
Abstract
The heat shock response (HSR) is characterized by the induction of molecular chaperones following a sudden increase in temperature. In eukaryotes, the HSR comprises the set of genes controlled by the transcription factor Hsf1. The HSR is induced by defects in co-translational protein folding, ribosome biogenesis, organellar targeting of nascent proteins, and protein degradation by the ubiquitin proteasome system. Upon heat shock, these processes may be endogenous sources of polypeptide ligands that activate the HSR. Mechanistically, these ligands are thought to titrate the chaperone Hsp70 away from Hsf1, releasing Hsf1 to induce the full arsenal of cellular chaperones to restore protein homeostasis. In metazoans, this cell-autonomous feedback loop is modulated by the microenvironment and neuronal cues to enable tissue-level and organism-wide coordination.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: The HSR in Health and Disease
The heat shock response (HSR) is conserved in all kingdoms of life and is characterized by the induction of molecular chaperones following a sudden increase in temperature. Initially observed as heat-induced chromosomal puffs in Drosophila, the HSR has long served as a model system for studying the molecular mechanisms of inducible transcription (Anckar and Sistonen 2011). In recent years, as protein homeostasis (proteostasis) has become increasingly implicated in cancer, neurodegenerative disease and aging, studies of the HSR have focused on its role as the regulatory nexus for the proteostasis network (Labbadia and Morimoto 2014).
In eukaryotes, the HSR is regulated by a conserved family of heat shock transcription factors (HSFs). HSFs are winged helix-loop-helix DNA binding proteins that trimerize and recognize a conserved motif found in the promoters of chaperone genes (Hentze et al. 2016; Littlefield and Nelson 1999; Neudegger et al. 2016; Sorger and Nelson 1989). Yeast and invertebrates have a single HSF – Hsf1 – while mammalian genomes encode Hsf1 along with Hsf2 and additional tissue-specific paralogs implicated in development (Anckar and Sistonen 2011). In the absence of Hsf1, mouse embryonic fibroblasts (MEFs) fail to induce chaperone genes following heat shock, indicating that Hsf1 is primarily responsible for regulating the canonical HSR in mammalian cells (Mahat et al. 2016; McMillan et al. 1998; Solís et al. 2016). However, Hsf2 can hetero-oligomerize with Hsf1, and MEFs lacking Hsf2 show increased basal expression and altered induction of HSR targets, suggesting that Hsf2 may modulate Hsf1 (Jaeger et al. 2016; Östling et al. 2007; Solís et al. 2016). Consistent with an antagonistic interaction, Hsf1 has been shown to promote survival and malignancy in cancer models, while Hsf2 suppresses tumor progression (Björk et al. 2016).
Cancer cells rely on the HSR to support rapid growth and to counteract the deleterious consequences of high mutational loads (Dai 2018; Dai and Sampson 2016). In many human tumor samples and cancer cell lines, Hsf1 is constitutively activated and required for proliferation (Dai et al. 2007; Santagata et al. 2011). High levels of Hsf1 in tumor samples – both in the tumor cells as well as in the supporting stromal cells – is correlated with poor prognosis in several cohorts of cancer patients (Santagata et al. 2011; Scherz-Shouval et al. 2014). Moreover, Hsf1 knock out mice are resistant to tumor growth (Dai and Sampson 2016; Dai et al. 2007; 2012; Min et al. 2007). Based on these observations, inhibiting Hsf1 has been proposed as an anticancer strategy (Whitesell and Lindquist 2009).
Conversely, activating Hsf1 has been proposed as a therapeutic strategy to combat neurodegenerative diseases (Neef et al. 2011). Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), Parkinson’s disease, frontotemporal dementia, and Huntington’s disease all share protein aggregation as a hallmark (Soto 2003). Chaperone levels are known to decline in the brain with age, and Hsf1 undergoes abnormal degradation in cell line and mouse models of Huntington’s disease (Brehme et al. 2014; Gomez-Pastor et al. 2017; Yang et al. 2014). Moreover, activation of Hsf1 by a natural product ameliorates phenotypes associated with polyglutamine expansion diseases in Drosophila (Nelson et al. 2014). Thus, enhancing Hsf1 activity could induce the HSR to prevent or reverse protein aggregation and slow neurodegeneration.
The central and opposing roles for Hsf1 in cancer and neurodegenerative diseases suggest that therapeutic intervention into the HSR may involve an inherent trade-off, so quantitative and dynamic control would be highly valuable. Understanding the mechanisms that regulate Hsf1 in healthy cells may reveal the processes that break down and are hijacked in disease.
2 Defining the Transcriptional Response to Heat Shock
Counterintuitively, the HSR comprises only a fraction of genes that change expression upon heat shock. In yeast, hundreds of genes are induced following an increase in temperature, and hundreds more are repressed (Gasch et al. 2000; Solís et al. 2016). However, Hsf1 controls expression of a dedicated proteostasis regulon containing fewer than 50 of the induced genes and has no role in transcriptional repression (Pincus et al. 2018; Solís et al. 2016). In addition to robust transcriptional activation, Hsf1 drives intergenic interactions among its target gene loci during heat shock and remodels the three-dimensional architecture of the yeast genome (Chowdhary et al. 2017; 2019). The remainder of the differentially expressed genes comprise the environmental stress response (ESR), a generic program activated by a variety of environmental perturbations. The induced ESR is controlled by the general stress response transcription factors Msn2/4, while the repressed ESR is enriched for highly expressed genes encoding factors involved in central metabolism and ribosome biogenesis (Gasch and Werner-Washburne 2002). Heat shock-dependent repression of highly expressed genes occurs in Drosophila and mammalian cells too and is also Hsf1-independent (Duarte et al. 2016; Mahat et al. 2016). Thus, an additional conserved aspect of the transcriptional response to elevated temperature is repression of highly expressed genes (Anckar and Sistonen 2011). As in yeast, Hsf1 only controls a fraction of the genes induced by heat shock in mammalian cells (Mahat et al. 2016; Solís et al. 2016). Nevertheless, the HSR has come to be defined as the set of genes induced by Hsf1. Using this definition, the HSR is highly conserved across eukaryotes and limited to genes encoding chaperones and other proteostasis factors.
However, the HSR is not so clearly defined in the context of multicellular development, metabolism and cancer. Hsf1 is known to drive transcriptional programs distinct from the canonical HSR during development and inflammation, in animal models of obesity, and in highly malignant cancer cells (Ali et al. 2018; Li et al. 2016; Ma et al. 2015; Mendillo et al. 2012). In addition to proteostasis factors, Hsf1 controls cell cycle and metabolic genes that promote oncogenesis in multiple tumor types (Mendillo et al. 2012). Hsf1 is also active in cancer associated fibroblasts in the stroma surrounding tumors and drives another distinct transcriptional program in these cells that promotes tumor growth via paracrine signaling (Scherz-Shouval et al. 2014). It is not yet clear how Hsf1 is directed to alternative sites in the genome to regulate these noncanonical transcriptional programs.
3 What Activates the HSR?
Despite its name, the HSR is not exclusively sensitive to temperature. Oxidative stress, glucose depletion and overexpression of a constitutively misfolded protein also activate Hsf1 and induce chaperones, as do several classes of small molecules including Hsp90 inhibitors, proteasome inhibitors, amino acid analogs, and ribosome biogenesis inhibitors (Alford and Brandman 2018; Geiler-Samerotte et al. 2011; Hahn and Thiele 2004; Kim et al. 1999a, b; Trotter et al. 2002; Tye et al. 2019; Yamamoto et al. 2007). At the molecular level, these inputs converge on Hsf1.
It is often presumed that heat shock – and by extension the set of other environmental and chemical perturbations that activate Hsf1 – causes a fraction of the mature proteome to denature and aggregate, and these aggregates serve as the molecular signals that induce the HSR (Lindquist 1986). At the biochemical level, differential centrifugation experiments have shown that proteins sediment in high molecular weight fractions during heat shock in a manner that is partially reversible by molecular chaperones, suggesting heat shock-dependent protein aggregation (Mogk et al. 1999). But it is now appreciated that enzymes in these aggregates can retain activity, suggesting that these assemblies do not contain denatured or misfolded proteins (Riback et al. 2015; Wallace et al. 2015). At the cell biological level, observations in yeast and mammalian cells revealed that in response to heat shock, chaperones form subcellular foci that colocalize with aggregation-prone reporter proteins, and chaperones form similar puncta during heat shock in the absence of reporters (Cherkasov et al. 2013; Kaganovich et al. 2008; Solís et al. 2016). These results were interpreted to suggest that endogenous metastable proteins denature and are also in these puncta. However, no such endogenous proteins have been identified.
Genome-wide deletion and RNAi screens were conducted in yeast and human cells to identify genes involved in regulating a reporter gene under the control of Hsf1. The yeast screen identified genes that when deleted altered reporter levels at 25 and 37 °C, while the human screen revealed factors that modulated induction of the reporter following heat shock and recovery, mostly identifying genes required for activation (Brandman et al. 2012; Raychaudhuri et al. 2014). Both screens implicated the proteasome in negatively regulating Hsf1 activity, while the top hits in the yeast screen also included chaperones, organelle targeting machinery, and the ribosome quality control complex (RQC) as negative regulators of Hsf1. Chemical genetic experiments revealed that acute depletion of ribosome biogenesis factors also results in potent Hsf1 activation due to the accumulation of orphan ribosomal proteins (Albert et al. 2019; Tye et al. 2019). Synthetic mutant proteins that clog ER and mitochondrial import pathways likewise activate Hsf1 (Boos et al. 2019; Shmidt et al. 2019). A unifying theme among these mutants is that proteins are likely to accumulate in the cell that are not present in wild type cells: ubiquitin proteasome system (UPS) intermediates, unbound chaperone clients, mistargeted organellar proteins, partially-translated 60S ribosome-bound nascent chains, and unincorporated ribosomal proteins (Fig. 3.1).

Sources of ligands for the HSR. The canonical HSR is induced by defects in a variety of cell biological processes. These include nascent protein folding and complex formation, ribosome biogenesis (leading to accumulation of orphan ribosomal proteins, oRPs), ribosome quality control (RQC), ER and mitochondrial targeting, tail anchored protein (TAP) insertion, degradation by the ubiquitin/proteasome system (UPS). A common theme among these processes is that their disruption results in the accumulation of proteins in the cytosol that are not supposed to be there
Do these genetic results support the protein aggregate model? Rather than implicating denaturation of the mature proteome, the genetics suggest that Hsf1 is sensitive to dynamic aspects proteostasis: nascent chain folding, protein complex formation, ribosome biogenesis, post-translational organelle targeting, and degradation. Consistent with this notion, pretreatment with cycloheximide to stop translation prior to heat shock abolishes HSR induction (Baler et al. 1992). Moreover, a small molecule screen in human cells for modulators of a reporter gene of Hsf1 activity revealed that broad classes of translation inhibitors prevent HSR activation (Santagata et al. 2013). Taken together, these biochemical, cell biological, genetic, and pharmacological experiments suggest that the HSR does indeed monitor proteostasis. However, it is unlikely to be the case that mature proteins denature en masse upon heat shock, and the resulting aggregates activate Hsf1. Rather, Hsf1 appears to respond to stalled ribosomes recognized by RQC, orphan ribosomal proteins that result from aborted ribosome biogenesis, clogged ER and mitochondrial import machinery, and an overtaxed UPS. Thus, the HSR seems tuned to surveil the early and late events in the life of proteins rather than the mature proteome.
4 Hsp70 and Hsf1 Constitute a Negative Feedback Loop That Controls the HSR
How are inefficiencies in protein complex formation, ribosome biogenesis, organelle targeting, and degradation communicated to Hsf1 to induce the HSR? Based on observations in Drosophila, the HSR is canonically thought to be an autoregulatory feedback loop for heat shock protein (HSP) expression (Didomenico et al. 1982; Solomon et al. 1991). In this model, often referred to as the chaperone titration model, excess HSPs bind to and repress a transcription factor. When conditions change and the cell needs more chaperones – i.e., when HSPs become limiting and are titrated away – the transcription factor is free to induce more HSPs until they are in excess again.
The mechanistic precedent for the chaperone titration model was established in E. coli. In this prokaryotic system, the homolog of the Hsp70 chaperone (DnaK) represses the heat shock transcription factor σ32 by binding and accelerating its degradation; when the levels of DnaK become limiting, σ32 accumulates and induces transcription of DnaK along with the rest of the HSR (Bukau and Walker 1990; Straus et al. 1990). In yeast, molecular genetic experiments also suggested that Hsp70 autoregulated its own expression: mutation of the two highly expressed Hsp70 paralogs (ssa1 ssa2) results in induction of a third Hsp70 paralog (SSA3), and this induction requires the heat shock element (HSE) – the conserved binding site for Hsf1 – in the SSA3 promoter (Boorstein and Craig 1990). In human cells, the genes encoding Hsp70 also contain HSEs in their promoters, and the Hsp70 protein directly binds to Hsf1 and impairs its ability associate with HSE-containing DNA (Shi et al. 1998). Taken together, these data support a model in which the HSR is an autoregulatory loop controlled by Hsp70 in both prokaryotic and eukaryotic cells.
Affinity purification experiments coupled to mass spectrometry using Hsf1 as bait revealed a specific and dynamic interaction between Hsp70 and Hsf1 in yeast cells (Zheng et al. 2016). Hsf1 has also been shown to directly crosslink to the Hsp70 substrate binding domain under nonstress conditions and dissociate during heat shock and other genetic and chemical perturbations to proteostasis (Masser et al. 2019). Under basal conditions, Hsf1 co-precipitates with Hsp70; the interaction is lost following acute heat shock; over sustained heat shock, the interaction is restored. The dynamics of the Hsp70:Hsf1 interaction are the mirror image of Hsf1-dependent transcription, which is transiently increased during heat shock (Zheng et al. 2016). Mutational analysis and biochemical binding assays revealed a specific Hsp70 binding site on Hsf1 known as conserved element 2 (CE2) (Krakowiak et al. 2018). CE2 is required for Hsf1 repression under basal conditions and deactivation of Hsf1 following heat shock. In addition, a second Hsp70 binding site has been identified in the N-terminal region of Hsf1 (Peffer et al. 2019). Transcriptional induction of Hsp70 is also required for appropriate regulation of the HSR, as HSE disruption in the promoters of the Hsp70 genes impairs Hsf1 deactivation following heat shock (Krakowiak et al. 2018). Thus, Hsp70 and Hsf1 form a negative feedback loop in which Hsf1 induces Hsp70 expression, and Hsp70 represses Hsf1 activity (Fig. 3.2).

Hsp70 and Hsf1 form a negative feedback loop that controls the HSR. Hsp70 binds and represses Hsf1 under basal conditions. Heat shock and other proteotoxic stress conditions generate unstable polypeptides (UPs) – the ligands of the HSR depicted in Fig. 3.1. UPs titrate Hsp70 away from Hsf1, leaving Hsf1 free to induce transcription of Hsp70 and the rest of the HSR target genes. Once the UPs have been cleared, Hsp70 is again in excess and can bind and deactivate Hsf1
5 Hsp90 Negatively Regulates Hsf1 Orthogonally to Hsp70
Pharmacological and genetic experiments also demonstrated that impaired Hsp90 function activates Hsf1 (Brandman et al. 2012; Kim et al. 1999b). Hsp90 was shown to bind to Hsf1 in mammalian cell lysate and has also been proposed to regulate Hsf1 via a titration model (Zou et al. 1998). In yeast, deletion of the highly expressed Hsp90 paralog induces Hsf1 activation, but no protein-protein interaction has been reported between Hsp90 and Hsf1. Recently, use of a novel reporter of Hsp90 availability revealed that Hsp90 represses Hsf1 in a manner that is independent of Hsp70 (Alford and Brandman 2018). The mechanism by which this orthogonal Hsp90 axis regulates Hsf1 remains unknown.
6 Phosphorylation Is Dispensable for Hsf1 Activity During Heat Shock
In addition to regulation by HSPs, Hsf1 is also post-translationally modified in response to heat shock. Hsf1 has been shown to be ubiquitylated by FBXW7, resulting in degradation by the proteasome, as well as SUMOylated and acetylated in human cells (Hong et al. 2001; Kourtis et al. 2015; Westerheide et al. 2009). Hsf1 has also been shown to be phosphorylated in diverse eukaryotes (Anckar and Sistonen 2011; Sorger and Pelham 1988). In yeast, there is evidence that Hsf1 can be phosphorylated on 73 distinct sites, and 15 phosphorylation sites were identified in the regulatory domain of human Hsf1 (Budzyński et al. 2015; Zheng et al. 2016). In both cases, however, mutational analysis revealed that simultaneous mutation of all sites to alanine – resulting in mutants that cannot be phosphorylated – had minimal effects on the ability of Hsf1 to activate the HSR in response to heat shock. In other words, phosphorylation is dispensable for activation of Hsf1. However, a mutant that mimics constitutive hyperphosphorylation (via substitution of negatively charged amino acids) is highly active under basal conditions in yeast (Zheng et al. 2016). Thus, while phosphorylation is not necessary for Hsf1 activation, negative charge is sufficient. Neither the phosphorylation-deficient mutant nor the phospho-mimetic mutant altered the interaction between Hsf1 and Hsp70 in yeast (Zheng et al. 2016). Thus, like Hsp90 inhibition, phosphorylation represents a regulatory axis orthogonal to the Hsp70 feedback loop.
While dispensable for activation in response to heat shock, single cell measurements revealed that Hsf1 phosphorylation promotes cell-to-cell variation in the HSR in yeast. Variation in the expression of Hsp90 driven by Hsf1 phosphorylation enables cells to acquire resistance to an antifungal drug (Zheng et al. 2018). Hsp90 is known to promote phenotypic diversity and has been proposed to play important roles in molecular evolution (Lindquist 2009). By generating variation in Hsp90 levels across a population, Hsf1 phosphorylation may be advantageous for cells in fluctuating environmental conditions.
Despite the minimal role of Hsf1 phosphorylation sites following heat shock, multiple kinases have been implicated in Hsf1 phosphorylation in yeast and mammalian cells. In human cells, MEK has been shown to promote Hsf1 activation, while AMPK inhibits Hsf1 (Dai et al. 2015; Tang et al. 2015). In addition, ERK, GSK3β and CK2α′ phosphorylate Hsf1 to target it to the UPS for degradation (Li et al. 2017). In yeast, the AMPK homolog Snf1 has also been shown to phosphorylate Hsf1 and modulate the HSR (Hahn and Thiele 2004). The discrepancy between these kinase-mediated regulatory events and the ability of Hsf1 to be activated in the absence of phosphorylation is unresolved. It is clear that Hsf1 becomes phosphorylated during heat shock, but it is not the simple case that phosphorylation is required to activate Hsf1 following heat shock. Phosphorylation may be an important mode of Hsf1 regulation in response to signals other than heat shock.
7 Coordination of the HSR Across Tissues
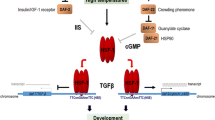
In multicellular animals, homeostasis is a property of the organism rather than the individual cell. As such, cell-autonomous Hsf1 regulatory mechanisms are augmented by intercellular coordination in metazoans (Fig. 3.3). For example, insulin/IGF-1 signaling is known to modulate Hsf1 activity in C. elegans to regulate lifespan via Hsf1-dependent control of cytoskeletal integrity (Baird et al. 2014; Hsu et al. 2003). Moreover, at the onset of reproductive maturity in C. elegans, signals from germ stem cells result in organism-wide inactivation of the HSR by epigenetic silencing of target gene promoters (Labbadia and Morimoto 2015). In mammals, as described above, the tumor microenvironment and organism-wide metabolic signaling subjugate Hsf1 and the transcriptional program it controls (Ma et al. 2015; Scherz-Shouval et al. 2014). These forms of non-autonomous regulation are mediated by hormones, cytokines and growth factors. In most cases, it is not yet understood how the signaling cascades activated by these ligands impinge on Hsf1, nor how Hsf1 is then deployed to regulate distinct target genes.

Non-autonomous regulation of Hsf1 results in non-canonical HSR induction. In addition to the cell-autonomous regulatory mechanisms that control Hsf1 activity to control the canonical HSR target genes (red arrows), metazoans can regulate Hsf1 and the HSR via non-autonomous signals. The tumor microenvironment, inflammatory cytokines, dietary hormones, and neuronal signaling has been shown to involve Hsf1. Once activated by these extracellular signals, Hsf1 can initiate distinct transcriptional programs that only partially overlap with the canonical HSR
In addition to paracrine and endocrine signaling, neuronal signaling in C. elegans is required for organism-wide induction of the HSR in response to temperature (Prahlad et al. 2008). Indeed, separate Hsf1-dependent neural signals have been shown to be responsible for mediating heat shock signaling and longevity (Douglas et al. 2015). Moreover, local ectopic expression of a misfolded protein in muscle cells was shown to induce the HSR across multiple tissue types, and local over-expression of Hsp90 in neurons or intestinal cells suppressed HSR induction arising from proteostasis defects in muscle cells (van Oosten-Hawle et al. 2013). These results indicate that both forward stress signaling and chaperone-mediated feedback control operate across tissues.
8 Conclusion
The HSR is both highly conserved and remarkably plastic. In eukaryotes from yeast to humans, the same core set of chaperone-encoding genes is induced by heat shock, and these genes harbor the same cis-acting motif in their promoters that is recognized by the same sequence-specific DNA binding protein, Hsf1. Approaches from genetics, chemical biology, biochemistry and cell biology reveal a coherent picture of the molecular consequences of heat shock and the regulatory mechanisms that govern Hsf1 activity. Rather than global protein denaturation, heat shock is likely to impair key biogenesis processes like protein complex formation and ribosome production, resulting in the accumulation of orphan subunits and other unstable polypeptides that are sequestered and/or degraded via chaperones. Heat shock, or disruption of these biogenesis and degradation pathways by genetic or pharmacological means, activates Hsf1 principally by titrating the chaperone Hsp70 away from its repressive interaction with Hsf1. Phosphorylation and Hsp90 also regulate Hsf1 activity, but the mechanisms remain unresolved. While these core cell-autonomous regulatory mechanisms are conserved over evolution, metazoans have expanded both the input signals that activate Hsf1 as well as the target genes that Hsf1 controls. While this plasticity in the HSR enables the proteostasis network to incorporate signals from other cells and allows Hsf1 to activate distinct transcriptional programs, this plasticity may also permit tumor cells to hijack the HSR. However, the deep conservation of the cell-autonomous regulatory mechanisms may allow for the development of targeted therapeutics that will allow us to take back control.
References
Albert B, Kos-Braun IC, Henras AK, Dez C, Rueda MP, Zhang X, Gadal O, Kos M, Shore D (2019) A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. Elife 8:e45002
Alford BD, Brandman O (2018) Quantification of Hsp90 availability reveals differential coupling to the heat shock response. J Cell Biol 217:3809–3816
Ali A, Biswas A, Pal M (2018) HSF1 mediated TNF-α production during proteotoxic stress response pioneers proinflammatory signal in human cells. FASEB J 33:2621–2635
Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 80:1089–1115
Baird NA, Douglas PM, Simic MS, Grant AR, Moresco JJ, Wolff SC, Yates JR, Manning G, Dillin A (2014) HSF-1–mediated cytoskeletal integrity determines thermotolerance and life span. Science 346:360–363
Baler R, Welch W, Voellmy R (1992) Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor. J Cell Biol 117:1151–1159
Björk J, Åkerfelt M, Joutsen J, Puustinen M, Cheng F, Sistonen L, Nees M (2016) Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene 35:1770
Boorstein W, Craig E (1990) Transcriptional regulation of SSA3, an HSP70 gene from Saccharomyces cerevisiae. Mol Cell Biol 10:3262–3267
Boos F, Krämer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zöller E, van der Laan M et al (2019) Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat Cell Biol 21(4):442–451
Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li G-W, Zhou S, King D, Shen PS, Weibezahn J et al (2012) A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151:1042–1054
Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M et al (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9:1135–1150
Budzyński MA, Puustinen MC, Joutsen J, Sistonen L (2015) Uncoupling stress-inducible phosphorylation of heat shock factor 1 from its activation. Mol Cell Biol 35:2530–2540
Bukau B, Walker G (1990) Mutations altering heat shock specific subunit of RNA polymerase suppress major cellular defects of E. coli mutants lacking the DnaK chaperone. EMBO J 9:4027–4036
Cherkasov V, Hofmann S, Druffel-Augustin S, Mogk A, Tyedmers J, Stoecklin G, Bukau B (2013) Coordination of translational control and protein homeostasis during severe heat stress. Curr Biol 23:2452–2462
Chowdhary S, Kainth AS, Gross DS (2017) Heat shock protein genes undergo dynamic alteration in their three-dimensional structure and genome organization in response to thermal stress. Mol Cell Biol 37:e00292-17
Chowdhary S, Kainth AS, Pincus D, Gross DS (2019) Heat shock factor 1 drives intergenic association of its target gene loci upon heat shock. Cell Rep 26:18–28.e5
Dai C (2018) The heat-shock, or HSF1-mediated proteotoxic stress, response in cancer: from proteomic stability to oncogenesis. Philos Trans R Soc Lond B Biol Sci 373:20160525
Dai C, Sampson S (2016) HSF1: guardian of proteostasis in cancer. Trends Cell Biol 26:17–28
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018
Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L, Lindquist S (2012) Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 122:3742–3754
Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, Dai C (2015) Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J 34:275–293
Didomenico BJ, Bugaisky GE, Lindquist S (1982) The heat shock response is self-regulated at both the transcriptional and posttranscriptional levels. Cell 31:593–603
Douglas PM, Baird NA, Simic MS, Uhlein S, McCormick MA, Wolff SC, Kennedy BK, Dillin A (2015) Heterotypic signals from neural HSF-1 separate thermotolerance from longevity. Cell Rep 12:1196–1204
Duarte FM, Fuda NJ, Mahat DB, Core LJ, Guertin MJ, Lis JT (2016) Transcription factors GAF and HSF act at distinct regulatory steps to modulate stress-induced gene activation. Genes Dev 30:1731–1746
Gasch AP, Werner-Washburne M (2002) The genomics of yeast responses to environmental stress and starvation. Funct Integr Genomics 2:181–192
Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11:4241–4257
Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond AD (2011) Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc National Acad Sci 108:680–685
Gomez-Pastor R, Burchfiel ET, Neef DW, Jaeger AM, Cabiscol E, McKinstry SU, Doss A, Aballay A, Lo DC, Akimov SS et al (2017) Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat Commun 8:14405
Hahn J-S, Thiele DJ (2004) Activation of the Saccharomyces cerevisiae heat shock transcription factor under glucose starvation conditions by Snf1 protein kinase. J Biol Chem 279:5169–5176
Hentze N, Breton L, Wiesner J, Kempf G, Mayer MP (2016) Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. Elife 5:e11576
Hong Y, Rogers R, Matunis M, Mayhew C, Goodson M, Park-Sarge O, Sarge K, Goodson M (2001) Regulation of heat shock transcription factor 1 by stress-induced SUMO-1 modification. J Biol Chem 276:40263–40267
Hsu A-L, Murphy CT, Kenyon C (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300:1142–1145
Jaeger AM, Charles PW, Sistonen L, Thiele DJ (2016) Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat Struct Mol Biol 23:147–154
Kaganovich D, Kopito R, Frydman J (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454:1088
Kim D, Kim S-H, Li GC (1999a) Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun 254:264–268
Kim H, Kang H, Kim H (1999b) Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life 48:429–433
Kourtis N, Moubarak RS, Aranda-Orgilles B, Lui K, Aydin IT, Trimarchi T, Darvishian F, Salvaggio C, Zhong J, Bhatt K et al (2015) FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol 17:322–332
Krakowiak J, Zheng X, Patel N, Feder ZA, Anandhakumar J, Valerius K, Gross DS, Khalil AS, Pincus D (2018) Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. Elife 7:e31668
Labbadia J, Morimoto RI (2014) The biology of proteostasis in aging and disease. Annu Rev Biochem 84:1–30
Labbadia J, Morimoto RI (2015) Repression of the heat shock response is a programmed event at the onset of reproduction. Mol Cell 59:639–650
Li J, Chauve L, Phelps G, Brielmann RM, Morimoto RI (2016) E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev 30:2062–2075
Li J, Labbadia J, Morimoto RI (2017) Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol 27:895–905
Lindquist S (1986) The heat-shock response. Annu Rev Biochem 55:1151–1191
Lindquist S (2009) Protein folding sculpting evolutionary change. Cold Spring Harb Symp Quant Biol 74:103–108
Littlefield O, Nelson H (1999) A new use for the “wing” of the “winged” helix-turn-helix motif in the HSF–DNA cocrystal. Nat Struct Mol Biol 6:464–470
Ma X, Xu L, Alberobello A, Gavrilova O, Bagattin A, Skarulis M, Liu J, Finkel T, Mueller E (2015) Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1α transcriptional Axis. Cell Metab 22:695–708
Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT (2016) Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell 62:63–78
Masser AE, Kang W, Roy J, Mohanakrishnan Kaimal J, Quintana-Cordero J, Friedländer MR, Andréasson C (2019) Cytoplasmic protein misfolding titrates Hsp70 to activate nuclear Hsf1. ELife 8. pii: e47791. PMID: 31552827
McMillan RD, Xiao X, Shao L, Graves K, Benjamin IJ (1998) Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem 273:7523–7528
Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562
Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF (2007) Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 26:5086–5097
Mogk A, Tomoyasu T, Goloubinoff P, Rüdiger S, Röder D, Langen H, Bukau B (1999) Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J 18:6934–6949
Neef DW, Jaeger AM, Thiele DJ (2011) Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov 10:930
Nelson VK, Ali A, Dutta N, Ghosh S, Jana M, Ganguli A, Komarov A, Paul S, Dwivedi V, Chatterjee S et al (2014) Azadiradione ameliorates polyglutamine expansion disease in Drosophila by potentiating DNA binding activity of heat shock factor 1. Oncotarget 5
Neudegger T, Verghese J, Hayer-Hartl M, Hartl UF, Bracher A (2016) Structure of human heat-shock transcription factor 1 in complex with DNA. Nat Struct Mol Biol 23:140–146
Östling P, Björk JK, Roos-Mattjus P, Mezger V, Sistonen L (2007) Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J Biol Chem 282:7077–7086
Peffer S, Goncalves D, Morano KA (2019) Regulation of the Hsf1-dependent transcriptome via conserved bipartite contacts with Hsp70 promotes survival in yeast. J Biol Chem 294(32):12191–12202. PMID: 31239354
Pincus D, Anandhakumar J, Thiru P, Guertin MJ, Erkine AM, Gross DS (2018) Genetic and epigenetic determinants establish a continuum of Hsf1 occupancy and activity across the yeast genome. Mol Biol Cell 29(26):3168–3182
Prahlad V, Cornelius T, Morimoto RI (2008) Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320:811–814
Raychaudhuri S, Loew C, Körner R, Pinkert S, Theis M, Hayer-Hartl M, Buchholz F, Hartl UF (2014) Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell 156:975–985
Riback J, Laskowski P, Scott JL, Wallace E, Rojek AE, Schwartz MH, Sosnick TR, Drummond AD (2015) Heat shock triggers assembly of tRNA synthetases into an active supercomplex. Biophys J 108:221a
Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S et al (2011) High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc National Acad Sci 108:18378–18383
Santagata S, Mendillo ML, Tang Y, Subramanian A, Perley CC, Roche SP, Wong B, Narayan R, Kwon H, Koeva M et al (2013) Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341:1238303
Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, Dias-Santagata D, Koeva M, Stemmer SM, Whitesell L et al (2014) The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158:564–578
Shi Y, Mosser DD, Morimoto RI (1998) Molecularchaperones as HSF1-specific transcriptional repressors. Genes Dev 12:654–666
Shmidt R, Schessner JP, Borner G, Schuck S (2019) The proteasome biogenesis regulator Rpn4 cooperates with the unfolded protein response to promote ER stress resistance. Elife 8:pii: e43244
Solís EJ, Pandey JP, Zheng X, Jin DX, Gupta PB, Airoldi EM, Pincus D, Denic V (2016) Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol Cell 63:60–71
Solomon J, Rossi J, Golic K, McGarry T, Lindquist S (1991) Changes in hsp70 alter thermotolerance and heat-shock regulation in Drosophila. New Biol 3:1106–1120
Sorger PK, Nelson H (1989) Trimerization of a yeast transcriptional activator via a coiled-coil motif. Cell 59:807–813
Sorger PK, Pelham H (1988) Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54:855–864
Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4(1):49–60
Straus D, Walter W, Gross C (1990) DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma 32. Genes Dev 4:2202–2209
Tang Z, Dai S, He Y, Doty RA, Shultz LD, Sampson S, Dai C (2015) MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160:729–744
Trotter EW, Kao C, Berenfeld L, Botstein D, Petsko GA, Gray JV (2002) Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J Biol Chem 277:44817–44825
Tye BW, Commins N, Ryazanova LV, Wühr M, Springer M, Pincus D, Churchman SL (2019) Proteotoxicity from aberrant ribosome biogenesis compromises cell fitness. Elife 8:43002
van Oosten-Hawle P, Porter RS, Morimoto RI (2013) Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153:1366–1378
Wallace E, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM et al (2015) Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162:1286–1298
Westerheide SD, Anckar J, Stevens SM, Sistonen L, Morimoto RI (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323:1063–1066
Whitesell L, Lindquist S (2009) Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets 13:469–478
Yamamoto A, Ueda J, Yamamoto N, Hashikawa N, Sakurai H (2007) Role of heat shock transcription factor in Saccharomyces cerevisiae oxidative stress response. Eukaryot Cell 6:1373–1379
Yang S, Huang S, Gaertig MA, Li X-J, Li S (2014) Age-dependent decrease in chaperone activity impairs MANF expression, leading to purkinje cell degeneration in inducible SCA17 mice. Neuron 81:349–365
Zheng X, Krakowiak J, Patel N, Beyzavi A, Ezike J, Khalil AS, Pincus D (2016) Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. Elife 5:e18638
Zheng X, Beyzavi A, Krakowiak J, Patel N, Khalil AS, Pincus D (2018) Hsf1 phosphorylation generates cell-to-cell variation in Hsp90 levels and promotes phenotypic plasticity. Cell Rep 22:3099–3106
Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–480
Acknowledgements
Thanks to A. Ali and G. Bushkin for critical reading of the manuscript. This work was supported by an Early Independence Award from the NIH (DP5 OD017941).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pincus, D. (2020). Regulation of Hsf1 and the Heat Shock Response. In: Mendillo, M.L., Pincus, D., Scherz-Shouval, R. (eds) HSF1 and Molecular Chaperones in Biology and Cancer. Advances in Experimental Medicine and Biology, vol 1243. Springer, Cham. https://doi.org/10.1007/978-3-030-40204-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-40204-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40203-7
Online ISBN: 978-3-030-40204-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)