
Abstract
The tumor microenvironment (TME) is rich in matrix components, growth factors, cytokines, and enzymatic modifiers that respond to changing conditions, to alter the fundamental properties of the tumor bed. Perlecan/HSPG2, a large, multi-domain heparan sulfate proteoglycan, is concentrated in the reactive stroma that surrounds tumors. Depending on its state in the TME, perlecan can either prevent or promote the progression of cancers to metastatic disease. Breast, prostate, lung, and renal cancers all preferentially metastasize to bone, a dense, perlecan-rich environment that is initially a “hostile” niche for cancer cells. Driven by inflammation, production of perlecan and its enzyme modifiers, which include matrix metalloproteinases (MMPs), sulfatases (SULFs), and heparanase (HPSE), increases in the reactive stroma surrounding growing and invading tumors. MMPs act upon the perlecan core protein, releasing bioactive fragments of the protein, primarily from C-terminal domains IV and V. These fragments influence cell adhesion, invasion, and angiogenesis. Sulfatases and heparanases act directly upon the heparan sulfate chains, releasing growth factors from reservoirs to reach receptors on the cancer cell surface. We propose that perlecan modifiers, by promoting the degradation of the perlecan-rich stroma, “flip the molecular switch” and convert the “hostile” stroma into a welcoming one that supports cancer dissemination and metastasis. Targeted therapies that prevent this molecular conversion of the TME should be considered as potential new therapeutics to limit metastasis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Perlecan/HSPG2
- Heparan sulfate proteoglycan
- Matrix metalloproteinase
- Sulfatase
- Heparanase
- Bone
- Inflammation
- Metastasis
- Glycocalyx
- Breast
- Prostate
- Lung
- Renal
- Reactive stroma
6.1 Introduction to the Tumor Microenvironment (TME)
6.1.1 Components of the TME
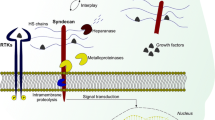
As the cancer field shifts toward a macroscopic view of the tumor microenvironment (TME), the need to understand the complexities of tumor-stromal interactions moves to the forefront [1]. Cellular and molecular interactions at the cancer cell surface relay a constant stream of signals that influence cancer growth and metastasis. The TME consists of the non-cancerous cells present in and around the tumor including fibroblasts, immune cells, and endothelial cells in conjunction with extracellular matrices (ECM) that can support growth, survival, and metastasis of cancer cells. These matrices, factors that bind to them, and their enzymatic modifiers can be produced by any of the cells present in the TME, working dynamically either to prevent or promote cancer cell dissemination (Fig. 6.1).

Dynamic interactions among various cell types present in the TME. Reactive stroma (left panel) includes perlecan that attempts to “wall off” the tumor, limiting invasion and preventing dissemination. Disseminating tumors remodel the ECM, including cleavage of perlecan, allowing for tumor dyscohesion and invasion
6.1.2 Role of TME in Cancer Progression
While the exact nature of the cancer-stromal interaction is still being defined, work in many labs has begun to illustrate the enormous impact the TME can have on cancer progression. For example, Yu-Lee et al. conducted a systematic study in mice, inoculating similar numbers of prostate cancer cells into two locations: subcutaneously in the back and intrafemorally. A comparison of the outcomes between these two groups revealed significantly less growth for tumors growing in the bone versus their skin counterparts. Specifically, cells in the bone, a perlecan-rich environment, became dormant, whereas those in the skin formed tumors within 3–5 weeks [2]. This finding can only be explained in the context of the cancer-stromal interface, highlighting the importance of localized TME, especially in bone, on key aspects of cancer cell behavior.
Breast, prostate, lung, and renal cancers represent about 75% of all cancers that preferentially metastasize to bone [3, 4]. The bone represents a unique TME that, once colonized, is associated with progressive metastasis and often lethal disease [5]. Given that the TME in bone is initially “hostile” to invading cancer cells, it is interesting to consider how an initially dormancy-inducing TME becomes one that fosters cancer cell growth and metastasis [6]. Cancers metastasizing to bone can be divided into two subtypes: osteolytic and osteoblastic/sclerotic. Osteolytic bone metastases are responsible for the destruction of bone, while those of the osteoblastic subtype are considered bone-forming. Prostate cancer bone metastases are most often osteoblastic, though some more neuroendocrine tumors can produce a mixed population of osteoblastic and osteolytic lesions [7]. Bone metastases from primary renal, lung, and breast tumors have a tendency to be more osteolytic, where osteoclasts are controlled by the invading cancer cells [8, 9]. In each case, the TME plays a vital role in determination of the type of lesion that will form and how the cancer will progress.
6.1.3 Extracellular Matrix in the TME
Researchers have begun to appreciate the impact that various ECM constituents in the TME can have on normal and disease biology [1]. In traditional wound healing responses, ECM remodeling and growth factor actions bring damaged tissue back to homeostasis. These normal processes are pathologically co-opted by cancer cells in the TME, leading to its description as the “wound that never heals” [10]. As recently defined in the Matrisome Project, the ECM is composed of 274 core proteins with 753 associated factors, proteins, and regulators, each functioning to maintain tissue integrity and to provide a reservoir of readily available factors to promote wound healing and regeneration [11]. Each tissue expresses a unique subset of these components that comprise the TME. In cancer, these ECM components in the TME can become major drivers or inhibitors of metastasis and disease progression. Proteoglycans are core components of the Matrisome that are hallmarked by their structural and functional diversity, play major roles in cancer cell fate.
Proteoglycans are defined as proteins containing one or more covalently attached glycosaminoglycan (GAG) chains. GAG chains are categorized into four major classes, heparan sulfate, chondroitin sulfate, keratan sulfate, and hyaluronate, the last of which is synthesized as a free glycan [12]. The composition of these GAG chains on proteoglycans varies greatly within different tissues, with some predominated by heparan sulfate and others by chondroitin or keratan sulfate. Among these GAGs, heparan sulfate plays an essential role in the binding of heparin-binding growth factors (HBGFs) and is composed of unbranched negatively charged disaccharide units with spatially organized sulfate groups to endow binding specificity for individual HBGFs [13]. Release of these HBGFs relies upon three key groups of extracellular enzymes that can modify heparan sulfate polymers and alter growth factor binding and local bioavailability: matrix metalloproteinases (MMPs), sulfatases (SULF1 and SULF2), and heparanase (HPSE) [14].
6.2 The TME, Glycocalyx, and Pericellular Matrix
6.2.1 TME and Cancer Cell Behavior
Tumors are not just masses of clustered malignant cells, but rather they can be considered as “disorganoids” that are composed of various cell types, including fibroblasts, stromal cells, immune cells, and cells from the vascular network that are encased by a dense ECM in the pericellular space. Cancer cells not only depend on driver oncogenes to survive, grow, and metastasize, but they also rely on pro-survival signals produced in the associated stroma [15]. Despite their growth persistence, highly aneuploid, genetically unstable cancer cells are often quite fragile and die rapidly when separated from the TME to which they have become accustomed.
6.2.2 Glycocalyx
The “glycocalyx,” another component of the TME, is a layer of glycans present on the surface of cancer cells as well as various normal cell types and tissue structures [12]. The glycocalyx serves a variety of functions that both protect cells and ensure their survival. Heparan sulfate proteoglycans (HSPGs), such as syndecan and glypican, are present at the cell surface where they often function as co-receptors for growth factor signaling complexes. For example, binding of fibroblast growth factor-2 (FGF-2/basic FGF) to its receptor is stabilized by heparan sulfate found on the GAG chains of the co-receptor, typically syndecan [16]. Acting at or near the cell surface, extracellular modifiers of heparan sulfate such as the SULFs and HPSE can play vital roles in modulation of growth factor signaling, cell survival, invasion, and metastasis.
6.2.3 Pericellular Matrix
As cancer progresses, normal tissue boundaries are disrupted and local ECM turnover prevails. Among these ECM components, heparan sulfate proteoglycan 2 (HSPG2)/perlecan, a major component of the basement membrane, is critically involved in patrolling tissue boundaries [17]. Perlecan can be produced by some cancer cells, but the majority of perlecan in the TME is made by cells in the reactive stroma where it co-localizes with smooth muscle actin, tenascin, and thrombospondins [17, 18]. Unlike the HSPGs syndecan and glypican that reside in the glycocalyx of cancer cells, perlecan is fully secreted and resides in the pericellular space [12]. Perlecan is present at high levels in the reactive stroma surrounding breast, lung, renal, and prostate cancer lesions (Fig. 6.2). Perlecan modification by SULF1, SULF2, or HPSE in the TME affects cancer cell proliferation, survival, invasion, and metastasis [12, 14]. Upon injury or invasion that penetrates the basement membrane, cancer cells come in contact with the cells in the stromal compartment of the TME. In stroma, bound HBGFs can be released enzymatically from perlecan bound and sequestered in the stroma. This occurs as a direct consequence of the activation of various matrix remodeling enzymes in the TME that include both proteases and glycosaminoglycanases. These degradative processes continue during metastasis, such as to bone, where they foster the development of secondary and tertiary metastases. In this chapter, we will focus specifically on three of these extracellular enzyme modifiers of perlecan: MMPs, SULFs, and HPSE. Each of these enzymes plays a role in the TME during initial cancer invasion and metastasis and then later in the metastatic niche of bone or other common sites of secondary cancer growth.

Immunofluorescence staining of perlecan-rich stroma in the TME surrounding a primary prostatic lesion. Dotted line indicates start of the non-permissive perlecan barrier adjacent to the basement membrane surrounding the lesion. Perlecan (green) and nuclei (blue). Note the intense staining of perlecan surrounding the blood vessels near the tumor (arrowheads)
6.3 Perlecan/HSPG2 in the Tumor Microenvironment
6.3.1 Perlecan Function in the TME
In the presence of transforming growth factor-β (TGF-β), tumor necrosis factor-α (TNF-α) is a major cytokine regulator of perlecan mRNA expression in cancer cells, normal stromal cells, and a subpopulation of bone marrow stromal cells [19]. In the context of breast cancer, the perlecan promoter can be positively regulated by TGF-β and negatively regulated by interferon-γ (INF-γ) [20]. TGF-β and TNF-α recruit and activate immune, endothelial, and stromal cells at the primary tumor or metastatic sites; this process, in turn, further triggers production of inflammatory cytokines and ECM, creating a positive feedforward loop. In normal tissues, perlecan possesses antitumoral activity by stabilizing tissue borders, decreasing cell motility, and favoring cell survival. Epithelial cells, epidermal cells, endothelial cells, smooth muscle cells, fibroblasts, osteocytes, and chondrocytes all can synthesize perlecan [17]. These various perlecan cellular sources contribute to the distribution of perlecan in the basement membrane, in the stromal matrix, and at other tissue borders including in bone [17]. It is well known that an intact epithelial basement membrane exists in benign tumors, whereas invasive tumors lack an intact basement membrane allowing cells to move into stroma [21]. Perlecan expression is highly regulated in the TME surrounding invasive and metastatic carcinomas, specifically in the desmoplastic stroma and at sites of bone metastasis [19, 22,23,24,25,26]. Also, perlecan expression is induced in various tumors, particularly those undergoing epithelial-to-mesenchymal transition (EMT) [27]. Studies have indicated that metastatic tumors might be detected by the host defense system, and these tumors are encapsulated with dense perlecan-rich matrix to prevent further dissemination of these tumor cells [19, 28]. Evidence suggests it is likely that tumor cells and cells in the stroma defeat this barrier function over time by expressing enzymes that participate in basement membrane degradation, such as MMPs, SULFs, and HPSE.
Until recently, the identity of a direct binding partner of perlecan at the cancer cell surface remained elusive. A recent study from our lab found that semaphorin 3A (SEMA3A) and the most C-terminal portion of the fourth domain of perlecan, domain IV-3, interact with one another to induce prostate cancer cell-cell cohesion and dissolution of focal adhesions [29]. Work done by Herman et al. showed the strong influence of SEMA3A in the TME surrounding prostate cancer cells, where it inhibits migration and invasion [30]. The recently described interaction of SEMA3A with perlecan may explain the similar phenotypes observed when both molecules are dynamically altered in the TME [29].
Perlecan in the TME not only acts as a physical barrier to restrict cell movement, but its heparan sulfate chains also sequester bioactive proteins such as HBGFs, chemokines, cytokines, and some enzymes, adding to the complexity of perlecan’s role in tissue remodeling and understanding of its role in tumor progression [27]. A wide variety of HBGFs form complexes with perlecan, such as members of the fibroblast growth factor family, vascular endothelial growth factor, heparin-binding EGF, and many cytokines (e.g., interleukin-3 (IL-3), granulocyte-macrophage colony-stimulating factor (GM-CSF), and INF-γ [31]. FGF2 is sequestered in complex with perlecan in the basement membrane and stroma of various tissues and by other HSPGs in the glycocalyx [32, 33]. Perlecan’s heparan sulfate chains typically sequester the FGF ligand, although interactions of other FGFs with the core protein have been reported [34]. The release of the FGF ligand from sequestration sites in stroma allows for diffusion to receptor binding sites in the glycocalyx to activate complex signaling cascades that control cell proliferation, motility, and adhesion [12, 35].
6.3.2 Perlecan and Angiogenesis
Neoangiogenesis, the development of new blood vessels from pre-existing vasculature, is required in early tumorigenesis to supply nutrients and oxygen to cancer cells [12, 36, 37]. Angiogenesis in the TME is a complex process, which involves the organized actions of pericytes, endothelial cells, and smooth muscle cells [20]. A group of major players needed for malignant angiogenesis is the family of MMPs (Table 6.1) [36, 38]. Proteolytic release of the C-terminal region of perlecan produces fragments with dramatic effects on angiogenesis [39]. These fragments, known variously as endorepellin, domain V, and C-terminal laminin-like globular domain (LG3), remain a very active area of study that may lead to production of novel classes of therapeutics for a variety of angiogenic-related disorders [40, 41].
6.4 Immune Cells in the TME
6.4.1 Immune Cells and Cytokines
Immune infiltration and resulting inflammation are hallmark features of a reactive stromal response. Chronic inflammation is a major driver of ECM deposition and catabolic enzyme upregulation, with a net overall effect of increased tissue turnover. This turnover digests the matrix-bound core protein and releases diffusible perlecan fragments that can have activities distinct from the intact proteoglycan. Peptide mapping showed the majority of these fragments are derived from the C-terminus and can be detected in the blood of patients with metastases [23]. Various studies found macrophages to represent a large portion of the diverse immune infiltration population in the TME. Tumor-associated macrophages (TAMs) in advanced disease phenotypically resemble M2 macrophages, often stimulating and promoting neovascularization and the induction of vascular network formation [42]. Interestingly, high levels of TAM infiltration are associated with poor patient survival and dim prognosis in patients with lung, breast, renal, or prostate cancers. The presence of these TAMs in the TME can exacerbate chronic inflammation and stimulate ECM remodeling, paralleling events that would occur in wound healing [43]. While TAMs are the most abundant immune cell type in the TME, several reports in prostate cancer show an increased presence of other immune cells including myeloid-derived suppressor cells and natural killer cells, all potentially conferring the innate TME immune response [44]. Ongoing work aims to determine how the presence of these various classes of immune cells in the TME contribute to the transition from a “hostile” TME to one that participates and accelerates metastasis and lethal progression.
6.4.2 Inflammation in the TME
TNF-α, a protein often present during inflammation, is present at high levels in the tumor microenvironment of prostate, breast, lung, and renal cancers [45, 46]. TNF-α can be produced by many cell types in the TME, but it is most commonly known as a factor released by TAMs. TNF-α released by immune cells in the prostate cancer microenvironment increases the expression and secretion of perlecan by both prostate cancer cells and bone stromal cells via TNF-α-induced nuclear factor kappa-light-chain-enhancer of activated B cell (NFκB) translocation to the nucleus. Once inside the nucleus, NFκB undergoes a unique binding step, where binding to the HSPG2 promoter region increases perlecan transcript levels [19]. In breast cancer, the tumor microenvironment demonstrates a similar phenomenon, where TNF-α released from reactive stroma in breast cancer signals for increased expression of ECM proteins and ECM remodeling enzymes [46]. Similarly, TNF-α, interleukin-1β (IL-1β) , and interleukin-6 (IL-6) all contribute to cancer metastasis through induced secretion of HPSE from endothelial cells in the TME. The presence of HPSE further favors EMT and enhances pro-metastatic signaling [47, 48].Along with HPSE, other remodeling enzymes (e.g., MMPs, SULFs) can be activated by resulting inflammation from infiltrating immune cells. Hagemann et al. published a co-culture study with TAMs and invasive breast cancer cell lines, observing an increase in MMP-2, MMP-3, MMP-7, and MMP-9 in a TNF-ɑ-dependent manner [49]. In the TME, this TAM-mediated inflammation plays various roles, upregulating not just the production of perlecan but also perlecan-modifying enzymes, ultimately helping to flip the “molecular switch.”
6.5 Perlecan Modifiers in the TME
The reactive TME is rich in perlecan and its enzyme modifiers whose expression is regulated by environmental factors such as inflammation, factors produced by the disseminated cancer cells themselves, tissue turnover, and the unique character of the tumor site. Table 6.1 provides a summary of some of the more common enzyme modifiers found in the TME that influence the molecular state of perlecan and that together comprise the molecular switch responsible for converting a TME from “hostile” to one that actively participates in tumor growth and metastasis. Thus, these modifiers can be considered to be the factors that “flip the switch” from conditions that limit progression to those that favor further metastasis and onset of lethal disease.
6.5.1 MMPs
It is well accepted that MMPs are upregulated in many cancers, especially in the presence of chronic inflammation. These MMPs have been studied for decades for their capacity to degrade and remodel surrounding matrix in the TME, fostering invasive and metastatic disease. MMP-1, MMP-3, and MMP-7 can digest perlecan in the TME, but for MMP-1 and MMP-3, the efficient removal of heparan sulfate or chondroitin sulfate chains first must occur [38, 53]. MMP-7 demonstrates a unique ability to degrade perlecan without prior removal of the GAG chains, a feature that contributes to its overall impact in the degradation of basement membranes and destruction of reactive stroma [53]. In 2009, work done by the Parks group demonstrated an interesting interplay between heparan sulfate chains and the proteolytic activity of MMP-7, showing that sulfated GAGs can drive activity and specificity of the MMP [60]. While each of these MMPs plays vital roles in contributing to the whole cancer TME landscape, their localization often differs. MMP-7 more frequently localizes to the luminal cancer cell compartment while MMP-1, MMP-2, and MMP-9 tend to localize specifically to the stromal cells [61]. MMP-7 status in renal cell carcinoma, and other cancers, is a major indicator of disease progression and prognosis [23, 62]. In renal cell carcinomas, MMP-2 and MMP-9 showed increased expression in relation to their normal counterparts [63]. Interestingly, in a mouse model of prostate cancer, mice without MMP-2 showed increased survival outcome measures, while those with deficient MMP-7 demonstrated no significant changes in survival outcomes but showed a reduction in both endothelial area coverage and vessel size. In this same study, mice with deficient MMP-9 showed similar numbers of vessels within the tumor as compared to the control but demonstrated a decrease in vessel size, with a more elongated and regular vessel shape, illustrating the impact that various MMPs can have on tumor angiogenesis and survival [61]. Considered together, in a survey of the breast, lung, prostate, and renal literature, it is generally true that elevated levels of MMPs correlate with poor prognosis for patients. Knowing this, it seems that targeting MMPs would be an effective method to control the progression of cancers to metastatic and, ultimately, lethal disease. Marimastat, a competitive MMP inhibitor, underwent clinical trial in both breast and small-cell lung cancer, failing in both settings. When compared to the placebo group, patients receiving Marimastat treatment showed no significant benefit in progression-free survival, and in some cases, treatment resulted in inferior overall patient health due to musculoskeletal toxicity [64, 65]. While we choose to highlight Marimastat in this chapter, other clinical trials with MMP inhibitors have yielded similar results, illustrating their ineffectiveness as a singular therapy [66]. These inhibitors have not yet been discarded as an option for treatment however, as combinatorial therapies with other compounds, such as Carboplatin, have yielded promising preliminary results [67].
6.5.2 SULFs
Studies of the molecular composition of the TME have shown that extracellular sulfatases frequently reside in the stroma surrounding growing tumors where they can act directly on perlecan deposited there. Sulfatase (SULF) expression patterns in the TME are complex, with different cancers demonstrating unique SULF signatures. It is interesting to think about the impact of SULF localization in explaining these unique signatures in the TME. SULFs localized at the cancer cell surface would have a large negative impact on the ability of cell surface heparan sulfate proteoglycans in the glycocalyx such as syndecan and glypican to act in their co-receptor roles. SULFs acting at this location release growth factors away from the cell surface, suppressing growth and creating growth factor reservoirs in the surrounding stroma. In contrast, those SULFs localized in the stromal compartment release available HBGFs from the stroma to diffuse and bind their specific receptors at the cell surface. These opposing actions ultimately promote localized growth of the cancer cells even in the presence of SULFs in the glycolax. Studies performed using various cancer cell types demonstrate the dynamic influence of SULFs on the invasion and growth potential for cancers. For example, in reports using breast cancer cells, SULF1 expression is reduced while SULF2 is upregulated in the localized tumor [68, 69]. Lung cancer SULF expression patterns match those of breast cancer, with SULF2 being upregulated in the tumor cells [70]. In prostate cancer, overexpression of SULF2 in the transfected prostate cancer cell lines DU-145 and PC3 presented an oncogenic phenotype, with prostate cancer cells showing greater viability and increased migration capacity [71]. In a study utilizing patient samples with renal cell carcinoma, high SULF2 expression in the tumor cells was correlated with a less invasive phenotype, with low SULF2 expression correlating with advanced invasive features [72]. While the utilization of each SULF by these cancer types remains under investigation, the known ability of these enzymes to modulate growth factor release in the TME makes them interesting enzymes to define. Currently, studies aim to investigate why the two SULF isoforms have variable regulation patterns among various cancer subtypes.
6.5.3 HPSE
Heparan sulfates on perlecan bind a wide variety of molecules in the TME, creating a reservoir of rich growth-promoting and angiogenic factors. Many HBGFs can bind simultaneously to a single heparan sulfate chain depending on its length and pattern of sulfation. As an endo-β-D-glucuronidase, heparanase specificity relies on the O-sulfation along the heparan sulfate chains on the full-length molecule and cleaves the GAG at specific undersulfated regions. These regions typically flank the highly sulfated sites to which most HBGFs are attached [73]. Growth factors such as FGF2/bFGF and VEGF released by HPSE provide an important mechanism supporting neovascularization in cancer, illustrating some of the influence of HPSE-released growth factors on cancer progression [74, 75].
In many cancers, elevated expression of HPSE is associated with poor prognosis, indicating its key role in the promotion of primary tumors to lethal disease [76,77,78]. HPSE expression in clear cell renal cell carcinomas positively correlates with patient outcomes, with those patients expressing higher HPSE experiencing higher levels of invasion and metastasis [79]. In the case of breast cancer, studies performed with cells overexpressing HPSE showed that tumors grew faster and showed increased vascularization [78]. One study examining prostate cancer clinical samples showed that HPSE levels were significantly higher in cancer tissue than in the corresponding normal tissues that were sampled [80]. In another study increased levels of HPSE were associated with increased metastasis and, in the case of breast cancer metastasis, bone resorption was observed [81]. Because of the correlation between HPSE and cancer aggressiveness, several HPSE inhibitors have been developed and tested in preclinical models with promising results, showing a reduction in tumor growth and reduced angiogenesis [82]. While these inhibitors have shown promising results, current thinking is that efficacy will be most enhanced as a combinatorial therapy. As these inhibitors progress through clinical trials, it will be interesting to see what combinatorial agents are most effective.
6.6 Conclusions, Perspectives, and Future Directions
Emerging evidence places perlecan as a border proteoglycan and signaling hub in the basement membrane and pericellular matrix, where it coordinates and integrates a myriad of cellular signals to maintain proper tissue homeostasis. In cancer, where normal tissue compartments are disrupted by tissue turnover, perlecan becomes a participant in aberrant signaling that fosters progression, invasion, and metastasis. Differences in perlecan’s ability to signal depend on context to explain its effects on tumor growth, angiogenesis, blood vessel integrity, endothelial cell proliferation, cancer cell adhesion, and motility [20]. In the context of the TME, cellular behavior can be modulated by the actions of perlecan’s modifiers that can change its structure. MMPs, particularly MMP-7, cleave perlecan producing fragments that can have very different bioactivities from the intact proteoglycan [53, 83]. MMP-7 stands out as it demonstrates a unique ability to degrade perlecan without prior removal of the GAG chains in reactive stroma [53]. One key “hot spot” for function-altering cleavage is domain IV, a key region of the core protein that functions to determine cell-cell versus cell-matrix interactions. Another key functional region of the core protein is evidenced by studies showing that C-terminal cleavage of perlecan produces fragments from domain V that modulate angiogenesis [38, 39, 84]. Thus, while intact perlecan can serve as a suppressor of invasion and angiogenesis, MMP-cleaved perlecan can support cell migration, enhancing tissue turnover, and triggering angiogenesis, a phenomenon that we have called the “molecular switch.” This switch can lead to diverse outcomes, either positive or negative, for tumor progression [20].
Other enzyme modifiers in the TME, SULFs and HPSE, work to influence localized bioavailability of HBGFs. Strategies to either restore or reduce sulfatase expression/activity, depending on cancer type, using small molecule inhibitors can help to create novel cancer treatments. HPSE cleaves heparan sulfate chains with associated growth factors. In cancers that preferentially metastasize to bone HPSE is elevated, correlating with poor prognosis [76,77,78]. One of the HBGFs bound on the heparan sulfate chains that is released is FGF2/bFGF, which participates in neovascularization in cancer [74]. An attractive approach to inhibit HPSE is the use of neutralizing antibodies, though some small molecule compounds have also been investigated [85]. Future work envisions studies of the effects of perlecan fragments on inflammation, recruitment of immune cells to tumor sites, production of circulating tumor cells, and formation of metastases and how the perlecan modifiers can be targeted to prevent cancer dissemination.
The perlecan-rich bone marrow is initially a “hostile” niche. Metastatic cancer cells adapt to this niche and thrive in the TME. Recent studies identify perlecan modification by MMPs and glycosaminoglycanases as main factors to trigger a desmoplastic reaction. In the TME, chronic inflammation and abnormal immune infiltration drive ECM deposition, catabolic enzyme production, and tissue turnover. It is interesting to consider that combining immunotherapy or existing chemotherapies with targeted therapies (e.g., MMP, SULF or HSPE inhibitors, or angiogenesis/inflammation blockers) might lead to new ways to limit bone metastases by stabilizing the tumor-suppressing properties of perlecan in the TME. The main challenge is to identify and tailor the treatment to the individual cancer type, a goal that can only be achieved by first thoroughly understanding the ways that perlecan and its modifiers interact in the context of the TME.
References
Welch DR, Hurst DR (2019) Defining the hallmarks of metastasis. Cancer Res 79(12):3011–3028. https://doi.org/10.1158/0008-5472.CAN-19-0458
Yu-Lee L-Y, Yu G, Lee Y-C et al (2018) Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFβRIII–p38MAPK–pS249/T252RB pathway. Cancer Res 78(11):2911–2924. https://doi.org/10.1158/0008-5472.CAN-17-1051
Tsuya A, Kurata T, Tamura K, Fukuoka M (2007) Skeletal metastases in non-small cell lung cancer: a retrospective study. Lung Cancer 57(2):229–232. https://doi.org/10.1016/J.LUNGCAN.2007.03.013
Tofe AJ, Francis MD, Harvey WJ (1975) Correlation of neoplasms with incidence and localization of skeletal metastases: an analysis of 1,355 diphosphonate bone scans. J Nucl Med 16(11):986–989. http://jnm.snmjournals.org/content/16/11/986.long
Macedo F, Ladeira K, Pinho F et al (2017) Bone metastases: an overview. Oncol Rev 11(1):321. https://doi.org/10.4081/oncol.2017.321
Zhang C, Soori M, Miles F et al (2011) Paracrine factors produced by bone marrow stromal cells induce apoptosis and neuroendocrine differentiation in prostate cancer cells. Prostate 71(2):157. https://doi.org/10.1002/PROS.21231
Keller ET, Brown J (2004) Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem 91:718–729. https://doi.org/10.1002/jcb.10662
Esposito M, Guise T, Kang Y (2018) The biology of bone metastasis. Cold Spring Harb Perspect Med 8(6). https://doi.org/10.1101/cshperspect.a031252
Logothetis CJ, Lin S-H (2005) Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer 5(1):21–28. https://doi.org/10.1038/nrc1528
Byun JS, Gardner K (2013) Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol 182(4):1055–1064. https://doi.org/10.1016/j.ajpath.2013.01.009
Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO (2016) The extracellular matrix: tools and insights for the “omics” era. Matrix Biol 49:10–24. https://doi.org/10.1016/J.MATBIO.2015.06.003
Kang H, Wu Q, Sun A, Liu X, Fan Y, Deng X (2018) Cancer cell glycocalyx and its significance in cancer progression. Int J Mol Sci 19(9):2484. https://doi.org/10.3390/ijms19092484
Kirkpatrick CA, Selleck SB (2007) Heparan sulfate proteoglycans at a glance. J Cell Sci 120(11):1829–1832. https://doi.org/10.1242/jcs.03432
Nagarajan A, Malvi P, Wajapeyee N (2018) Heparan sulfate and Heparan Sulfate Proteoglycans in cancer initiation and progression. Front Endocrinol (Lausanne) 9(AUG):1–11. https://doi.org/10.3389/fendo.2018.00483
Owusu BY, Galemmo R, Janetka J, Klampfer L (2017) Hepatocyte growth factor, a key tumor-promoting factor in the tumor microenvironment. Cancers (Basel) 9(4):1–16. https://doi.org/10.3390/cancers9040035
Kelly R, Editor G, Tkachenko E, Rhodes JM, Simons M (2005) New kids on the signaling block. Circ Res 96:488–500. https://doi.org/10.1161/01.RES.0000159708.71142.c8
Farach-Carson MC, Warren CR, Harrington DA, Carson DD (2014) Border patrol: insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol 34:64–79. https://doi.org/10.1016/J.MATBIO.2013.08.004
S C, G I, S F et al (2015) Spatial organization of the tenascin-C microenvironment in experimental and human cancer. Cell Adhes Migr 9(1–2):4–13. https://doi.org/10.1080/19336918.2015.1005452
Warren CR, Grindel BJ, Francis L, Carson DD, Farach-Carson MC (2014) Transcriptional activation by NFκB increases perlecan/HSPG2 expression in the desmoplastic prostate tumor microenvironment. J Cell Biochem 115(7):1322–1333. https://doi.org/10.1002/jcb.24788
Poluzzi C, Iozzo RV, Schaefer L (2016) Endostatin and endorepellin: a common route of action for similar angiostatic cancer avengers. Adv Drug Deliv Rev 97:156–173
Nerlich AG, Lebeau A, Hagedorn HG, Sauer U, Schleicher ED (1998) Morphological aspects of altered basement membrane metabolism in invasive carcinomas of the breast and the larynx. Anticancer Res 18:3515–3520
Nackaerts K, Verbeken E, Deneffe G, Vanderschueren B, Demedts M, David G (1997) Heparan sulfate proteoglycan expression in human lung-cancer cells. Int J Cancer 74(3):335–345. https://doi.org/10.1002/(SICI)1097-0215(19970620)74:3<335::AID-IJC18>3.0.CO;2-A
Grindel B, Li Q, Arnold R et al (2016) Perlecan/HSPG2 and matrilysin/MMP-7 as indices of tissue invasion: tissue localization and circulating perlecan fragments in a cohort of 288 radical prostatectomy patients. Oncotarget 7(9):10433–10447. https://doi.org/10.18632/oncotarget.7197
Iozzo RV, Cohen IR, Grässel S, Murdoch AD (1994) The biology of perlecan: the multifaceted heparan sulphate proteoglycan of basement membranes and pericellular matrices. Biochem J 302(Pt 3):625–639. https://doi.org/10.1042/bj3020625
Melrose J, Smith S, Cake M, Read R, Whitelock J (2005) Comparative spatial and temporal localisation of perlecan, aggrecan and type I, II and IV collagen in the ovine meniscus: an ageing study. Histochem Cell Biol 124(3–4):225–235. https://doi.org/10.1007/s00418-005-0005-0
Gbormittah FO, Lee LY, Taylor K, Hancock WS, Iliopoulos O (2014) Comparative studies of the proteome, glycoproteome, and N-glycome of clear cell renal cell carcinoma plasma before and after curative nephrectomy. J Proteome Res 13(11):4889–4900. https://doi.org/10.1021/pr500591e
Mongiat M, Taylor K, Otto J et al (2000) The protein core of the proteoglycan perlecan binds specifically to fibroblast growth factor-7. J Biol Chem 275(10):7095–7100. https://doi.org/10.1074/jbc.275.10.7095
Gubbiotti MA, Neill T, Iozzo RV (2017) A current view of perlecan in physiology and pathology: a mosaic of functions. Matrix Biol 57–58:285–298
Grindel BJ, Martinez JR, Tellman TV et al (2018) Matrilysin/MMP-7 cleavage of perlecan/HSPG2 complexed with semaphorin 3A supports FAK-mediated stromal invasion by prostate cancer cells. Sci Rep 8(1):7262. https://doi.org/10.1038/s41598-018-25435-3
Herman JG, Meadows GG (2007) Increased class 3 semaphorin expression modulates the invasive and adhesive properties of prostate cancer cells. Int J Oncol 30(5):1231–1238. http://www.ncbi.nlm.nih.gov/pubmed/17390026. Accessed June 8, 2018
Farach-Carson MC, Carson DD (2007) Perlecan a multifunctional extracellular proteoglycan scaffold. Glycobiology 17(9):897–905. https://doi.org/10.1093/glycob/cwm043
Aviezer D, Hecht D, Safran M, Eisinger M, David G, Yayon A (1994) Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 79(6):1005–1013. https://doi.org/10.1016/0092-8674(94)90031-0
Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC (1993) Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem 268(32):23906–23914. http://www.jbc.org.eu1.proxy.openathens.net/content/268/32/23906. Accessed July 15, 2019
Smith SM, West LA, Hassell JR (2007) The core protein of growth plate perlecan binds FGF-18 and alters its mitogenic effect on chondrocytes. Arch Biochem Biophys 468(2):244–251
Zhou Z, Wang J, Cao R et al (2004) Impaired angiogenesis, delayed wound healing and retarded tumor growth in Perlecan heparan sulfate-deficient mice. Cancer Res 64(14):4699–4702. https://doi.org/10.1158/0008-5472.CAN-04-0810
Deryugina EI, Quigley JP (2010) Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: contrasting, overlapping and compensatory functions. Biochim Biophys Acta, Mol Cell Res 1803(1):103–120. https://doi.org/10.1016/j.bbamcr.2009.09.017
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M (2006) Angiogenesis in cancer. Vasc Heal Risk Manag 2(3):213–219
Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA (1996) The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem 271(17):10079–10086. https://doi.org/10.1074/JBC.271.17.10079
Bix G, Iozzo RV (2008) Novel interactions of perlecan: unraveling perlecan’s role in angiogenesis. Microsc Res Tech 71(5):339–348. https://doi.org/10.1002/jemt.20562
Bix G, Fu J, Gonzalez EM et al (2004) Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through α2β1 integrin. J Cell Biol 166(1):97–109. https://doi.org/10.1083/jcb.200401150
Lee B, Clarke D, Al Ahmad A et al (2011) Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest 121(8):3005–3023. https://doi.org/10.1172/JCI46358
Guo C, Buranych A, Sarkar D, Fisher PB, Wang X-Y (2013) The role of tumor-associated macrophages in tumor vascularization. Vasc Cell 5(1):20. https://doi.org/10.1186/2045-824X-5-20
Gonzalez H, Hagerling C, Werb Z (2018) Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32(19–20):1267–1284. https://doi.org/10.1101/gad.314617.118
Lin D, Wang X, Choi SYC, Ci X, Dong X, Wang Y (2016) Immune phenotypes of prostate cancer cells: evidence of epithelial immune cell-like transition? Asian J Urol 3(4):195–202. https://doi.org/10.1016/j.ajur.2016.08.002
Yoshida N, Ikemoto S, Narita K et al (2002) Interleukin-6, tumour necrosis factor α and interleukin-1β in patients with renal cell carcinoma. Br J Cancer 86(9):1396–1400. https://doi.org/10.1038/sj.bjc.6600257
Miles DW, Happerfield LC, Naylor MS, Bobrow LG, Rubens RD, Balkwwill FR (1994) Expression of tumour necrosis factor (TNFα) and its receptors in benign and malignant breast tissue. Int J Cancer 56(6):777–782. https://doi.org/10.1002/ijc.2910560603
Chen G, Wang D, Vikramadithyan R et al (2004) Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry 43(17):4971–4977. https://doi.org/10.1021/BI0356552
Shang G-S, Liu L, Qin Y-W (2017) IL-6 and TNF-α promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol Lett 13(6):4657–4660. https://doi.org/10.3892/ol.2017.6048
Hagemann T, Robinson SC, Schulz M, Trümper L, Balkwill FR, Binder C (2004) Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-dependent up-regulation of matrix metalloproteases. Carcinogenesis 25(8):1543–1549. https://doi.org/10.1093/carcin/bgh146
Chintala SK, Tonn JC, Rao JS (1999) Matrix metalloproteinases and their biological function in human gliomas. Int J Dev Neurosci 17:495
Eddy A (1996) Insights into renal interstitial fibrosis. J Am Soc Nephrol 7:2495–2508
Knox SM, Whitelock JM (2006) Review Perlecan : how does one molecule do so many things ? Cell Mol Life Sci 63:2435–2445. https://doi.org/10.1007/s00018-006-6162-z
Grindel BJ, Martinez JR, Pennington CL et al (2014) Matrilysin/matrix metalloproteinase-7(MMP7) cleavage of perlecan/HSPG2 creates a molecular switch to alter prostate cancer cell behavior. Matrix Biol 36:64–76. https://doi.org/10.1016/j.matbio.2014.04.005
Quanting B, Murphy G, Breathnach R (1989) Pump-1 cDNA codes for a protein with characteristics similar to those of classical collagenase family members. Biochemistry 28:5327–5334. https://doi.org/10.1021/bi00439a004
Wilson L, Matrisian LM (1996) Matrilysin : an epithelial matrix metalloproteinase with potentially novel functions. Int J Biochem Cell Biol 28(2):123–136
Wilson CL, Heppner KJ, Labosky PA, Hogan BLM, Matrisian LM (1997) Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci U S A 94:1402–1407
El Masri R, Seffouh A, Lortat-Jacob H, Vivès RR (2017) The “in and out” of glucosamine 6-O-sulfation: the 6th sense of heparan sulfate. Glycoconj J 34(3):285–298. https://doi.org/10.1007/s10719-016-9736-5
Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CLR, Dierks T (2007) The heparanome-the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol 129(2):290–307. https://doi.org/10.1016/j.jbiotec.2007.01.022
Okada Y, Yamada S, Toyoshima M, Dong J, Nakajima M, Sugahara K (2002) Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis: hierarchical sulfate groups with differential effects and the essential target disulfated trisaccharide sequence. J Biol Chem 277(45):42488–42495. https://doi.org/10.1074/jbc.M206510200
Ra H-J, Harju-Baker S, Zhang F, Linhardt RJ, Wilson CL, Parks WC (2009) Control of promatrilysin (MMP7) activation and substrate-specific activity by sulfated glycosaminoglycans. J Biol Chem 284(41):27924–27932. https://doi.org/10.1074/jbc.M109.035147
Littlepage LE, Sternlicht MD, Rougier N et al (2010) Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res 70(6):2224–2234. https://doi.org/10.1158/0008-5472.can-09-3515
Miyata Y, Iwata T, Ohba K, Kanda S, Nishikido M, Kanetake H (2006) Expression of matrix metalloproteinase-7 on cancer cells and tissue endothelial cells in renal cell carcinoma: prognostic implications and clinical significance for invasion and metastasis. Clin Cancer Res 12(23):6998–7003. https://doi.org/10.1158/1078-0432.CCR-06-1626
Kugler A, Hemmerlein B, Thelen P, Kallerhoff M, Radzun H, Ringert R (1998) Expression of metalloproteinase 2 and 9 and their inhibitors in renal cell carcinoma. J Urol 160:1914–1918. https://www.ncbi.nlm.nih.gov/pubmed/9783985. Accessed June 10, 2019
Sparano JA, Bernardo P, Stephenson P et al (2004) Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J Clin Oncol 22(23):4683–4690. https://doi.org/10.1200/JCO.2004.08.054
Shepherd FA, Giaccone G, Seymour L et al (2002) Prospective, randomized, double-blind, placebo-controlled trial of marimastat after response to first-line chemotherapy in patients with small-cell lung cancer: a trial of the National Cancer Institute of Canada-Clinical Trials Group and the European Organization for Research and Treatment of Cancer. J Clin Oncol 20(22):4434–4439. https://doi.org/10.1200/JCO.2002.02.108
Hoekstra R, Eskens FA, Verweij J (2001) Matrix metalloproteinase inhibitors: current developments and future perspectives. Oncologist 6(5):415–427. https://doi.org/10.1634/theoncologist.6-5-415
Liu J, Tsao MS, Pagura M et al (2003) Early combined treatment with carboplatin and the MMP inhibitor, prinomastat, prolongs survival and reduces systemic metastasis in an aggressive orthotopic lung cancer model. Lung Cancer 42(3):335–344. https://doi.org/10.1016/S0169-5002(03)00355-6
Morimoto-Tomita M, Uchimura K, Bistrup A et al (2005) Sulf-2, a proangiogenic heparan sulfate endosulfatase, is upregulated in breast cancer. Neoplasia 7(11):1001–1010. http://www.ncbi.nlm.nih.gov/pubmed/16331886. Accessed May 7, 2019
Narita K, Chien J, Mullany SA et al (2007) Loss of HSulf-1 expression enhances autocrine signaling mediated by amphiregulin in breast cancer. J Biol Chem 282(19):14413–14420. https://doi.org/10.1074/jbc.M611395200
Lemjabbar-Alaoui H, van Zante A, Singer MS et al (2010) Sulf-2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene 29(5):635–646. https://doi.org/10.1038/onc.2009.365
Vicente CM, Lima MA, Nader HB, Toma L (2015) SULF2 overexpression positively regulates tumorigenicity of human prostate cancer cells. J Exp Clin Cancer Res 34(1):25. https://doi.org/10.1186/s13046-015-0141-x
Kumagai S, Ishibashi K, Kataoka M et al (2016) Impact of Sulfatase-2 on cancer progression and prognosis in patients with renal cell carcinoma. Cancer Sci 107(11):1632. https://doi.org/10.1111/CAS.13074
Pikas DS, Li JP, Vlodavsky I, Lindahl U (1998) Substrate specificity of heparanases from human hepatoma and platelets. J Biol Chem 273(30):18770–18777. https://doi.org/10.1074/jbc.273.30.18770
Ishai-Michaeli R, Eldor A, Vlodavsky I (1990) Heparanase activity expressed by platelets, neutrophils, and lymphoma cells releases active fibroblast growth factor from extracellular matrix. Cell Regul 1(11):833–842. http://www.ncbi.nlm.nih.gov/pubmed/2088528. Accessed May 29, 2019
Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N (2006) Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res 66(3):1455–1463. https://doi.org/10.1158/0008-5472.CAN-05-1811
Sun X, Zhang G, Nian J et al (2017) Elevated heparanase expression is associated with poor prognosis in breast cancer: a study based on systematic review and TCGA data. Oncotarget 8(26):43521–43535. https://doi.org/10.18632/oncotarget.16575
Cohen E, Doweck I, Naroditsky I et al (2008) Heparanase is overexpressed in lung cancer and correlates inversely with patient survival. Cancer 113(5):1004–1011. https://doi.org/10.1002/cncr.23680
Cohen I, Pappo O, Elkin M et al (2006) Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer 118(7):1609–1617. https://doi.org/10.1002/ijc.21552
Mikami S, Oya M, Shimoda M et al (2008) Expression of heparanase in renal cell carcinomas: implications for tumor invasion and prognosis. Clin Cancer Res 14(19):6055–6061. https://doi.org/10.1158/1078-0432.CCR-08-0750
Lerner I, Baraz L, Pikarsky E et al (2008) Function of heparanase in prostate tumorigenesis: potential for therapy. Clin Cancer Res 14(3):668–676. https://doi.org/10.1158/1078-0432.ccr-07-1866
Kelly T, Suva LJ, Huang Y et al (2005) Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res 65(13):5778–5784. https://doi.org/10.1158/0008-5472.CAN-05-0749
Heyman B, Yang Y (2016) Mechanisms of heparanase inhibitors in cancer therapy. Exp Hematol 44(11):1002–1012. https://doi.org/10.1016/j.exphem.2016.08.006
Farach-Carson MC, Brown AJ, Lynam M, Safran JB, Carson DD (2008) A novel peptide sequence in perlecan domain IV supports cell adhesion, spreading and FAK activation. Matrix Biol 27(2):150–160. https://doi.org/10.1016/j.matbio.2007.09.007
Douglass S, Goyal A, Iozzo RV (2015) The role of perlecan and endorepellin in the control of tumor angiogenesis and endothelial cell autophagy. Connect Tissue Res 56(5):381. https://doi.org/10.3109/03008207.2015.1045297
Ilan N, Elkin M, Vlodavsky I (2006) Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol 38(12):2018–2039. https://doi.org/10.1016/j.biocel.2006.06.004
Acknowledgments
The authors would like to thank Lynn Opdenaker for her contribution in staining and acquiring the image seen in Fig. 6.2. This work was supported partially by NIH/NCI grant P01CA098912. T.V.T is a Dr. John J. Kopchick Fellow awarded through the University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cruz, L.A., Tellman, T.V., Farach-Carson, M.C. (2020). Flipping the Molecular Switch: Influence of Perlecan and Its Modifiers in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1245. Springer, Cham. https://doi.org/10.1007/978-3-030-40146-7_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-40146-7_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40145-0
Online ISBN: 978-3-030-40146-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)