited macrophages [113]. The primary function of Nrf2 is to maintain redox homeostasis by activating expression of antioxidant and xenobiotic-detoxifying genes under exposure to different toxic electrophilic compounds [17, 83]. More detailed functions of Nrf2 in obesity will be discussed below. FoxO proteins, in particular FoxO1, are also transcriptional regulators of adaptive stress responses and they induce the expression of genes coding both intra- and extracellular antioxidant proteins [115]. In addition, FoxO 1 regulates of adipocyte size and adipose tissue-specific gene expression in response to excessive calorie intake [116]. Thus, on short-term scale, complex adaptive responses are induced in a cell against damaging effects of obesity-associated oxidative stress to prevent obesity progression.
When accumulation of storage lipids in adipose tissue progresses further, hypertrophied adipocytes and other cells present in the adipose tissue produce large amounts of pro-inflammatory adipokines, which stimulate infiltration of macrophages and T-lymphocytes. In the inflammatory process, macrophages in adipose tissue release chemoattractants for macrophages that induces inflammation to become chronic [11]. Chronic inflammation aggravates ROS production that, in turn, leads to chronic activation of stress-responsive transcription factors that finally causes depletion of protective antioxidant mechanisms and increase in oxidation of macromolecules.
5 Nrf2, Oxidative Stress, Obesity, and Inflammation
Recent data suggest that redox-sensitive transcriptional factors, which participate in maintenance of a balanced redox state, play important roles in obesity. It is not surprising because of the developing oxidative stress, which accompanies fat accumulation and obesity progression. Hence, the manipulations with molecular pathways that produce or eliminate ROS are becoming a popular tool for potential intervention for obesity and related metabolic syndrome treatment. In this context, Nrf2 transcriptional factor came under the spotlight in obesity research because it regulates the adaptive response to endogenous and exogenous oxidative or electrophilic stresses [14, 16,-21].
5.1 Regulation of Nrf2 Activity: General Concepts
Nrf2 belongs to the family of the cap ‘n’ collar transcription (CNC) factors with a basic leucine zipper (bZIP). The homologs of Nrf2 found in the invertebrates, nematode Caenorhabditis elegans (SKN-1 protein) and Drosophila melanogaster (CncC protein), were found to share the same protective functions [17, 117]. In mammals, Nrf2 protein consists of seven functional domains called Nrf2-ECH homology (Neh) 1–7 domains. Neh1 domain is a CNC-bZIP dimerization domain that allows Nrf2 to form heterodimer with small Maf protein, DNA, and other transcription partners [18, 118]. The highly conserved Neh2 domain at N-terminal region contains two important motifs known as DLG and ETGE, which are involved in the interaction between Nrf2 and its negative regulator Keap1 protein [119, 120]. The Neh3 domain is located at the C-terminus of the protein and is essential for the transactivation of responsive genes by Nrf2. The Neh4 and Neh5 domains are considered as transactivation domains that cooperatively bind to cAMP response element binding (CREB) protein. Finally, Neh6 and Ne7domains, which are located in the middle of Nrf2, have been reported to be associated with redox-insensitive suppression and degradation of Nrf2 protein [18, 121].
The activity of Nrf2 is controlled through a complex transcriptional/epigenetic and post-translational network that ensures an increase in its activity during redox perturbation, inflammation, growth factor stimulation, and nutrient/energy fluxes, thereby allowing the factor to orchestrate adaptive responses to diverse forms of stress [17, 18, 91, 121,122,123].
Nrf2 is regulated at the transcriptional level by itself [124] and other transcription factors including PPARγ [125] and NF-κB [126]. Epigenetic mechanisms such as methylation of the Nrf2 promoter in CpG islands, H3 histone methylation and H4 histone acetylation, and inhibition of Nrf2 synthesis at the post-transcriptional level due to interference with miRNAs [127] were found to be involved in the regulation of Nrf2 synthesis [118, 122].
The main regulation of Nrf2 activity occurs at post-translational level and can be Keap1-dependent and Keap1-independent (Fig. 3) [20, 118, 122]. Keap1-dependent Nrf2 regulation is the most studied.

Regulation of Nrf2 activity under physiological conditions and overnutrition. Under physiological conditions, Nrf2 is sequestered with Keap1 in cytoplasm that leads to its CUL3-mediated ubiquitination followed by proteasome degradation. In addition, glycogen kinase 3 (GSK3) phosphorylates Nrf2, and this facilitates the recognition of Nrf2 by β-TrCP protein for CUL1-mediated ubiquitination and subsequent proteasome degradation. Overnutrition causes intensification mitochondrial respiration followed by ROS increase. Enhanced ROS levels induce oxidation of SH-groups in Keap1 protein, allowing Nrf2 to dissociate from Keap1 and to translocate to the nucleus, where it forms the complex with Maf protein and activates a number of ARE-genes. High glucose also stimulates insulin signaling resulting in activation of kinase PKB/Akt, which, in turn, phosphorylates and inactivates GSK-3
Keap1 (Kelch-like ECH-associated protein 1) is a main repressor of Nrf2 activity and is a cysteine-rich protein which exists in the form of homodimer. Under physiological conditions, the KELCH domains of the Keap1 homodimer bind to the DLG and ETGE motifs of the Neh2 domain of Nrf2 in the cytosol, where ETGE acts as a hinge with higher affinity and DLG acts as a latch [20, 120]. Keap1 serves a substrate adaptor for cullin-based E3 (Cul3) ubiquitin ligase, which performs ubiquitination of lysine residues in the Neh2 domain of Nrf2 protein [128,129,130]. Ubiquitinated Nrf2 becomes a target for 26S proteasome complex and undergoes proteolytic degradation with a t1/2 of less than 20 min [129, 130]. Therefore, Keap1 protein is an inhibitor of Nrf2 by blocking Nrf2 translocation to the nucleus and promoting its degradation. The rapid turnover of Nrf2 prevents the unnecessary expression of Nrf2 target genes [18, 118].
Under oxidative stress or upon exposure to Nrf2 activators, Nrf2 dissociates from Keap1 due to oxidation of SH-groups of cysteine residues in Keap1 protein. Because of disturbance of the Keap1-Nrf2-Cul3 complex, Nrf2 is stabilized (t1/2 of up to 200 min) and can be translocated to the nucleus. In the nucleus, Nrf2 protein forms a heterodimer with Maf proteins and binds to promoters of genes containing the so-called ARE (Antioxidant Response Element) sequence followed by activating their transcription [120, 121, 131]. In Nrf2, Neh4 and Neh5 domains also act as transactivation domains, but bind to another transcriptional co-activator known as CBP (cAMP response element-binding protein-binding protein) [132]. Neh5 domain has a redox-sensitive nuclear-export signal, which is crucial for the regulation and cellular localization of Nrf2 [133].
Keap1-independent regulation occurs via repression of Nrf2 by β-transducin repeat-containing protein (β-TrCP), which binds to two motifs in the serine-rich Neh6 domain of Nrf2 [20, 134]. β-TrCP is a substrate receptor for the Skp1-Cul1-Rbx1/Roc1 ubiquitin ligase complex that targets Nrf2 for ubiquitination and proteasomal degradation [135]. Notably, the repression of Nrf2 by β-TrCP is increased when serine/threonine kinase glycogen synthase kinase (GSK)-3 is activated. GSK-3 was found to phosphorylate Nrf2 in the Neh6 domain to facilitate the recognition of Nrf2 by β-TrCP and subsequent protein degradation [134, 135]. Inhibition of Nrf2 by GSK-3 is antagonized by protein kinase B (PKB)/Akt, which is activated by phosphoinositide-dependent kinase (PDK)1. In turn, PDK1 is activated by phosphatidylinositol-(3,4,5)-triphosphate (PIP3) produced by phosphatidylinositide 3-kinase (PI3K) by phosphorylation of phosphatidylinositol (4,5)-diphosphate (PIP2) [18]. Elevated insulin signaling, in response to feeding, activates PI3K followed by PKB/Akt activation. In turn, PKB/Akt inhibits GSK-3β through phosphorylation of Ser9 on GSK-3β [136]. Several other non-canonical regulators of Nrf2 activity were also revealed, and they are described in detail elsewhere [18, 118, 122].
5.2 Role of Nrf2 in Redox Homeostasis and Energy Metabolism
The transcriptional factor Nrf2 is considered as the master regulator of the redox cellular state [117, 121, 123, 137]. Nrf2 regulates the expression of over 1000 genes [138], whose products are involved in cell protection against oxidants, electrophiles, and genotoxic compounds, as well in cellular proliferation, intermediary metabolism, and immune responses [18, 118]. Besides regulation of the expression of drug-metabolizing enzymes (e.g., NADPH quinone oxidoreductase (NQO1), glutathione S-transferase, ABC-binding cassette proteins, and P450 subunits), Nrf2 simultaneously controls key components of endogenous antioxidant system. In particular, it controls the expression of enzymes of glutathione synthesis, glutathione peroxidase (GPX)2, which reduces of peroxides with forming oxidized glutathione (GSSG), and glutathione reductase (GSR)1, which reduces GSSG [18, 139]. Thus, Nrf2 ensures maintenance of intracellular pool of reduced glutathione (GSH). Besides that, Nrf2 also activates the expression of cytosolic thioredoxin, thioredoxin reductase, and sulfiredoxin, which all reduce oxidized protein thiols [18, 138]. Many drug-metabolizing enzymes and antioxidant systems require NADPH as a cofactor. The enzymes, which generate NADPH, such as glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, isocitrate dehydrogenase, and malic enzyme, are all regulated by Nrf2 [137, 140]. Ntr2 also regulates expression of proteasome subunits and enzymes of lipid metabolism (e.g., acetyl-Coa oxidase, lipases); in particular, Nrf2 inhibits lipogenesis and supports β-oxidation of fatty acids [18, 141]. Moreover, NRf2 induces synthesis of proteins with anti-inflammatory functions (e.g., heme-oxygenase) and inhibits the expression of pro-inflammatory cytokines [18, 20]. Two transcriptional factors involved in adipogenesis are regulated by Nrf2, CCAAT/enhancer-binding protein β (CEBPβ) and peroxisome proliferator-activated receptor γ (PPARγ) [18]. Thus, in addition of stress-stimulated induction of antioxidant and detoxification genes, Nrf2 contributes to adaptation by modulating cell proliferation and metabolism of proteins, carbohydrates, and lipids and by modulating immune responses.
5.3 Effects of Nrf2 Deficiency and Activation on Obesity
Many studies had been performed to investigate the detailed role of Nrf2 under physiological and pathological conditions using various experimental approaches such as Nrf2 gene deletion, Nrf2 pharmacological activators, and Nrf2 gene overexpression. In addition to liver, intestine, lung, and kidney, where detoxification reactions routinely occur, Nrf2 was found to be abundantly expressed or highly inducible in human and mouse adipocytes. Data suggest that in the adipose tissue Nrf2 functions include not only detoxification and antioxidant defense, but also regulation of metabolism, immune response, and cell proliferation [16, 17, 81, 142].
Obesity and its related metabolic complications had already been associated with the increased oxidative stress; researchers made, therefore, the plausible hypothesis that Nrf2, being a central coordinator of antioxidant gene expression, may participate in the pathophysiological mechanisms of these diseases. Many cellular models and Nrf2 knock-out (Nrf2-KO, Nrf2-null) or Keap1 knock-out mice were used to testing this hypothesis. In support of the hypothesis, Nrf2-KO mice have been shown to have increased susceptibility to chemicals inducing hepatotoxicity [143, 144] and inflammation [145]. However, experimental data regarding role of Nrf2 in adipogenesis and adipocyte function are conflicting. In study of Tanaka et al. [146], wild-type and Nrf2-KO mice fed a high-fat diet for 4 weeks did not differ in weight gain and hepatic TAG levels, but Nrf2-KO mice had higher levels of hepatic free fatty acids and malondialdehyde. Hence, that study did not reveal clearly if Nrf2 signaling is implicated in obesity. The study by Pi et al. [147] showed Nrf2-KO mice gain less weight than the wide type mice when were fed a high-fat diet for a long-term period (3 months). To identify the relationship between Nrf2 and adipogenesis, the authors compared the adipocyte differentiation capacity of mouse fibroblasts isolated from wild-type embryos with those from Nrf2-deficient embryos. In the absence of Nrf2, adipocyte differentiation was suppressed, resulting from the down-regulation of PPAR𝛾 (peroxisome proliferator-activated receptor gamma), a transcription factor with a central role in the expression of the adipogenic program. The authors also found, that Nrf2 deficiency suppressed adipogenesis in 3T3L1 pre-adipocytes and human subcutaneous pre-adipocytes, whereas of Keap1-KO in 3 T3-L1 cells resulted in the activation of Nrf2 and an enhanced adipogenesis [147]. These findings are also supported by other studies demonstrating that Nrf2 deletion leads to the inhibition of adipogenesis and the protection of mice from high-fat diet-induced obesity and its complications. In particular, Nrf2-KO mice had a higher sensitivity to insulin and altered metabolic profile with lower circulating glucose, HDL, and leptin concentrations [148, 149]. Moreover, the constant activation of Nrf2 due to Keap1 knockdown increased markers of metabolic syndrome [17, 150, 151] and Nrf2 deficiency improved glucose tolerance [151] in mice fed a high-fat diet.
Conflicting results have also been reported showing that the loss of Nrf2 is associated with increased differentiation capacity of adipocytes [152, 153]. In the study of Shin et al. [152], Nrf2-deficient mouse embryonic fibroblast cells had markedly accelerated adipogenic differentiation compared with wild-type cell lines. This phenotype was reversed in Keap1-KO cells, which constitutively express higher levels of Nrf2, resulting in a more delayed differentiation [152]. The enhanced activity of Nrf2 was shown to repress hepatic gluconeogenic and lipogenic program to prevent adipogenesis and onset of diabetes mellitus in mice on a high-fat diet [153, 154, 155, 156]. Other studies showed that the deletion of Nrf2 from adipocytes results in severe systemic metabolic dysfunction, including enhanced oxidative stress, inflammation, and insulin resistance, in mice with high-fat diet-induced obesity [81, 142, 157].
In addition to genetic manipulations, the effects of Nrf2 on obesity and metabolic syndrome were explored using its pharmacological activators, e.g., oltipraz, sulforaphane, curcumin, and 1-[2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oyl] imidazole. These compounds have been identified as the specific activators of Nrf2 expression in vitro and in vivo [17]. A number of studies [156, 158,159,160] suggest that the Nrf2 pharmacological activators could protect mice against obesity, improve insulin-resistant phenotype, and suppress oxidative stress, supposedly through the Nrf2 pathway [16, 17].
These discrepancies of Nrf2 effects on obesity may be due to differences in cell lines, animals used, diet composition, feeding period, etc. Nonetheless, it is clear that Nrf2 plays critical roles in adipose tissue. We can suppose that intensity of oxidative stress play an important role in manifestation of effects of Nrf2. At short-term scale, low intensity of oxidative stress can lead to activation of Nrf2 resulting in the induction of adaptive response and the inhibition of proliferation/differentiation of adipocytes. When the intensity of oxidative stress is enhanced, the capacity of protective systems reduces and adipogenic effects of Nrf2 stimulation are observed [5]. Pharmacological activators of Nrf2 can moderately decrease the levels of ROS that ameliorates obesity phenotypes. However, the chronic Nrf2 activation may not be sufficient to cope with increased ROS levels that stimulates adipocyte differentiation, adipose tissue enlargement, and decreases insulin sensitivity.
5.4 Anti-Inflammatory Effects of Nrf2 in Obesity
5.4.1 Nrf2 and Keap1 Deficiency Effects
Activation of Nrf2 transcription factor has been linked to cytoprotection via improving redox sate [18]. In addition, pharmacological activation of Nrf2 inhibits inflammation and impairs degenerative disease providing an interface between redox and anti-inflammatory responses [22]. In consistent with this, Nrf2-KO mice are more susceptible to the pro-inflammatory effects of allergens, LPS, and a high-fat diet [145, 161, 162]. The increased sensitivity of Nrf2-KO mice to inflammation may be connected with a loss of PRARγ protein activity because it is known that Nrf2 positively regulates PRARγ [125] and that PRARγ exerts strong anti-inflammatory effects [163]. Nrf2 deficiency can exacerbate inflammation and promote atherosclerosis and liver injury in a variety of murine models [164,165,166,167]. At the same time, it was reported that Nrf2 deficiency prevents the early onset and development of atherosclerosis and promotes lipid metabolism in ApoE knock-out mice [168] and prevents diet-induced obesity and obesity-associated chronic inflammation in mouse adipose tissue [142, 147, 149, 151, 169].
Controversial functions of Nrf2 in the regulation of inflammation and metabolism have also been observed in numerous Nrf2-activated cellular and mouse models. For example, genetic activation of Nrf2 via Keap1 knockdown [154, 155] or using pharmacological inducers of Nrf2 [21, 158, 159, 160, 170] represses inflammation and prevents the onset and development of diabetes mellitus in mice. However, constant enhanced activity Nrf2 has been reported to aggravate inflammation, worsen insulin resistance and promote hepatic steatosis and lipid accumulation in mice [153, 171]. Several studies suggest that regulatory effects of Nrf2 in inflammation may occur independent of its classic function in redox regulation [165, 167]. One may suppose that the intensity of oxidative stress is a contributing factor to the effects of Nrf2 in obesity. Both Nrf2 deficiency and its constitutive activation was found to inhibit mitochondrial respiration and ROS production in mitochondria [123]. Increased ROS levels can result in the overproduction of cytokines, which induces oxidative stress in target cells. Pro-inflammatory oxidative stress may cause further activation of NF-ĸB and the overproduction of cytokines aggravating inflammation. At the same time, modest activation of Nrf2 by pharmacological compounds or at obesity onset may have beneficial effects, since it allowing maintain stable-state low ROS levels which are sufficient to activation of effective adaptive responses and to control proliferation and differentiation. This can explain the increased sensitivity of both Nrf2-KO mice and Keap1-KO to inflammation.
5.4.2 Protective Effects of Nrf2: Inhibition of Synthesis of Pro-Inflammatory Mediators and Stimulation of Anti-Inflammatory Proteins
The protective mechanisms of Nrf2 in inflammation could be connected with the regulation of expression of proteins participating in immune responses. Heme oxygenase-1 (HO-1) is a well-known target of Nrf2. Nrf2 induces the HO-1 gene expression and it is one of the classic Nrf2 regulated genes, which is widely used in numerous in vitro and in vivo studies. HO-1 is rate-limiting enzyme that catalyzes the degradation of heme into carbon monoxide (CO) and free iron, and biliverdin to bilirubin. Enzymatic degradation of pro-inflammatory free heme as well as the production of anti-inflammatory compounds such as CO and bilirubin play major roles in maintaining the protective effects of HO-1 [20, 172]. Several studies have demonstrated that HO-1 and its metabolites have anti-inflammatory effects in obesity [21, 160, 173,]. Flavonoid quercetin and triterpene celastrol were shown recently to reduce obesity-induced hepatic inflammation by inducing HO-1, which promotes macrophage phenotype switching in adipose tissue from pro-inflammatory M1 to anti-inflammatory M2 macrophages [21, 173].
In addition to induction of HO-1, Nrf2 may inhibit many inflammatory mediators and enzymes [20]. The activation of Nrf2 was reported to prevent LPS-induced transcriptional up-regulation of pro-inflammatory cytokines, including IL-6 and IL-1β [165]. IL-1β and IL-6 production is also increased in Nrf2-KO mice with dextran sulfate-induced colitis [174]. Many plant-derived compounds such as quercetin, celastrol, butein, curcumin, and broccoli extract display anti-inflammatory effects in high-fat diet-induced obesity. These effects were supposed to be mediated by NRf2 and include reduction of M1 macrophage infiltration and release of chemoattractant protein MCP-1, inhibition of synthesis of intercellular adhesion molecule-1(ICAM-1) and pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β), as well as increase of anti-inflammatory cytokines (IL-10) in liver [173], adipose tissue, and adipocyte 3 T3-L1 cell cultures [21, 142].
5.4.3 Crosstalk Between Nrf2 and NF-ĸB Pathways
In the inflammation process, a key player is NF-κB-mediated signaling pathway. NF-κB is normally an inactive cytoplasmic complex, linked to an inhibitory protein, Iκ-B, which masks its nuclear localization signal. External pro-inflammatory stimuli and oxidative stress cause rapid Iκ-B phosphorylation. This causes dissociation of I-κB from NF-κB and subsequent nuclear translocation. In the nucleus, NF-κB induces the transcription of a number of inflammatory chemokines and cytokines. These effects of NF-κB were found in most metabolic tissues, including hepatocytes, adipocytes, neurons, and β cells [1, 22]. Production of pro-inflammatory cytokines intensifies oxidative stress causing further activation of NF-ĸB and the overproduction of cytokines. Activation of the Nrf2 plays an important role in disrupting this cycle [20]. The in vivo studies suggest that Nrf2 negatively regulates the NF-kB signaling pathway [22, 175]. In response to bacterial LPS, Nrf2 knockdown significantly increases NF-ĸB-dependent gene transcription [162]. In addition, increased expression of Nrf2-dependent downstream HO-1 inhibits NF-ĸB activity [176]. Moreover, long-term overnutrition leads to inactivation of Nrf2 and activation of NF-ĸB [21, 177]. Pharmacological inductors of Nrf2 suppress NF-ĸB activation, but promote an increase in Nrf2 activity [21, 177]. We can suppose that low and moderate increase in ROS levels at obesity onset can promote Nrf2 activation followed by induction of protective adaptive response. However, higher increase in oxidative stress intensity leads to activation of NF-ĸB, which triggers chronic inflammatory response inducing further oxidative stress. Because of high intensity oxidative stress, Nrf2 can undergo inactivation [91, 92]. This allows us to suppose that redox-sensitive Nrf2 signaling could be a potential target for anti-obesity interventions at early stages of the disease.
6 Conclusions
Collectively, current data suggest that obesity is associated with the development of both oxidative stress and inflammation, which are considered to be main culprits of obesity-related metabolic complications. However, there is no clear correlation between metabolic disturbances, inflammation, and oxidative stress in obese individuals. In particular, enhanced oxidative stress can have both anti-obesity and obesity-promoting effects, including inflammation progression. Furthermore, an obesity-like phenotype does not always show a reduced life span in invertebrate and mammalian models [72–74, 79,] and there is still a question of whether obesity leads to shortening of life span in humans [178]. Emerging data propose the redox-sensitive regulator of oxidative stress response Nrf2 to be a major molecular player in energy metabolism and a potential target for anti-obesity interventions. However, controversial effects of Nrf2 in obesity progression and prevention were observed, possibly due to using preferentially Nrf2-null mice or Keap-null mice/cells displaying constant Nrf2-activation, which seem to have their own phenotypes distinct from that of wild type. Pharmacological activators Nrf2 seems to be promising for further clarification of the effects of graduated activation Nrf2 in the treatment of obesity and related metabolic disorders. An important gap in current knowledge is the inadequate understanding of the tissue-specific effects of Nrf2, and this is particularly important for metabolic and inflammatory diseases.
Abbreviations
- AGEs:
-
Advanced glycation end products
- BAT:
-
Brown adipose tissue
- BMI:
-
Body mass index
- CEBPβ:
-
CCAAT/enhancer-binding protein β
- ETC :
-
Electron transport chain
- FFAs:
-
Free fatty acids
- FoxO :
-
Forkhead box O family of proteins
- GSK3:
-
Glycogen synthase kinase 3
- HO-1:
-
Heme oxygenase-1
- IL:
-
Interleukin
- Keap1:
-
Kelch-like ECH-associated protein (Nrf2 repressor protein)
- Keap1-KO:
-
Keap1 knock-out
- LDL :
-
Low-density lipoprotein
- LPS :
-
Lipopolysaccharide
- MCP-1:
-
Monocyte chemoattractant protein
- MetS:
-
Metabolic syndrome
- Neh :
-
Nrf2-ECH homology functional domains of nuclear-related factor 2
- NF-κB :
-
Nuclear factor-κB
- NOX :
-
NADPH oxidase
- Nrf2:
-
Nuclear-related factor 2
- Nrf2-KO:
-
Nrf2 knock-out
- PAI-1:
-
Plasminogen activator inhibitor
- PPARγ :
-
Peroxisome proliferator-activated receptor γ
- RCS :
-
Reactive carbonyl species
- ROS :
-
Reactive oxygen species
- TAG:
-
Triacylglycerides
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor alpha
- WAT:
-
White adipose tissue
- β-TrCP:
-
β-transducin repeat-containing protein
References
Catrysse L, Van LG. Inflammation and the metabolic syndrome: the tissue-specific functions of NF-kB. Trends Cell Biol. 2017;27:417–29.
WHO Global Health Observatory Data Repository. Geneva, World Health Organization, http://apps.who.int/gho/data/view.main. Accessed 21 May 2015.
Savini I, Catani MV, Evangelista D, Gasperi V, Avigliano L. Obesity-associated oxidative stress: strategies finalized to improve redox state. Int J Mol Sci. 2013;14:10497–538.
Nikolopoulou A, Kadoglou NPE. Obesity and metabolic syndrome as related to cardiovascular disease. Expert Rev Cardiovasc Ther. 2012;10:933–9.
Bayliak MM, Abrat OB, Storey JM, Storey KB, Lushchak VI. Interplay between diet-induced obesity and oxidative stress: comparison between Drosophila and mammals. Comp Biochem Physiol Part A Mol Integr Physiol. 2019;228:18–28.
Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347–55.
Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González Á, Esquivel-Chirino C, Durante-Montiel I, Sánchez-Rivera G, Valadez-Vega C, Morales-González JA. Inflammation, oxidative stress, and obesity. Int J Mol Sci. 2011;12:3117–32.
Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:1–12.
Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;9:191–200.
Muñoz A, Costa M. Nutritionally mediated oxidative stress and inflammation. Oxidative Med Cell Longev. 2013;2013:610950.
Castro AM, Macedo-de la Concha LE, Pantoja-Meléndez CA. Low-grade inflammation and its relation to obesity and chronic degenerative diseases. Rev Médica del Hosp Gen México. 2017;80:101–5.
Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract. 2013;7:1–12.
Drehmer DL, de Aguiar AM, Brandt AP, Petiz L, Cadena SMSC, Rebelatto CK, Brofman PRS, Filipak Neto F, Dallagiovanna B, Abud APR. Metabolic switches during the first steps of adipogenic stem cells differentiation. Stem Cell Res. 2016;17:413–21.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61.
Bryan S, Baregzay B, Spicer D, Singal PK, Khaper N. Redox-inflammatory synergy in the metabolic syndrome. Can J Physiol Pharmacol. 2013;91:22–30.
Seo H-A, Lee I-K. The role of Nrf2: adipocyte differentiation, obesity, and insulin resistance. Oxidative Med Cell Longev. 2013;2013:184598.
Zhang Z, Zhou S, Jiang X, Wang Y-H, Li F, Wang Y-G, Zheng Y, Cai L. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev Endocr Metab Disord. 2015;16:35–45.
Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218.
Chartoumpekis DV, Kensler TW. New player on an old field; the Keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr Diabetes Rev. 2013;9:137–45.
Ahmed SMU, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol basis Dis. 2017;1863:585–97.
Luo D, Guo Y, Cheng Y, Zhao J, Wang Y, Rong J. Natural product celastrol suppressed macrophage M1 polarization against inflammation in diet-induced obese mice via regulating Nrf2/HO-1, MAP kinase and NF-kappaB pathways. Aging (Albany NY). 2017;9:2069–82.
Lampiasi N, Montana G. An in vitro inflammation model to study the Nrf2 and NF-kappaB crosstalk in presence of ferulic acid as modulator. Immunobiology. 2018;223:349–55.
Saklayen MG. Hypertension and obesity the global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;9:1–8.
Dereń K, Nyankovskyy S, Nyankovska O, Łu E (2018) The prevalence of underweight, overweight and obesity in children and adolescents from Ukraine. 1–7
Heerwagen MJR, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol. 2010;299:R711–22.
Vaiserman A, Koliada A, Lushchak O. Developmental programming of aging trajectory. Ageing Res Rev. 2018;47:105–22.
Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26:968–76.
Kylin E. Studien ueber das Hypertonie-Hyperglyka" mie-Hyperurika" miesyndrom. Zentralblatt fuer Inn Medizin. 1923;44:105–27.
Vague J. La differentiation sexuelle facteur determinant des formes de l’obesite. Presse Med. 1947;30:339–40.
Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–43.
Simon GE, Von Korff M, Saunders K, Miglioretti DL, Crane PK, Van Belle G, Kessler RC. Association between obesity and psychiatric disorders in the US adult population. Arch Gen Psychiatry. 2006;63:824–30.
Ferrannini E. Metabolic syndrome: a solution in search of a problem. J Clin Endocrinol Metab. 2007;92:396–8.
Bruce KD, Hanson MA. The developmental origins, mechanisms, and implications of metabolic syndrome. J Nutr. 2010;140:648–52.
Palaniappan LP, Wong EC, Shin JJ, Fortmann SP, Lauderdale DS. Asian Americans have greater prevalence of metabolic syndrome despite lower body mass index. Int J Obes. 2011;35:393–400.
Van Eenige R, van der Stelt M, Rensen PCN, Kooijman S. Regulation of adipose tissue metabolism by the endocannabinoid system. Trends Endocrinol Metab. 2018;29:326–37.
Gustafson B. Adipose tissue, inflammation and atherosclerosis. J Atheroscler Thromb. 2010;17:332–41.
Baer PC. Adipose-derived stem cells and their potential to differentiate into the epithelial lineage. Stem Cells Dev. 2011;20:1805–16.
Smith U, Kahn BB. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J Intern Med. 2016;280:465–75.
Gastaldelli A, Gaggini M, DeFronzo RA. Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: results from the San Antonio metabolism study. Diabetes. 2017;66:815–22.
Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10.
Morigny P, Houssier M, Mouisel E, Langin D. Adipocyte lipolysis and insulin resistance. Biochimie. 2016;125:259–66.
Kahn BB, Flier JS. Obesity and insulin resistance find the latest version: obesity and insulin resistance. J Clin Invest. 2000;106:473–81.
Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–6.
Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80.
Bastien M, Poirier P, Lemieux I, Després JP. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Dis. 2014;56:369–81.
Compher C, Badellino KO. Obesity and inflammation: lessons from bariatric surgery. J Parenter Enter Nutr. 2008;32:645–7.
Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K. Systematic review of metabolic syndrome biomarkers: a panel for early detection, management, and risk stratification in the west virginian population. Int J Med Sci. 2016;13:25.
Itoh M, Suganami T, Hachiya R, Ogawa Y. Adipose tissue remodeling as homeostatic inflammation. Int J Inflam. 2011;2011:1–8.
Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010;316:129–39.
Lee SW, Jo HH, Kim MR, You YO, Kim JH. Association between metabolic syndrome and serum leptin levels in postmenopausal women. J Obstet Gynaecol. 2012;32:73–7.
Yun JE, Kimm H, Jo J, Jee SH. Serum leptin is associated with metabolic syndrome in obese and nonobese Korean populations. Metabolism. 2010;59:424–9.
Laclaustra M, Corella D, Ordovas JM. Metabolic syndrome pathophysiology: the role of adipose tissue. Nutr Metab Cardiovasc Dis. 2007;17:125–39.
Kaser S, Tatarczyk T, Stadlmayr A, Ciardi C, Ress C, Tschoner A, Sandhofer A, Paulweber B, Ebenbichler CF, Patsch JR. Effect of obesity and insulin sensitivity on adiponectin isoform distribution. Eur J Clin Investig. 2008;38:827–34.
Yamauchi T, Iwabu M, Okada-Iwabu M, Kadowaki T. Adiponectin receptors: a review of their structure, function and how they work. Best Pract Res Clin Endocrinol Metab. 2014;28:15–23.
Tanaka T, Narazaki M, Kishimoto T. IL-6 in immunity, inflammation, disease. Cold Spring Harb Perspect Biol. 2014;6:16295–6.
Pal M, Febbraio MA, Whitham M. From cytokine to myokine: the emerging role of interleukin-6 in metabolic regulation. Immunol Cell Biol. 2014;92:331–9.
Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obes. 2004;28:1357–64.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa KI, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505.
Panee J. Monocyte chemoattractant protein 1 (MCP-1) in obesity and diabetes. Cytokine. 2012;60:1–12.
Gotoh K, Inoue M, Masaki T, et al. A novel anti-inflammatory role for spleen-derived interleukin-10 in obesity-induced inflammation in white adipose tissue and liver. Diabetes. 2012;61:1994–2003.
Gormez S, Demirkan A, Atalar F, Caynak B, Erdim R, Sozer V, Gunay D, Akpinar B, Ozbek U, Buyukdevrim AS. Adipose tissue gene expression of adiponectin, tumor necrosis factor-α and leptin in metabolic syndrome patients with coronary artery disease. Intern Med. 2011;50:805–10.
Kosteli A, Zechner R, Ferrante AW Jr, Sugaru E, Haemmerle G, Martin JF, Lei J. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120:3466–79.
Castoldi A, De Souza CN, Saraiva Câmara NO, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol. 2016;6:1–11.
Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–70.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86.
Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014;19:512–26.
Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2014;177:7303–11.
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–85.
Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol. 2008;3:545–56.
Carrillo JLM, Campo, JOM, Coronado OGC, Gutiérrez PTV, Cordero FC, Juárez JV (2018) Adipose tissue and inflammation IntechOpen. https://doi.org/10.5772/intechopen.74227
Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, Di Rosa G, Gitto E, Arrigo T. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2015;16:378–400.
Alcalá M, Calderon-Dominguez M, Bustos E, Ramos P, Casals N, Serra D, Viana M, Herrero L. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci Rep. 2017;7:1–12.
Amirkhizi F, Siassi F, Djalali M, Hamedi S. Impaired enzymatic antioxidant defense in erythrocytes of women with general and abdominal obesity. Obes Res Clin Pract. 2014;8:e26–34.
Brown LA, Kerr CJ, Whiting P, Finer N, McEneny J, Ashton T. Oxidant stress in healthy normal-weight, overweight, and obese individuals. Obesity (Silver Spring). 2009;17:460–6.
D’Archivio M, Annuzzi G, Vari R, Filesi C, Giacco R, Scazzocchio B, Santangelo C, Giovannini C, Rivellese AA, Masella R. Predominant role of obesity/insulin resistance in oxidative stress development. Eur J Clin Investig. 2012;42:70–8.
Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Ichi MK, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57:1071–7.
Masania J, Malczewska-Malec M, Razny U, Goralska J, Zdzienicka A, Kiec-Wilk B, Gruca A, Stancel-Mozwillo J, Dembinska-Kiec A, Rabbani N, Thornalley PJ. Dicarbonyl stress in clinical obesity. Glycoconj J. 2016;33:581–9.
Semchyshyn H. Is part of the fructose effects on health related to increased AGE formation? In: Uribarry J, editor. Dietary AGEs and their role in health and disease. Boca Raton: CRC Press; 2017. p. 119–28.
Tinahones FJ, Murri-Pierri M, Garrido-Sánchez L, García-Almeida JM, García-Serrano S, García-Arnés J, García-Fuentes E. Oxidative stress in severely obese persons is greater in those with insulin resistance. Obesity. 2009;17:240–6.
Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, Szalai G, Sonntag WE, Ungvari Z, Csiszar A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–26.
Tarantini S, Valcarcel-Ares MN, Yabluchanskiy A, Tucsek Z, Hertelendy P, Kiss T, Gautam T, Zhang XA, Sonntag WE, de Cabo R, Farkas E, Elliott MH, Kinter MT, Deak F, Ungvari Z, Csiszar A. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression, and cognitive decline in mice, mimicking the aging phenotype. J Gerontol A Biol Sci Med Sci. 2018;73:853–63.
Rani V, Deep G, Singh RK, Palle K, Yadav UCS. Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sci. 2016;148:183–93.
Schneider KS, Chan JY. Emerging role of Nrf2 in adipocytes and adipose biology. Adv Nutr. 2013;4:62–6.
Pihl E, Zilmer K, Kullisaar T, Kairane C, Magi A, Zilmer M. Atherogenic inflammatory and oxidative stress markers in relation to overweight values in male former athletes. Int J Obes. 2006;30:141–6.
Chrysohoou C, Panagiotakos DB, Pitsavos C, Skoumas I, Papademetriou L, Economou M, Stefanadis C. The implication of obesity on total antioxidant capacity in apparently healthy men and women: the ATTICA study. Nutr Metab Cardiovasc Dis. 2007;17:590–7.
Rovenko BM, Kubrak OI, Gospodaryov DV, Perkhulyn NV, Yurkevych IS, Sanz A, Lushchak OV, Lushchak VI. High sucrose consumption promotes obesity whereas its low consumption induces oxidative stress in Drosophila melanogaster. J Insect Physiol. 2015;79:42–54.
Rovenko BM, Kubrak OI, Gospodaryov DV, Yurkevych IS, Sanz A, Lushchak OV, Lushchak VI. Restriction of glucose and fructose causes mild oxidative stress independently of mitochondrial activity and reactive oxygen species in Drosophila melanogaster. Comp Biochem Physiol Part A Mol Integr Physiol. 2015;187:27–39.
Rovenko BM, Perkhulyn NV, Gospodaryov DV, Sanz A, Lushchak OV, Lushchak VI. High consumption of fructose rather than glucose promotes a diet-induced obese phenotype in Drosophila melanogaster. Comp Biochem Physiol Part A Mol Integr Physiol. 2015;180:75–85.
Bray GA, Champagne CM. Beyond energy balance: there is more to obesity than kilocalories. J Am Diet Assoc. 2005;105:17–23.
Garaschuk O, Semchyshyn HM, Lushchak VI. Healthy brain aging: interplay between reactive species, inflammation and energy supply. Ageing Res Rev. 2018;43:26–45.
Lushchak VI. Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp Biochem Physiol Part C Toxicol Pharmacol. 2011;153:175–90.
Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact. 2014;224:164–75.
Semchyshyn HM. Reactive carbonyl species in vivo: generation and dual biological effects. ScientificWorldJournal. 2014;2014:417842.
Vankoningsloo S, Piens M, Lecocq C, Gilson A, De Pauw A, Renard P, Demazy C, Houbion A, Raes M, Arnould T. Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. J Lipid Res. 2005;46:1133–49.
Den Hartigh LJ, Omer M, Goodspeed L, Wang S, Wietecha T, O’Brien KD, Han CY. Adipocyte-specific deficiency of NADPH-oxidase 4 delays the onset of insulin resistance and attenuates adipose tissue inflammation in obesity. Arterioscler Thromb Vasc Biol. 2017;37:466–75.
Han CY, Umemoto T, Omer M, Den Hartigh LJ, Chiba T, Leboeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED, Chait A. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J Biol Chem. 2012;287:10379–93.
Cleary MP, Zisk JF. Anti-obesity effect of two different levels of dehydroepiandrosterone in lean and obese middle-aged female Zucker rats. Int J Obes. 1986;10:193–204.
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45.
Moraru A, Wiederstein J, Pfaff D, Fleming T, Miller AK, Nawroth P, Teleman AA. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metab. 2018;27:926–934.e8.
Semchyshyn HM. Fructation in vivo: detrimental and protective effects of fructose. Biomed Res Int. 2013;2013:343914.
Semchyshyn HM, Lozinska LM, Miedzobrodzki J, Lushchak VI. Fructose and glucose differentially affect aging and carbonyl/oxidative stress parameters in Saccharomyces cerevisiae cells. Carbohydr Res. 2011;346:933–8.
Semchyshyn HM, Miedzobrodzki J, Bayliak MM, Lozinska LM, Homza BV. Fructose compared with glucose is more a potent glycoxidation agent in vitro, but not under carbohydrate-induced stress in vivo: potential role of antioxidant and antiglycation enzymes. Carbohydr Res. 2014;384:61–9.
Drougard A, Fournel A, Valet P, Knauf C. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front Neurosci. 2015;9:56–61.
Lee H, Lee YJ, Choi H, Ko EH, Kim J-W. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J Biol Chem. 2009;284:10601–9.
Kanda Y, Hinata T, Kang SW, Watanabe Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. 2011;89:250–8.
Turker I, Zhang Y, Zhang Y, Rehman J. Oxidative stress as a regulator of adipogenesis. FASEB J. 2007;21:A1053.
Schröder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29:239–45.
Li Y, Mouche S, Sajic T, Veyrat-Durebex C, Supale R, Pierroz D, Ferrari S, Negro F, Hasler U, Feraille E, Moll S, Meda P, Deffert C, Montet X, Krause K-H, Szanto I. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes. 2012;36:1503–13.
De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am J Pathol. 2009;175:927–39.
Stolarczyk E. Adipose tissue inflammation in obesity: a metabolic or immune response? Curr Opin Pharmacol. 2017;37:35–40.
Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45.
Wolowczuk I, Verwaerde C, Viltart O, Delanoye A, Delacre M, Pot B, Grangette C. Feeding our immune system: impact on metabolism. Clin Dev Immunol. 2008;2008:639803.
Monteiro R, Azevedo I (2010) Chronic inflammation in obesity and the metabolic syndrome. Mediat Inflamm. pii:289645 https://doi.org/10.1155/2010/289645.
Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21:1443–55.
Klotz LO, Sánchez-Ramos C, Prieto-Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72.
Nakae J, Cao Y, Oki M, Orba Y, Sawa H, Kiyonari H, Iskandar K, Suga K, Lombes M, Hayashi Y. Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes. 2008;57:563–76.
Pitoniak A, Bohmann D. Mechanisms and functions of Nrf2 signaling in Drosophila. Free Radic Biol Med. 2015;88:302–13.
Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol Res. 2018;134:92–9.
Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88:101–7.
Abed DA, Goldstein M, Albanyan H, Jin H, Hu L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B. 2015;5:285–99.
Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–46.
Huang Y, Li W, Su Z, Kong A-NT. The complexity of the Nrf2 pathway: beyond the antioxidant response. J Nutr Biochem. 2015;26:1401–13.
Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179–88.
Kwak M-K, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–92.
Cho H-Y, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, Kleeberger SR. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. 2010;182:170–82.
Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood. 2012;120:5188–98.
Kurinna S, Werner S. NRF2 and microRNAs: new but awaited relations. Biochem Soc Trans. 2015;43:595–601.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86.
Kobayashi A, Kang M-I, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9.
Villeneuve NF, Lau A, Zhang DD. Regulation of the Nrf2–Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010;13:1699–712.
Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–5.
Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–68.
Li W, Yu S-W, Kong A-NT. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–63.
Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–81.
Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–33.
Pearl LH, Barford D. Regulation of protein kinases in insulin, growth factor and Wnt signalling. Curr Opin Struct Biol. 2002;12:761–7.
Lee J-M, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–38.
Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38:5718–34.
Walsh J, Jenkins RE, Wong M, Olayanju A, Powell H, Copple I, O’Neill PM, Goldring CEP, Kitteringham NR, Park BK. Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: biochemical, pharmacological and toxicological implications. J Proteome. 2014;108:171–87.
Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123:590–600.
Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, Goldring CEP, Powell H, Sanderson C, Williams S, Higgins L, Yamamoto M, Hayes J, Park BK. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J Proteome. 2010;73:1612–31.
Xue P, Hou Y, Chen Y, Yang B, Fu J, Zheng H, Yarborough K, Woods CG, Liu D, Yamamoto M, Zhang Q, Andersen ME, Pi J. Adipose deficiency of Nrf2 in Ob/Ob mice results in severe metabolic syndrome. Diabetes. 2013;62:845–54.
Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, Harada T, Yamamoto M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–77.
Randle LE, Goldring CEP, Benson CA, Metcalfe PN, Kitteringham NR, Park BK, Williams DP. Investigation of the effect of a panel of model hepatotoxins on the Nrf2-Keap1 defence response pathway in CD-1 mice. Toxicology. 2008;243:249–60.
Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59.
Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, Klaassen CD. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther. 2008;325:655–64.
Pi J, Leung L, Xue P, Wang W, Hou Y, Liu D, Yehuda-Shnaidman E, Lee C, Lau J, Kurtz TW, Chan JY. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem. 2010;285:9292–300.
Kim B-R, Lee GY, Yu H, Maeng HJ, Oh TJ, Kim KM, Moon JH, Lim S, Jang HC, Choi SH. Suppression of Nrf2 attenuates adipogenesis and decreases FGF21 expression through PPAR gamma in 3T3-L1 cells. Biochem Biophys Res Commun. 2018;497:1149–53.
Xu J, Donepudi AC, More VR, Kulkarni SR, Li L, Guo L, Yan B, Chatterjee T, Weintraub N, Slitt AL. Deficiency in Nrf2 transcription factor decreases adipose tissue mass and hepatic lipid accumulation in leptin-deficient mice. Obesity (Silver Spring). 2015;23:335–44.
More VR, Xu J, Shimpi PC, Belgrave C, Luyendyk JP, Yamamoto M, Slitt AL. Keap1 knockdown increases markers of metabolic syndrome after long-term high fat diet feeding. Free Radic Biol Med. 2013;61:85–94.
Zhang L, Dasuri K, Fernandez-Kim S-O, Bruce-Keller AJ, Keller JN. Adipose-specific ablation of Nrf2 transiently delayed high-fat diet-induced obesity by altering glucose, lipid and energy metabolism of male mice. Am J Transl Res. 2016;8:5309–19.
Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, Kensler TW. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol. 2007;27:7188–97.
Xu J, Kulkarni SR, Donepudi AC, More VR, Slitt AL. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes. 2012;61:3208–18.
Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW, Yamamoto M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol. 2013;33:2996–3010.
Slocum SL, Skoko JJ, Wakabayashi N, Aja S, Yamamoto M, Kensler TW, Chartoumpekis DV. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch Biochem Biophys. 2016;591:57–65.
Sampath C, Rashid MR, Sang S, Ahmedna M. Specific bioactive compounds in ginger and apple alleviate hyperglycemia in mice with high fat diet-induced obesity via Nrf2 mediated pathway. Food Chem. 2017;226:79–88.
Chartoumpekis DV, Palliyaguru DL, Wakabayashi N, Fazzari M, Khoo NKH, Schopfer FJ, Sipula I, Yagishita Y, Michalopoulos GK, O’Doherty RM, Kensler TW. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am J Physiol Endocrinol Metab. 2018;315:E180–95.
He H-J, Wang G-Y, Gao Y, Ling W-H, Yu Z-W, Jin T-R. Curcumin attenuates Nrf2 signaling defect, oxidative stress in muscle and glucose intolerance in high fat diet-fed mice. World J Diabetes. 2012;3:94–104.
Yu Z, Shao W, Chiang Y, Foltz W, Zhang Z, Ling W, Fantus IG, Jin T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia. 2011;54:922–34.
Tuzcu Z, Orhan C, Sahin N, Juturu V, Sahin K. Cinnamon polyphenol extract inhibits hyperlipidemia and inflammation by modulation of transcription factors in high-fat diet-fed rats. Oxidative Med Cell Longev. 2017;2017:1583098.
Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon RJ, Dinkova-Kostova AT, Dillon JF, Hayes JD, Ashford MLJ. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol. 2014;34:3305–20.
Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–95.
Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–63.
Yuan X, Huang H, Huang Y, Wang J, Yan J, Ding L, Zhang C, Zhang L. Nuclear factor E2-related factor 2 knockdown enhances glucose uptake and alters glucose metabolism in AML12 hepatocytes. Exp Biol Med (Maywood). 2017;242:930–8.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624.
Collins AR, Gupte AA, Ji R, Ramirez MR, Minze LJ, Liu JZ, Arredondo M, Ren Y, Deng T, Wang J, Lyon CJ, Hsueh WA. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler Thromb Vasc Biol. 2012;32:2839–46.
Ding L, Yuan X, Yan J, Huang Y, Xu M, Yang Z, Yang N, Wang M, Zhang C, Zhang L. Nrf2 exerts mixed inflammation and glucose metabolism regulatory effects on murine RAW264.7 macrophages. Int Immunopharmacol. 2019;71:198–204.
Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 2008;3:e3791.
Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Sykiotis GP, Habeos IG. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes. 2011;60:2465–73.
Xu L, Nagata N, Ota T. Glucoraphanin: a broccoli sprout extract that ameliorates obesity-induced inflammation and insulin resistance. Adipocytes. 2018;7:218–25.
Gerstgrasser A, Melhem H, Leonardi I, Atrott K, Schafer M, Werner S, Rogler G, Frey-Wagner I. Cell-specific activation of the Nrf2 antioxidant pathway increases mucosal inflammation in acute but not in chronic colitis. J Crohns Colitis. 2017;11:485–99.
Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73:3221–47.
Kim C-S, Choi H-S, Joe Y, Chung HT, Yu R. Induction of heme oxygenase-1 with dietary quercetin reduces obesity-induced hepatic inflammation through macrophage phenotype switching. Nutr Res Pract. 2016;10:623–8.
Osburn WO, Karim B, Dolan PM, Liu G, Yamamoto M, Huso DL, Kensler TW. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer. 2007;121:1883–91.
Jiang T, Tian F, Zheng H, Whitman SA, Lin Y, Zhang Z, Zhang N, Zhang DD. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-kappaB-mediated inflammatory response. Kidney Int. 2014;85:333–43.
Bellezza I, Tucci A, Galli F, Grottelli S, Mierla AL, Pilolli F, Minelli A. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. J Nutr Biochem. 2012;23:1583–91.
Illesca P, Valenzuela R, Espinosa A, Echeverria F, Soto-Alarcon S, Ortiz M, Videla LA. Hydroxytyrosol supplementation ameliorates the metabolic disturbances in white adipose tissue from mice fed a high-fat diet through recovery of transcription factors Nrf2, SREBP-1c, PPAR-gamma and NF-kappaB. Biomed Pharmacother. 2019;109:2472–81.
Walls HL, Backholer K, Proietto J, McNeil JJ. Obesity and trends in life expectancy. J Obes. 2012;2012:107989.
Acknowledgments
The work was partially supported by the Ministry of Education and Science of Ukraine (#0118 U00347).
Conflicts of Interest: The authors declare that there are no conflicts of interests regarding the publication of this article.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bayliak, M.M., Abrat, O.B. (2020). Role of Nrf2 in Oxidative and Inflammatory Processes in Obesity and Metabolic Diseases. In: Deng, H. (eds) Nrf2 and its Modulation in Inflammation. Progress in Inflammation Research, vol 85. Springer, Cham. https://doi.org/10.1007/978-3-030-44599-7_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-44599-7_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-44597-3
Online ISBN: 978-3-030-44599-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)