
Abstract
Rubber tree is an important crop species grown over an expansive area in southern Thailand to harvest natural latex. The most commonly grown clone here is RRIM 600, but other clones are also grown in some areas, including BPM 24 which is a cytoplasmic male sterile elite clone. The organellar genomes, from both mitochondria and chloroplast of BPM 24 have been analyzed. The chloroplast sequence of BPM 24 is nearly identical to that published for the clone RRIM 600 with the exception of 5 SNPs, which were either non-coding or silent. The mitochondrial genome is remarkably different from that of other rubber tree clones such as RRIM 600, and the cause of cytoplasmic sterility has been identified as a novel transcript containing a portion of ATPase subunit 9 (atp9). The nuclear genome of BPM 24 was assembled using a combination of Illumina and PacBio sequence data and Chicago HiC scaffolding, which produced an assembly containing a relatively small number of fragmented contigs. A high-density SNP map was used to anchor almost one-third of the sequence into 18 linkage groups that captured two-thirds of the protein coding sequences. Comparing orthologous genes with other members of the Euphorbiaceae family identified cassava as the closest relative with a shared whole genome duplication event that took place following a split from a common ancestor with castor bean.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Rubber tree genome
- De novo assembly
- Hi-C assembly
- Genome anchoring
- SNP markers
- Genome duplication
- Gene synteny
- Organellar genomes
- Cytoplasmic male sterility
4.1 Introduction
Hevea brasiliensis, or rubber tree, is a deciduous perennial tree crop indigenous to the rain forests of the Amazonian basin in South America. Of the 10 Hevea species, H. brasiliensis is the only one which produces enough high-quality natural rubber on a commercial scale, this accounting for more than 98% of total global production (Priyadarshan and Clément-Demange 2004). Rubber tree is an outbreeding species with a long breeding cycle which may take around 30–40 years. Thus, its genome sequence is important for high-throughput marker discovery and subsequent identification of important QTLs, as well as the underlying genes that control important commercial traits. Only then can marker-assisted breeding be employed effectively, allowing large numbers of desirable traits to be introduced into a single clone using a gene pyramiding approach. For such a strategy, it is important to have markers that are either the causative mutation of the trait of interest, or those which are tightly linked with the causative genes.
Rubber farming in Thailand began in 1899 when Phraya Ratsadanupradit Mahison Phakdi, the commissioner of Monthon Phuket (consisting of the provinces Phuket, Thalang, Ranong, Phang Nga, Takua Pa, Krabi, Kelantan and Terengganu with Satun added later), visited Malaya (now Malaysia) and observed the rubber plantations and farming methods employed there. He did not obtain any of the young rubber trees from that visit, but later visited Indonesian rubber farms and brought some young rubber tree seedlings back to Thailand, which were planted in Kantang District, Trang Province. These initial plantations were used to show the profitability of rubber trees and to produce seeds for distribution to local small-scale farmers (Stifel 1973). Since around 1960, the most popular clone in Thailand has been RRIM 600. Additionally, some smallholder farms planted BPM 24, a clone recommended for small-scale planting. The clone BPM 24 has good latex production and response to wounding, and is best suited to cut tapping.
One of the rubber tree clones with a sequenced genome is BPM 24 (BPM = Balai Penelitian Perkebunan Medan) (Pootakham et al. 2017). The clone BPM 24 is a cytoplasmic male sterile descendant of a GT 1 (female) x AVROS 1734 (male) cross that was developed in the 1990s and is considered a notable clone (Priyadarshan and Clément-Demange 2004). The clone GT 1 (GT = Gondang Tapen, Indonesia) is a primary clone that can be traced back to the 1920s. GT 1 itself is male sterile, as is its offspring BPM 24, and the offspring of BPM 24. The male parent, AVROS 1734 (AVROS = Algemeene Vereeniging van Rubberplanters ter Oostkust van Sumatra), is a clone which originated from Indonesia that has high yield and good resistance to common rubber tree diseases. References to AVROS 1734 in the literature go as far back as the 1920s. Both parental strains tend to have higher yields than BPM 24 across a range of environments; yet BPM 24 is considered an elite clone and was recommended as a preferred clone for small-scale planting because it has good response to cut tapping and exhibits a high degree of resistance to two major fungal pathogens (Phytophthora and Corynespora) found throughout Southeast Asia.
4.2 Genome Assembly
Plants have three distinct genomes: nuclear, mitochondrial and plastid. Mitochondria and chloroplasts are thought to have originated from separate endosymbiotic events where the cell incorporated a proteobacterium or a cyanobacterium, respectively (Zimorski et al. 2014). Since these ancient events, the three genomes have undergone significant co-evolution involving gene transfer between them, primarily in the form of genes moving into the nuclear genome (Lloyd and Timmis 2011). Mitochondria and chloroplasts have mRNA transcript processing not seen in the nuclear genome, including polycistronic cleavage, mRNA editing and trans-splicing (Wicke et al. 2011).
4.2.1 Chloroplast Genome
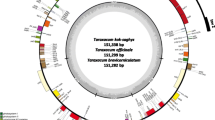
The chloroplast genomes of land plants are quite conserved, they generally have a cyclic structure with two single copy regions separated by two copies of an inverted repeat, a large single copy region and a small single copy region (Wicke et al. 2011). The rubber tree chloroplast is no exception to this (Tangphatsornruang et al. 2011). The rubber tree RRIM 600 chloroplast genome was assembled de novo into a 161,191 bp circular DNA strand using 454 shotgun sequencing and Newbler (Tangphatsornruang et al. 2011, Fig. 4.1). The genome contains 112 unique genes including 30 transfer RNA (tRNA) genes, 4 ribosomal RNA (rRNA) genes and 78 predicted protein coding genes (Table 4.1). The large single copy region is 89,209 bp in size and the small single copy region is 18,362 bp in size, separated by the inverted repeats which are 26,810 bp each. Phylogenetic analysis of the chloroplast genes identified cassava (Manihot esculenta) as the closest relative. The rubber tree chloroplast genome has a 30 kb inversion compared to the cassava genome. Comparing the chloroplast genomic sequence to chloroplast transcript sequences revealed 51 RNA editing sites that occur in 26 protein coding genes and 3 introns. The BPM 24 chloroplast sequence is identical to the RRIM 600 published sequence, with the exception of four synonymous coding SNPs in the gene ndhF and one non-coding SNP (Table 4.2).

The rubber tree chloroplast genome (RRIM 600). The inner circle delineates the single-copy regions flanked by the inverted repeats. The outer circle shows the exons of each gene with plus/minus strand coding indicated by the exon box appearing outside or inside the circle, respectively. Introns are represented by a grey box and * in the name. Pseudogenes are marked with an #. Arrows indicate the position of a 30 kb inversion compared to the cassava chloroplast genome (Tangphatsornruang et al. 2011)
4.2.2 Mitochondrial Genome
Plant mitochondrial genomes range in size from 200 kb in Brassica hirta (Palmer and Herbon 1987) to 11.3 Mb in Silene conica (Sloan et al. 2012). Mitochondrial genomes have expanded in land plants and accumulated large numbers of repeat sequences which cause frequent recombination events and dynamic genome rearrangements within a species, leading to the generation of multiple circular DNA strands with overlapping sequence and different copy number (Turmel et al. 2003; Bullerwell and Gray 2004; Allen et al. 2007; Chang et al. 2011).
The mitochondria of BPM 24 was investigated in order to obtain a reference mitochondrial genome and also to investigate the cause of male sterility (Shearman et al. 2014). Maternal inheritance of male sterility in BPM 24 and the lack of causative mutations in the chloroplast genome suggested that the cause of male sterility in BPM 24 could be within the mitochondrial genome. The mitochondrial genome was assembled into 37 contigs using 454 sequence data, including 8 kb paired-end libraries and Illumina paired-end sequencing data. The scaffold graph, which shows joins between contigs, was complex with many contigs joining to multiple other contigs. This complex scaffold graph suggests that the genome exists not as a single circle, but as many subgenomic circles with partial sequence overlap. A master circle mitochondrial sequence was generated by traversing the contig graph in such a way that all contigs were used at least once with as little duplication as possible. This resulted in a mitochondrial sequence that was 1,325,823 bp in size with approximately 350 kb of duplicated sequence (Fig. 4.2).

The BPM 24 mitochondria master circle. Grey arches represent Illumina paired-end sequence mapping data, blue arches show direct repeats, and orange arches show inverted repeats (inner circle). The outer circle shows the exons of each gene with plus/minus strand coding indicated by the exon box appearing outside or inside the circle, respectively (Shearman et al. 2014)
The mitochondrial genome was annotated using Illumina RNA-seq data which allowed for 65 open reading frames to be identified (Table 4.3; Shearman et al. 2014). There was group II trans-splicing in three genes, nad1, nad2 and nad5, as has been found in multiple plant species (Bonen 2008). Group II trans-splicing is the splicing of introns from two or more separate mRNA molecules that are encoded on opposite strands. Each of the trans-spliced nad genes identified in rubber tree are also trans-spliced in other species (Bonen 2008). There were 19 tRNA genes identified and five of them occurred twice in the assembled mitochondrial master circle (Table 4.3). Seven tRNA genes are from gene transfer events, largely consistent with previously identified chloroplast gene transfer events (Wang et al. 2007). A notable difference is that BPM 24 lacks a copy of trnS-GGA which is common in species closely related to rubber tree, including Ricinus communis (castor bean), suggesting that the chloroplast-derived trnS-GGA was lost in the rubber tree after the split from R. communis.
The BPM 24 mitochondrial contigs have 11 rearrangements compared to RRIM 600, with five of these occurring within 1 kb of a gene. The mitochondrial genome was found to contain multiple structural arrangements within a single individual for each of the six varieties that were sequenced (BPM 24, RRIM 600, RRII 105, RRIC 110, RRIT 251, PB 235), consistent with the existence of many subgenomic circles. One of these 11 rearrangements was unique to BPM 24 and includes 240 bp of sequence not shared by any other clone, making it a good candidate for the cause of male sterility. This additional sequence is expressed, according to RNA-seq data, and encodes an mRNA of 51 amino acids, 33 of which are identical to the tail end of ATPase subunit 9 (atp9). An entire trans-membrane domain is included in this additional sequence which likely allows it to compete with the functional copy of atp9 in the ATP synthase complex, and reduce the efficiency of ATP production sufficiently to cause apoptosis in the high-energy demanding anthers. A novel or fusion transcript is a common occurrence in cytoplasmic male sterile plants and often involves a portion of, or is near an ATP synthase subunit gene (Carlsson et al. 2008). To conclude, the cause of cytoplasmic male sterility in BPM 24 should be the expression of this additional mRNA that resulted from a genomic rearrangement.
4.2.3 Nuclear Genome
The cost and difficulty of obtaining a de novo whole genome sequence depend on the type of sequencing technology used to interrogate each nucleotide. Short read shotgun sequencing using platforms such as Illumina, BGI and Ion Torrent, alone can generate a fragmented genome assembly that contains the majority of unique sequence. Such a genome assembly is heavily fragmented because the short read lengths cannot span repeat sequences. Long read shotgun sequencing platforms such as PacBio and Nanopore now allow for genomes to be less fragmented, but still far from complete. A method currently gaining traction is the use of short read sequencing with high-throughput chromosome conformation capture (HiC) to explore DNA linkage in chromatin (Putnam et al. 2016). Segments of DNA that are in close proximity are more likely to ligate than distant segments and this information can be used to scaffold contigs generated through shotgun sequencing. There are two methods employed; one involves using high molecular weight DNA to reconstitute chromatin in vitro followed by proximity ligation and shotgun sequencing, and the other uses in vivo chromatin to generate proximity ligation allowing for longer range capture than the in vitro method.
The rubber tree nuclear genome consists of 18 chromosomes and is roughly 2.15 Gb in size. The rubber tree genome is one of the more difficult genomes to sequence because of its size and high repeat content, with repeat sequences accounting for around 78% of the genome (Rahman et al. 2013). A draft sequence of the rubber tree genome, utilising a whole genome shotgun sequencing approach, became available in 2013 (Rahman et al. 2013). The large number of repeat sequences caused a heavily fragmented genome assembly, despite the genome assembly using data from three different sequencing platforms (454, Illumina and SOLiD), and multiple library types (single end, and 200 bp, 500 bp, 8 kb and 20 kb paired-end reads). Prior to the scaffolding step, the library consisted of 1,223,366 contigs ranging from 200 to 46,694 bp. Scaffolding reduced the total assembly to 1,150,326 scaffolds and contigs ranging from 200 to 531,465 bp. Linkage map information (Le Guen et al. 2011) was incorporated into the rubber tree genome allowing for 143 scaffolds to be anchored (Rahman et al. 2013). Additional attempts to use linkage maps to place scaffolds made incremental improvements, but for the most part, these only joined a small number of the larger contigs (Shearman et al. 2015; Pootakham et al. 2015).
The BPM 24 nuclear genome was assembled as an ongoing process over several years, combining data from multiple platforms as sequencing technology continued to advance (Pootakham et al. 2017). In total, there were ten separate libraries with insert sizes ranging from 350 bp to 12 kb consisting of: 6 Gb of single end and 1 Gb of 8 to 20 kb mate-pair libraries from 454 GS FLX + runs, 79 Gb of Illumina HiSeq 2000 paired-end reads, and 56 Gb of 10 to 12 kb insert PacBio reads. In addition to this, scaffolding was performed by Dovetail Genomics using Capture Hi-C Analysis of Genome Organization (Chicago) technology to construct long-range linkage libraries based on proximity ligation of in vitro reconstituted chromatin (Putnam et al. 2016). A total of six Chicago libraries were constructed and sequenced on an Illumina HiSeq system which produced 769 million paired-end reads of DNA fragments between 1 and 50 kb, yielding approximately 44 fold coverage. As a result of this approach, multiple assemblies of the genome were produced allowing for comparison of the different assembly methods and data types (Table 4.4). The first assembly used SOAPdenovo to assemble the Illumina reads into contigs, followed by assembly with the 454 data using Newbler which resulted in 989,097 contigs totalling 869 Mb with an N50 of 1,316 bp. Adding the long read PacBio data to this for scaffolding using DBG2OLC software reduced the number of contigs to 658,583 and increased the total sequence to 1,249 Mb with an N50 of 51,412 bp. This assembly was then used as input for Chicago Hi-C, which reduced the number of contigs to 592,580 and increased the total sequence to 1,256 Mb and the N50 to 96,825 bp. The long reads of the PacBio platform allowed many repeat sequences to be traversed in a single read which greatly improved the assembly quality compared to short read sequences. The addition of Chicago Hi-C technology roughly doubled the N50 size of the assembly, thus representing a significant increase in assembly quality.
Adding linkage map information to the assembly allowed for 1,568 scaffolds totalling 363 Mb to be anchored onto 18 linkage groups. These anchored scaffolds included 68.4% of the predicted protein-coding genes and thus represents a large portion of the unique sequences in the rubber tree genome. The markers used to anchor these scaffolds in a genetic map were from genotype-by-sequencing which utilized methylation-sensitive enzymes, giving the resulting SNPs a high chance to be in euchromatic regions. The sequences which remain unanchored contain roughly 80% repetitive sequence, making it difficult to anchor.
Repeat sequences account for 69.2% of the assembled genome and are composed of 3.5 Mb (0.28%) of low complexity elements, 1.3 Mb (0.1%) of small RNA structures, 13 Mb (1.11%) of simple sequence repeats, 21 Mb (1.72%) of DNA transposons, 160 Mb (12.73%) of unclassified elements and nearly 670 Mb (53.31%) of retrotransposons (Table 4.5). The majority of retrotransposons are long terminal repeats (LTR; 51.55%), and include Copia-like (10.23%) and Gypsy-like (37.75%) elements, which are the most abundant LTR subfamilies in other sequenced Euphorbia genomes (Chan et al. 2010; Sato et al. 2011; Wang et al. 2014).
The protein-coding sequences were annotated using multiple ab initio gene prediction tools, homology searches against sequence databases and the transcript alignment program, PASA. Sequencing data included PacBio Iso-seq libraries (46,129 non-redundant transcripts) and published RNA-seq data obtained from leaf, bark, latex and root (Rahman et al. 2013; Chao et al. 2015; Li et al. 2016; Tang et al. 2016). The RNA-seq data was incorporated with outputs from gene prediction programs and homology searches using the program EvidenceModeler, which produced a consensus set of 43,868 protein-coding genes. In the rubber tree, the average gene length (2,747 bp), average exon length (223 bp), and average number of exons per gene (4) were comparable to those in cassava (Wang et al. 2014) and castor bean (Chan et al. 2010).
Of the 43,868 predicted genes, 30,232 (68.92%) were assigned InterPro motifs, 20,107 received GO annotations, and 33,718 (76.9%) and 16,794 (38.3%) had significant BLASTP matches to proteins in the Genbank non-redundant protein and the SwissProt databases, respectively. The long PacBio reads facilitated the identification of alternative splicing in several genes, consistent with Maize 58 and Sorghum 59. In addition to protein-coding genes, there were 623 tRNA, 274 (rRNA), 282 small nucleolar RNA (snoRNA), 164 small nuclear RNA (snRNA) and 193 micro RNA (miRNA) genes in the BPM 24 assembly. A large number of resistance-related genes, 1,275 putative pattern-recognition receptor genes, were identified in the rubber tree genome. Resistance genes tended to be grouped in clusters indicative of tandem duplication followed by function diversification.
A major evolution mechanism that is common in eudicots that can give rise to new genes is whole genome duplication followed by gene loss and diploidization. The rubber tree appears to have experienced a paleotetraploidization event based on 164 syntenic blocks, of ten or more genes each, containing 2,951 paralogous gene pairs distributed across the 18 linkage groups.
4.3 Synteny with Euphorbiaceae
Comparison of the BPM 24 rubber tree genome with other Euphorbiaceae species reaffirms that cassava is the closest ancestor (Pootakham et al. 2017). The evolutionary divergence of the rubber tree from other Euphorbiaceae was analysed by comparing the transversion rate at fourfold degenerate sites (4DTv) of orthologous gene pairs identified in syntenic blocks between the rubber tree and its close relatives. A whole genome duplication event took place after the ancestor of castor bean split from the other lineages and this is reflected in the 4DTv values from the transversion rate comparison. Both the rubber tree and cassava also have 18 chromosomes while physic nut (Jatropha curcas) has 11 and castor bean has 10, also supporting a whole genome duplication event before the rubber tree and cassava diverged (Fig. 4.3a). Speciation times were estimated based on orthologues of single gene families in each of the species, with black cottonwood as an outgroup (Fig. 4.3b). The origin of the Euphorbiaceae family has been estimated to be at 58 million years ago (mya) (Magallon et al. 1999). The maximum likelihood tree suggested a speciation time of around 36 mya for Hevea and Manihot, and a divergence time of the Ricinus lineage from other Euphorbia around 60 mya, this in agreement with the estimates from previous phylogenetic analyses (Bredeson et al. 2016).

a Distribution of 4DTv distances between syntenic gene pairs among rubber tree, cassava and castor bean. b A maximum likelihood phylogenetic tree of Euphorbiaceae species, with black cottonwood as an outgroup (Pootakham et al. 2017)
Comparison of the gene sets of the rubber tree with other sequenced Euphorbiaceae species (M. esculenta, J. curcas and R. communis) identified 15,605 orthologous gene clusters within the rubber tree. The comparison again confirms that the rubber tree and cassava shared the most recent ancestor, with 13,680 gene clusters in common between the two species. The rubber tree shares 13,040 gene clusters with J. curcas and 13,028 with R. communis and a total of 11,759 gene clusters were common to all four Euphorbiaceae species considered. The rubber tree also had 934 gene families that were not found in the other species, where these were significantly enriched with genes related to molecular function categories such as catalytic and binding activities.
4.4 Conclusion
As we obtain whole genome sequence data from multiple individuals of each species, it has become increasingly clear that significant structural variation can exist between different individuals of the same species, including genome rearrangement and the presence/absence variations of entire genes (Hu et al. 2018; Sun et al. 2018). Thus, sequencing only a single individual results in a high chance of missing a large portion of the variation that exists within that species and limits the potential of identifying variants for QTLs of interest. Having whole genome assemblies of multiple rubber tree clones is important since marker-assisted selection for QTLs is desirable for rubber tree. We now have whole genome assemblies for BPM 24, RRIM 600 and Reyan 7-33-97, including chloroplast and mitochondrial genomes. These will serve as valuable resources for marker development, QTL mapping, and functional variant discovery. As the cost of long-read sequencing becomes more affordable, it is possible to obtain high-quality whole genome assemblies from additional rubber tree clones to investigate existing structural variations that may be associated with agronomic traits of interest.
References
Allen JO, Fauron CM, Minx P, Roark L, Oddiraju S, Lin GN, Meyer L, Sun H, Kim K, Wang C, Du F, Xu D, Gibson M, Cifrese J, Clifton SW, Newton KJ (2007) Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics 177:1173–1192
Bonen L (2008) Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion 8:26–34
Bredeson JV, Lyons JB, Prochnik SE, Wu GA, Ha CM, Edsinger-Gonzales E, Grimwood J, Schmutz J, Rabbi IY, Egesi C, Nauluvula P, Lebot V, Ndunguru J, Mkamilo G, Bart RS, Setter TL, Gleadow RM, Kulakow P, Ferguson ME, Rounsley S, Rokhsar DS (2016) Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nat Biotechnol 34:562–570
Bullerwell CE, Gray MW (2004) Evolution of the mitochondrial genome: protist connections to animals, fungi and plants. Curr Opin Microbiol 7:528–534
Carlsson J, Leino M, Sohlberg J, Sundström JF, Glimelius K (2008) Mitochondrial regulation of flower development. Mitochondrion 8:74–86
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, Cahoon EB, Gedil M, Stanke M, Haas BJ, Wortman JR, Fraser-Liggett CM, Ravel J, Rabinowicz PD (2010) Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28:951–956
Chang S, Yang T, Du T, Huang Y, Chen J, Yan J, He J, Guan R (2011) Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in Brassica. BMC Genom 12:497
Chao J, Chen Y, Wu S, Tian WM (2015) Comparative transcriptome analysis of latex from rubber tree clone CATAS8-79 and PR107. Genom Data 5:120–121
Hu Z, Wang W, Wu Z, Sun C, Li M, Lu J, Fu B, Shi J, Xu J, Ruan J, Wei C, Li Z (2018) Novel sequences, structural variations and gene presence variations of Asian cultivated rice. Sci Data 5:180079
Le Guen V, Garcia D, Doaré F, Mattos CRR, Condina V, Couturier C, Chambon A, Weber C, Espéout S, Seguin M (2011) A rubber tree’s durable resistance to Microcyclus ulei is conferred by a qualitative gene and a major quantitative resistance factor. Tree Genet Genomes 7:877–889
Li D, Wang X, Deng Z, Liu H, Yang H, He G (2016) Transcriptome analyses reveal molecular mechanism underlying tapping panel dryness of rubber tree (Hevea brasiliensis). Sci Rep 6:23540
Lloyd AH, Timmis JN (2011) The origin and characterization of new nuclear genes originating from a cytoplasmic organellar genome. Mol Biol Evol 28:2019–2028
Magallon S, Crane PR, Herendeen PS (1999) Phylogenetic pattern, diversity, and diversification of eudicots. Ann Mo Bot Gard 86:297–372
Palmer JD, Herbon LA (1987) Unicircular structure of the Brassica hirta mitochondrial genome. Curr Genet 11:565–570
Pootakham W, Ruang-Areerate P, Jomchai N, Sonthirod C, Sangsrakru D, Yoocha T, Theerawattanasuk K, Nirapathpongporn K, Romruensukharom P, Tragoonrung S, Tangphatsornruang S (2015) Construction of a high-density integrated genetic linkage map of rubber tree (Hevea brasiliensis) using genotyping-by-sequencing (GBS). Front Plant Sci 6:367
Pootakham W, Sonthirod C, Naktang C, Ruang-Areerate P, Yoocha T, Sangsrakru D, Theerawattanasuk K, Rattanawong R, Lekawipat N, Tangphatsornruang S (2017) De novo hybrid assembly of the rubber tree genome reveals evidence of paleotetraploidy in Hevea species. Sci Rep 7:41457
Priyadarshan PM, Clément-Demange A (2004) Breeding Hevea rubber: formal and molecular genetics. Adv Genet 52:51–115
Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, Haussler D, Rokhsar DS, Green RE (2016) Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res 26:342–350
Rahman AY, Usharraj AO, Misra BB, Thottathil GP, Jayasekaran K, Feng Y, Hou S, Ong SY, Ng FL, Lee LS, Tan HS, Sakaff MK, Teh BS, Khoo BF, Badai SS, Aziz NA, Yuryev A, Knudsen B, Dionne-Laporte A, Mchunu NP, Yu Q, Langston BJ, Freitas TA, Young AG, Chen R, Wang L, Najimudin N, Saito JA, Alam M (2013) Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genom 14:75
Sato S, Hirakawa H, Isobe S, Fukai E, Watanabe A, Kato M, Kawashima K, Minami C, Muraki A, Nakazaki N, Takahashi C, Nakayama S, Kishida Y, Kohara M, Yamada M, Tsuruoka H, Sasamoto S, Tabata S, Aizu T, Toyoda A, Shin-i T, Minakuchi Y, Kohara Y, Fujiyama A, Tsuchimoto S, Kajiyama S, Makigano E, Ohmido N, Shibagaki N, Cartagena JA, Wada N, Kohinata T, Atefeh A, Yuasa S, Matsunaga S, Fukui K (2011) Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res 18:65–76
Shearman JR, Sangsrakru D, Ruang-Areerate P, Sonthirod C, Uthaipaisanwong P, Yoocha T, Poopear S, Theerawattanasuk K, Tragoonrung S, Tangphatsornruang S (2014) Assembly and analysis of a male sterile rubber tree mitochondrial genome reveals DNA rearrangement events and a novel transcript. BMC Plant Biol 14:45
Shearman JR, Sangsrakru D, Jomchai N, Ruang-Areerate P, Sonthirod C, Naktang C, Theerawattanasuk K, Tragoonrung S, Tangphatsornruang S (2015) SNP identification from RNA sequencing and linkage map construction of rubber tree for anchoring the draft genome. PLoS One 10:e0121961
Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, Palmer JD, Taylor DR (2012) Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol 10:e1001241
Stifel SD (1973) The growth of the rubber economy of Southern Thailand. J Southeast Asian Stud 4:107–132
Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, Song W, Zhang M, Cui Y, Dong X, Liu H, Ma X, Jiao Y, Wang B, Wei X, Stein JC, Glaubitz JC, Lu F, Yu G, Liang C, Fengler K, Li B, Rafalski A, Schnable PS, Ware DH, Buckler ES, Lai J (2018) Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat Genet 50:1289–1295
Tang C, Yang M, Fang Y, Luo Y, Gao S, Xiao X, An Z, Zhou B, Zhang B, Tan X, Yeang HY, Qin Y, Yang J, Lin Q, Mei H, Montoro P, Long X, Qi J, Hua Y, He Z, Sun M, Li W, Zeng X, Cheng H, Liu Y, Yang J, Tian W, Zhuang N, Zeng R, Li D, He P, Li Z, Zou Z, Li S, Li C, Wang J, Wei D, Lai CQ, Luo W, Yu J, Hu S, Huang H (2016) The rubber tree genome reveals new insights into rubber production and species adaptation. Nat Plants 2:16073
Tangphatsornruang S, Uthaipaisanwong P, Sangsrakru D, Chanprasert J, Yoocha T, Jomchai N, Tragoonrung S (2011) Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 475:104–112
Turmel M, Otis C, Lemieux C (2003) The mitochondrial genome of Chara vulgaris: insights into the mitochondrial DNA architecture of the last common ancestor of green algae and land plants. Plant Cell 15:1888–1903
Wang D, Wu YW, Shih AC, Wu CS, Wang YN, Chaw SM (2007) Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol Biol Evol 24:2040–2048
Wang W, Feng B, Xiao J, Xia Z, Zhou X, Li P, Zhang W, Wang Y, Møller BL, Zhang P, Luo M-C, Xiao G, Liu J, Yang J, Chen S, Rabinowicz PD, Chen X, Zhang H-B, Ceballos H, Lou Q, Zou M, Carvalho LJCB, Zeng C, Xia J, Sun S, Fu Y, Wang H, Lu C, Ruan M, Zhou S, Wu Z, Liu H, Kannangara RM, Jørgensen K, Neale RL, Bonde M, Heinz N, Zhu W, Wang S, Zhang Y, Pan K, Wen M, Ma P-A, Li Z, Hu M, Liao W, Hu W, Zhang S, Pei J, Guo A, Guo J, Zhang J, Zhang Z, Ye J, Ou W, Ma Y, Liu X, Tallon LJ, Galens K, Ott S, Huang J, Xue J, An F, Yao Q, Lu X, Fregene M, López-Lavalle LAB, Wu J, You FM, Che, M, Hu S, Wu G, Zhong S, Ling P, Chen Y, Wang Q, Liu G, Liu B, Li K, Peng M (2014) Cassava genome from a wild ancestor to cultivated varieties. Nat Commun 5:5110
Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D (2011) The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol 76(3–5):273–297
Zimorski V, Ku C, Martin WF, Gould SB (2014) Endosymbiotic theory for organelle origins. Curr Opin Microbiol 22:38–48
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Shearman, J.R., Pootakham, W., Tangphatsornruang, S. (2020). The BPM 24 Rubber Tree Genome, Organellar Genomes and Synteny Within the Family Euphorbiaceae. In: Matsui, M., Chow, KS. (eds) The Rubber Tree Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-030-42258-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-42258-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-42257-8
Online ISBN: 978-3-030-42258-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)