
Abstract
Immune inhibition orchestrated by multiple negative checkpoint receptors (NCRs) is required to control aberrant immune responses and maintain homeostasis. An imbalance between immune activation and inhibition is believed to be an important mechanism driving the development of a number of diseases. Several tumors hijack this delicate balance and are able to increase the expression of various inhibitory receptors on cytotoxic T cells (CTLs) resulting in ineffective anti-tumor CTL responses. T cell immunotherapies that blocks NCR signaling have shown remarkable clinical success and extend median progression-free survival as well as overall survival in a wide range of cancer settings. In chronic infections such as HIV and Hepatitis B and C, CTLs have been found to overexpress various NCRs resulting in an “exhausted” CTL phenotype, which is associated with disease progression and the development of co-morbidities. On the other hand, downregulation of NCRs is thought to lead to autoimmune diseases such as multiple sclerosis and other neuroinflammatory conditions. In this chapter, we primarily focus on advances in our current understanding of NCRs and the role they play in human health and disease as well as the ongoing efforts to develop novel immunotherapies that target these receptors and reverse immune perturbations impacted by NCR dysregulation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Negative checkpoint receptors
- Immune regulation
- Cancer
- Neuroinflammation
- Infectious disease
- Immunotherapy
1 Introduction to Negative Immune Checkpoint Receptors
The human immune system employs a number of mechanisms for maintaining balance between immune activation and suppression. Infection, cancers, and injury trigger inflammatory processes that need to be “checked” in order to avoid aberrant responses. This complex process involves multiple cellular components that must work in concert to achieve an appropriate level of inflammation. Under homeostatic conditions, immune cells are designed so that they are not constitutively active which could lead to damage of healthy tissues or disease. One of the ways in which the immune system moderates this balance is through the binding of ligand on antigen presenting cells (APCs) to corresponding negative immune checkpoint receptors (NCRs) on T cells [1, 2]. Specifically, these immune checkpoints are proteins that act as T cell receptor (TCR) co-signaling partners that deliver either positive or negative signals to T lymphocytes. Ligand binding results in down regulation of T cell activation and suppression of effector functions. However, in many disease states, this system is hijacked, consequently leading to T cell dysfunction and ineffective responses. Chronic infection and cancers have been shown to do just that.
Much effort has been made towards better understanding these NCRs and the role they play in health and disease. Moreover, identifying ways in which these NCRs can be blocked to restore effector function and better position T cells to fight cancerous tumors and infections has been a major focus of research. Harnessing this system in order to enhance T cell function has been groundbreaking for the field of immunology, so much so that the 2018 Nobel prize in physiology and medicine went to the two men who discovered these important immune molecules, James Allison and Tasuku Honjo. The idea of mobilizing our own immune systems to fight disease is not a new idea. However, the discovery of these immune modulatory receptors has made this tantalizing possibility feasible as a promising clinical treatment. Among the most prominent immune checkpoint receptor-ligand combinations are CTLA-4/CD80, PD-1/PD-L1, TIGIT/PVR and Tim-3/Galectin 9. In this chapter, we discuss the role of NCRs in health and disease, particularly in cancer, infection and neuroinflammatory conditions, as well as attempt to highlight advances being made in the field of immunotherapy.
2 Negative Immune Checkpoint Receptors in Cancer
Under normal homeostatic conditions, immune checkpoint receptors are critical for self-tolerance . In various cancer settings, however, these proteins are overexpressed allowing tumor cells to evade the immune response [3,4,5]. Blocking these receptor-ligand interactions is able to produce robust anti-tumor responses in some patients. Although several mechanisms have been proposed as to how NCR blockade leads to sustained clinical benefit, the main mechanism is thought to be by reinvigoration of cytotoxic CD8+ T cells (CTLs). CTLs require co-stimulation by ligation of co-stimulatory receptors expressed on T cells and co-stimulatory ligand on antigen presenting cells (APC) [6]. CD28 is one of the most studied co-stimulatory receptors that binds to CD80/CD86. Cytotoxic T-lymphocyte antigen 4 (CTLA-4) , which is expressed on both regulatory T cells (Tregs) and activated T cells, binds to CD80/CD86 with higher affinity than CD28 and induces trans-endocytosis of the ligands which competitively limits the availability of CD80/86 to CD28 [7, 8]. Consequently, CTLA-4 is able to limit the activation of CTLs and prevent autoimmunity and tissue damage due to hyper-inflammatory responses. However, in cancer settings, overexpression of CTLA-4 on CTLs in the tumor microenvironment limits their anti-tumor effector function and allows for immune evasion. Inhibition of CTLA-4 is thought to work through interruption of the CTLA-4-CD80/CD86 axis, which clears the path for CD28-CD80/CD86 binding and subsequent CTL activation Table 1.
Programmed cell death-1 (PD-1) is a member of the CD28 superfamily . It delivers negative signals upon interaction with its two ligands, PD-L1 and PD-L2 [9]. Known to be important in peripheral tolerance and protection from autoimmune attacks, PD-1 and its ligands are also responsible for attenuated immunity to infection and cancers. Furthermore, PD-1 has been shown to facilitate chronic infection and tumor progression. Expression of programmed PD-1 on CTLs is enhanced by TCR stimulation and cytokine stimulation including common-gamma chain cytokines and inhibition of PD-1 occurs via multiple costimulatory pathways including ZAP70, PI3K and RAS. PD-1 can also activate basic zipper transcriptional factor ATF-like (BATF), which further interrupts T cell activation [10]. In the tumor microenvironment, both PD-1 and PD-L1 expression on CTLs and tumor cells are elevated due to chronic antigen and inflammatory cytokine stimulation [5]. Furthermore, the promotor region of PD-1 is often hypomethylated, resulting in further upregulation of PD-1 expression [11]. Ligation of PD-1 by PD-L1 attenuates TCR and co-stimulatory signals resulting in increased apoptosis, reduced cytokine production and cellular proliferation, as well as decreased tumor killing activity of CTLs. PD-1 blockade is able to reinvigorate functionally “exhausted” CTLs and reverse their dysfunctional state leading to enhanced effector function. PD-1 is expressed on many cell types and its expression on melanoma cells has been shown to promote tumor growth via activation of mTOR signaling [12]. On tumor-associated macrophages, PD-1 expression is negatively correlated with phagocytic activity against tumor cells in vivo. PD-1/PD-L1 blockade enhances phagocytic activity and extends median survival in mouse models of cancer [13].
Since the introduction of Ipilimumab , a monoclonal antibody (mAb) that targets the CTLA-4 receptor and the first NCR inhibitor approved for clinical use, the field of cancer immunotherapy has experienced an unprecedented expansion and vastly improved cancer treatment options [14, 15]. Clinical trials using inhibitors targeting PD-1 and its cognate ligand, PD-L1, for treatment of melanoma as well as other malignancies have experienced marked success [16,17,18,19,20,21,22]. The number of NCR inhibitors that have successfully attained FDA approval for clinical use has grown substantially in recent years. Moreover, several of these immune checkpoint inhibitors have shown ever-growing for a wide variety of cancer types including non-small cell lung cancer, urothelial carcinoma, Hodgkin lymphoma, head and neck squamous cell carcinoma, and most recently cervical cancer.
The ability of NCR-targeted immunotherapies to extend median progression-free survival as well as overall survival in cancer patients has been remarkable [17,18,19,20,21,22]. Randomized clinical trials with treatment-naïve patients with metastatic melanoma demonstrated superior efficacy of PD-1 blockade by pembrolizumab, a first-line treatment for cancers that overexpress PD-L1, over cytotoxic dacarbazine chemotherapy with a response rate of 27% vs 10% and median overall survival of 37.5 vs 11.2 months [23, 24]. Unfortunately, responsiveness to NCR inhibitors can vary quite substantially between individuals. While complete response, defined as the disappearance of all signs of cancer after treatment, has been observed in some patients, majority of individuals exhibit only partial response. The complicated nature and relationship between the cancer microenvironment and negative checkpoint receptors are believed to be responsible, at least in part, for the observed variation.
Despite the success of clinical trials using single NCR blockade, inhibition of one NCR alone may not be sufficient enough to reinvigorate all functionally exhausted T cells. One strategy for enhancing therapeutic efficacy of NCR-targeted therapy is combinatory blockade. In a clinical trial comparing the combination of CTLA-4 and PD-1 blockade against CTLA-4 single blockade in melanoma patients, combinatory therapy exhibited a higher response rate (58% vs 19%), median progression free survival (11.5 vs 6.9 months) and 4-year survival rate (37% vs 9%) [25]. There was no statistical significance between CTLA-4/PD-1 combination and PD-1 single blockade, but descriptive analyses showed a trend toward superior outcomes. Unfortunately, treatment related side effects have been known to increase in combinatory therapy and must be taken into consideration when initiating these therapies.
CTLA-4 and PD-1 , which have served as vanguard molecules in many early immunotherapy studies, are just two molecules on a growing list of NCRs that have been found to have immunomodulatory activity [3,4,5, 26,27,28]. Current research is now focused on exploring blockade of other NCRs either alone or in combination with PD-1 and/or CTLA-4 [29, 30].
3 Immunotheapeutic Targets in Preclinical Development and in Ongoing Clinical Trials
Lymphocyte Activation Gene 3 (LAG-3) is expressed on activated CD4+ and CD8+ T cells and polarizes CD4+ T cells to a regulatory phenotype [26, 31, 32]. LAG-3 inhibits proliferation and cytokine production of effector T cells, and it is overexpressed on tumor-infiltrating CTLs [33]. Although the effect of anti-LAG-3 monotherapy has been found to be limited, dual blockade with PD-1 shows synergistic effects that results in better survival and complete response rates than either monotherapy in mouse models of B16 melanoma and MC38 colorectal cancer [34]. The efficacy of LAG-3 blockade also has been assessed in mouse models in the context of ovarian cancer, lymphoma, and multiple myeloma [35]. Its signaling pathway is still largely unknown but it is thought to be different from that of PD-1 due to the synergetic effects observed in PD-1/LAG-3 dual-blockade [36]. LAG-3 is highly expressed on Tregs and is correlated with IL-10 production. Structurally, LAG-3 resembles CD4 and consequently binds to MHC class II with higher affinity, thus inhibiting CD4+ T cell priming by dendritic cells (DCs) via competitive inhibition. Galectin-3 and LSECtin, which is highly expressed on tumor cells, are also binding partners for LAG-3 and are implicated in the disruption of effective anti-tumor responses [37, 38]. Several phase 1–2 clinical trials using dual blockade are currently ongoing.
B and T Lymphocyte Attenuator (BTLA) , also known as CD272, shares functional similarity to PD-1 and CTLA-4 and is often co-expressed with PD-1 on tumor infiltrating CTLs [39]. Mainly expressed on B, T and mature lymphocytes, BTLA binds specifically to herpes virus entry mediator (HVEM) and inhibits T cell activation and cytokine production [40]. BTLA is downregulated with progressive differentiation of CTLs, but cancer-specific cells have been observed to maintain high levels of BTLA expression. BTLA has also been found to be a marker of a less cytotoxic T cell subset in diffuse large B-cell lymphoma [41]. Reversal of BTLA signaling is achievable ex vivo, which restores CTL function. Currently, no clinical trials are ongoing with regard to this molecule, but interest remains.
V-domain Immunoglobulin-containing Suppressor of T-cell Activation (VISTA) is unique due to its dual role as a receptor on T cells and a ligand on APCs [42, 43]. The highest levels of VISTA expression are found in myeloid cells. Within the CD4 compartment, VISTA expression is highest on naïve and Foxp3+ Tregs [44], but is also detected at lower levels on CD8+ T cells and NK cells. Molecularly, it is very similar to PD-1 but appears to have non-overlapping function. Current data shows that antagonist anti-VISTA antibodies serve to increase effector function of tumor-reactive T cells within the tumor microenvironment, as well as decrease the presence of monocytic myeloid-derived suppressor cells. Furthermore, VISTA blockade has also been shown to reduce the emergence of tumor-specific Foxp3+ Tregs [44, 45]. Whether or not VISTA blockade enables expansion of the T cell repertoire, decreased exhaustion or T cell reinvigoration remains unclear. In fact, increased effector function may be an indirect consequence of VISTA blocking on myeloid cells. If so, cancers characterized by high infiltration of myeloid-derived suppressor cells may be particularly responsive to VISTA-targeted therapies. There are currently two phase 1 clinical trials that are ongoing (NCT02671955, NCT02812875).
T cell Immunoreceptor with Ig and ITIM Domains (TIGIT) is expressed on NK cells, Tregs, and CD4+ and CD8+ T cells, particularly activated, memory and follicular helper T cells. TIGIT is a negative regulator of the CD226-CD112/CD155 axis. CD226, which is expressed on NK cells, monocytes and activated T cells, induces activation upon ligation with CD112 or CD155, which are expressed on APCs. TIGIT binds to CD155 and CD112 with higher affinity to competitively inhibit CD226 ligation. TIGIT is highly expressed on CTLs and NK cells in the tumor microenvironment and inhibits cytokine production and cytotoxic activity [46,47,48]. Blockade with anti-TIGIT mAb has been shown to partially restore normal immune function to CD4+ and CD8+ T cells. TIGIT signaling in Tregs has been found to stabilize the expression of signature Treg genes and promote Treg function. When TIGIT binds to CD155 on DCs IL-12 production decreases and IL-10 production increases [46,47,48]. TIGIT blockade could disrupt the immunosuppressive activity of Tregs and DCs, thus indirectly reinvigorating CTLs and NK. Currently, two phase 1 (NCT03119428, NCT03628677) and one phase 2 (NCT03563716) study targeting TIGIT in a melanoma model are ongoing.
T cell Immunoglobulin and Mucin-containing Domain 3 (Tim-3) is expressed on a wide variety of immune cells and binds to multiple ligands including galectin-9, phosphatidylserine, HMGB-1 and CECAM-1. Tim-3 has multifactorial roles depending on cell type and context, but in general works as an inhibitory regulator of T cell function [26]. Tim-3 is expressed on terminally exhausted T cells and CD8+ T cells expressing Tim-3 have been found to accumulates in tumors. Tim-3+ CD8+ T cells have been found to secrete a lesser variety of cytokines and in reduced amounts as compared to Tim-3− CD8+ T cells. Interestingly, Tim-3 upregulation during anti-PD-1 therapy is reported to be a mechanism of adaptive resistance to therapy both in mice and humans. Consequently, dual-blockade of PD-1 and Tim-3 successfully improves survival length in a mouse model of lung cancer [49]. Tim-3 is also expressed on Tregs. Tumor-infiltrating Tregs highly express Tim-3 and exhibit superior suppressive function compared to Tim-3− Tregs. Blockade of Tim-3 in combination with PD-L1 has been shown to markedly reduce the suppressive function of Tregs [50]. A recent study showed that Tim-3 blockade in combination with paclitaxel, a chemotherapy drug, enhanced production of chemokine CXCL9 from CD103+ conventional DCs and enhanced CD8+ T cell response, which ultimately slowed the progression of cancer in a murine breast cancer model [51]. In contrast, monotherapy with anti-Tim-3 has been found to be suboptimal [50, 52]. NK cells also express high level of Tim-3 and can be used as a marker of cytokine producing and cytotoxic NK cells. However, cross-linking of Tim-3 by antibody suppresses NK cell-mediated cytotoxicity [53]. Accordingly, NK cell dysfunction in melanoma patients is reversed by blockade of Tim-3 [54]. Monocytes, macrophages, and dendritic cells express Tim-3 and have been shown to exhibit both immune-enhancing and inhibitory functions depending on the disease context and ligand binding partner [51, 55, 56]. Tim-3 is known to interrupt toll-like receptor (TLR) 2/4 signaling via NF-κB inhibition on these cells [57].
Tim-3 blockade has shown promising results in multiple preclinical studies and there are several phase 1 studies are currently underway (NCT03489343, NCT02817633, NCT03099109, NCT02608268, NCT03652077, NCT03066648, NCT030680508, NCT03311412, NCT03744468, NCT03708328, NCT03446040) [58]. Most of these studies are focused on dual PD-1/Tim-3-targeted therapies. Since the expression of Tim-3 on T cell is mostly limited to terminally exhausted states, as opposed to PD-1 and CTLA-4 , which are expressed on all activated T cells, the side effect of anti-Tim-3 therapy is expected to be less severe than anti-PD-1 and anti-CTLA-4 therapies.
3.1 Checkpoint Blockade in Combination with Other Immunomodulatory Agents
The targeting of positive immune checkpoint receptors in combination with NCR blockade has also been an appealing therapeutic strategy that may be able to overcome the immunosuppressive tumor microenvironment. Among these receptors is the tumor necrosis factor receptor superfamily (TNFRSF), which includes OX-40, 4-1BB, ICOS, GITR and CD40. These receptors all serve as co-stimulatory molecules for T cell activation. Some are well studied and agonistic antibodies against these molecules have been tested in multiple clinical trials. One of these studies highlighted the importance of timing in the administration of anti-PD-1 and anti-OX40 mAbs [59]. Concurrent administration of the two antibodies was expected to enhance the efficacy of PD-1 blockade by providing costimulatory signals via OX-40. However, no synergetic effect was observed. Instead, increased apoptosis of CTLs occurred. Interestingly, sequential administration of anti-OX40 followed by PD-1 blockade showed increased cell proliferation, reduced cell death and decreased expression of other NCRs on CTLs. Because NCR-mediated immune suppression is not the only mechanism by which the immune system controls for aberrant responses, combining NCR blockade together with other co-stimulatory agents may better enhance immune function.
Future Directions
Immunotherapies focused on NCR blockade have shown remarkable potential in the cancer arena, though complete response to PD-1/PD-L1 blockade has been limited to a subset of patients receiving immunotherapy [60, 61]. Designing combination NCR blockades that include different mechanisms of action may improve efficacy by disrupting the immunosuppressive tumor microenvironment while boosting immune cell functioning. Furthermore, combinatorial strategies may be able to improve response rates while also curbing undesirable side effects.
4 Negative Immune Checkpoint Receptors in Infectious Diseases
Chronic antigen-driven immune activation and inflammation are known to expand NCRs on CTLs. Just as we see in many cancers, these exhausted CTLs are enriched in patients with chronic infectious diseases including viral, bacterial and parasitic infections. Persistent antigen exposure and stimulation drives gene expression that is distinct from naïve, memory and activated T cells and causes pathogen-specific T cells to become functionally inactive culminating in T cell exhaustion. Exhausted T cells is now recognized as a general feature of most chronic viral infections and the expression of inhibitory receptors is implicated in the pathogenesis of many infection-associated diseases. The focus of much research is restoring function to T cells in order to effectively clear infection.
4.1 Human Immunodeficiency Virus (HIV)
HIV is a chronic viral infection, that if left untreated leads to rapid depletion of CD4+ T cells and acquired immunodeficiency syndrome (AIDS). HIV can be controlled with highly active antiretroviral treatment (HAART); however, low level viral replication and chronic inflammation are thought to cause HIV-related comorbidities even despite viral suppression. The PD-1/PDL-1 axis has been well studied in the context of HIV infection and PD-1 has been found to be highly expressed on CTLs in HIV-infected individuals [62, 63]. CTLA-4 expression, on the other hand, is not increased on CTLs in HIV infection [63, 64]. Increased numbers of PD-1-expressing CTLs correlates with higher viral loads and impaired cytotoxic and cytokine-producing functions. However, this impairment seems to be reversible via PD-1 blockade as observed in ex vivo studies. In addition, in vivo PD-1 blockade in simian models of HIV (SIV) have been shown to suppress viral load to some extent even in the absence of antiretroviral therapy (ART). It is believed that this is achieved, at least in part, through enhanced cell-mediated immunity by CTLs [65].
CD4+ T helper cells in HIV have been shown to express high levels of CTLA-4 and PD-1 and expression of these receptors correlates with HIV disease progression [62, 63, 66]. In an SIV rhesus macaque model, inhibition of CTLA-4 or PD-1 reinvigorated CD4+ T cells and induced antibody production. Of note, CD4+ T cell subsets that are able to harbor latent HIV have been shown to express multiple NCRs. Therefore, it is thought that NCRs may serve as reliable markers of viral persistence [67,68,69]. Likewise, CTLA-4+PD-1− CD4+ T cells in lymph nodes have been identified as a possible reservoir in SIV infection models. In humans, PD-1+ follicular helper T cells found in the periphery have been proposed as one of several possible viral reservoirs [66, 70]. Consequently, CTLA-4 or PD-1 blockade has been shown to cause transient increases in viral load in simian models.
The effects of CTLA-4 or PD-1 blockade in humans has been studied in cancer patients who were also infected with HIV [71, 72]. A case report for the use of ipilimumab, a CTLA-4 inhibitory, in combination with ART reported increases in viral load followed by subsequent decline. HIV DNA, a surrogate marker of HIV reservoir size, was not shown to change during the course of treatment with ipilimumab. On the other hand, in patients who received nivolumab, a PD-1 inhibitor, HIV DNA levels decreased profoundly compared to their pre-treatment status. This is thought to be due to the reactivation of HIV reservoirs in latently infected T cells via NCR blockade and attack of these infected cells by reinvigorated immune cells, specifically virus-specific CTLs [73]. Because treatment outcome seems to fluctuate among treated individuals, further study is warranted to overcome this challenge. Furthermore, the expression of multiple NCRs, namely LAG-3, TIGIT and Tim-3 on CD4+ T cells and CTLs are known to be significantly increased in HIV-infected individuals [67, 69, 74,75,76,77]. Given the increased expression of multiple NCRs on T cells, combinatorial blockade of these receptors in additional to PD-1 blockade may exhibit improved restoration of T cell function compared to single blockade and should be explored in preclinical models.
4.2 Human T Cell Lymphotropic Virus (HTLV-1)
HTLV-1 causes a wide spectrum of disease, most notably adult T cell leukemia (ATL) or HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) [78]. Only a small proportion of individuals develop one of the two diseases and only after many years of chronic infection. In this section, we will discuss NCR expression in the context of ATL. HAM/TSP will be discussed in the next section.
HTLV-1 preferentially infects CD4+CD25+ Tregs but is known to also infect CD8+ T cells as well as B cells and monocytes. In HTLV-1 infection, individuals who develop ATL have been found to have fewer HTLV-1-specific CTLs as compared to asymptomatic carriers (AC) [79, 80]. PD-1 expression on HTLV-1-specific CTLs did not differ between the two groups and was more commonly found on CD25+ and CD25− helper T cells rather than CTLs. In contrast, PD-L1 expression on helper T cells, which includes HTLV-1 infected cells, is higher in ATL patients than in AC and blockade of PD-L1 has been shown to enhance IFN-γ production from HTLV-1 specific CTLs. Interestingly, PD-1 expression has been associated with poor proliferation and cytokine production. Therefore, the PD-1/PD-L1 axis not only facilitates immune evasion of HTLV-1 infected cells but may also play a protective role in controlling hyperproliferation of infected cells.
A phase 2 clinical trial using a PD-1 inhibitor was conducted in 3 ATL patients but resulted in rapid progression of disease after a single dose [81]. Patients exhibited leukocytosis, increased viral loads and increased peripheral atypical cells. This increase in viral load was also observed in HIV infected individuals treated with the same PD-1 inhibitory antibody, nivolumab. PD-1 expression may be a means of evading immune surveillance for virus-infected cells by limiting viral replication. Upon PD-1 blockade, virus reactivation in the case of HIV, and hyperproliferation in the case of HTLV-1 may be taking place leading to viral spread and disease progression.
4.3 Hepatitis B Virus (HBV)
Every year, nearly 650,000 people die of HBV-associated end-stage liver diseases . The pathogenic mechanism of HBV is not completely understood. However, NCRs have been implicated in HBV pathogenesis. The overall CTL response in HBV infection is weak and can be undetectable. It has been suggested that the chronicity of HBV infection and lack of CTL response may be related to increased expression of NCRs. PD-1, LAG-3, TIGIT and Tim-3 expression on CTLs has been shown to be elevated in HBV infection, and has also been shown to correlate with disease progression [82]. PD-1 blockade in HBV murine models show reinvigoration of CTLs and decreases in viral load [83]. The efficacy of PD-1 blockade in human HBV infection has been studied in cancer patients infected with HBV. However, similarly to HIV, several case reports have reported increased HBV viral loads and acute hepatitis after treatment with PD-1 blockade [84]. BTLA expression on CD8+ T cells are observed at increased levels on specific T cell subsets, particularly central memory T cells in the periphery and effector memory in the liver [85]. It is believed that this increased expression of BTLA during homing of T cells into tissues and prevents the transition of CD8+ T cells from a central memory phenotype to effector phenotype further allows virus-infected cells to evade immune clearance [86]. Studies targeting BTLA would likely reveal immune mechanisms of HBV pathogenesis as well as expand treatment options for chronic HBV infection.
4.4 Hepatitis C Virus (HCV)
HCV , another chronic viral infection affecting the liver, is prone to dysregulated T cell function and subsequent pathology. Like many chronic infections, the PD-1, TIGIT, LAG-3 and Tim-3 pathways are manipulated in order to favor viral persistence [74]. PD-1 expression is found at elevated levels on virus-specific T cells and correlates directly with viremia [6]. Furthermore, T cells displayed skewed cytokine production, predominantly producing suppressive IL-10. Antiviral treatment results in normalization of cytokine production, immune reactivation and decreases in PD-1 expression. NCR blockade is less likely to be used as a curative strategy for HCV infection due to the development of drugs like Harvoni, a combination of highly effective Direct-Acting Antivirals (DAA), which is able to clear infection in up to 95% of patients. Even so, PD-1 blockade has been approved for treatment of hepatocellular carcinoma. Synergistic effects have been observed with the use of immune checkpoint inhibitors together with molecular targeted agents or local therapy. Many phase III trials are underway and researchers are awaiting the outcome of these trials with high expectations [87].
4.5 Mycobaceterium tuberculosis (TB)
Cytokines such as IFN-γ and TNF-α secreted from CD4+ T cells and CTLs play a central role in the immune response against TB , which are essential for the activation of macrophages. PD-1, LAG-3 and Tim-3 levels increase during TB infection and expression of these molecules are linked to decreased production of cytokines and poorer outcome in several murine and human studies [88,89,90]. Blockade of these NCRs show restoration of T cell function and decreased bacterial loads. However, a study with PD-1 knockout mice showed aberrant T cell activation and high levels of inflammatory cytokine production that lead to decreased survival rate compared to wild type controls [91]. This result suggests that there exists a fine balance between immune restoration and over-activation with the use of novel therapeutic strategies that employ immunotherapies.
4.6 Malaria
Malaria is a global problem with over half a million people dying each year, most of these individuals being children and pregnant women. With no vaccine available and resistance to antimalarial drugs on the rise, new therapeutic approaches are much needed [92]. During malaria infection, CD4+ T cell activation and subsequent antibody production from B cells are responsible for developing protective immunity. However, it has been shown that Plasmodium-specific T cells show features of T cell exhaustion [93]. The identification of exhausted T cells in malaria, therefore, provides an avenue for novel therapeutic approaches. Therefore, new therapeutic approaches and prevention strategies need to be explored. Expression of CTLA-4, PD-1, LAG-3, TIGIT and Tim-3 on CD4+ T cells increases in infected individuals and correlates with disease severity in both human and murine studies [93,94,95,96]. Blockade of PD-1 results in increased cytokine production from CD4+ T cells and decreased parasite burdens in murine models. Dual blockade of PD-L1 and LAG-3 has been shown to increase levels of protective IgG antibodies, follicular helper T cells, and Plasmodium-specific CD4+ and CD8+ T cells resulting in enhanced parasite clearance in mouse models [93]. PD-1/LAG-3 dual-blockade may be able to attribute its synergy to the upregulation of IFN-γ upon PD-L1 blockade, which in turn leads to higher expression of MHC class II, the ligand for LAG-3, and consequently increased LAG-3 inhibitory signaling.
4.7 Future Directions
T cell exhaustion driven by negative immune checkpoint receptor signaling is a common mechanism of immune evasion in chronic infection and cancer. The approval of inhibitory blockade therapies for various cancers opens the door for these strategies to be used in the treatment of chronic infections. Blockade of these pathways alone or in combination offers great hope for better treating and perhaps curing many infection-associated diseases.
5 Negative Immune Checkpoint Receptors in Neuroinflammation
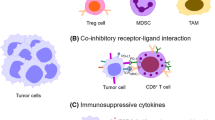
As discussed earlier, expression of NCRs is linked to impaired tumor suppression and T cell exhaustion. Under steady-state conditions, these inhibitory signals play an important role in preventing aberrant cellular activation and the development of autoimmune diseases [26, 97]. Furthermore, the CNS can be a target of many acute infections, as well as a reservoir of latent and persistent pathogens. The brain is considered an immune-privileged tissue site, therefore swift resolution of infection and associated proinflammatory responses, such as through NCR signaling, is ideal to limit CNS complications. Conversely, premature termination of the immune response through NCR signaling prior to rectifying the mediator of neuroinflammation could be detrimental. Here, we discuss current studies that implicate NCRs as important players in neuroinflammation, particularly in the context of neurodegenerative (multiple sclerosis and Alzheimer’s disease) and infectious diseases (HIV, HTLV-1 and viral encephalitis) Fig. 1.

Schematic overview of the role NCRs play in the settings of cancer, infection and neuroinflammation
Under homeostatic conditions, engagement of NCRs with cognate ligand results in decreased T cell activation and is one of the ways in which the body naturally curbs aberrant immune responses. However, persistent immune activation, as seen in cancer and chronic infection, can lead to increased expression of these NCRs resulting in inhibition of effective CTL function and “exhausted” T cell phenotypes. In neuroinflammatory settings, decreased NCR expression leads to dysregulated T cell responses, further inflammation and disease progression
5.1 Multiple Sclerosis (MS)
MS is a chronic inflammatory neurodegenerative disease and pathogenesis is characterized by blood brain barrier (BBB) impairment and infiltration of peripheral immune cells into the CNS, particularly CD4+ effector T cells [98]. The protective function of NCRs has been well investigated using various mouse models of experimental autoimmune encephalitis (EAE), which are widely used in MS studies. Deletion or blockade of CTLA-4 , PD-1, VISTA, TIGIT or Tim-3 causes exaggeration of EAE or other autoimmune-related diseases, which may be a result of the accumulation of activated T cells [43, 99,100,101,102].
In humans, NCRs expression has been observed at higher levels on T cells in cerebrospinal fluid (CSF) relative to peripheral blood [79]. This is most likely the result of tighter control of T cell activation in the CNS as a means of controlling inflammation in the brain. Accordingly, polymorphisms in CTLA-4, PD-1, LAG-3 or Tim-3 have been shown to correlate with the progression of MS [103,104,105,106]. Unlike what we see in HIV, lower expression of these receptors is observed on T cells in both the CSF and in the periphery of MS patients compared to healthy donors. Furthermore, individuals with MS exhibit enhanced T cell function following stimulation ex vivo. For instance, the ex vivo stimulation of TIGIT on Tregs from MS patients has been shown to reverse T cell suppression and reduce the production of IFN-γ in effector T cells [107]. Of note, stimulation with IFN-ß or glatiramer acetate, which are both used for treatment of MS, induces mRNA expression of these negative checkpoint receptors [106, 108]. In vitro experiments show that human brain endothelial cells maintain the integrity of the BBB and can express PD-L1 or PD-L2 to modulate T cell transmigration and immune responses, but this protective function might be impaired in MS due to the decreased levels of PD-L2 on brain endothelium [109].
5.2 Alzheimer’s Disease (AD)
AD , an age-related neurodegenerative disease, is the most common form of dementia. Evidence shows that the pathogenesis of AD is not restricted to neuronal dysfunction but strongly linked to inflammation and alterations in immunological mechanisms in the brain [110]. The production of proinflammatory cytokines and other inflammatory mediators in the CNS have been observed in AD, which leads to the recruitment of peripheral immune cells, further promoting neuroinflammation [111]. The PD1/PD-L1 pathway has been implicated as an important means of regulating neuroinflammatory responses in AD. It has been shown that PD-1 and PD-L1 expression is decreased on CD4+ T cells and CD14+ monocytes and macrophages, respectively. An increase in the frequency of PD-1+ Tregs has also been observed in AD patients [112, 113]. Interestingly, PD-1 blockade was found to reduce disease in mouse models of AD through the mobilization of monocyte-derived macrophages to the brain [114].
5.3 Viral Encephalitis
Viral encephalitis is characterized by severe acute inflammation of the brain parenchyma. Often times, inflammation extends to the meninges as well. Serious cases of encephalitis can result in death. Some examples of viral infections that can result in encephalitis are herpes simplex viruses (HSV), varicella-zoster (VSV), cytomegalovirus (CMV), Epstein-Barr (EBV), influenza A, a number of flaviviruses (West Nile, Dengue, Yellow Fever), paramyxoviruses (rubella, measles), and polyomaviruses [115]. Although studies investigating NCRs in viral encephalitis are limited, those that are available found associations between decreased NCR responses and favorable outcomes as measured as by decreased neuroinflammation and CNS damage. For example, in a polyomavirus CNS infection model in mice, the absence or blockade of PD-1 signaling was shown to limit the severity of neuroinflammation during persistent infection and increase the number of virus-specific, tissue-resident CD8+ memory T cells in the brain [116]. Additionally, in a murine study of CMV CNS infection, it was demonstrated that the PD-1/PD-L1 pathway plays a role in the generation of CNS-resident memory T cells following viral infection [117].
5.4 HTLV-1 Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP)
HAM/TSP is a demyelinating disease caused by HTLV-1 infection, which resembles MS in several ways. Like MS, HAM/TSP is associated with over-activation of the immune system [100, 118]. It is reported that PD-1 expression on HTLV-1-specific CTLs from HAM/TSP patients is comparable to levels seen in asymptomatic carriers (AC). However, PD-L1 induction on HTLV-1-infected helper T cells after antigen-specific stimulation is profoundly reduced in HAM/TSP patients as compared to AC. Furthermore, T cells from HAM/TSP patients exhibit increased cytokine production following peptide stimulation ex vivo [100]. In contrast to PD-1, Tim-3 is expressed to a lesser extent on HTLV-1 specific CTLs in HAM/TSP patients compared to AC. However, these CTLs display greater cytokine production upon peptide stimulation [118]. With no vaccine or treatments available for HAM/TSP, NCR blockade may prove to be an effective means of reinvigorating immune cells to better combat and clear infection.
5.5 HIV-Associated Neurocognitive Disorders (HAND)
Complications regarding cognitive performance is a concern for many individuals living with HIV in the era of highly active antiretroviral therapy (HAART) [119]. The disease pathogenesis HAND has not been fully elucidated; however, chronic CNS inflammation is regarded as a key component of the development of this disease. The brain harbors HIV in macrophages and microglia cells and low levels of HIV RNA is detected even after long-term administration of HAART [120]. Low-level viral replication may induce chronic inflammation of the brain and consequently lead to neurocognitive impairment. However, the relationship between NCR expression and HAND severity still remains unclear. The brain is an immune privileged site and because HIV infected cells likely serve as reservoirs in the CNS, therapies that overcome the physical barriers that exist to protect this site are currently being developed. A clinical trial of pembrolizumab treatment in HIV-infected adults on suppressive ART is currently ongoing (NCT03239899) to investigate the safety and efficacy of PD-1 blockade, particularly measuring outcomes impacting HIV-1 biology and immune function in the CNS.
5.6 Other CNS Infections
In contrast to the previously discussed neuroinflammatory diseases, upregulation of NCRs is observed in some cerebral infections. Peripheral helper T cells from patients with cerebral malaria showed higher expression of CTLA-4 and PD-1 compared to those from patients with uncomplicated malarial infection [96]. In malaria infected mice, soluble PD-L2 treatment that inhibits PD-1 and PD-L1 ligation ameliorates cerebral malaria symptoms and increases survival [121]. In a mouse model of Toxoplasma gondii infection, CTL in the brain exhibited high levels of PD-1 expression. Blockade of PD-1 was found to restore CTL function, diminish brain cysts and improve survival rates [122, 123]. Furthermore, upregulation of the Tim-3 NCR has been observed in the brain of mice infected with Toxoplasma gondii and is hypothesized to potentially regulate Th1-biased responses [124].
It is known that various cells of the CNS including astrocytes, microglia and neurons express PD-1. Chen et al. recently showed that PD-1 expression on neurons suppressed nociceptive neuronal activity during acute and chronic pain stimulation [125]. However, the role of PD-1 in these cells is still largely unknown. However, engagement of these inhibitory receptors could be utilized for the treatment of immune-mediated neuroinflammatory episodes.
5.7 Future Directions
The process of neuroinflammation is complex and an increased understanding of contributing mechanisms will lead to improved means of intervention. Examination of recent literature shows that NCRs likely play a role in neuroinflammation, driven either by neurodegenerative or infectious disease processes. While results from animal studies and patient cohort studies are promising, further investigation, particularly in the safety and efficacy of manipulating these pathways, is warranted to posit NCRs and their respective ligands as additional therapeutic targets in the context of neuroinflammation.
6 Summary
Advances in the fields of immunology and immunotherapy have yielded promising immunotherapeutic strategies for previously difficult-to-treat diseases. Specifically, research focused on immune checkpoint blockade in various cancer settings and in chronic infections are showing great promise in restoring functionality to exhausted T cells. Further research that characterizes the expression of NCRs and the dynamics of their expression in various disease states is crucial for developing novel therapeutic strategies and improving treatment outcomes. As new immune checkpoints are discovered and our understanding of these important molecules advances, the prospect of curing cancers, viral infections and neurological diseases will continue to push the field of immunotherapy forward and serve as an impetus for continued research.
References
McKinney EF, Smith KG. T cell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr Opin Immunol. 2016;43:74–80.
McKinney EF, et al. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523(7562):612–6.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5.
Alfei F, Zehn D. T cell exhaustion: an epigenetically imprinted phenotypic and functional makeover. Trends Mol Med. 2017;23(9):769–71.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–42.
Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;36(2):63–70.
Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–3.
Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol. 2011;350:17–37.
Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18(3):153–67.
Sasidharan Nair V, et al. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin Epigenetics. 2018;10:78.
Kleffel S, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162(6):1242–56.
Gordon SR, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9.
Baumeister SH, et al. Coinhibitory pathways in immunotherapy for Cancer. Annu Rev Immunol. 2016;34:539–73.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Gong J, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8.
Albiges L, et al. Efficacy of targeted therapies after PD-1/PD-L1 blockade in metastatic renal cell carcinoma. Eur J Cancer. 2015;51(17):2580–6.
Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9.
Brahmer J, et al. Nivolumab versus Docetaxel in advanced squamous-cell non-small-cell lung Cancer. N Engl J Med. 2015;373(2):123–35.
Ferris RL, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–67.
Kranawetter M, et al. Activity of Pembrolizumab in recurrent cervical Cancer: case series and review of published data. Int J Gynecol Cancer. 2018;28(6):1196–202.
Long GV, et al. Nivolumab for patients with advanced melanoma treated beyond progression: analysis of 2 phase 3 clinical trials. JAMA Oncol. 2017;3(11):1511–9.
Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30.
Ascierto PA, et al. Survival outcomes in patients with previously untreated BRAF wild-type advanced melanoma treated with Nivolumab therapy: three-year follow-up of a randomized phase 3 trial. JAMA Oncol. 2019;5(2):187–94.
Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in untreated melanoma. N Engl J Med. 2015;373(13):1270–1.
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44(5):989–1004.
Ni L, Dong C. New checkpoints in cancer immunotherapy. Immunol Rev. 2017;276(1):52–65.
Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–33.
Wolchok JD, et al. Overall survival with combined Nivolumab and Ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–56.
Hellmann MD, et al. Nivolumab plus Ipilimumab in Lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–104.
Zhang Q, et al. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci Immunol. 2017;2(9)
Grosso JF, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117(11):3383–92.
Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37.
Wierz M, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood. 2018;131(14):1617–21.
Andrews LP, et al. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. 2017;276(1):80–96.
Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. 2002;169(10):5392–5.
Xu F, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74(13):3418–28.
Kouo T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of Plasmacytoid dendritic cells. Cancer Immunol Res. 2015;3(4):412–23.
Watanabe N, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4(7):670–9.
Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev. 2009;229(1):244–58.
Quan L, et al. BTLA marks a less cytotoxic T-cell subset in diffuse large B-cell lymphoma with high expression of checkpoints. Exp Hematol. 2018;60:47–56.. e1
Lines JL, et al. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2(6):510–7.
Lines JL, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74(7):1924–32.
Nowak EC, et al. Immunoregulatory functions of VISTA. Immunol Rev. 2017;276(1):66–79.
Le Mercier I, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74(7):1933–44.
Zhang Q, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Guillerey C, et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood. 2018;132(16):1689–94.
Kurtulus S, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053–62.
Koyama S, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Sakuishi K, et al. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2(4):e23849.
de Mingo Pulido A, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast Cancer. Cancer Cell. 2018;33(1):60–74.. e6
Liu JF, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37(1):44.
Ndhlovu LC, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119(16):3734–43.
da Silva IP, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014;2(5):410–22.
Anderson AC, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318(5853):1141–3.
Chiba S, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–42.
Yang X, et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J Immunol. 2013;190(5):2068–79.
Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2019;
Messenheimer DJ, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with Anti-OX40. Clin Cancer Res. 2017;23(20):6165–77.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14.
Sharma P, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Velu V, et al. Role of PD-1 co-inhibitory pathway in HIV infection and potential therapeutic options. Retrovirology. 2015;12:14.
Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–4.
Steiner K, et al. Enhanced expression of CTLA-4 (CD152) on CD4+ T cells in HIV infection. Clin Exp Immunol. 1999;115(3):451–7.
Velu V, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206–10.
McGary CS, et al. CTLA-4(+)PD-1(-) memory CD4(+) T cells critically contribute to viral persistence in antiretroviral therapy-suppressed, SIV-infected rhesus macaques. Immunity. 2017;47(4):776–88.. e5
Fromentin R, et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog. 2016;12(7):e1005761.
Hatano H, et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J Infect Dis. 2013;208(1):50–6.
Chew GM, et al. TIGIT Marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog. 2016;12(1):e1005349.
Noto A, et al. CD32(+) and PD-1(+) Lymph Node CD4 T cells support persistent HIV-1 transcription in treated Aviremic individuals. J Virol. 2018;92(20)
Guihot A, et al. Drastic decrease of the HIV reservoir in a patient treated with nivolumab for lung cancer. Ann Oncol. 2018;29(2):517–8.
Wightman F, et al. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. AIDS. 2015;29(4):504–6.
Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and Kill”: strategies to eliminate latent HIV. Cell Host Microbe. 2018;23(1):14–26.
Hashimoto M, et al. CD8 T cell exhaustion in chronic infection and Cancer: opportunities for interventions. Annu Rev Med. 2018;69:301–18.
Fujita T, et al. Expansion of dysfunctional Tim-3-expressing effector memory CD8+ T cells during simian immunodeficiency virus infection in rhesus macaques. J Immunol. 2014;193(11):5576–83.
Jones RB, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205(12):2763–79.
Tian X, et al. The upregulation of LAG-3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J Immunol. 2015;194(8):3873–82.
Hermine O, Ramos JC, Tobinai K. A review of new findings in adult T-cell Leukemia-lymphoma: a focus on current and emerging treatment strategies. Adv Ther. 2018;35(2):135–52.
Kozako T, et al. Programmed death-1 (PD-1)/PD-1 ligand pathway-mediated immune responses against human T-lymphotropic virus type 1 (HTLV-1) in HTLV-1-associated myelopathy/tropical spastic paraparesis and carriers with autoimmune disorders. Hum Immunol. 2011;72(11):1001–6.
Shimauchi T, et al. Augmented expression of programmed death-1 in both neoplastic and non-neoplastic CD4+ T-cells in adult T-cell leukemia/lymphoma. Int J Cancer. 2007;121(12):2585–90.
Ratner L, et al. Rapid progression of adult T-cell Leukemia-lymphoma after PD-1 inhibitor therapy. N Engl J Med. 2018;378(20):1947–8.
Ye B, et al. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015;6:e1694.
Tzeng HT, et al. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS One. 2012;7(6):e39179.
Koksal AS, et al. HBV-related acute hepatitis due to immune checkpoint inhibitors in a patient with malignant melanoma. Ann Oncol. 2017;28(12):3103–4.
Yu X, et al. BTLA/HVEM Signaling: milestones in research and role in chronic hepatitis B virus infection. Front Immunol. 2019;10:617.
Wang H, et al. Hepatic expansion of virus-specific CD8(+)BTLA(+) T cells with regulatory properties in chronic hepatitis B virus infection. Cell Immunol. 2017;311:36–45.
Okusaka T, Ikeda M. Immunotherapy for hepatocellular carcinoma: current status and future perspectives. ESMO Open. 2018;3(Suppl 1):e000455.
Khan N, et al. T-cell exhaustion in tuberculosis: pitfalls and prospects. Crit Rev Microbiol. 2017;43(2):133–41.
Jayaraman P, et al. TIM3 mediates T cell exhaustion during Mycobacterium tuberculosis infection. PLoS Pathog. 2016;12(3):e1005490.
Phillips BL, et al. LAG-3 potentiates the survival of Mycobacterium tuberculosis in host phagocytes by modulating mitochondrial signaling in an in-vitro granuloma model. PLoS One. 2017;12(9):e0180413.
Fujita K, Terashima T, Mio T. Anti-PD1 antibody treatment and the development of acute pulmonary tuberculosis. J Thorac Oncol. 2016;11(12):2238–40.
Shankar EM, Vignesh R, Dash AP. Recent advances on T-cell exhaustion in malaria infection. Med Microbiol Immunol. 2018;
Butler NS, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2011;13(2):188–95.
Abel A, et al. Differential expression pattern of co-inhibitory molecules on CD4(+) T cells in uncomplicated versus complicated malaria. Sci Rep. 2018;8(1):4789.
Costa PA, et al. Induction of inhibitory receptors on T cells during Plasmodium vivax malaria impairs cytokine production. J Infect Dis. 2015;212(12):1999–2010.
Mackroth MS, et al. Acute malaria induces PD1+CTLA4+ effector T cells with cell-extrinsic suppressor function. PLoS Pathog. 2016;12(11):e1005909.
Zhang Q, Vignali DA. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity. 2016;44(5):1034–51.
Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747.
Hurwitz AA, et al. Cytotoxic T lymphocyte antigen-4 (CTLA-4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)-resistant BALB/c mice. Proc Natl Acad Sci U S A. 2002;99(5):3013–7.
Salama AD, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198(1):71–8.
Monney L, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536–41.
Joller N, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. 2011;186(3):1338–42.
Kroner A, et al. A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann Neurol. 2005;58(1):50–7.
Liu J, Zhang HX. CTLA-4 gene and the susceptibility of multiple sclerosis: an updated meta-analysis study including 12,916 cases and 15,455 controls. J Neurogenet. 2014;28(1–2):153–63.
Lundmark F, et al. Association analysis of the LAG3 and CD4 genes in multiple sclerosis in two independent populations. J Neuroimmunol. 2006;180(1–2):193–8.
Yang L, et al. Lack of TIM-3 immunoregulation in multiple sclerosis. J Immunol. 2008;180(7):4409–14.
Lucca LE, et al. TIGIT signaling restores suppressor function of Th1 Tregs. JCI Insight. 2019;4(3)
Schreiner B, et al. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol. 2004;155(1–2):172–82.
Pittet CL, et al. Human brain endothelial cells endeavor to immunoregulate CD8 T cells via PD-1 ligand expression in multiple sclerosis. J Neuroinflammation. 2011;8:155.
Liu YH, et al. Immunity and Alzheimer’s disease: immunological perspectives on the development of novel therapies. Drug Discov Today. 2013;18(23–24):1212–20.
Dansokho C, Heneka MT. Neuroinflammatory responses in Alzheimer’s disease. J Neural Transm (Vienna). 2018;125(5):771–9.
Saresella M, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010;21(3):927–38.
Saresella M, et al. A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer’s disease. Neurobiol Aging. 2012;33(3):624.. e11–22
Baruch K, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22(2):135–7.
George BP, Schneider EB, Venkatesan A. Encephalitis hospitalization rates and inpatient mortality in the United States, 2000–2010. PLoS One. 2014;9(9):e104169.
Shwetank, et al. PD-1 dynamically regulates inflammation and development of brain-resident memory CD8 T cells during persistent viral encephalitis. Front Immunol. 2019;10:783.
Prasad S, et al. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J Neuroinflammation. 2017;14(1):82.
Abdelbary NH, et al. Reduced Tim-3 expression on human T-lymphotropic virus type I (HTLV-I) Tax-specific cytotoxic T lymphocytes in HTLV-I infection. J Infect Dis. 2011;203(7):948–59.
Saylor D, et al. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12(4):234–48.
Marban C, et al. Targeting the brain reservoirs: toward an HIV cure. Front Immunol. 2016;7:397.
Karunarathne DS, et al. Programmed Death-1 Ligand 2-mediated regulation of the PD-L1 to PD-1 Axis is essential for establishing CD4(+) T cell immunity. Immunity. 2016;45(2):333–45.
Bhadra R, et al. Control of toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc Natl Acad Sci U S A. 2011;108(22):9196–201.
Xiao J, et al. PD-1 immune checkpoint blockade promotes brain leukocyte infiltration and diminishes cyst burden in a mouse model of toxoplasma infection. J Neuroimmunol. 2018;319:55–62.
Wu B, et al. Upregulated expression of Tim-3 involved in the process of toxoplasmic encephalitis in mouse model. Parasitol Res. 2013;112(7):2511–21.
Chen G, et al. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat Neurosci. 2017;20(7):917–26.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Fujita, T., Clements, D.M., Premeaux, T.A., Ndhlovu, L.C. (2020). Perspectives on the Role of T Cell Negative Immune Checkpoint Receptors in Health and Disease. In: Jain, P., Ndhlovu, L. (eds) Advanced Concepts in Human Immunology: Prospects for Disease Control. Springer, Cham. https://doi.org/10.1007/978-3-030-33946-3_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-33946-3_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-33945-6
Online ISBN: 978-3-030-33946-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)