
Abstract
This chapter introduces to the book part “Molecular Basis of Liver Stiffness and Cell Biology.” The molecular basis of liver stiffness (LS) is complex and multifactorially controlled at the systemic, cellular, and intracellular level. In addition, all cells including liver cells sense and respond to their environmental stiffness through mechano-signaling processes. Principally, stiffness is modulated either by structural conditions, e.g., matrix deposition such as collagen, the degree of perfusion or pressure-related conditions that are all inter-related. Pressure itself is controlled in a complex manner. While the static component of pressure is mainly affected by the vascular bed with its elastic properties and water filling status, the dynamic component is related to blood flow, hepatic resistance, and the hemorheology. At the intracellular level, stiffness is affected by intracellular pressure and stretch forces on the cellular membranes and intermediary filaments. The intracellular pressure is likewise controlled by many conditions including transport proteins to control osmotic pressure, or water influx, e.g., by aquaporins. We are still at the beginning to understand how liver cells, fibroblasts and hepatic stellate cells sense environmental stiffness. Quantitative in vitro studies of cells adhering to gels of varying adjustable stiffness are a useful tool in order to elucidate these mechanisms. Other chapters of this book part will introduce to the role of stiffness for hepatocyte function in vitro, to liver mechanics and the profibrotic response at the cellular level and, finally, to the role of sinusoidal pressure and hepatic arterialization in driving hepatic fibrosis. It is expected that biomechanic concepts will be instrumental for our future understanding of stiffness, fibrosis development and liver diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Liver cirrhosis
- Sinusoidal pressure hypothesis
- Liver stiffness
- Liver congestion
- Arterialization
- Stretch force
- Hepatic stellate cells
- Fibroblasts
- Cellular and intercellular mechano-signaling
- Biomechanics
- Integrins
- Talin
- Vinculin
- Mechanosensitive ion channels
- Extracellular matrix
- Actin
- Focal adhesions
Introduction
Liver stiffness (LS) appears to be a rather complex parameter that is modulated by many factors at the systemic, organ-, cellular, and intracellular level. This is primarily the matrix composition of the liver itself such as collagen deposition. Second, pressure-related factors contribute largely to LS and, third, liver perfusion. The dynamic component of pressure is associated with blood flow and eventually with an intact heart action. However, in combination with blood flow, the hepatic resistance and hemorheology also contribute to LS. Finally, there is also a static pressure component mainly derived from the vascular filling, e.g., through water retention but also characteristics of the vascular wall including muscle action and elastic properties. Figure 54.1 highlights all organ systems that are engaged in the control of LS. The following paragraphs are far from being complete but are thought to describe important aspects to be considered for a better understanding of LS in the future.

Organ systems and liver stiffness
Hepatic Blood Flow, Resistance, and Hemorheology
Many hemodynamic aspects of LS have been already discussed elsewhere in this book, e.g., in the chapter introducing the “Sinusoidal Pressure Hypothesis.” Some aspects listed in Fig. 54.2, however, are new and deserve some additional discussions. More details are also listed in Table 54.1. Thus, capillary pressure is expected to contribute to LS, similar to its role in lungs. Capillary pressure is the pressure between two immiscible fluids in a thin tube, resulting from the interactions of forces between the fluids and solid walls of the tube. Capillary pressure can serve as both an opposing and driving force for fluid transport. The role of capillary pressure in liver sinusoids is still largely unexplored, so its role for LS and molecular factors. However, it can be assumed that both blood constituents and wall properties contribute to capillary pressure.

Liver stiffness at the systemic level. Liver stiffness is primarily modulated by matrix, blood flow, hepatic resistance, hemorheology, and static pressure
This links to hemorheology or blood rheology which is the study of flow properties of blood and its elements of plasma and cells. Proper tissue perfusion can occur only when blood’s rheological properties are within certain levels and it has been well conceived for a long time that alterations of these properties play a significant role in disease processes. Blood viscosity is determined by plasma protein concentration, hematocrit (volume fraction of red blood cells), temperature, and mechanical properties of red blood cells. These mechanic properties include erythrocyte deformability and erythrocyte aggregation. Blood can be considered as a non-Newtonian fluid as the viscosity of blood varies with shear rate. Blood becomes less viscous at high shear rates like those experienced with increased flow such as during exercise or in peak-systole. Contrarily, blood viscosity increases when shear rate goes down with increased vessel diameters or with low flow, such as downstream from an obstruction or in diastole. This decrease of blood viscosity in capillaries is called Fåhraeus–Lindqvist effect [1].
Plasma viscosity is determined by water-content and macromolecular components. Nevertheless, hematocrit has the strongest impact on whole blood viscosity. One unit increase in hematocrit can cause up to a 4% increase in blood viscosity. This relationship becomes even stronger with increasing hematocrit. Thus, when the hematocrit rises from 40 to 60%, relative viscosity of the blood rises from 4 to 8, which is an increase by 100% [2]. In polycythemia, the blood viscosity can become as great as 10 times that of water, and its flow through blood vessels is greatly retarded because of increased resistance to flow.
Stiffness at the Cellular Level
As already discussed in book Part IV in the chapter of “Histological Confounders,” several cellular conditions are known to be associated with LS. Fibrosis or collagen deposition shows the closest association with LS. Fibrosis is followed by features of hepatocyte injury including ballooning, lobular and portal inflammation, Mallory’s hyaline in the now called Mallory Denk bodies, and apoptosis. Inflammation is followed by other histological features that are positively and significantly associated with LS: microgranulomas, acidophil bodies, megamitochondria, glycogenated nuclei, and large lipo-granulomas. These mostly intracellular histological parameters are all features of apoptotic cell damage or death. Notably, steatosis itself such as lipid droplets are not significantly correlated with LS, in some cohorts even slightly negatively. The role of hemodynamic pressure is visualized in Fig. 54.3. It demonstrates how vascular pressure or sinusoidal pressure causes stretching of peri-vascular or perisinusoidal aligned structures or cells. These stretch forces will further elevate stiffness or LS but also engage in biomechanical signaling [3,4,5,6]. There will be also bidirectional interactions between pressure and peri-vascular structures. For instance, inflammation and ballooned hepatocytes will increase vascular resistance, increase pressure, and further stretch the surrounding.

Modulation of tissue stiffness by vascular or sinusoidal pressure. A liver sinus is shown schematically. Hepatocyte cell death, inflammation, or congestion all lead to increased sinusoidal pressure that causes stretching of, e.g., hepatic stellate cells (HSC), liver sinus enothelial cells (LSEC) or hepatocytes (HC)
Intracellular Components and Stiffness
Figure 54.4 now briefly highlights cellular and intracellular structures that can affect cellular stiffness and organ stiffness such as LS. It should be noted, however, that our knowledge about these intracellular constiutents are still largely unexplored and poorly validated. Apart from cellular “matrix constituents,” intracellular pressure will control wall tension and tension of intermediate filaments such as cytokeratin 18 (CK18). Many other cellular proteins are involved in anchoring cells to ECM or between cells. Thus, adherens junctions (AJ) are not only involved in anchoring the cell to the ECM but are also actively involved in transducing mechanical forces. AJ contain cadherins (such as E-Cadherin and N-Cadherin) that are linked to the cytoskeleton (F-actin) via linker proteins β-catenin and α-catenin [7]. Cadherin-based cellular adhesions signal by a broad range of extra-, inter-, and intracellular mechanisms, which involve several kinases and phosphatases [8]. Tight junctions (TJ) are found at the apical membrane of all epithelia, thereby acting as barriers for lipids and proteins by preventing diffusion between apical and basolateral intramembrane domains (Fig. 54.5). TJ consist of transmembrane proteins including occludin, claudins, tricellulin, and junctional adhesion molecules (JAMs) as well as cytosolic proteins acting as scaffolding proteins that anchor membrane components to the actin cytoskeleton, e.g., ZO-1 to -3 or include signaling molecules and transcription factors (e.g., ZONAB) [9]. Their elevated expression, namely, occludin, claudin 1, 2, 4 and 7, has been observed in liver cirrhosis and HCC [10, 11]. Desmosomes are adhesive junctions consisting of transmembrane proteins (desmoglein and desmocollin) that interact with linker molecules of the armadillo family (plakoglobin, plakophilins, and desmoplakin) [12], thereby providing resistance to mechanical forces through direct interactions with cytokeratins, major proteins of the keratin-containing intermediate filaments (IF) [13, 14]. A recent study investigating mechanical pressure (BDL rat model) and IF changes in liver demonstrated a disappearance of pericanalicular sheath and rearrangement of IF at the hepatocyte periphery [15]. IF in hepatocytes are mainly composed of CK18 and form a meshwork extending from desmosomes at the lateral cell membrane throughout the cytoplasm (Fig. 54.5). Desmosomal cadherins interact with each other and facilitate IF attachment. Furthermore, desmosomes are extremely stable and may play a role in reorganization of gap junctions (GJ) [16] that are important for intercellular communication. GJ are formed by hemichannels (connexons) of adjacent cells and are built up by six connexin proteins (Cx), which allow passive diffusion of small and hydrophilic molecules (<1 kDa) into neighboring cells. The most abundant connexins found in the liver are Cx 26, 32, 36, 40, and 43 [17]. GJ may contribute to modulation of portal pressure and intrahepatic vascular relaxation [17].

Stiffness and intracellular components. Stiffness is also affected by intracellular pressure (P) and stretch forces (red arrows) on the cellular membranes and intermediary filaments. Several intercellular junctions are schematically shown such as tight junctions (TJ), gap junctions (GJ), and adherence junctions (AJ). Intermediary filaments such as CK18 play an important role in liver disease. CK18 is interacting with intercellular junctions such as AJ. Finally, the intracellular pressure (P) is likewise controlled by many conditions including transport proteins (T) to control osmotic pressure, protein shuttles, or water influx, e.g., by aquaporins. Below, the blood flow direction and sinusoidal pressure are shown. We are only at the beginning to understand the role of all these cellular factors in defining stiffness and biomechanic signaling. Further abbreviations: ECM extracellular matrix, SP sinusoidal pressure

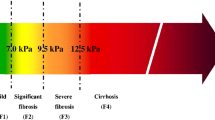
Similar stiffness values are found under pro-fibrogenic conditions in cellular and human studies. (a) Stiffness scale with cutoff values for normal, F3 and F4 fibrosis. (b) Known stiffness conditions to activate fibroblasts using atomic force microscopy (see [4]). (c) Known human LS values in various pathological conditions that ultimately cause liver fibrosis (see [45])
In summary, intracellular pressure in association with intercellular junctions, anchoring proteins, and intermediary filaments seem to play an important role in defining cellular stiffness and all these conditions are still largely unexplored.
How Do Cells Respond to Mechanical Stress?
Mechano-sensing has been studied for many decades and various underlying mechanisms seem to be involved. Cells must adhere to a solid. However, an understanding of how tissue cells—including fibroblasts, myocytes, neurons, and other cell types—sense matrix stiffness is just emerging with quantitative studies of cells adhering to gels (or to other cells) with which elasticity can be tuned to approximate that of tissues.
Key roles in molecular pathways are played by adhesion complexes and the actin-myosin cytoskeleton, whose contractile forces are transmitted through transcellular structures [18]. Potential sensing mechanisms include cation channels of the transient receptor potential (TRP) family, the actin-interacting protein zyxin and G protein-coupled receptors that are activated in response to stretch [19, 20] while ion channel activation and alterations in cytoskeletal stability are part of the response to hydrostatic pressure [21]. Members of TRP family of cation channels are emerging as important players in mechanotransduction pathways. Localized within mechanosensory structures, they are activated by mechanical deformations/stretching and trigger fast as well as sustained cytoskeletal remodeling responses [20]. In HSCs, these channels have been shown to be upregulated during fibrosis development and if blocked, myofibroblast differentiation was attenuated, thus suggesting an important role in HSC activation [22]. Likewise, the stress fiber network within these cells structurally reinforces and provides tension to tissues such as those found in healing wounds. Stress fibers have been observed to polymerize in response to mechanical forces which involves calcium-signaling [23]. Furthermore, liver sinus endothelial cells (LSECs) are highly specialized endothelial cells, which line liver sinusoids and are likely to be the first to sense shear stress due to changes in sinusoidal pressure or elevated blood flow. Moreover, the cells contain fenestrae allowing passage of soluble factors smaller than 100–150 nm between the sinusoidal blood and parenchymal cells. A contractile cytoskeleton ring composed of actin and myosin supports the fenestrae. The size and density of fenestrae is affected by portal pressure and shear stress, as well as soluble factors [24,25,26]. A recent study suggests that the lack of fenestration plays an important role in fibrosis development and a restoration of LSEC differentiation was shown to promote HSC quiescence, enhances regression of fibrosis and prevents progression of cirrhosis in vivo [27]. Therefore, the role of LSECs in mechano-sensing and fibrosis development requires further investigation.
Role of Myofibroblasts and ECM in Mechano-signaling
Myofibroblasts are regarded as major matrix generating cells in the liver but also in other tissues. In fact, besides HSCs, a large panel of cells can develop this phenotype upon activation including chondrocytes, osteoblasts, smooth muscle cells, pericytes, fibrocytes, or epithelial cells undergoing epithelial-to-mesenchymal transition. Neo-expression of the alpha isoform of smooth muscle actin (α-SMA) is used as marker for activated myofibroblasts [28]. Fibroblasts without a contractile apparatus form only very small and immature adhesions with the ECM [29, 30]. During mechanic stress, these focal complexes mature into focal adhesions (FA) [4]. HSCs undergo myofibroblast transformation if coated on stiff matrices even in the absence of the pro-fibrogenic cytokine TGF-β [31]. Most importantly, however, it was also shown that an increase in LS precedes histological matrix deposition in a rodent model [32]. In these concepts, the HSCs are described as sensing cells that respond to a stiff matrix by producing more matrix proteins. Indeed, HSCs have been known for a long time to be contractile and respond to changes in their environment [33]. In myofibroblasts, activated TGF-β results in increased α-SMA, which interacts with cellular myosin to contract and produce increased tension. TGF-β is a common factor downstream of many mechanical forces; in addition to tension, other forces including shear forces mediated by interstitial fluid flow and stretch have been implicated in TGF-β activation and release [34, 35]. It is quite striking to see that comparable stiffness values have been observed in patients with various liver diseases and confounders (inflammation, cholestasis, congestion) obtained by transient elastography in humans and in cellular studies analyzing the pro-fibrogenic response of HSC and fibroblasts (α-SMA activation and TGF-β release) under culturing conditions with exactly defined stiffness as assessed by atomic force microscopy (for details, see Fig. 54.5a–c). The identical levels of stiffness and pro-fibrogenic conditions both in clinical and cellular studies are a strong argument for the role of sinusoidal pressure and pressure-mediated stiffness elevation in fibrosis progression [5]. Thus, pressure could be one of the long seeked physiological correlates that modulate tissue stiffness (see Fig. 54.3 and Appendix Fig. A.14).
Principles of Mechano-sensing: Lessons from Pressure-Sensing in Vessels and Cells
Physical forces of gravity, hemodynamic stresses, and movement play a critical role in tissue development and have been studied for a long time [36]. Yet, little is known about how cells convert these mechanical signals into a chemical response. In a model presented by Ingber in 1997, it was postulated that cells are hard-wired to respond immediately to mechanical stresses transmitted over cell surface receptors that physically couple the cytoskeleton to extracellular matrix (e.g., integrins) or to other cells (cadherins, selectins, CAMs). Many signal transducing molecules that are activated by cell binding to growth factors and extracellular matrix associate with cytoskeletal scaffolds within focal adhesion complexes. Mechanical signals, therefore, may be integrated with other environmental signals and transduced into a biochemical response through force-dependent changes in scaffold geometry or molecular mechanics. Myofibroblasts are regarded as major matrix generating cells in the liver but also in other tissues. In fact, besides HSCs, a large panel of cells can develop this phenotype upon activation including chondrocytes, osteoblasts, smooth muscle cells, pericytes, fibrocytes, or epithelial cells undergoing epithelial-to-mesenchymal transition. As already discussed above, neo-expression of the alpha isoform of smooth muscle actin (α-SMA) is used as marker for activated myofibroblasts.
An important concept includes cell–matrix interactions such as focal adhesions (FAs). The cellular actin-myosin cytoskeleton exerts tension on ECM proteins via integrin attachments located within FAs that link the cell’s actin cytoskeleton and plasma membrane to the underlying ECM (Fig. 54.6a). FAs change protein composition and dynamics and grow in size in response to tension [37, 38]. Mechano-sensing by focal adhesions during cell adhesion to the ECM can be, for instance, mediated by talin (Fig. 54.6a), a connecting protein between ECM-binding integrin receptors and the actin cytoskeleton. In response to this increased tension, vinculin can bind to talin resulting in a force- and direction-dependent focal adhesion reinforcement [39]. Another example is mechano-sensing through the Latency Associated Peptide (LAP) complex. Thus, TGF-β that is stored in the LAP complex of the ECM can undergo activation as a direct result of mechanical tension. Through integrin attachments, cells are able to exert tension on the LAP. In a soft environment, it deforms in response to tension and the complex remains intact. If the matrix is stiff, however, resistance to cell-generated tension results in deformation of the LAP and the concomitant release of active TGF-β [40, 41]. A third example is mechanosensitive ion channels that perceive changes in plasma membrane tension, which can be modulated by the actin network [39]. Mechanical forces are thought to gate ion channels by inducing a conformational switch resulting in pore opening and ion flux.

Molecular examples of mechano-sensing. These already established mechanisms could all contribute to sensing the sinusoidal pressure via intercellular or ECM-cellular stretch forces ultimately causing matrix deposition to withstand the pressure. Many cells including HSC and fibroblasts have tactile properties and can sense the rigidity of the pressure-modulated tissues stiffness. (a) Mechano-sensing by focal adhesions during cell adhesion to the ECM. For instance, stretch forces free cryptic binding sides of talin, a connecting protein between ECM-binding integrin receptors and the actin cytoskeleton. In response this increased tension, vinculin can bind to talin resulting in a force- and direction-dependent focal adhesion reinforcement. (b) Stretch-sensing and pro-fibrogenic response by latent TGF-β activation. Integrin binding to a specific RGD site in LAP transmits intracellular force to the latent TGF-β complex consisting of LTBP1, TGF-β, and LAP. In case of, e.g., pressure-induced stretch forces, RGD-linked ECM will pull LAP away and this conformation change will release TGF-β. (c) Stretch-sensing by mechanosensitive ion channels (MIC). MIC perceive changes in plasma membrane tension, which can be modulated by the actin network. Mechanical forces are thought to gate ion channels by inducing a conformational switch resulting in pore opening and ion flux. Abbreviations: ECM extracellular matrix, LAP latency associated protein, LTBP1 latent transforming growth factor β binding protein 1, TGF-β transforming growth factor β
Many lessons on mechano- and pressure-mediated signaling pathways and sensing have been learnt from vascular biology [3, 6, 42,43,44]. It is also interesting to note that mechano-induced gene expression profiles include hypoxia-regulated genes such as HIF1alpha [6]. This could be a further hint that pressure changes are always associated with oxygen changes. For example, elevation of vascular pressure in response to vascular resistance will be followed by a decrease in oxygen. Under extreme conditions of a complete blockage of blood flow, pressure will be maximized while oxygen rapidly decreases. Figure 54.7 schematically depicts potentially involved pathways in sinusoidal pressure and LS induced mechano-sensing in the liver. These should be addressed in future studies.

Potential sinusoidal pressure-induced pathways ultimately leading to liver fibrosis. Abbreviations: AngII Angiotensin II, AP-1 Activator protein-1, CAT catalase, Egr-1 early growth response gene-1, ERK-1/2 extracellular signal regulated kinases 1 and 2, FAK focal adhesion kinase, GADD-153 growth arrest and DNA damage-inducible gene 153, JNK c-Jun N-terminal kinase, HSC hepatic stellate cell, HSP-60 heat shock protein-60, LSEC liver sinus endothelial cells, MCA monocyte chemotactic antigen, MIC Mechanosensitive ionic channels, NO nitric oxide, NOX NADPH-dependent oxidases, PCNA proliferating cell nuclear antigen, REB response element binding protein, SGK serum-glucocorticoid-induced protein kinase (a serine/threonine protein kinase), SMC smooth muscle cells, SM22-α smooth muscle cells specific protein, TGF-β transforming growth factor β, THA-2 thromboxane synthase-A2, TNF-α tumor necrosis factor-α
References
Fåhraeus R. The suspension stability of the blood. Physiol Rev. 1929;9(2):241–74.
Wells RE Jr, Merrill EW. Influence of flow properties of blood upon viscosity-hematocrit relationships. J Clin Invest. 1962;41(8):1591–8.
Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116(8):1448–61.
Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11(2):120–6.
Mueller S. Does pressure cause liver cirrhosis? The sinusoidal pressure hypothesis. World J Gastroenterol. 2016;22(48):10482.
Anwar MA, Shalhoub J, Lim CS, Gohel MS, Davies AH. The effect of pressure-induced mechanical stretch on vascular wall differential gene expression. J Vasc Res. 2012;49(6):463–78.
Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91(2):691–731.
Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol. 1997;13:119–46.
Balda MS, Matter K. Tight junctions and the regulation of gene expression. Biochim Biophys Acta. 2009;1788(4):761–7.
Holczbauer A, Gyongyosi B, Lotz G, Torzsok P, Kaposi-Novak P, Szijarto A, et al. Increased expression of claudin-1 and claudin-7 in liver cirrhosis and hepatocellular carcinoma. Pathol Oncol Res. 2014;20(3):493–502.
Yeh TH, Krauland L, Singh V, Zou B, Devaraj P, Stolz DB, et al. Liver-specific beta-catenin knockout mice have bile canalicular abnormalities, bile secretory defect, and intrahepatic cholestasis. Hepatology. 2010;52(4):1410–9.
Hatzfeld M. Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion? Biochim Biophys Acta. 2007;1773(1):69–77.
Moll R, Franke WW. Intermediate filaments and their interaction with membranes. The desmosome-cytokeratin filament complex and epithelial differentiation. Pathol Res Pract. 1982;175(2–3):146–61.
Kroger C, Loschke F, Schwarz N, Windoffer R, Leube RE, Magin TM. Keratins control intercellular adhesion involving PKC-alpha-mediated desmoplakin phosphorylation. J Cell Biol. 2013;201(5):681–92.
Song JY, Van Noorden CJ, Frederiks WM. Alterations of hepatocellular intermediate filaments during extrahepatic cholestasis in rat liver. Virchows Arch. 1997;430(3):253–60.
Windoffer R, Beile B, Leibold A, Thomas S, Wilhelm U, Leube RE. Visualization of gap junction mobility in living cells. Cell Tissue Res. 2000;299(3):347–62.
Hernandez-Guerra M, Gonzalez-Mendez Y, de Ganzo ZA, Salido E, Garcia-Pagan JC, Abrante B, et al. Role of gap junctions modulating hepatic vascular tone in cirrhosis. Liver Int. 2014;34(6):859–68.
Discher DE. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310(5751):1139–43.
Suresh Babu S, Wojtowicz A, Freichel M, Birnbaumer L, Hecker M, Cattaruzza M. Mechanism of stretch-induced activation of the mechanotransducer zyxin in vascular cells. Sci Signal. 2012;5(254):ra91.
Kuipers AJ, Middelbeek J, van Leeuwen FN. Mechanoregulation of cytoskeletal dynamics by TRP channels. Eur J Cell Biol. 2012;91(11–12):834–46.
Myers KA, Rattner JB, Shrive NG, Hart DA. Hydrostatic pressure sensation in cells: integration into the tensegrity model. Biochem Cell Biol. 2007;85(5):543–51.
Abhilash PA, Harikrishnan R, Indira M. Ascorbic acid suppresses endotoxemia and NF-kappaB signaling cascade in alcoholic liver fibrosis in guinea pigs: a mechanistic approach. Toxicol Appl Pharmacol. 2014;274(2):215–24.
Nekouzadeh A, Pryse KM, Elson EL, Genin GM. Stretch-activated force shedding, force recovery, and cytoskeletal remodeling in contractile fibroblasts. J Biomech. 2008;41(14):2964–71.
Morsiani E, Aleotti A, Ricci D. Haemodynamic and ultrastructural observations on the rat liver after two-thirds partial hepatectomy. J Anat. 1998;192(Pt 4):507–15.
Morsiani E, Mazzoni M, Aleotti A, Gorini P, Ricci D. Increased sinusoidal wall permeability and liver fatty change after two-thirds hepatectomy: an ultrastructural study in the rat. Hepatology. 1995;21(2):539–44.
Gatmaitan Z, Varticovski L, Ling L, Mikkelsen R, Steffan AM, Arias IM. Studies on fenestral contraction in rat liver endothelial cells in culture. Am J Pathol. 1996;148(6):2027–41.
DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740–6.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63.
Tamariz E, Grinnell F. Modulation of fibroblast morphology and adhesion during collagen matrix remodeling. Mol Biol Cell. 2002;13(11):3915–29.
Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60(1):24–34.
Wells RG. The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J Clin Gastroenterol. 2005;39(4 Suppl 2):S158–61.
Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293(6):G1147–54.
Kawada N, Tran-Thi TA, Klein H, Decker K. The contraction of hepatic stellate (Ito) cells stimulated with vasoactive substances. Possible involvement of endothelin 1 and nitric oxide in the regulation of the sinusoidal tonus. Eur J Biochem. 1993;213(2):815–23.
Ng CP, Hinz B, Swartz MA. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci. 2005;118(Pt 20):4731–9.
Sakata R, Ueno T, Nakamura T, Ueno H, Sata M. Mechanical stretch induces TGF-beta synthesis in hepatic stellate cells. Eur J Clin Invest. 2004;34(2):129–36.
Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997;59:575–99.
Sagir A, Erhardt A, Schmitt M, Haussinger D. Transient elastography is unreliable for detection of cirrhosis in patients with acute liver damage. Hepatology. 2008;47(2):592–5.
Millonig G, Friedrich S, Adolf S, Fonouni H, Golriz M, Mehrabi A, et al. Liver stiffness is directly influenced by central venous pressure. J Hepatol. 2010;52(2):206–10.
Kanoldt V, Fischer L, Grashoff C. Unforgettable force—crosstalk and memory of mechanosensitive structures. Biol Chem. 2019;400(6):687–98.
Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, et al. The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol. 2011;21(24):2046–54.
Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179(6):1311–23.
Humphrey JD. Mechanisms of arterial remodeling in hypertension: coupled roles of wall shear and intramural stress. Hypertension. 2008;52(2):195–200.
Atta HM. Varicose veins: role of mechanotransduction of venous hypertension. Int J Vasc Med. 2012;2012:538627.
Pfisterer L, Konig G, Hecker M, Korff T. Pathogenesis of varicose veins—lessons from biomechanics. Vasa. 2014;43(2):88–99.
Mueller S, Sandrin L. Liver stiffness: a novel parameter for the diagnosis of liver disease. Hepat Med. 2010;2:49–67.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mueller, S. (2020). Introduction to the Molecular Basis of Liver Stiffness and Its Relation to Mechano-signaling. In: Mueller, S. (eds) Liver Elastography. Springer, Cham. https://doi.org/10.1007/978-3-030-40542-7_54
Download citation
DOI: https://doi.org/10.1007/978-3-030-40542-7_54
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40541-0
Online ISBN: 978-3-030-40542-7
eBook Packages: MedicineMedicine (R0)