
Abstract
In recent years, gene expression data combined with machine learning methods revolutionized cancer classification which had been based solely on morphological appearance. However, the characteristics of gene expression data have very-high-dimensional and small-sample-size which lead to over-fitting of classification algorithms. We propose a novel gene expression classification model of multiple classifying algorithms with synthetic minority oversampling technique (SMOTE) using features extracted by deep convolutional neural network (DCNN). In our approach, the DCNN extracts latent features of gene expression data, then the SMOTE algorithm generates new data from the features of DCNN was implemented. These models are used in conjunction with classifiers that efficiently classify gene expression data. Numerical test results on fifty very-high-dimensional and small-sample-size gene expression datasets from the Kent Ridge Biomedical and Array Expression repositories illustrate that the proposed algorithm is more accurate than state-of-the-art classifying models and improve the accuracy of classifiers including non-linear support vector machines (SVM), linear SVM, k nearest neighbors and random forests.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Synthetic over sampling
- Enhancing data
- Deep convolutional neural network
- Support vector machines
- Classification
- Gene expression data
1 Introduction
In recent decades, cancer has become a major public health issue in the world. According to the World Health Organization (WHO), the cancer patient rises to 18.1 million new cases and 9.6 million cancer deaths in 2018. Therefore, more and more studies have been done finding effective solutions to diagnose and treat this disease in recent years. However, there are still many challenges in cancer treatment because possible causes of cancer are genetic disorders or epigenetic alterations in the somatic cells [1]. Moreover, cancer could be known as a disease of altered gene expression. There are many proteins are turned on or off and they dramatically change the basic activity of the cell. Microarray technology enables researchers to investigate and address issues which is once thought to be impractical for the simultaneous measurement of the expression levels of thousands of genes in a single experiment [2]. Information of gene expression profile may be used to find and diagnose diseases or to see how well the body responds to treatment, so many algorithms have been done to analyse gene expression data. During the past decade, many classification algorithms have been used to classify gene expression data, which include support vector machines (SVM) used by [3], neural network in [4], k nearest neighbors (kNN) in [5], C4.5 decision trees (C4.5) in [6], random forests (RF) in [7], decision trees based bagging and boosting style algorithm in [8], bagging of oblique decision stumps (Bag-RODS) and boosting of oblique decision stumps (Boost-RODS) in [9].
In spite of many classification algorithms for gene expression data have risen during recent years but these algorithms remain a critical need to improve classifying accuracy. There are two main research challenges that most state-of-the-art classification algorithms are facing when dealing with gene expression data including very-high-dimensional and small-samples-size. These challenges mean that a characteristic of microarray gene expression data is that the number of variables (genes) n far exceeds the number of samples m, commonly known as “curse of dimensionality” problem. These issues lead to statistical and analytical challenges and conventional statistical methods give improper result due to the high dimension of gene expression data with a limited number of patterns [10]. In practice, it isn’t feasible when to build machine learning model due to the extremely large feature sets with millions of features and high computing cost. In addition, another challenge of gene expression classification model is that training data sample size is relatively small compared to features vector size, therefore the classification models may give poor classification performance due to over-fitting.
In order to solve the issues of classifying gene expression, people often use dimension reduction and enhancing data methods. In recent, the problem very-high-dimensional data can be solve by feature extraction with DCNN [11,12,13,14]. The main advantage of this network is the ability to extract new latent features from the gene expression data, then send them to the classifiers. To tackle the small-sample-size task, many studies have used enhancing methods to improve classification accuracy. SMOTE [15] is a very popular over-sampling method that generates new samples in by interpolation from the minority class. A benefit of using SMOTE is that this algorithm can generate new data from original data and it can use to enhance training sample size. However, this algorithm often use on low-dimensional data [16] because it seems beneficial but less effective for very-high-dimensional data. The cause of this problem is that the interpolation process using k nearest neighbors algorithm. For this reason, it could suffer over-fitting problem for very-high-dimensional data. In practical, this over-sampling algorithm for kNN without variable selection shouldn’t be used because it strongly biases the classification towards the minority class [17]. Therefore, it often is combined with data preprocessing methods including feature selection or feature extraction.
In this paper, we proposed a new learning algorithms for the precise classification of gene expression data of non-linear support vector machine (SVM), linear SVM (LSVM), kNN, RF with SMOTE using features extracted by DCNN (call DCNN-SMOTE-[SVM, LSVM, kNN, RF]). The algorithms perform the training task with three main steps. First of all, we use new DCNN model to extract new features from gene expression data. The new features can improve the dissimilarity power of gene expression representations and thus obtain a higher accuracy rate than original features. Secondly, we propose SMOTE algorithm to enhance gene expression data using new features extracted by extraction model. Two new algorithms are used in conjunction with the classifiers learn to classify gene expression data efficiently. Results of 50 low-sample-size and very-high-dimensional microarray gene expression datasets from Kent Ridge Biomedical [18] and Array Expression repositories [19] illustrate that the proposed DCNN-SMOTE-SVM are more accurate the state-of-the-art classifying models including: SVM [20], LSVM [21], kNN [22], RF [23], C4.5 [24, 25]. In addition, DCNN and SMOTE also improve accurate of classifiers including linear SVM, RF and kNN.
The paper is organized as follows. Section refrela gives a brief overview of DCNN, SMOTE, and our proposed. Section 4 discusses about related works. Section 3 shows the experimental results, and the conclusions are presented in the final section.
2 Related Works
Our proposal is in some aspects related to classification approaches for gene expression data. The first approach is the popular frameworks for gene expression classification involve the main steps as follows: the feature extraction or the feature selection of gene expression, and learning classifiers. [26] applied GA/KNN method to generate the subset of the features and then use kNN algorithm to classify. In recent years, deep convolutional neural network has achieved remarkable results in computer vision [27], text classification [28]. In addition, DCNN is also used for omics, biomedical imaging and biomedical signal processing [29]. The paper of [30] proposed to use deep learning algorithm based on the deep convolutional neural network, for classification of gene expression data. Lyu et al. use DCNN [31] to predict over 11,000 tumors from 33 most prevalent forms of cancer. These algorithms aim reduce dimension of data. More recent DCNN-SVM [13] propose to classify gene expression data. In addition, many other methods have been implemented for extracting only the important information from the gene expression data thus reducing their size [32,33,34,35]. Feature extraction creates new variables as combinations of others to reduce the dimensionality of the selected features.
The second approach use enhancing data methods, then use in conjunction with SVM that efficiently classify small-sample-size data. The paper [36] propose enhancing the gene expression classification of support vector machines with generative adversarial networks. SynTReN algorithm generate gene expression data using network topology method [37]. Moreover, there have been several applications of the enhancing data in bioinformatics, such as [38, 39]. SMOTE is a enhancing data method to generate new data with equal probabilities [15]. In spire of, its behaviour on high-dimensional data has not been thoroughly investigated [40]. The paper [41] is shown in the high-dimensional setting only kNN algorithm based on the Euclidean distance seem to benefit substantially from the use of SMOTE, provided that feature selection is performed before using SMOTE.
In our algorithm, we take advantages of both approaches DCNN and SMOTE to solve two main issues of classifying gene expression data. Firstly, we use the benefits of DCNN to extract new latent features from gene expression data. This measure is proposed in previous our paper [13] that can address issue very-high-dimension of gene expression. However, we upgrade new architecture of DCNN for gene expression data classification in our approach. The new feature vector size is only approximately 10% compare with origin size. Secondly, we propose SMOTE algorithms to generate new samples from new features extracted by DCNN. The advantages of DCNN for classifying gene expression data are taken advantage when using SMOTE algorithm to generate new data as well as to tackle limit of this algorithm for gene expression data. In addition, in this paper we also apply our model for other algorithms including support vector machine [42], linear SVM [21], k nearest neighbors [22], random forests [23] and decision trees C4.5 [24, 25].

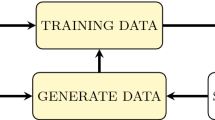
The workflow of our method
3 Methods
In our study, we use multiple classifying algorithms, SMOTE and DCNN for the precise classification of gene expression data. Our learning approach is composed of three phases that is illustrated in Fig. 1. Firstly, the new DCNN is used to extract new features from gene expression data. Secondly, we use SMOTE algorithm to enhance gene expression data using new features extracted by DCNN. Finally, these algorithms are used in conjunction with the various classifiers learn to classify gene expression data efficiently.
3.1 Feature Extraction Gene Expression Data by DCNN

A new DCNN architecture for feature extraction in processing gene expression data.
DCNN plays a dominant role in the community of deep learning models [43]. It is a multi-layer neural network architecture that is directly inspired by the visual cortex of the human brain [44]. In network structure, the successive layers are designed to learn progressively higher-level features, until the last layer which produces categories. Once training processing is completed, the last layer, which is a linear classified operating on the features extracted by the previous layers. Although DCNN is the most widely used method in the field of image processing. However, it is a algorithms that is rarely used in gene expression classification.
In order to develop a powerful classifier which can implicitly extract sparse feature relations from an extremely large feature space, we propose to a extraction model based on DCNN, which is one of the state-of-the-art learning techniques. The architecture of this model consists of two convolutional layers, two pooling layers, and a fully connected layer which is shown in Fig. 2. The layers are respectively named CONV1, POOLING1, CONV2, POOLING2, and output (numbers indicate the sequential position of the layers). The input layer receives the gene expression in the 2-D matrix format. We embedded each high-dimensional vector expression data into a 2-D image by adding some zeros at the last line of the image. The first CONV1 layer contains 4 feature maps and kernel size (\(3\times 3\)). The second layer, POOLING1 layer, is taken as input of the average pooling output of the first layer and filter with (\(2\times 2\)) sub-sampling layer. CONV2 uses convolution kernel size (\(3\times 3\)) to output 2 feature maps POOLING2 is a (\(2\times 2\)) sub-sampling layer. We propose to use the Tanh activation function as neurons. The final layer has a variable number of maps that combine inputs from all map in POOLING2. The feature maps of the final sub sampling layer are then fed into the actual classifier consisting of an arbitrary number of fully connected layers. The output layer uses to extract new features from original gene expression data.
3.2 Enhancing Gene Expression Data by SMOTE
Synthetic Minority Over-sampling Technique was first introduced by [15] that is an over-sampling approach. The main idea of this algorithm is that the minority class is over-sampled by creating synthetic examples rather than by oversampling with replacement. It begins with define k nearest neighbors algorithm for each minority sample, than generating synthetic examples duplication through its neighbors as many as the desired percentage among minority class observations. However, this algorithm is often experimented on low-dimensional data [16] in most situations. In fact, it seems beneficial but less effective for very-high-dimensional data. Therefore, it often is combined with data preprocessing methods including feature selection or feature extraction. These methods aim reduce dimension of origin data.
We propose a new SMOTE algorithm (1) that generates new synthetically gene expression data from new features extracted of DCNN. Our algorithm generates synthetic data which has almost similar characteristics of the training data points. Synthetic data points (\(x_{new}\)) are generated in the following way. Firstly, the algorithm takes the feature vectors and its nearest neighbors, computes the distance between these vectors. Secondly, the difference is multiplied by a random number (\(\lambda \)) between 0 and 1, and it is added back to feature vector. This causes the selection of a random point along the line segment between two specific features. Then, linear support vector machine is used to set label for generating samples with constant \(C = 10^3\). An amount of new samples (\(p\%\)) and k nearest neighbors are hyper parameters of the algorithm.

3.3 Gene Expression Classification of SVM with SMOTE Using Features Extracted from DCNN
The original SVM algorithm was invented by Vapnik [42]. SVM algorithm is systematic and properly motivated by the statistical learning theory. This algorithm is a supervised learning model that is widely applied for classifications and regressions [45].
The SVM algorithm find the best separating plane furthest from the different classes. To achieve this purpose, the SVM tries to maximize the distance between two boundary hyperplanes to reduce the probability of misclassification. The optimal hyperplane found by SVM is maximally distant from the two classes of labeled points located on each side (Fig. 3). In practice, the SVM algorithm gives good accuracy in classifying very-high-dimensional data. Although the SVM is well-known as an efficient model for classifying gene expression data, the small-sample-size training datasets degrade the classification performance of any model [46]. In addition to performing linear classification, the algorithm has been very successful in building highly non-linear classifiers by means of kernel-based learning methods [47]. Kernel-based learning methods aim to transform the input space into higher dimensions, such as a radial basis function (RBF), sigmoid function, and polynomial function. In the proposed approach, a non-linear SVM with an RBF kernel is used for classifying gene expression after extraction feature and enhancing data.

SVM for binary classification
The proposed algorithm is effective combination of three algorithms DCNN, SMOTE and SVM. The algorithm performs the training task with three main phases (Fig. 1).
First of all, we implement a new DCNN that extract new features from origin gene expression data. Our model has take advantage of DCNN is that this model can learn latent features from very-high-dimensional input spaces. This process can be viewed as projection of data from higher dimensional space to a lower dimensional space. Moreover, these new features we can improve the dissimilarity power of gene expression representations and thus obtain higher accuracy rate than original features.
Although the data dimension has reduced but training data sample size is relatively diminutive compared to feature vector size, so that classifiers may give poor classification performance due to over-fitting. In a second order phase training, our model use SMOTE generates new sample from features extracted by DCNN model. In our approach, in the very-high-dimensional data setting only kNN classifiers based on the Euclidean distance seem to benefit substantially from the use of over-sampling, provided that feature extraction by extraction model is performed before using this algorithm. For traditional over-sampling algorithm, it is not effective for very-high-dimensional data and this problem has tackled by DCNN model in our approach.
Last but not least, our model generates new training data following which the classifiers learns to classify gene expression data efficiently in this phase. The classifiers consist non-linear SVM, linear SVM, kNN, RF and C4.5 that are used to classify new data. In our approach, we propose to use RBF kernel type in SVM model because it is general and efficient [45]. Moreover, the combination of DCNN and SMOTE can improve accuracy classification of linear SVM, RF and kNN.
4 Evaluation
We are interested in the classification performance of our proposal for gene expression data classification. Therefore, we report the comparison of the classification performance obtained by our model and the best state-of-the-art algorithms including support vector machine (SVM) [42], linear SVM (LSVM) [21], decision trees (C4.5) [25], k nearest neighbors (kNN) [22], random forests (RF) [23].
In addition, we also compare various version of DCNN-SMOTE (DCNN-SMOTE \(\rightarrow \) [SVM, LSVM, RF, kNN, C4.5]) with SVM, LSVM, RF, kNN, C4.5. These results are used so as to evaluate performance of classifiers after using DCNN-SMOTE. Moreover, we interested in the effective of enhancing model, therefore we also evaluate the algorithms classification using features extracted by extraction model (DCNN \(\rightarrow \) [SVM, LSVM, RF, C4.5, kNN]), then they are compared to our proposed.
In order to evaluate the effectiveness in classification tasks, we have implemented DCNN-SMOTE-SVM and its version in Python using Scikit [48] and TensorFlow [49] libraries. Other algorithms like RF, C4.5 in Scikit library. We use the highly efficient standard SVM algorithm LibSVM [21] with one-versus-one strategy for multi-class. We used the Student’s test to assess classification results of learning algorithms.
All tests were run under Linux Mint on a 3.07 GHz Intel(R) Xeon(R) CPU PC with 8 GB RAM.
4.1 Experiments Setup
Experiments are conducted with fifty very-high-dimensional datasets from the Biomedical [18] and Array Express repositories [19]. The characteristics of datasets are summarized in Table 1.
The evaluation protocols are illustrated in the last column of Table 1. With datasets having training set (trn) and testing set (tst) available, we use the training data to tune the parameters of the algorithms for obtaining a good accuracy in the learning phase. Then the obtained model is evaluated on the test set. With a datasets having less than 300 data points, the test protocol is leave-one-out cross-validation (loo). For the others, we use 10-fold cross-validation protocols remains the most widely to evaluate the performance [50]. The total classification accuracy measure is used to evaluate the classification models.
As for training model, we tune the parameters for three algorithms including DCNN, SMOTE and the parameters of classifiers.
In order to train network, we use Adam for optimization [51] with batch size is 8 to 32. We start to train with a learning rate of 0.00002 for all layers, and then rise it manually every time when the validation error rate stopped improving. Cross entropy is used to define a loss function in DCNN. The number of epochs is 200.
In our algorithm, the number of neighbors (k) is chosen in 1, 3, 5, 7, 9. The samples were over-sampled (p) at 100%, 200% and 300% of its original samples size. We tune the hyper-parameter \(\gamma \) of RBF kernel and the cost C (a trade-off between the margin size and the errors) to obtain the best correctness. The cost C is chosen in \({1, 10, 10^2, 10^3, 10^4, 10^5}\), and the hyper-parameter \(\gamma \) of RBF kernel is tried among \({1.E-1, 1.E-2, 1.E-3, 1.E-4, 1.E-5}\). All the optimal parameters is show in Table 2.
With other algorithms, the cost constant C of linear SVM is set to \(10^3\). In non-linear SVM, we adjust the hyper-parameter \(\gamma \) and the cost C to get the best result. RF learns 200 decision trees to classify all datasets. kNN tries to use k among \(\{1, 3, 5, 7\}\).
4.2 Classification Results
Table 3 gives results of classifying algorithms on 50 gene expression datasets. The best results are bold faces and the second ones are italic. The plot charts in Figs. 4, 5 and 6 also visualise classification results. Table 4 summarizes results of these statistical tests with paired Student ratio test present the mean accuracy of these models.
First and foremost, we evaluate feature extraction and enhancing data algorithms. We compare the accuracy of classifying algorithms (SVM, LSVM, kNN and RF) and various versions of DCNN-SMOTE including (DCNN-SMOTE-SVM, DCNN-SMOTE-LSVM, DCNN-SMOTE-kNN and DCNN-SMOTE-RF).
At first sight, Tables 3, 4 and Fig. 6 show that DCNN-SMOTE-SVM, DCNN-SMOTE-LSVM, DCNN-SMOTE-kNN, DCNN-SMOTE-RF significantly increases the mean accuracy of 4.83, 3.37, 2.9, 2.08% points compared to SVM, LSVM, kNN and RF respectively. All p-values are less than 0.05. In detail, DCNN-SMOTE-SVM has good performances compared to SVM with 29 wins, 11 ties, 10 defeats, p-value = 1.33E-03 as well as DCNN-SMOTE-LSVM has 34 wins, 7 ties, 9 defeats (p-value = 8.72E-03) compared to LSVM. In the comparison to kNN, DCNN-SMOTE-kNN outperforms 29 out of 50 datasets (29 wins, 4 ties, 17 defeats, p-value = 2.26E-03). Besides, DCNN-SMOTE-RF has 29 wins, 12 ties, 9 defeats (p-value = 2.78E-02) compared to RF. These results show effective of DCNN and SMOTE that improve accuracy of SVM, LSVM, RF and kNN classifiers. In the comparison between DCNN-SMOTE-C4.5 with C4.5, DCNN-SMOTE-C4.5 slightly superior to decision tree of C4.5 with 27 wins, 2 tie, 21 defeat, p-value = 1.06E-01 (not significant different).
In addition, it is clear that DCNN-SMOTE-SVM shows the best performance. Tables 3 and 4 show that it significantly improves the mean accuracy of 4.82, 3.53, 9.40, 5.55, 12.48% points compared to SVM, LSVM, kNN, RF and C4.5 respectively. All p-values are less than 0.05. In detail, it has 29 wins, 10 ties, 11 defeats (p-value = 1.33E-03) against SVM and 33 wins, 11 ties, 6 defeats (p-value = 4.68E-04) compared to LSVM. This model has also 48 wins, 1 tie, 1 defeat (p-value = 5.57E-09) compared to kNN and 41 wins, 5 ties, 4 defeats (p-value = 6.01E-09) compared to RF. In the comparison to C4.5, our model outperforms 46 out of 50 datasets (46 wins, 1 ties, 3 defeat, p-value = 3.12E-12).
Moreover, DCNN-SMOTE-SVM model efficiently classify more than various versions including DCNN-SMOTE\(\rightarrow \)[LSVM, kNN, RF and C4.5]. In detail, this model gives good performances compared to DCNN-SMOTE\(\rightarrow \)[LSVM, kNN, RF and C4.5] which improves the mean accuracy of 0.63, 5.67, 3.47, 9.7 respectively.
Furthermore, DCNN, SMOTE models enhance the accuracy of classifiers compared to the algorithms classifications using the features extraction from DCNN. It is clear that DCNN-SMOTE\(\rightarrow \)[SVM, LSVM, kNN, RF, C4.5] increase the mean accuracy of 0.98, 1.09, 3.15, 1.06, 1.00% points compared to DCNN \(\rightarrow \)[SVM, LSVM, kNN, RF, C4.5]. These results show using DCNN and SMOTE is effectively more than our paper previous [13].
The running time of a our model includes three parts: the time to train the deep convolutional networks for extracting the features, the time to generate new samples and the training time for the classifier on the new data. The average time of the first part on 50 datasets is 48.26 s. The average time of the second part is 29.37 s. Finally, the average time of the second part for SVM, kNN, LSVM, RF and C4.5 in the our model are, respectively, 0.91, 0.08, 1.83, 2.75 and 1.37 s. While the running time of SVM, kNN, LSVM, RF and C4.5 are 8.48, 0.31, 54.85, 10.76 and 6.3 s.
These experiments allow us to believe that our approach efficiently handle gene expression data with the small sample size in very-high-dimensional input. Moreover, the combination of DCNN and SMOTE is not only improve performance of non-linear SVM and but also linear SVM, kNN and random forests.

Comparison the accuracy of DCNN-SMOTE-SVM and SVM, DCNN-SMOTE-LSVM and LSVM on 50 datasets (%).

Comparison the accuracy of DCNN-SMOTE-RF and RF, DCNN-SMOTE-kNN and kNN on 50 datasets (%).

Comparison of the mean accuracy of the classification models
5 Conclusion and Future Works
We have presented a new classification algorithm of multiple classifiers with SMOTE using features extracted by DCNN that tackle with the very-high-dimensional and small-sample-size issues of gene expression data classification. A new DCNN model extract new features from origin gene expression data, then a SMOTE algorithm generates new data from the features of DCNN was implemented. These models are used in conjunction with classifiers that efficiently classify gene expression data. From the obtained results, it is observed that DCNN-SMOTE can improve performance of SVM, linear SVM, random forests and k nearest neighbors algorithms. In addition, the proposed DCNN-SMOTE-SVM approach has the most accurate, when compared to the than the-state-of-the-art classification models in consideration.
In the near future, we intend to provide more empirical test on large benchmarks and compare to other algorithms. A promising future research aims at automatically tuning the hyper-parameters of our algorithms.
References
Chakraborty, S., Rahman, T.: The difficulties in cancer treatment. Ecancermedicalscience 6, ed16 (2012)
Schena, M., Shalon, D., Davis, R.W., Brown, P.O.: Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270(5235), 467–470 (1995)
Furey, T.S., Cristianini, N., Duffy, N., Bednarski, D.W., Schummer, M., Haussler, D.: Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 16(10), 906–914 (2000)
Khan, J., et al.: Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 7(6), 673 (2001)
Li, L., Weinberg, C.R., Darden, T.A., Pedersen, L.G.: Gene selection for sample classification based on gene expression data: study of sensitivity to choice of parameters of the GA/KNN method. Bioinformatics 17(12), 1131–1142 (2001)
Netto, O. P., Nozawa, S. R., Mitrowsky, R. A. R., Macedo, A. A., Baranauskas, J. A., Lins, C.: Applying decision trees to gene expression data from DNA microarrays: a leukemia case study. In: XXX Congress of the Brazilian Computer Society, X Workshop on Medical Informatics, p. 10 (2010)
Díaz-Uriarte, R., De Andres, S.A.: Gene selection and classification of microarray data using random forest. BMC Bioinform. 7(1), 3 (2006)
Tan, A.C., Gilbert, D.: Ensemble machine learning on gene expression data for cancer classification. Appl. Bioinform. 2(3 Suppl.), S75–S83 (2003)
Huynh, P.H., Nguyen, V.H., Do, T.N.: Random ensemble oblique decision stumps for classifying gene expression data. In: Proceedings of the Ninth International Symposium on Information and Communication Technology, SoICT 2018, pp. 137–144. ACM, New York (2018)
Pinkel, D., et al.: High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20(2), 207 (1998)
Singh, R., Lanchantin, J., Robins, G., Qi, Y.: Deepchrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics 32(17), i639–i648 (2016)
Liu, J., Wang, X., Cheng, Y., Zhang, L.: Tumor gene expression data classification via sample expansion-based deep learning. Oncotarget 8(65), 109646 (2017)
Huynh, P.-H., Nguyen, V.-H., Do, T.-N.: A coupling support vector machines with the feature learning of deep convolutional neural networks for classifying microarray gene expression data. In: Sieminski, A., Kozierkiewicz, A., Nunez, M., Ha, Q.T. (eds.) Modern Approaches for Intelligent Information and Database Systems. SCI, vol. 769, pp. 233–243. Springer, Cham (2018). https://doi.org/10.1007/978-3-319-76081-0_20
Huynh, P.H., Nguyen, V.H., Do, T.N.: Novel hybrid DCNN-SVM model for classifying RNA-sequencing gene expression data. J. Inf. Telecommun. 3(4), 533–547 (2019). https://doi.org/10.1080/24751839.2019.1660845
Chawla, N.V., Bowyer, K.W., Hall, L.O., Kegelmeyer, W.P.: Smote: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357 (2002)
Van Hulse, J., Khoshgoftaar, T.M., Napolitano, A.: Experimental perspectives on learning from imbalanced data. In: Proceedings of the 24th International Conference on Machine Learning, pp. 935–942. ACM (2007)
Blagus, R., Lusa, L.: Smote for high-dimensional class-imbalanced data. BMC Bioinform. 14(1), 106 (2013)
Jinyan, L., Huiqing, L.: Kent ridge bio-medical data set repository. Technical report (2002)
Brazma, A., et al.: ArrayExpress a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 31(1), 68–71 (2003)
Vapnik, V.N.: The Nature of Statistical Learning Theory. Springer, New York (1995)
Chang, C.C., Lin, C.J.: LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2(3), 27 (2011)
Fix, E., Hodges, J.: Discriminatory analysis-nonparametric discrimination: small sample performance. Technical report, California University, Berkeley (1952)
Breiman, L.: Random forests. Mach. Learn. 45(1), 5–32 (2001)
Breiman, L., Friedman, J. H., Olshen, R., Stone, C. J.: Classification and Regression Trees, vol. 8, pp. 452–456. Wadsworth International Group (1984)
Quinlan, J.R.: C4.5: Programs for Machine Learning. Morgan Kaufmann Publishers Inc., San Francisco (1993)
Li, Y., et al.: A comprehensive genomic pan-cancer classification using the cancer genome atlas gene expression data. BMC Genom. 18(1), 508 (2017)
Krizhevsky, A., et al.: Imagenet classification with deep convolutional neural networks. In: Advances in Neural Information Processing Systems, pp. 1097–1105 (2012)
Kim, Y.: Convolutional neural networks for sentence classification. arXiv preprint arXiv:1408.5882 (2014)
Min, S., Lee, B., Yoon, S.: Deep learning in bioinformatics. Briefings. Bioinformatics 18, bbw068 (2016)
Zeebaree, D.Q., Haron, H., Abdulazeez, A.M.: Gene selection and classification of microarray data using convolutional neural network. In: 2018 International Conference on Advanced Science and Engineering (ICOASE), pp. 145–150. IEEE (2018)
Lyu, B., Haque, A.: Deep learning based tumor type classification using gene expression data. In: Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, BCB 2018, pp. 89–96. ACM, New York (2018)
Ambroise, C., McLachlan, G.J.: Selection bias in gene extraction on the basis of microarray gene-expression data. Proc. Nat. Acad. Sci. 99(10), 6562–6566 (2002)
Vert, J.P., Kanehisa, M.: Graph-driven feature extraction from microarray data using diffusion kernels and kernel CCA. In: Advances in Neural Information Processing Systems, pp. 1449–1405 (2003)
Wang, A., Gehan, E.A.: Gene selection for microarray data analysis using principal component analysis. Stat. Med. 24(13), 2069–2087 (2005)
Sun, G., Dong, X., Xu, G.: Tumor tissue identification based on gene expression data using DWT feature extraction and PNN classifier. Neurocomputing 69(4–6), 387–402 (2006)
Huynh, P.H., Nguyen, V., Do, T.N.: Enhancing gene expression classification of support vector machines with generative adversarial networks. J. Inf. Commun. Converg. Eng. 17, 14–20 (2019)
Van den Bulcke, T., et al.: SynTReN: a generator of synthetic gene expression data for design and analysis of structure learning algorithms. BMC Bioinform. 7, 43 (2006)
Costa, P., et al.: End-to-end adversarial retinal image synthesis. IEEE Trans. Med. Imaging 37(3), 781–791 (2018)
Moeskops, P., Veta, M., Lafarge, M.W., Eppenhof, K.A.J., Pluim, J.P.W.: Adversarial training and dilated convolutions for brain MRI segmentation. In: Cardoso, M.J., et al. (eds.) DLMIA/ML-CDS -2017. LNCS, vol. 10553, pp. 56–64. Springer, Cham (2017). https://doi.org/10.1007/978-3-319-67558-9_7
Lusa, L., et al.: Class prediction for high-dimensional class-imbalanced data. BMC Bioinform. 11(1), 523 (2010)
Fernández, A., García, S., Herrera, F., Chawla, N.V.: Smote for learning from imbalanced data: progress and challenges, marking the 15-year anniversary. J. Artif. Int. Res. 61(1), 863–905 (2018)
Vapnik, V.N.: An overview of statistical learning theory. IEEE Trans. Neural Netw. 10(5), 988–999 (1998)
Hinton, G.E., Salakhutdinov, R.R.: Reducing the dimensionality of data with neural networks. Science 313(5786), 504–507 (2006)
Hubel, D.H., Wiesel, T.: Shape and arrangement of columns in cat’s striate cortex. J. Physiol. 165(3), 559–568 (1963)
Burges, C.J.: A tutorial on support vector machines for pattern recognition. Data Min. Knowl. Disc. 2(2), 121–167 (1998)
Popovici, V., et al.: Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 12(1), R5 (2010)
Cristianini, N., Shawe-Taylor, J.: An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods. Cambridge University Press, Cambridge (2000)
Pedregosa, F., et al.: Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011)
Abadi, M., Agarwal, A., Barham, P., Brevdo, E., Chen, Z.: TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems (2015)
Wong, T.T.: Performance evaluation of classification algorithms by k-fold and leave-one-out cross validation. Pattern Recogn. 48(9), 2839–2846 (2015)
Kingma, D.P., Ba, J.: Adam: a method for stochastic optimization. In: Proceedings of the 3rd International Conference on Learning Representations (ICLR) (2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Huynh, PH., Nguyen, VH., Do, TN. (2019). A Combined Enhancing and Feature Extraction Algorithm to Improve Learning Accuracy for Gene Expression Classification. In: Dang, T., Küng, J., Takizawa, M., Bui, S. (eds) Future Data and Security Engineering. FDSE 2019. Lecture Notes in Computer Science(), vol 11814. Springer, Cham. https://doi.org/10.1007/978-3-030-35653-8_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-35653-8_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-35652-1
Online ISBN: 978-3-030-35653-8
eBook Packages: Computer ScienceComputer Science (R0)