
Abstract
Blood cells in the peripheral circulation originate from hematopoietic stem cells in the bone marrow. The majority of hematopoietic stem cells distribute in close proximity to the endosteal surfaces, form specific microenvironment, called niche, and achieve self-renewal and differentiation, communicating with cells involved in bone metabolism. PTH plays important roles not only in differentiation of osteoblastic lineage but also in blood cell production in the bone marrow. Persistently high circulating levels of PTH are known to cause anemia, immunosuppression, and thrombocytopenia mainly through myelofibrosis that limits the site of hematopoiesis. On the other hand, recent investigations have also revealed that PTH accelerates proliferation and mobilization of hematopoietic cells, which may contribute to the improvement of the outcome in bone marrow transplantation. These beneficial effects of PTH require support of marrow stromal cells and bone cells including osteoblasts. Thus, PTH is a key mediator for crosstalk among bone cells, hematopoietic stem cells, and marrow stromal cells and plays a crucial role in normal hematopoiesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Bone consists of cells and extracellular matrices composed of an inorganic component such as calcium phosphate and an organic substance such as collagen. Bone matrix formation is mainly regulated by coordinated action of three kinds of cells, osteoblasts, osteoclasts, and osteocytes. Osteoblasts are derived from mesenchymal stem cells, regulate osteogenesis, and ultimately transform into osteocytes. Osteoclasts are formed by differentiation and fusion of bone marrow-derived monocyte/macrophage progenitor cells.
Bone marrow (BM) is composed of hematopoietic stem cells (HSCs) and marrow stromal cells. HSCs are defined as cells which possess the ability of self-renewal and of differentiation into all blood cell lineages including leukocytes, erythrocytes, and platelets (Table 1) [1]. A specific microenvironment consisting of HSCs and surrounding supportive tissues is called HSCs niche, in which crosstalk among HSCs and other numerous cells is involved in normal hematopoiesis [2]. Marrow stromal cells, also called mesenchymal stem cells, are nonhematopoietic stem cells, and can differentiate into osteoblasts, chondrocytes, adipocytes, or skeletal muscle cells.
It has been reported that PTH not only regulates bone metabolism but also affects blood cell production and function. Hyperparathyroidism (HPT) suppresses normal hematopoiesis via myelofibrosis, while PTH has been shown to have a potential to improve survival of HSCs by acting directly or indirectly on these cells. Furthermore, recent studies focused on the interaction between osteoblasts and HSCs, revealing that HSCs niche lines selectively on the surface of the endosteum similarly to osteoblasts [3] and that PTH influences the interaction between HSCs and osteoblastic lineage. In this chapter, we summarize the pathological effects of PTH on the proliferation and viability of erythrocytes, leukocytes, and platelets, and also outline recent reports on the physiological roles of PTH in hematopoiesis.
PTH and Erythropoiesis
Immature erythroid progenitor cells in the BM are stimulated by erythropoietin (EPO), differentiate into mature erythrocytes, and are released into peripheral circulation. Normal mature erythrocytes have a lifespan of approximately 120 days [4], and old erythrocytes are destroyed in the liver and spleen. Decreased production or increased breakdown of normal erythrocytes could result in decreased hemoglobin concentrations and insufficient oxygen supply. This condition is called anemia.
End stage renal disease (ESRD) is associated with a high incidence of anemia and secondary HPT (SHPT). Renal anemia is mainly caused by an impaired production of EPO in the kidney for maintaining normal hemoglobin levels. Besides, many other factors could be involved in the pathogenesis of anemia, including iron deficiency, inflammation, bleeding, and malnutrition. In addition, several studies have suggested that SHPT may also play a role in the pathogenesis renal anemia.
Pathogenesis of Anemia Associated with Hyperparathyroidism
Myelofibrosis
Myelofibrosis is characterized by extensive fibrosis of the BM, resulting in anemia, hepatosplenomegaly, and extramedullary hematopoiesis [5]. Primary myelofibrosis is observed in patients with myeloproliferative diseases due to gene mutations in HSCs, whereas secondary myelofibrosis develops in patients suffering from solid tumors. SHPT is also known as a possible cause of such secondary myelofibrosis [6]. Recent studies investigated the pathogenetic mechanisms of myelofibrosis due to persistently high serum levels of PTH.
Lotinun et al. performed a microarray analysis on 5531 genes using mRNA extracted from the bone of rats receiving continuous administration of 1–34 PTH and found an increased expression of platelet-derived growth factor-A (PDGF-A) [7]. The investigators also confirmed that triazolopyrimidine (Trapidil), a PDGF-A inhibitor, suppresses myelofibrosis. Immunohistochemical staining revealed that PDGF-A is localized to mast cells, which suggests the interaction between myelofibrosis and mast cells. The follow-up study by Lowry et al., the same research group, showed that both the receptor tyrosine kinase inhibitor Gleevec and the phosphoinositide 3 (IP3)-kinase inhibitor wortmannin attenuated myelofiborosis in rats induced by continuous 1–34 PTH infusion. These results indicate that PDGF-A accelerates PTH-induced myelofibrosis through IP3 signaling pathways [8]. This study also demonstrated that mast cells redistribute from BM to the bone surface after PTH infusion, supporting the possibility that mature mast cells play crucial roles in the pathogenesis of myelofibrosis.
In line with these experimental studies, several clinical studies suggested that persistently high serum PTH levels are associated with myelofibrosis. Zingraff et al. performed bone biopsy before and after parathyroidectomy (PTx) in patients with SHPT and compared the extent of myelofibrosis [9]. They showed that hematocrit levels increased in patients who experienced improved myelofibrosis, suggesting that the severity of SHPT is related to the extent of myelofibrosis, that myelofibrosis is a reversible disorder, and that improvement of fibrosis might improve anemia. Rao et al. performed bone biopsy in hemodialysis patients receiving recombinant human EPO (rhEPO) and revealed that patients with EPO hyporesponsiveness had higher intact PTH levels, higher osteoclast number and eroded surface, and greater extent of myelofibrosis compared to good responders to rhEPO [10]. Taken together, these data suggest that SHPT may cause myelofibrosis and impair normal erythropoiesis, resulting in worsening of anemia and EPO hyporesponsiveness.
Inhibited Production of Erythroid Progenitor
Burst-forming unit-erythroid (BFU-E) and colony-forming unit-erythroid (CFU-E) are erythroid progenitors and are shown to express PTH/PTH-related protein receptor (PPR). This fact supports the possibility that PTH directly acts on these cells. Meytes et al. cultured BFU-E collected from human peripheral blood and showed that bovine 1–84 PTH inhibits BFU-E colony formation and that inactivation of PTH abolishes this effect [11]. However, these results were not reproduced when they used 1–34 bovine PTH, and the reasons for the discrepant results have not been elucidated so far. Taniguchi et al. found that the density of BFU-E and CFU-E in BM of dialysis patients were lower than healthy volunteers [12]. The researchers also obtained CFU-E from BM of healthy volunteers and showed that addition of uremic sera to culture medium inhibited CFU-E colony formation. This effect was more pronounced with higher PTH levels in the uremic sera. They further showed that treatment with 1–34 human PTH suppressed CFU-E colony formation in a dose-dependent manner. These results indicate that PTH suppresses proliferation of early erythroid progenitor CFU-E in vitro.
However, several subsequent studies have produced conflicting results. Dunn et al. showed that addition of 1–84 bovine PTH at concentrations 10 to 100 times normal levels failed to inhibit erythropoiesis or synthesis of heme in cultured fetal mouse liver cells [13]. Komatsuda et al. examined the effect of 1–34 human PTH (maximum concentrations of 300 ng/mL) or 1–84 human PTH (maximum concentrations of 5000 pg/mL) on CFU-E or BFU-E collected from healthy volunteers and found that neither erythropoiesis nor granulomonopoiesis were suppressed by human PTH [14]. The findings of these two studies do not support the hypothesis that PTH inhibits erythropoiesis. Thus, taken together, it remains unclear whether PTH acts on erythroid progenitor and exert inhibitory effects on erythropoiesis.
Inhibition of Erythropoietin Synthesis
EPO is mainly secreted from the kidney and plays crucial roles in differentiation of HSCs into erythroid lineage [15, 16]. EPO production is strongly related to hypoxia inducible factor (HIF) [17]. Under normoxic conditions, hydroxylation by prolyl hydroxylases (PHD) and ubiquitination by E3 ubiquitin ligase, such as von Hippel-Lindau protein, triggers proteasome-dependent degradation of HIF and thereby suppresses transcriptional activity of HIF. By contrast, under hypoxic conditions, suppression of PHD activity inhibits HIF degradation and enhances translocation of HIF into nucleus, wherein HIF forms dimers and initiates gene expression by binding at consensus sequence, hypoxia responsive element (HRE). HIF is known to regulate the expression of more than 800 genes, including EPO, vascular endothelial growth factor (VEFG), and platelet-derived growth factor beta polypeptide (PDFGB) [18]. Binding with HRE in the FOXD-1 expressing stroma-derived cells including peritubular interstitial fibroblast-like cells, renin producing cells, or vascular smooth muscle cells leads EPO secretion [19, 20]. These hypoxic reactions are impaired in CKD patients, leading to insufficient EPO secretion and renal anemia. Administration of erythropoiesis stimulating agents (ESAs) is required to compensate for relative or absolute endogenous EPO deficiency, but hemoglobin levels do not elevate sufficiently in some dialysis patients, which is called ESA hyporesponsiveness. This condition is caused by multiple factors, including iron deficiency, inflammation, malnutrition, and SHPT.
Several studies assessed the effect of PTx on endogenous EPO concentrations. One observational study of patients with primary HPT (PHPT) demonstrated that PTx did not alter the levels of endogenous EPO [21]. However, several studies of ESRD with SHPT demonstrated significant increases in circulating EPO levels after PTx [21, 22]. Thus, elevated PTH levels might contribute to decreased endogenous EPO production in ESRD patients, although it is unclear whether this is a direct effect on EPO-producing cells. Nonetheless, in a cross-sectional study of dialysis patients by Borawski et al., serum intact PTH levels were not associated with hemoglobin or circulating EPO levels [23]. Similarly, McGonigle et al. reported that there was no association between serum intact PTH levels and hematocrit in ESRD patients receiving hemodialysis or continuous ambulatory peritoneal dialysis and that PTx did not affect serum EPO concentrations or hematocrit [24]. Collectively, it remains unclear whether PTH causes decreased EPO production or ESA hyporesponsiveness in ESRD patients.
Interestingly, a recent experimental study by Wong et al. demonstrated that in UMR106.01 mature osteoblasts, human 1–34 PTH reduced the expression of HIF-1α levels and HIF signaling under normoxic conditions [25]. This result raises the hypothesis that PTH may also inhibit HIF signaling in EPO-producing cells in the kidney and thereby suppress EPO production, and further research would be needed to test this possibility.
Shortened Erythrocyte Survival
The lifetime of human erythrocytes is approximately 120 days [4]. ATP plays crucial roles in maintenance of erythrocyte morphology. Hyperosmolarity, oxidative stress, energy depletion, hyperthermia, and a wide variety of xenobiotics and endogenous substances can trigger influx of calcium into erythrocytes and thereby decrease intracellular ATP, which causes eryptosis [26,27,28,29]. In uremic patients, PTH has been considered to shorten the lifetime of erythrocytes and several studies have examined this possibility.
Bogin et al. cultured erythrocytes collected from healthy volunteers and showed that both 1–84 bovine PTH and 1–34 bovine PTH accelerated influx of calcium into erythrocytes and increased osmotic fragility of erythrocytes [30]. In another study, Akmal et al. harvested 51Cr-labelled erythrocytes from 5/6 nephrectomized dogs, those with 5/6 nephrectomy and thyroparathyroidectomy, and control animals, and compared lifetime of erythrocytes among three groups in vitro [31]. While the lifetime of erythrocytes from 5/6 nephrectomized dogs was significantly shorter than control group, the dogs undergoing 5/6 nephrectomy and thyroparathyroidectomy showed comparable lifetime of erythrocytes to control animals, thus suggesting that PTH shortens the lifetime of erythrocytes in uremic animals.
Treatment for Hyperparathyroidism and Anemia
Treatment options for SHPT include vitamin D receptor activator (VDRA), calcimimetics, percutaneous ethanol injection therapy, and PTx, whereas PTx is the standard and definitive treatment of PHPT. The major purpose of lowering PTH levels in PHPT and SHPT is amelioration of disturbed bone and mineral metabolism, but several studies have suggested that PTH-lowering therapy may also improve anemia.
Parathyroidectomy
PTx is the definitive therapy for PHPT and SHPT. Several previous studies have shown that hemoglobin levels decrease transiently after PTx but then increase in the long term. A seminal early study by Zingraff et al. showed that mean hematocrit levels increased from 24.4% to 30.9% after PTx in dialysis patients with SHPT. Furthermore, they performed bone biopsy 6 to 9 months after PTx and revealed that the rise in hematocrit was more prominent among patients showing improvement of myelofibrosis [9]. Another group also reported that improvement of anemia after PTx for SHPT was more marked among patient with more severe myelofibrosis and lower hematocrit levels at baseline [32]. These data suggest that myelofibrosis associated with severe HPT contributes to anemia and that PTx could partially reverse the myelofibrosis and resultant anemia.
However, myelofibrosis is not always associated with renal anemia in ESRD patients. Mandolfo et al. performed bone biopsy in patients with severe SHPT and found that there was no association between the extent of myelofibrosis and severity of anemia [33]. Nonetheless, they found that patients receiving ESA administrations showed significant increases in hematocrit levels and reductions in the dose of ESA after PTx, indicating improved ESA responsiveness. Similarly, Coen et al. showed that hemoglobin levels increased after PTx in 45 dialysis patients with SHPT, and this increase was consistent across different surgical procedures (i.e., subtotal PTx, and total PTx with or without autotransplantation) [34]. This study included 16 patients receiving ESA and the researchers found that hemoglobin levels increased even though the mean ESA doses were decreased. Yasunaga et al. also demonstrated a rise in hemoglobin levels 1 year after PTx in patients with SHPT. Interestingly, they also found an increase in endogenous EPO levels from 22.6 ± 6.3 mU/mL to 143.8 ± 170.1 mU/mL and serum albumin levels from 3.9 ± 0.3 g/dL to 4.2 ± 0.4 g/dL after PTx, which may explain the improved anemia after surgery [35]. Finally, Trunzo et al. demonstrated that in dialysis patients with SHPT, PTx led to elevations in hemoglobin levels and reductions in the dose of rhEPO [36]. Taken together, PTx for SHPT may improve renal anemia, and this effect may be mediated not only by amelioration of myelofibrosis but also by increased renal or extra-renal production of EPO and improved nutritional status.
Several studies have also examined the effect of PTx on anemia in PHPT. Bhadada et al. reported that approximately half of PHPT patients were complicated with anemia and 75% of these anemic patients showed myelofibrosis on bone biopsy. The investigators observed an improvement of anemia after PTx among those with preexisting myelofibrosis [37]. Thus, PTx may improve anemia in patients with severe HPT, regardless of either primary or secondary.
Calcimimetics
PTH secretion is mainly regulated by the calcium-sensing receptor (CaSR) expressed in chief cells of the parathyroid gland, which senses the change of ionized calcium concentration. Calcimimetics bind to the CaSR and allosterically inhibit the secretion of PTH. Cinacalcet was clinically applied for PHPT and SHPT, whereas newly developed etelcalcetide and evocalcet can be used exclusively for uremic patients with SHPT at present [38,39,40]. Cinacalcet has been used clinically for a long time and has been suggested to decrease the risk of mortality and cardiovascular disease [41, 42]. In addition, several recent reports suggest an improvement of anemia after cinacalcet prescription.
A retrospective study of 40 dialysis patients demonstrated that the doses of darbepoetin significantly decreased 1 year after the initiation of cinacalcet prescription, while hemoglobin levels remained unchanged [43]. Among responders who achieved more than 30% reduction in intact PTH levels, the changes in intact PTH levels were associated with the dose reduction of darbepoetin. Tanaka et al. performed a secondary analysis of a 3-year, multicenter, prospective cohort study on 3,201 dialysis patients with SHPT to assess whether cinacalcet use is associated with improvement of anemia. The investigators showed that cinacalcet was associated with 1.1-fold increase in the odds of achieving the target hemoglobin levels after adjustment with potential confoundings [44]. These studies suggest that cinacalcet can increase the response to ESA and thereby improve renal anemia.
Vitamin D Receptor Activator and Nutritional Vitamin D
The vitamin D receptors are expressed in chief cells of the parathyroid gland and play a role in the regulation of PTH secretion. VDRA administration and supplementation of nutritional vitamin D are thus therapeutic options for SHPT especially among patients with vitamin D deficiency. Several studies have examined whether the use of VDRA or nutritional vitamin D is associated with improvement of anemia or ESA hyporesponsiveness in dialysis patients.
Albitar et al. demostrated that in dialysis patients with SHPT, administration of alfacalcidol led to elevations in hemoglobin concentrations and reticulocyte count together with reductions in intact PTH levels [45]. Another study showed that dialysis patients receiving intravenous calcitriol showed increased hemoglobin levels along with decreased PTH levels regardless of ESA use [46]. Of note, the association of calcitriol administration with increased hemoglobin levels was found only in patients who achieved significant suppression of PTH levels. Thus, it is suggested that the improvement of anemia after VDRA administration is mediated through the reduction in PTH levels.
As for vitamin D status, it is reported that vitamin D insufficiency, defined as serum levels of 25-hydroxyvitamin D (25(OH)D) lower than 30 ng/mL, is present in more than half of dialysis patients [47]. Vitamin D deficiency has been shown to be associated with anemia [48], and several studies have examined whether supplementation of nutritional vitamin D improves ESA hyporesponsiveness.
In a small randomized study by Rianthavorn et al. comparing ergocalciferol and placebo in children with CKD and vitamin D deficiency, the ergocalciferol group showed a significant improvement of ESA hyporesponsiveness without changes in PTH levels [49]. Miskulin et al. performed a double-blind, placebo-controlled, randomized clinical trial to examine the effect of ergocalciferol on ESA responsiveness [50]. The primary outcome was the change in rhEPO dose over 6 months. The study population included 276 dialysis patients with vitamin D insufficiency, and 80% or more were concomitantly treated with VDRAs. The researchers demonstrated that intact PTH levels were comparable between ergocalciferol and placebo arms and there were no significant changes in rhEPO doses in both arms. The lack of response in intact PTH levels to ergocalciferol is consistent with the results of another randomized, placebo-controlled study showing that cholecalciferol supplementation decreased intact PTH levels in non-hemodialysis patients but not in hemodialysis patients [51]. Therefore, nutritional vitamin D supplementation has little or no effect on intact PTH levels in ESRD patients, which may explain the lack of improvement of anemia in this population.
Taken together, VDRA may improve anemia or ESA hyporesponsiveness by decreasing PTH levels, but the effect of nutritional vitamin D is much less marked in ESRD patients. The expression of vitamin D receptor is detectable in non-classical target organs including immune system (T and B cells, macrophages, and monocytes), reproductive system (uterus, testis, ovary, prostate, placenta, and mammary glands), endocrine system (pancreas, pituitary, thyroid, and adrenal cortex), muscles, brain, skin, and liver, suggesting the pleiotropic effects of vitamin D or VDRA [52]. Furthermore, a recent study demonstrated the presence of vitamin D receptor in HSCs [53]. These data support the possibility of the direct effect of vitamin D or VDRA on hematopoiesis. However, current clinical evidence in ESRD patients does not support this possibility and suggest that vitamin D or VDRA could provide improvement of anemia only when PTH levels were lowered.
Stimulatory Action of PTH for Hematopoiesis
Osteoblasts are bone-forming cells derived from mesenchymal stem cells. These cells line the endosteal surfaces and regulate calcification by producing collagen, non-collagenous protein such as osteocalcin and osteopontin, and proteoglycan such as decorin. The PPR that expresses in osteoblasts is an important mediator of bone formation and bone resorption, which is evidenced by the fact that overexpression of constitutively active PPR in transgenic mice leads to increase of both osteoblasts and osteoclasts in trabecular bone [54].
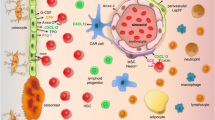
Importantly, HSCs are not distributed randomly in the BM but are more abundant near endosteal surfaces [55], and the interaction between HSCs and osteoblasts has been investigated. G-CSF and hepatocyte growth factor secreted from osteoblasts have been shown to play an important role in differentiation of HSCs [3, 56]. Furthermore, recent studies revealed that Jagged1/Notch signaling is involved in the interaction of osteoblasts and HSCs, and that PTH affects the interaction.
Notch is a transmembrane receptor that plays an essential role in cell differentiation and function and regulates the cell-fate specification by binding of its ligand Jagged1. Osteoblasts are one of the cells that produces Jagged1 whose expression is increased by PTH [57, 58]. Jagged1 produced by osteoblasts not only regulates cell-fate determination by activating Notch 1 in an autocrine manner but also affects HSCs proliferation by activating Notch 1 in these cells (Fig. 1) [57, 58]. Furthermore, in mices undergoing allogenic BM transplantation, 1–34 rat PTH administration, given 5 times a week for four weeks followed by lethal irradiation, was beneficial for survival of grafted HSCs [57].

Schematic of PTH-involved signaling leading to self-renewal and efflux into peripheral circulation of HSCs. HSC, hematopoietic stem cell; PTH, parathyroid hormone; PPR, PTH/PTH-related protein receptor 1
In addition to the Jagged1/Notch signaling, cadherin-11 is also shown to be involved in the interaction of osteoblasts and HSCs regulated by PTH (Fig. 1). Cadherin-11 is an adhesion molecule that is selectively expressed by stromal cell-derived cells, especially fibroblasts, and is thought to be one of the important inflammatory mediators [59]. Yao et al. isolated and expanded marrow stromal cells obtained from healthy volunteers and performed coculture of these cells and isolated CD34+ HSCs to investigate the effect of PTH on proliferation of CD34+ HSCs [60]. The ability of culture-expanded human marrow stromal cells to expand cocultured CD34+ HSCs was enhanced when cocultured with PTH, but their ability was totally eliminated when cocultured with Transwell system, suggesting that direct interaction via adhesion molecules between HSCs and marrow stromal cells are necessary for the enhancement. The researchers also demonstrated that PTH increased the expression of cadherin-11 in marrow stromal cells and that depletion of cadherin-11 expression in marrow stromal cells by small interfering RNA abolished the enhancement of HSC expansion by PTH-treated marrow stromal cells. This study suggests that cadherin-11 is an important molecule for the adhesion of marrow stromal cells to HSCs and that PTH supports proliferation of HSCs through increased expressions of cadherin-11. Furthermore, they assessed the effect of 1–34 human PTH treatment following allogenic BM transplant in lethally irradiated mice and demonstrated that PTH administration increased the expression of cadherin-11 on marrow stromal cells and improved survival rate of the mice. Collectively, these experiment data support the possibility that PTH promotes the proliferation of HSCs.
Recent studies also suggested that PTH accelerates mobilization of HSCs into peripheral circulation. One clinical study of PHPT patients showed that the HSC count in peripheral blood was significantly higher in PHPT patients than healthy volunteers and that PTx lead to reductions in the peripheral HSCs [61]. In this study population, the levels of cytokines such as vascular endothelial growth factor, which enhance mobilization of BM cells into peripheral blood, were comparable to those of healthy volunteers, suggesting that the effect of PTH to promote HSC mobilization was independent of these cytokines. Another study by Yu et al. evaluated the effect of teriparatide (1–34 human PTH) for up to 24 months on peripheral HSCs and other hematologic markers in patients with postmenopausal osteoporosis [62]. Only HSC count was significantly increased as early as 3 months, while the number of leukocytes, T cells, B cells, erythrocytes, and platelets did not change significantly throughout the study period. Thus, teriparatide may enhance the mobilization of HSCs into peripheral blood, although the mechanism remains largely unknown. The mobilization of HSCs into peripheral blood is known to play a role in cancer cell growth and post‐infarction cardiac remodeling but is also suggested to support autologous stem cell collection.
The putative effects of PTH to increase HSCs and enhance their mobilization into peripheral circulation raise the possibility that PTH may support the engraftment after BM transplantation with a complete recovery of donor-derived hematopoiesis in leukocytes, erythrocytes, and thrombocytes. Based on these possibilities, accumulating studies have investigated the effect of PTH pretreatment on hematologic systems after BM transplantation. Ballen et al. performed phase I trial to assess the safety and efficacy of teriparatide administration for mobilization of HSCs into peripheral blood. A total of 20 patients who had 1 or 2 unsuccessful peripheral blood stem cell collections received escalating doses of 40 to 100 µg teriparatide for 14 days and 10 µg/kg filgrastim, a recombinant G-CSF agent, after myeloablative therapy [63]. Teriparatide administration was well-tolerated and successfully mobilized HSCs into peripheral blood in 40% and 47% of patients who failed 1 and 2 prior mobilization attempts, respectively. The success rate of achieving engraftment increases as more HSCs are harvested from peripheral blood. These data suggest that teriparatide is a promising agent for supporting hematopoiesis in BM transplantation.
It is known that the number of HSCs collected from umbilical cord blood (UCB) is limited, and even with the use of 2 UCB units, engraftment and immune reconstitution are often slow, leading to an elevated risk of infection and second malignancy [64, 65]. Thus, several clinical trials have been performed in an attempt to achieve early engraftment with the use of HSCs of UCB. However, previous studies have shown that ex vivo expansion of HSCs harvested from UCB did not the improve the survival of these cells after transfer to recipients [66, 67]. On the basis of accumulating evidence that PTH supports hematopoiesis and the results of the phase I trial showing that PTH pretreatment successfully mobilized HSCs into peripheral blood, phase II trial was conducted to examine the efficacy of PTH pretreatment for UCB transplantation [68]. This trial assessed whether 100 µg/day subcutaneous teriparatide administrations after a myeloablative or a reduced-intensity double UCB transplantation improves the rate of engraftment in patients indicated for UCB transplantation. All 13 patients achieved engraftment of neutrophil and platelet, but 4 patients died before day 100 due to transplant-associated complications, which led to early study closure although these deaths were not considered to be caused by teriparatide-related adverse effects. The researchers concluded that there was no evidence that PTH influenced blood count recovery. Although this study does not preclude the possibility that PTH supports engraftment after HSC transplantation, no additional trials have been performed until now and it remains unclear whether systemic administration of teriparatide after HSC transplantation is clinically beneficial.
Because teriparatide strongly induces bone formation, many clinical trials have assessed the efficacy of intermittent administration of teriparatide for postmenopausal osteoporosis. When we look into these results more closely, hematology-associated side effects were rarely reported; we found leukocytosis just in one study conducted in Japan, but the incidence rate was less than 1% [62, 69, 70]. These results suggest that the impact of teriparatide administration on mature blood cells in peripheral circulation is limited at least in postmenopausal women with osteoporosis.
Collectively, PTH administration may augment the production of HSCs and exert favorable effects on survival of HSCs. Moreover, teriparatide could induce mobilization of HSCs into peripheral blood. However, the effect of teriparatide on hematologic status in humans has not been well documented and the reason for the discrepant findings remains unknown. Further studies are needed to assess whether PTH administration increases proliferation of HSCs after transplantation and could be a novel strategy to support engraftment in this setting.
PTH and Immunity
In mammals, the immune system comprises two branches; innate immunity and acquired immunity. Both humoral and cellular factors are considered important in the immunity process. Humoral factors of innate immunity include inflammation and complement, and cellular factors include neutrophils, eosinophils, basophils, mast cells, macrophages, NK cells, and dendritic cells. As for acquired immunity, B cells engage in humoral immunity and T cells in cellular immunity.
ESRD patients are known to be immune-compromised, and it is reported that the annual mortality rate due to infectious diseases is 100 times or higher in dialysis patients than the general population [71]. Higher age, diabetes mellitus, lower albumin levels, catheter insertion, reuse of dialyzer, hyperphosphatemia, and elevated alkaline phosphatase (ALP) levels have been shown to be associated with higher infection-related mortality risk [72,73,74]. In addition, higher PTH levels are also shown to be associated with infectious death in dialysis patients [75]. Basic researches have shown that PTH adversely affects the function of leukocytes, which suggests that HPT has a negative impact on human immunity.
PTH and Polymorphonuclear Leukocytes
Innate immunity is the first line of defense against the invasion of foreign body or microbes into the body, wherein neutrophils, eosinophils, and basophils play essential roles. Several studies compared the function of polymorphonuclear leukocytes (PMNLs) in dialysis patients to that in healthy volunteers to estimate the effect of PTH on immune-related cell function. Massry et al. showed that elastase release from PMNLs was significantly decreased in dialysis patients [76]. Doherty et al. demonstrated that the migration of PMNLs was decreased in dialysis patients, particularly in those with higher PTH levels [77]. The same group also reported that patients with SHPT had higher resting levels of calcium and lower ATP contents in the cytosol of PMNLs. They also showed that phagocytic function was suppressed in these patients and this suppression was recovered by the calcium channel blocker verapamil [78,79,80]. These results raised the possibility that PTH disturbs phagocytosis or random migration of PMNLs, which could lead to impaired innate immunity. However, it should be stressed that the above studies compared PMNLs collected from patients with SHPT to those from healthy volunteers, which cannot dissociate the effect of PTH from that of other uremic toxins and thus cannot confirm the effect of PTH on the immune system. Future studies should compare the function of PMNLs among dialysis patients with different PTH levels.
PTH and Macrophages
Macrophages are mononuclear cells of the myeloid lineage and are originally derived from HSCs. Macrophages are not only circulating in the peripheral blood but also reside as Kupffer cells in the liver, as Langerhans cells in the lung, and as microglia in the brain. For decades, osteoclasts have been considered as macrophages that reside in bone. However, osteoclasts do not express F4/80 antigen that can be detected in almost all the other local resident macrophages. Indeed, osteoclasts were not diminished in macrophage Fas-induced apoptosis (MAFIA) transgenic mice, which allow for conditional depletion of macrophages, indicating that osteoclasts originate from myeloid progenitor that are different from other resident macrophages [81, 82]. Of note, several researchers confirmed the existence of F4/80 positive cells in BM that are distinct from osteoclasts [83]. These cells are called osteal macrophages and have been shown to control the function of osteoblasts in the vicinity of those cells.
Chang et al. demonstrated that the MAFIA transgenic mice do not have mature osteoblasts and show alterations in bone mineralization [83]. The researchers further performed coculture of osteoblasts and osteal macrophages and demonstrated that osteoblasts induce calcification only when cocultured with osteal macrophages, suggesting that osteal macrophages play an important role in mineralization induced by osteoblasts. Cho et al. also showed that intermittent PTH injection to mice increased osteal macrophages in endosteum and periosteum areas [81]. They also demonstrated that the MAFIA mice showed decreased cortical and trabecular bone volume and that PTH injection did not stimulate bone formation. These data suggest that osteal macrophages play a crucial role in the PTH-induced acceleration of bone formation.
Taken together, it can be considered that osteal macrophages play a key role in the regulation of bone metabolisms through crosstalk with osteoblasts. Much remains to be known about the nature and the mechanism of osteoclast differentiation, and further researches are needed to determine the role of PTH in this process.
PTH and B Cells
B cells are central mediators in humoral immunity and recognize bacteria and viruses and assist in the immune process through synthesis and release of antibodies. Human and bovine lymphocytes are known to express the PPR, and PTH has been shown to affect the function of B cells by activating adenylate cyclase [84].
Alexiewicz et al. obtained peripheral blood from healthy volunteers and dialysis patients and assessed the effects of PTH on the functions of isolated B cells [85]. The researchers confirmed the expression of the PPR in B cells and demonstrated that PTH acts on B cells directly and inhibits their differentiation by increasing intracellular production of cAMP. Gaciong et al. showed that bovine 1–84 PTH and synthetic bovine 1–34 PTH inhibited Staphylococcus aureus-stimulated production of immunoglobulin by cultured monocytes that were obtained either from dialysis patients or normal subjects [86]. They also showed that in 5/6 nephrectomized rats, PTx led to reductions in the amount of anti-Staphylococcus aureus antibody [87]. These findings suggest that PTH inhibits the differentiation and function of B cells at least in the setting of SHPT.
Kotzmann et al. examined 12 PHPT patients before and 6 months after PTx and 9 sex- and age-matched control subjects to determine the impact of PTH on serum immunoglobulin levels and immunophenotype of peripheral blood lymphocytes [88]. They found that immunoglobulin levels in PHPT patients did not change after PTx and were comparable to control subjects. Similarly, levels of T lymphocytes (CD3), B lymphocytes (CD19), NK cells (CD16/56) and monocytes (CD16) also did not change after PTx. Thus, the impact of high PTH on immune systems is relatively small in PHPT patients compared to that in SHPT patients.
The effect of PTH on B cells has not been explored in previous clinical trials of teriparatide for osteoporotic patients. It remains thus unknown whether intermittent administration of 1–34 PTH affects B cell differentiation and function.
PTH and T Cells
T cells are another class of lymphocytes and play an important role in immunity. T cells undergo an instructional process of positive and negative selection in the thymus. Mature T cells can be divided into different subgroups, namely CD4+ T cells which produce cytokines and interact with B cells, CD8+ T cells which exert cytotoxic effects on infected cells or cancer cells, and regulatory T cells which are considered to protect against autoimmune diseases [89]. In experimental studies, researchers often use phytohemagglutinin (PHA), which is one of the plant lectins and stimulates T cell blastoid transformation, differentiation, and proliferation through T cell receptors. It is well accepted that the extent of PHA-induced lymphocyte proliferation is an indicator of T cell-mediated immunity.
Interleukin 2 (IL2) produced by T cells plays crucial roles in proliferation and differentiation of T and B lymphocytes. The expression of IL2 depends on cytosolic calcium concentrations of T cells. PTH induces influx of calcium into variety of cells and, for that reason, may affect the synthesis of IL2 by T cells. Klinger et al. isolated lymphocytes from healthy subjects and examined whether addition of 1–34 bovine PTH or 1–84 bovine PTH affects IL2 production and PHA-induced T cell proliferation [90]. The investigators found that both 1–34 PTH and 1–84 PTH accelerated PHA-induced T cell proliferation dose-dependently, while inactivated 1–84 PTH did not produce a similar effect. Of note, PTH is known to increase intracellular cAMP through the PPR, but in this study, forskolin, a stimulator of intracellular cAMP levels, did not induce T cell proliferation, suggesting that PTH stimulates T cell proliferation through the cAMP-independent pathways. They also demonstrated that PHA-induced IL2 production was enhanced with 1–84 PTH treatment. These results indicate that PTH stimulates both T cell proliferation and IL2 production.
However, these experimental results were not confirmed in clinical studies. Tzanno-Martins et al. examined T cell function in patients with severe SHPT (average intact PTH 1,425 ± 623 pg/mL) who underwent PTx [91]. The investigators showed that lymphoproliferative response to PHA was significantly increased 4 months after PTx, but there were no significant changes in IL2.
Kotzmann et al. examined PHPT patients and showed increased proportion of CD4+ T cells and decreased proportion of CD8+ T cells compared to normal subjects [88]. However, PTx for these patients did not change the number of CD4+ T cells and CD8+ T cells, suggesting the impact of PTH on CD4/CD8 ratio was limited. Taken together, it remains unclear whether PTH affects T cell function and cellular immunity, and additional research is needed to confirm this interaction.
PTH and Bone Cell-Immune Cell Crosstalk
HSCs form specific microenvironment in BM, called as niche, and crosstalk between HSCs and adjacent cells is quite important for differentiation and proliferation of HSCs. Immune cells are derived from HSCs and released into peripheral circulation after differentiation from HSCs. Increasing evidence suggests that PTH plays an important role in crosstalk between HSCs and adjacent cells for differentiation of immune-related cells, especially B cells and T cells.
Tokoyoda et al. first demonstrated the importance of BM niche in B cell development by showing that early B cell progenitors directly contact marrow stromal cells expressing C-X-C motif chemokine 12 (CXCL12) and IL-7 [92]. Several studies subsequently revealed that marrow stromal cells in the BM niche are important for B cell differentiation and that osteoblast lineage supports hematopoiesis. Furthermore, the PPR signaling, which is essential for differentiation of osteoblast lineage, has been shown to accelerate differentiation and maturation of lymphocytes.
Zhu et al. demonstrated that B cell differentiation require their attachment to osteoblasts, which is mediated via osteoblast-expressed vascular cell adhesion molecule 1 (VCAM1), stromal-cell-derived factor 1 (SDF1), and IL-7 signalling induced by PTH [93]. The researchers also revealed that addition of c-Kit ligand, IL-6, and IL-3 produced by nonosteoblastic stromal cells induce myelopoiesis. Furthermore, transgenic mice with selective elimination of osteoblasts showed severely depleted pre-pro-B and pro-B cells from BM. These results reinforced the importance of osteoblasts in B cell commitment and maturation. Based on the fact that the G protein α subunit, Gsα, is one of the downstream mediators of PPR signaling, Wu et al. generated mice with osteoblast lineage-specific deletion of Gsα using Osx1-Cre to examine the effect of Gsα on B cell differentiation and explore chemokines involved in this process [94]. As expected, the osteoblast-specific Gsα-KO mice showed marked reductions in B cell precursors both in the BM and in the peripheral blood. These mice also showed decreased serum IL-7 levels, and subcutaneous injection of IL-7 partially restored the number of pro-B cells and pre-B cells. These data suggest that Gsα-dependent signaling pathways in osteoblast lineage cells regulate differentiation and proliferation of B cells, at least partially in an IL-7-dependent manner.
Panaroni et al. examined the differences in B lymphopoiesis between mice with Osx1-Cre-mediated deletion of PPR in osteoblast lineage cells, those with osteocalcin-Cre-mediated deletion in mature osteoblasts, and those with Dmp1-Cre-mediated deletion in osteocytes [95]. Compared to control mice, the transgenic mice with PPR deletion in osteoblast lineage cells showed a decreased number of B cells, comparable expression of CXCL12, and reduced expression of IL-7 in BM. By contrast, B cell precursors were increased in BM in mice with either mature osteoblast- or osteocyte-specific deletion of the PPR. These results suggest that PTH acts on immature osteoblast lineage cells and stimulates IL-7 production, which in turn induces differentiation of B cell precursors, but such effects do not occur in more mature osteoblasts or osteocytes. Collectively, it can be concluded that direct contact with osteoblast lineage cells in BM niche is important for B cell differentiation and proliferation, and this effect is mediated through PTH-induced IL-7 production by osteoblast lineage cells.
While studies have shown the roles of osteoblast lineage cells in B cell differentiation and proliferation, T cells have been shown to play a role in differentiation and proliferation of osteoblast lineage cells.
Gao et al. investigated the effects of continuous injection of 1–34 human PTH on bone metabolism using mice lacking T cells generated by injection of anti-CD4/CD8 antibodies [96]. Bone histomorphometry revealed that two weeks of PTH injection increased the number of osteoclasts and induced bone absorption in control mice, but not in T cell-depleted mice. The investigators further demonstrated that PTH treatment failed to increase osteoclast formation in BM derived from T cell-depleted mice, whereas the addition of CD4+ or CD8+ cells to T cell-depleted BM dose dependently increased the osteoclastogenic activity of PTH, suggesting that both CD4+ or CD8+ cells could promote osteoclast formation induced by PTH. The researchers also demonstrated that deletion of T cell-expressed CD40 ligand blunts the bone catabolic activity of PTH by decreasing bone marrow stromal cell number, the RANKL/OPG ratio, and osteoclastogenic activity. These data indicate that T cells play an essential role in increased bone resorption associated with HPT by regulating stromal cell proliferation and function through CD40L.
T cells play important roles not only in bone resorption but also in bone formation. Terauchi et al. intermittently administered 1–34 human PTH to T cell deficient mice and control mice and demonstrated that T cell null mice showed attenuation of bone formation compared to control mice [97]. The researchers also revealed that PTH increases the production of Wnt10b by CD8+ T cells in BM and thereby induces these lymphocytes to activate canonical Wnt signaling in preosteoblasts. Furthermore, intermittent PTH administration decreased apoptosis of preosteoblasts, which was less evident in T cell null mice compared to control mice. These effects could explain the decreased differentiation and proliferation of osteoblasts and reduced bone formation in the T cell null mice. These studies indicate that PTH and T cells play important roles in the differentiation from osteoblastic progenitors to mature osteoblast lineage cells. The close interaction between bone cells and immune cells in BM suggests that a full understanding of this crosstalk provide novel pharmacological targets for osteoporosis and other bone diseases.
PTH and Hemostasis
Platelets and coagulation factors are two major players in hemostasis. Primary hemostasis in the setting of bleeding due to vascular injury is achieved by contraction of blood vessels and aggregation of platelets. Arachidonic acid (AA) and adenosine diphosphate (ADP) are engaged in platelet aggregation. Circulating coagulation factors are inactive under normal circumstances, but could be sequentially activated in secondary hemostasis that occurs simultaneously with the primary hemostasis.
Thrombosis is a pathological process in which the hemostatic system is excessively activated and causes the development of platelet-aggregates that prevent normal blood circulation. This process contributes to the development of cardiovascular diseases (CVD), which is the major cause of death in ESRD patients. Many observational studies have shown associations between high PTH and elevated risk of CVD in predialysis and dialysis patients (see Chap. 9). Vascular calcification and endothelial dysfunction have been shown to mediate this association, and recent studies also suggest the involvement of platelet aggregation and coagulation in this process.
PTH and Platelets
Several clinical and experimental data suggest that PTH affects both the number and function of platelets. In one case report of a patient with PHPT, PTx led to improvement of anemia and thrombocytopenia along with improved myelofibrosis [98]. As such, the association between high PTH and myelofibrosis has been well documented, and it can be argued that elevated PTH may decrease the number of platelets through the development of myelofibrosis and thereby cause abnormal bleeding.
As for the association between PTH and the function of platelet, Ortega et al. confirmed that platelets express the PPR and showed that 1–36 PTHrP induces platelet aggregation by increasing the intracellular calcium concentration via MAP kinase pathway [99]. Because PTHrP and PTH share the same receptor (PPR), similar phenomenon can be expected with PTH. Verdoia et al. collected peripheral blood from 362 patients who received dual antiplatelet therapy of aspirin plus ADP antagonist (clopidogrel or ticagrelor) for acute coronary syndrome or after percutaneous coronary intervention for stable CVD, and examined AA-mediated and ADP-mediated platelet aggregation by measuring high residual-on-treatment platelet reactivity (HRPR) [100]. The researchers found that high intact PTH levels were associated with increased ADP-mediated platelet aggregation in patients with clopidogrel but not those with ticagrelor. These findings may suggest that PTH modulates the effect of ADP antagonist, but the reason for the discrepancy between clopidogrel and ticagrelor is unknown. Collectively, PTH may induce abnormal platelet aggregation, but it remains unclear whether the effect of PTH on platelet aggregation mediates the association between HPT and CVD.
PTH and Coagulopathy
While excessive platelet aggregation can cause thrombosis in artery, activation of coagulation factors can lead to venous thrombosis, such as deep vein thrombosis and pulmonary thromboembolism. Hepatocytes, which produce coagulation factors, are known to express the PPR [101], raising the possibility that PTH affects the production of coagulation factors.
Several case reports have demonstrated the occurrence of venous thrombus in PHPT patients. Pringle et al. reported two cases of renal venous thrombosis associated with PHPT [102]. Manosroi et al. reported a case of subclavian venous thrombosis and pulmonary embolisms associated with parathyroid carcinoma [103]. However, it should be mentioned that hypercalcemia associated with PHPT can cause renal diabetes insipidus, and the resultant hemoconcentration might have led to thrombus formation in these patients. Thus, it remains unknown whether PTH directly affects coagulation or thrombus formation.
Several researchers examined the association between PTH levels and coagulation factors in patients with PHPT or SHPT. Erem et al. showed that PHPT patients had higher tissue plasminogen activator (t-PA) and plasminogen activator inhibitor (PAI)-1 and lower tissue factor pathway inhibitor (TFPI) compared to healthy volunteer, suggesting a hypercoagulability and hypofibrinolytic state in these patients [104]. However, another research group performed a case-control study and revealed that PTx did not change plasma coagulation factors such as PAI-1 in PHPT patients [105]. Another cohort study demonstrated that in patients with HPT secondary to vitamin D deficiency, vitamin D supplementation did not change coagulation factors, such as prothrombin time, activated partial thromboplastin time, factors VII, VIII, and X, even though these patients showed a significant reduction in PTH levels [106]. Thus, it is unclear whether PTH affects the production of coagulation factors.
Conclusion
Over the past decades, many studies have focused on the adverse effects of PTH, which can cause myelofibrosis, anemia, and immunosuppression particularly in patients with PHPT and SHPT. However, recent investigations have also uncovered that PTH mediates crosstalk between HSCs and marrow stromal cells including osteoblasts, osteoclasts, and osteal macrophages, and represents an important player in differentiation and proliferation of these cells. Despite the accumulation of basic research showing the potential abilities of PTH to support hematopoiesis, there are only a few evidences supporting the clinical benefit of PTH in terms of hematopoiesis. Future research should examine whether PTH administration supports hematopoiesis and improves the outcome of BM transplantation.
References
Metcalf D. Concise review: hematopoietic stem cells and tissue stem cells: current concepts and unanswered questions. Stem Cells. 2007;25(10):2390–5.
Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci. 2016;1370(1):82–96.
Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood. 1996;87(2):518–24.
Dagg JH, Horton PW, Orr JS, Shimmins J. A direct method of determining red cell lifespan using radioiron: an application of the occupancy principle. Br J Haematol. 1972;22(1):9–19.
Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342(17):1255–65.
Albright F, Aub JC, Bauer W. Hyperparathyroidism: A common and polymorphic condition as illustrated by seventeen proved cases from one clinic. JAMA. 1934;102(16):1276–87.
Lotinun S, Sibonga JD, Turner RT. Triazolopyrimidine (trapidil), a platelet-derived growth factor antagonist, inhibits parathyroid bone disease in an animal model for chronic hyperparathyroidism. Endocrinology. 2003;144(5):2000–7.
Lowry MB, Lotinun S, Leontovich AA, Zhang M, Maran A, Shogren KL, et al. Osteitis fibrosa is mediated by Platelet-Derived Growth Factor-A via a phosphoinositide 3-kinase-dependent signaling pathway in a rat model for chronic hyperparathyroidism. Endocrinology. 2008;149(11):5735–46.
Zingraff J, Drueke T, Marie P, Man NK, Jungers P, Bordier P. Anemia and secondary hyperparathyroidism. Arch Intern Med. 1978;138(11):1650–2.
Rao DS, Shih MS, Mohini R. Effect of serum parathyroid hormone and bone marrow fibrosis on the response to erythropoietin in uremia. N Engl J Med. 1993;328(3):171–5.
Meytes D, Bogin E, Ma A, Dukes PP, Massry SG. Effect of parathyroid hormone on erythropoiesis. J Clin Invest. 1981;67(5):1263–9.
Taniguchi S, Shibuya T, Harada M, Niho Y. Prostaglandin-mediated suppression of in vitro growth of erythroid progenitor cells. Kidney Int. 1989;36(4):712–8.
Dunn CD, Trent D. The effect of parathyroid hormone on erythropoiesis in serum-free cultures of fetal mouse liver cells. Proc Soc Exp Biol Med. 1981;166(4):556–61.
Komatsuda A, Hirokawa M, Haseyama T, Horiuchi T, Wakui H, Imai H, et al. Human parathyroid hormone does not influence human erythropoiesis in vitro. Nephrol Dial Transplant. 1998;13(8):2088–91.
Jacobson LO, Goldwasser E, Fried W, Plzak L. Role of the kidney in erythropoiesis. Nature. 1957;179(4560):633–4.
Naets JP. The role of the kidney in erythropoiesis. J Clin Invest. 1960;39:102–10.
Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–54.
Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117(23):e207–17.
Koury MJ, Haase VH. Anaemia in kidney disease: harnessing hypoxia responses for therapy. Nat Rev Nephrol. 2015;11(7):394–410.
Kobayashi H, Liu Q, Binns TC, Urrutia AA, Davidoff O, Kapitsinou PP, et al. Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Invest. 2016;126(5):1926–38.
Urena P, Eckardt KU, Sarfati E, Zingraff J, Zins B, Roullet JB, et al. Serum erythropoietin and erythropoiesis in primary and secondary hyperparathyroidism: effect of parathyroidectomy. Nephron. 1991;59(3):384–93.
Washio M, Iseki K, Onoyama K, Oh Y, Nakamoto M, Fujimi S, et al. Elevation of serum erythropoietin after subtotal parathyroidectomy in chronic haemodialysis patients. Nephrol Dial Transplant. 1992;7(2):121–4.
Borawski J, Pawlak K, Mysliwiec M. Inflammatory markers and platelet aggregation tests as predictors of hemoglobin and endogenous erythropoietin levels in hemodialysis patients. Nephron. 2002;91(4):671–81.
McGonigle RJ, Wallin JD, Husserl F, Deftos LJ, Rice JC, O’Neill WJ, et al. Potential role of parathyroid hormone as an inhibitor of erythropoiesis in the anemia of renal failure. J Lab Clin Med. 1984;104(6):1016–26.
Wong A, Loots GG, Yellowley CE, Dose AC, Genetos DC. Parathyroid hormone regulation of hypoxia-inducible factor signaling in osteoblastic cells. Bone. 2015;81:97–103.
Dunn MJ. Red blood cell calcium and magnesium: effects upon sodium and potassium transport and cellular morphology. Biochim Biophys Acta. 1974;352(1):97–116.
White JG. Effects of an ionophore, A23187, on the surface morphology of normal erythrocytes. Am J Pathol. 1974;77(3):507–18.
Weed RI, LaCelle PL, Merrill EW. Metabolic dependence of red cell deformability. J Clin Invest. 1969;48(5):795–809.
Lang E, Qadri SM, Lang F. Killing me softly - suicidal erythrocyte death. Int J Biochem Cell Biol. 2012;44(8):1236–43.
Bogin E, Massry SG, Levi J, Djaldeti M, Bristol G, Smith J. Effect of parathyroid hormone on osmotic fragility of human erythrocytes. J Clin Invest. 1982;69(4):1017–25.
Akmal M, Telfer N, Ansari AN, Massry SG. Erythrocyte survival in chronic renal failure. Role of secondary hyperparathyroidism. J Clin Invest. 1985;76(4):1695–8.
Barbour GL. Effect of parathyroidectomy on anemia in chronic renal failure. Arch Intern Med. 1979;139(8):889–91.
Mandolfo S, Malberti F, Farina M, Villa G, Scanziani R, Surian M, et al. Parathyroidectomy and response to erythropoietin therapy in anaemic patients with chronic renal failure. Nephrol Dial Transplant. 1998;13(10):2708–9.
Coen G, Calabria S, Bellinghieri G, Pecchini F, Conte F, Chiappini MG, et al. Parathyroidectomy in chronic renal failure: short- and long-term results on parathyroid function, blood pressure and anemia. Nephron. 2001;88(2):149–55.
Yasunaga C, Matsuo K, Yanagida T, Matsuo S, Nakamoto M, Goya T. Early effects of parathyroidectomy on erythropoietin production in secondary hyperparathyroidism. Am J Surg. 2002;183(2):199–204.
Trunzo JA, McHenry CR, Schulak JA, Wilhelm SM. Effect of parathyroidectomy on anemia and erythropoietin dosing in end-stage renal disease patients with hyperparathyroidism. Surgery. 2008;144(6):915–8; discussion 9.
Bhadada SK, Bhansali A, Ahluwalia J, Chanukya GV, Behera A, Dutta P. Anaemia and marrow fibrosis in patients with primary hyperparathyroidism before and after curative parathyroidectomy. Clin Endocrinol (Oxf). 2009;70(4):527–32.
Block GA, Bushinsky DA, Cunningham J, Drueke TB, Ketteler M, Kewalramani R, et al. Effect of Etelcalcetide vs. Placebo on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: Two Randomized Clinical Trials. JAMA. 2017;317(2):146–55.
Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA. 2017;317(2):156–64.
Fukagawa M, Yokoyama K, Shigematsu T, Akiba T, Fujii A, Kuramoto T, et al. A phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of etelcalcetide (ONO-5163/AMG 416), a novel intravenous calcimimetic, for secondary hyperparathyroidism in Japanese haemodialysis patients. Nephrol Dial Transplant. 2017;32(10):1723–30.
Chertow GM, Block GA, Correa-Rotter R, Drueke TB, Floege J, Goodman WG, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367(26):2482–94.
Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, et al. Cinacalcet, fibroblast growth Factor-23, and cardiovascular disease in hemodialysis: the evaluation of cinacalcet HCl therapy to lower cardiovascular events (EVOLVE) Ttrial. Circulation. 2015;132(1):27–39.
Battistella M, Richardson RM, Bargman JM, Chan CT. Improved parathyroid hormone control by cinacalcet is associated with reduction in darbepoetin requirement in patients with end-stage renal disease. Clin Nephrol. 2011;76(2):99–103.
Tanaka M, Yoshida K, Fukuma S, Ito K, Matsushita K, Fukagawa M, et al. Effects of secondary hyperparathyroidism treatment on improvement in anemia: results from the MBD-5D study. PLoS ONE. 2016;11(10):e0164865.
Albitar S, Genin R, Fen-Chong M, Serveaux MO, Schohn D, Chuet C. High-dose alfacalcidol improves anaemia in patients on haemodialysis. Nephrol Dial Transplant. 1997;12(3):514–8.
Goicoechea M, Vazquez MI, Ruiz MA, Gomez-Campdera F, Perez-Garcia R, Valderrabano F. Intravenous calcitriol improves anaemia and reduces the need for erythropoietin in haemodialysis patients. Nephron. 1998;78(1):23–7.
Singer RF. Vitamin D in dialysis: defining deficiency and rationale for supplementation. Semin Dial. 2013;26(1):40–6.
Kiss Z, Ambrus C, Almasi C, Berta K, Deak G, Horonyi P, et al. Serum 25(OH)-cholecalciferol concentration is associated with hemoglobin level and erythropoietin resistance in patients on maintenance hemodialysis. Nephron Clin Pract. 2011;117(4):c373–8.
Rianthavorn P, Boonyapapong P. Ergocalciferol decreases erythropoietin resistance in children with chronic kidney disease stage 5. Pediatr Nephrol. 2013;28(8):1261–6.
Miskulin DC, Majchrzak K, Tighiouart H, Muther RS, Kapoian T, Johnson DS, et al. Ergocalciferol supplementation in hemodialysis patients with vitamin D deficiency: a randomized clinical trial. J Am Soc Nephrol. 2016;27(6):1801–10.
Marckmann P, Agerskov H, Thineshkumar S, Bladbjerg EM, Sidelmann JJ, Jespersen J, et al. Randomized controlled trial of cholecalciferol supplementation in chronic kidney disease patients with hypovitaminosis D. Nephrol Dial Transplant. 2012;27(9):3523–31.
Verstuyf A, Carmeliet G, Bouillon R, Mathieu C. Vitamin D: a pleiotropic hormone. Kidney Int. 2010;78(2):140–5.
Cortes M, Chen MJ, Stachura DL, Liu SY, Kwan W, Wright F, et al. Developmental vitamin D availability impacts hematopoietic stem cell production. Cell Rep. 2016;17(2):458–68.
Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107(3):277–86.
Lord BI, Testa NG, Hendry JH. The relative spatial distributions of CFUs and CFUc in the normal mouse femur. Blood. 1975;46(1):65–72.
Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med. 1994;179(5):1677–82.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–6.
Weber JM, Forsythe SR, Christianson CA, Frisch BJ, Gigliotti BJ, Jordan CT, et al. Parathyroid hormone stimulates expression of the Notch ligand Jagged1 in osteoblastic cells. Bone. 2006;39(3):485–93.
Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, et al. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci USA. 2011;108(20):8402–7.
Yao H, Miura Y, Yoshioka S, Miura M, Hayashi Y, Tamura A, et al. Parathyroid hormone enhances hematopoietic expansion via upregulation of cadherin-11 in bone marrow mesenchymal stromal cells. Stem Cells. 2014;32(8):2245–55.
Brunner S, Theiss HD, Murr A, Negele T, Franz WM. Primary hyperparathyroidism is associated with increased circulating bone marrow-derived progenitor cells. Am J Physiol Endocrinol Metab. 2007;293(6):E1670–5.
Yu EW, Kumbhani R, Siwila-Sackman E, DeLelys M, Preffer FI, Leder BZ, et al. Teriparatide (PTH 1-34) treatment increases peripheral hematopoietic stem cells in postmenopausal women. J Bone Miner Res. 2014;29(6):1380–6.
Ballen KK, Shpall EJ, Avigan D, Yeap BY, Fisher DC, McDermott K, et al. Phase I trial of parathyroid hormone to facilitate stem cell mobilization. Biol Blood Marrow Transplant. 2007;13(7):838–43.
Ballen KK, Cutler C, Yeap BY, McAfee SL, Dey BR, Attar EC, et al. Donor-derived second hematologic malignancies after cord blood transplantation. Biol Blood Marrow Transplant. 2010;16(7):1025–31.
Cahu X, Rialland F, Touzeau C, Chevallier P, Guillaume T, Delaunay J, et al. Infectious complications after unrelated umbilical cord blood transplantation in adult patients with hematologic malignancies. Biol Blood Marrow Transplant. 2009;15(12):1531–7.
de Lima M, McMannis J, Gee A, Komanduri K, Couriel D, Andersson BS, et al. Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase I/II clinical trial. Bone Marrow Transplant. 2008;41(9):771–8.
Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med. 2010;16(2):232–6.
Ballen K, Mendizabal AM, Cutler C, Politikos I, Jamieson K, Shpall EJ, et al. Phase II trial of parathyroid hormone after double umbilical cord blood transplantation. Biol Blood Marrow Transplant. 2012;18(12):1851–8.
Fujita T, Inoue T, Morii H, Morita R, Norimatsu H, Orimo H, et al. Effect of an intermittent weekly dose of human parathyroid hormone (1-34) on osteoporosis: a randomized double-masked prospective study using three dose levels. Osteoporos Int. 1999;9(4):296–306.
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–41.
Sarnak MJ, Jaber BL. Mortality caused by sepsis in patients with end-stage renal disease compared with the general population. Kidney Int. 2000;58(4):1758–64.
Powe NR, Jaar B, Furth SL, Hermann J, Briggs W. Septicemia in dialysis patients: incidence, risk factors, and prognosis. Kidney Int. 1999;55(3):1081–90.
Plantinga LC, Fink NE, Melamed ML, Briggs WA, Powe NR, Jaar BG. Serum phosphate levels and risk of infection in incident dialysis patients. Clin J Am Soc Nephrol. 2008;3(5):1398–406.
Blayney MJ, Pisoni RL, Bragg-Gresham JL, Bommer J, Piera L, Saito A, et al. High alkaline phosphatase levels in hemodialysis patients are associated with higher risk of hospitalization and death. Kidney Int. 2008;74(5):655–63.
Young EW, Albert JM, Satayathum S, Goodkin DA, Pisoni RL, Akiba T, et al. Predictors and consequences of altered mineral metabolism: the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2005;67(3):1179–87.
Massry SG, Schaefer RM, Teschner M, Roeder M, Zull JF, Heidland A. Effect of parathyroid hormone on elastase release from human polymorphonuclear leucocytes. Kidney Int. 1989;36(5):883–90.
Doherty CC, LaBelle P, Collins JF, Brautbar N, Massry SG. Effect of parathyroid hormone on random migration of human polymorphonuclear leukocytes. Am J Nephrol. 1988;8(3):212–9.
Alexiewicz JM, Smogorzewski M, Fadda GZ, Massry SG. Impaired phagocytosis in dialysis patients: studies on mechanisms. Am J Nephrol. 1991;11(2):102–11.
Massry S, Smogorzewski M. Dysfunction of polymorphonuclear leukocytes in uremia: role of parathyroid hormone. Kidney Int Suppl. 2001;78:S195–6.
Horl WH, Haag-Weber M, Mai B, Massry SG. Verapamil reverses abnormal [Ca2+]i and carbohydrate metabolism of PMNL of dialysis patients. Kidney Int. 1995;47(6):1741–5.
Cho SW, Soki FN, Koh AJ, Eber MR, Entezami P, Park SI, et al. Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. Proc Natl Acad Sci USA. 2014;111(4):1545–50.
Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55.
Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181(2):1232–44.
Perry HM 3rd, Chappel JC, Bellorin-Font E, Tamao J, Martin KJ, Teitelbaum SL. Parathyroid hormone receptors in circulating human mononuclear leukocytes. J Biol Chem. 1984;259(9):5531–5.
Alexiewicz JM, Klinger M, Pitts TO, Gaciong Z, Linker-Israeli M, Massry SG. Parathyroid hormone inhibits B cell proliferation: implications in chronic renal failure. J Am Soc Nephrol. 1990;1(3):236–44.
Gaciong Z, Alexiewicz JM, Linker-Israeli M, Shulman IA, Pitts TO, Massry SG. Inhibition of immunoglobulin production by parathyroid hormone. Implications in chronic renal failure. Kidney Int. 1991;40(1):96–106.
Gaciong Z, Alexiewicz JM, Massry SG. Impaired in vivo antibody production in CRF rats: role of secondary hyperparathyroidism. Kidney Int. 1991;40(5):862–7.
Kotzmann H, Koller M, Abela C, Clodi M, Riedl M, Graninger W, et al. Effects of parathyroid hormone and serum calcium on the phenotype and function of mononuclear cells in patients with primary hyperparathyroidism. Eur J Clin Invest. 1998;28(5):353–8.
Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19(7):665–73.
Klinger M, Alexiewicz JM, Linker-Israeli M, Pitts TO, Gaciong Z, Fadda GZ, et al. Effect of parathyroid hormone on human T cell activation. Kidney Int. 1990;37(6):1543–51.
Tzanno-Martins C, Futata E, Jorgetti V, Duarte AJ. Restoration of impaired T-cell proliferation after parathyroidectomy in hemodialysis patients. Nephron. 2000;84(3):224–7.
Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20(6):707–18.
Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109(9):3706–12.
Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A. 2008;105(44):16976–81.
Panaroni C, Fulzele K, Saini V, Chubb R, Pajevic PD, Wu JY. PTH signaling in osteoprogenitors is essential for B-lymphocyte differentiation and mobilization. J Bone Miner Res. 2015;30(12):2273–86.
Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, et al. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab. 2008;8(2):132–45.
Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. 2009;10(3):229–40.
Bhadada SK, Sridhar S, Ahluwalia J, Bhansali A, Malhotra P, Behera A, et al. Anemia and thrombocytopenia improves after curative parathyroidectomy in a patient of primary hyperparathyroidism (PHPT). J Clin Endocrinol Metab. 2012;97(5):1420–2.
Ortega A, Perez de Prada MT, Mateos-Caceres PJ, Ramos Mozo P, Gonzalez-Armengol JJ, Gonzalez Del Castillo JM, et al. Effect of parathyroid-hormone-related protein on human platelet activation. Clin Sci (Lond). 2007;113(7):319–27.
Verdoia M, Pergolini P, Rolla R, Nardin M, Barbieri L, Schaffer A, et al. Parathyroid hormone levels and high-residual platelet reactivity in patients receiving dual antiplatelet therapy with acetylsalicylic acid and clopidogrel or ticagrelor. Cardiovasc Ther. 2016;34(4):209–15.
Watson PH, Fraher LJ, Hendy GN, Chung UI, Kisiel M, Natale BV, et al. Nuclear localization of the type 1 PTH/PTHrP receptor in rat tissues. J Bone Miner Res. 2000;15(6):1033–44.
Pringle A, Smith EK. Renal vein thrombosis in acute hyperthyroidism. Br Med J. 1964;2(5410):675–6.
Manosroi W, Wannasai K, Phimphilai M. Pulmonary embolism and subclavian vein thrombosis in a patient with parathyroid carcinoma: case report and review of literature. J Med Assoc Thai. 2015;98(9):925–33.
Erem C, Kocak M, Nuhoglu I, Yilmaz M, Ucuncu O. Increased plasminogen activator inhibitor-1, decreased tissue factor pathway inhibitor, and unchanged thrombin-activatable fibrinolysis inhibitor levels in patients with primary hyperparathyroidism. Eur J Endocrinol. 2009;160(5):863–8.
Farahnak P, Larfars G, Sten-Linder M, Nilsson IL. Mild primary hyperparathyroidism: vitamin D deficiency and cardiovascular risk markers. J Clin Endocrinol Metab. 2011;96(7):2112–8.
Elbers LPB, Wijnberge M, Meijers JCM, Poland DCW, Brandjes DPM, Fliers E, et al. Coagulation and fibrinolysis in hyperparathyroidism secondary to vitamin D deficiency. Endocr Connect. 2018;7(2):325–33.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hamano, N., Komaba, H., Fukagawa, M. (2020). Effect of PTH on the Hematologic System. In: Covic, A., Goldsmith, D., Ureña Torres, P. (eds) Parathyroid Glands in Chronic Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-43769-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-43769-5_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-43768-8
Online ISBN: 978-3-030-43769-5
eBook Packages: MedicineMedicine (R0)