
Abstract
Cyclic di-AMP is a vital second messenger other than cyclic di-GMP that regulates diverse cellular physiological processes in many bacteria. Its cellular level is controlled by the counter-actions of diadenylate cyclases (DAC) and phosphodiesterases (PDE). Three kinds of PDEs have been identified to date that contain either a DHH–DHHA1 domain, an HD domain, or a metallo-phosphoesterase domain, respectively. The DHH–DHHA1 PDEs are of special interest because of their functional diversity. They can be further subdivided into either membrane-bound GdpP or stand-alone Rv2837c phosphodiesterase, which degrade cyclic di-AMP into linear 5′-pApA and AMP, respectively. The DHH–DHHA1 PDEs can also hydrolyze other cyclic di-NMPs (cyclic di-GMP or cGAMP) with low activity. In this chapter, we review the structures and functions of the DHH–DHHA1 domain of GdpP and Rv2837c that we reported in recent years. According to detailed structural and enzymatic analyses, we have summarized a unified molecular mechanism for the DHH–DHHA1 PDEs and systematically analyzed the catalytic activities of DHH–DHHA1 PDEs on other cyclic di-NMPs (cyclic di-GMP and cGAMP).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Recent research works have revealed that cyclic di-NMPs (cyclic di-GMP, cyclic di-AMP, and cGAMP) are widely exploited as second messengers in bacteria to serve crucial roles in both bacterial physiology and host–pathogen interactions. Cyclic di-GMP, the first discovered cyclic di-NMP signaling molecule, has been extensively studied since the late 1980s. It is now known to regulate many physiological processes such as biofilm formation, virulence, and motility in a wide variety of organisms [1, 2]. However, other than cyclic di-GMP, other cyclic di-nucleotides were also discovered. For example, cyclic di-AMP was unexpectedly uncovered by Karl-Peter Hopfner et al. in 2008 [3]. Different from cyclic di-GMP that is widely found in most bacteria, cyclic di-AMP is primarily discovered in Gram positive bacteria, some archaea, as well as in Gram negative bacteria to a limited extent. Until now, cyclic di-AMP has been known to get involved in many cellular processes such as sporulation, fatty acid synthesis, cell wall homeostasis, potassium transport, and virulence [4, 5]. Furthermore, a hybrid cyclic dinucleotide cGAMP (3′–3′) was also identified in 2012 to regulate the chemotaxis and colonization in Vibrio cholera [6]. Subsequently, 3′3′-cGAMP was also found to serve as the signaling molecule for regulating exoelectrogenesis in numerous deltaproteobacteria [7]. Intriguingly, cyclic di-NMP generation is not limited to microbes; mammalian cells can also synthesize 2′3′-cGAMP to activate the immune system in response to pathogen-derived DNA in the cytoplasm [3]. Compared to cyclic di-GMP, cyclic di-AMP, and 3′3′-cGAMP that all incorporate two 3′–5′ phosphodiester bonds, 2′3′-cGAMP exhibits mixed 2′–5′ and 3′–5′ phosphodiester bonds [8, 9]. Significantly, cyclic di-GMP and cyclic di-AMP from bacteria and 2′3′-cGAMP from mammalian cells have all been recognized by STING in the mammalian immune cells to trigger type 1 interferon production during infection [10, 11].
Since the cellular levels of cyclic di-NMPs vary widely and directly impact the cellular physiological state, the discovery of enzymes that synthesize and degrade cyclic di-NMPs has become one of the most studied topics of research. Past research has confirmed that cyclic di-GMP is cyclized from two molecules of GTP by diguanylate cyclases (DGCs) containing a GGDEF domain; it is hydrolyzed into pGpG or GMP by phosphodiesterases (PDEs) containing an EAL or HD-GYP domain, respectively [12]. Similar to the metabolism of cyclic di-GMP, the cellular level of cyclic di-AMP is also controlled by the counter-active enzymes of DAC and PDE, which contain either a DHH/DHHA1 (Asp-His-His and Asp-His-His-associated) domain, a His-Asp (HD) domain, or a metallo-phosphodiesterase domain. The PgpH PDE domain was identified in L. monocytogenes, and comprises an extracellular 7TM receptor-like domain and a cytoplasmic HD domain that can hydrolyze cyclic di-AMP into linear dinucleotide 5′-pApA [13], while the PDE domain of CdnP was recently discovered in group B Streptococcus to degrade cyclic di-AMP into two molecules of AMP [14]. Compared to the two PDEs just mentioned, the PDE DHH–DHHA1 domains seem to exhibit more functional diversity. According to the final product produced (pApA or AMP), the DHH–DHHA1 PDEs can be further divided into two subfamilies. The first subfamily contains homologs of membrane-bound GdpP PDE (GGDEF domain-containing proteins) that degrade cyclic di-AMP to linear pApA by hydrolyzing one of the two phosphodiester bonds, while the second subfamily includes the standalone DHH–DHHA1 PDEs such as Rv2837c from Mycobacterium tuberculosis, which degrades both cyclic di-AMP and pApA into two molecules of AMP.
All such cyclic di-AMP PDEs have been extensively studied, and the crystal structures of the HD domain of PgpH, the DHH–DHHA1 domain of GdpP, and Rv2837c have all been solved. These structures, especially those in complex with related nucleotides, have greatly advanced our understanding of the catalytic mechanism of these enzymes. In this chapter, by focusing on the structures and functions of GdpP and Rv2837c reported by our laboratory, we have summarized a unified catalytic mechanism for cyclic di-NMP hydrolysis by the PDE DHH–DHHA1 domain.
2 Cyclic di-AMP Conformation
Cyclic di-AMP comprises two AMP moieties cyclized by two 3′–5′ phosphodiester bonds. When crystallized in isolation, a mutually stacked cyclic di-AMP dimeric structure was observed with each cyclic di-AMP adopting a U-shaped conformation (closed conformation, Fig. 5.1a) [15]. The U-shaped cyclic di-AMP was also observed in the structure of STING–cyclic di-AMP complex (Fig. 5.1b) [9]. In contrast, cyclic di-AMP was found to adopt an extended conformation when bound to the DHHA1 domain of a GdpP PDE (Fig. 5.1c). Interestingly, when cyclic di-AMP binds to the active site of the HD domain of PgpH, it adopts a C-shaped conformation (Fig. 5.1d). It seems that cyclic di-AMP tends to adopt an open conformation when bound to PDEs. It is also interesting to learn that a new U-shaped cyclic di-AMP conformation was found when bound to PDE. In fact, two C-shaped cyclic di-AMPs (Fig.5.1e) were also observed to bind at two separated positions in a ydaO riboswitch [16]. To date, the structures of cyclic di-AMP in complex with different receptors suggest that cyclic di-AMP tends to form a monomer for cyclic di-AMP signaling. Although a conformation with two cyclic di-AMPs bridged by a third one was also observed in the pyruvate carboxylase obtained from Listeria monocytogenes (Fig. 5.1f), it has been reported as an artifact [17].

Unveiled cyclic di-AMP conformations. (a) A stacked asymmetric dimeric U-shaped conformation observed in the crystal structure of cyclic di-AMP. (b) A U-shaped cyclic di-AMP observed in the STING–cyclic di-AMP complex structure (PDB code: 5CFN). (c) An extended conformation of cyclic di-AMP bound to the DHH–DHHA1 domain of GdpP phosphodiesterase (PDB code: 5XSN). (d) A C-shaped conformation of cyclic di-AMP bound to the HD domain of PgpH phosphodiesterase (PDB code: 4S1B). (e) The structure of cyclic di-AMP bound to a ydaO riboswitch (PDB code: 4QLM). (f) Two cyclic di-AMPs bridged by a third one bound to pyruvate carboxylase (PDB code: 4QSH)
3 The DHH–DHHA1 Domain Containing Phosphodiesterases
The DHH–DHHA1 subfamily belongs to the DHH phosphoesterase superfamily, which shares four conserved N-terminal motifs and is named after the characteristic Asp-His-His sequence in the motif III. The DHH phosphoesterases can hydrolyze various substrates ranging from inorganic pyrophosphate to single-stranded (ss) DNA in eukaryotes, bacteria, and archaea [18, 19]. According to the difference of C-terminal sequences, the DHH superfamily can be further divided into two subfamilies of DHH–DHHA1 and DHH–DHHA2. The DHH–DHHA1 subfamily is more widespread in bacteria and archaea, including bacterial RecJ-exonuclease, NrnA-oligoribonuclease, YybT cyclic nucleotide phosphodiesterases, archaeal GAN, and HAN [18, 20]. On the contrary, the DHH–DHHA2 subfamily is more restricted in its distribution, and mainly comprises type II inorganic pyrophosphatase, yeast cytosol exopolyphosphatase, Drosophila prune protein, and pApase families [18, 20]. The catalytic domains of GdpP and Rv2837c homologs belong to the DHH–DHHA1 subfamily since they share a conserved GGGH motif at the C-terminus.
GdpP is the first characterized cyclic di-AMP PDE containing two transmembrane helical domains, a PAS (Per-Arnt-Sim) domain, a degenerate GGDEF domain, and a DHH–DHHA1 catalytic domain [21]. GdpP and its homologs are mostly found to exist in the Firmicutes and Tenericutes phyla, including Streptococcus pneumoniae, Listeria monocytogenes, and Staphylococcus aureus [22,23,24]. The GdpP family seems to exhibit specific PDE activity mainly on cyclic dinucleotides. Previous studies have reported that GdpP family was capable of degrading cyclic di-AMP or cyclic di-GMP by hydrolyzing one of the two 3′–5′ phosphodiester bonds to generate a linear 5′-pApA or 5′-pGpG product, respectively, but exhibited a much higher K m than cyclic di-AMP [21]. In addition, the hydrolysis activity of GdpP required the presence of Mn2+ ion and is competitively inhibited by the signaling molecule (p)ppGpp or its product 5′pApA [21, 25].
Unlike GdpP with a more restricted PDE activity on cyclic di-NMP, Rv2837c homologs, which contain only the standalone catalytic DHH–DHHA1 domain, were found to be less specific and exhibit high versatility in substrate choice. These proteins can function either as a nano-RNase (NrnA) with exonuclease activity on short single-stranded nucleic acids or as a CysQ-like phosphatase to dephosphorylate 3′-phosphoadenosine 5′-phosphate (pAp) to AMP [26, 27]. Rv2837c hydrolyzes cyclic di-AMP and linear 5′-pApA directly into two AMPs; it also hydrolyzes cyclic di-GMP and linear 5′-pGpG into two GMPs. Similar to GdpP, Rv2837c has lower hydrolysis activity toward cyclic di-GMP and requires the presence of Mn2+ ion for efficient catalysis [28]. Surprisingly, Rv2837c was also found to be capable of degrading 2′3′-cGAMP to linear dinucleotides 2′5′-pGpA. Compared to the relatively more limited distribution of GdpP homologs, Rv2837c homologs were found to be present in almost all strains containing a cyclic di-AMP signaling system [24].
4 Structure of DHH–DHHA1 Domain with a Binuclear Metal Center
To date, many crystal structures of the DHH–DHHA1 domains in PDE have been determined [15, 25, 29, 30], including those of Rv2837c and the DHH–DHHA1 catalytic domain of GdpP (GdpP-C) that were determined in our lab, as well as several reported by other groups. Interestingly, they all seem to share some common features, with a larger DHH domain at the amino-terminus and a smaller DHHA1 domain at the carboxy-terminus, which are connected by a long flexible loop to form a cleft in between (Fig. 5.2a, b). In the Rv2837c structure, the DHH subunit exhibits a five-stranded antiparallel β-sheet (β1–β5) that packs against ten α-helices (with 1, 2, 3, 4, 8, 9, and 10 on one side and 5, 6, and 7 on the other). Similar to DHH, the DHHA1 domain structure also forms a three-layer α–β–α sandwich configuration consisting of an antiparallel β-sheet (β6–β10) and five α-helices (with 12–14 on one side and 15, 16 on the other) (Fig. 5.2a). Structurally speaking, both DHH and DHHA1 domain structures of GdpP-C have a three-layer α–β–α sandwich architecture (Fig. 5.2b).

DHH–DHHA1 phosphodiesterases containing a binuclear metal center. (a) Schematic representation of the Rv2837c monomer, with the DHH domain colored in cyan and the DHHA1 domain colored in light orange. (b) Schematic representation of the GdpP-C monomer, with the DHH domain colored in green and the DHHA1 domain colored in orange. (c, d) The two Mn2+ coordination sites of the Rv2837c and GdpP-C phosphodiesterases, respectively. Residues in contact with the metal ions are shown in sticks, Mn1 and Mn2 are drawn in magenta spheres, and water molecules in red spheres
Based on reported structures, we found two Mn2+ ions (Mn1 and Mn2) that were well coordinated by several highly conserved His and Asp residues, as well as a crucial water molecule (W1) to bridge the two metal ions in the DHH domain of Rv2837c and GdpP (Fig. 5.2c,d). Mn1 is coordinated by Asp47, Asp106, His131, Asp181, W1, and W3 to form an octahedron, while Mn2 is coordinated by His41, Asp45, Asp106, W1, and W2 in the active site of Rv2837c (Fig. 5.2c) to form another octahedron. Similarly, in GdpP, Mn1 is coordinated by Asp349, Asp418, His442, Asp497, W1, and W2 to form an octahedron, while Mn2 is coordinated by His343, Asp347, Asp418, W1, and W3 to form another octahedron (Fig. 5.2d). These structures were also confirmed by mutation studies, and we found that mutation of any of these coordinating residues could almost eliminate the cyclic di-AMP hydrolysis activity. The crystal structures of Rv2837c, GdpP-C, as well as the available biochemical data suggest that both Rv2837c and GdpP assume a two-metal ion catalytic mechanism. Meanwhile, the PgpH HD domain was also found to contain two metal ions in the active site [13]. It is thus possible that all cyclic di-AMP phosphodiesterases employ a two-metal ion catalytic mechanism.
5 Hydrolysis of the 3′–5′ Phosphodiester Bond by the DHH–DHHA1 Domain
For the 3′–5′ phosphodiester bond to break apart during hydrolysis of cyclic di-AMP to 5′-pApA, the cyclic di-AMP molecule must adopt a certain conformation in the active site of the DHH–DHHA1 domain. The crucial information on this issue comes from the structures of GdpP-C in complex with cyclic di-AMP, cyclic di-GMP, and 5′-pApA. The overall structure and the coordination of the two Mn2+ ions remain unchanged in the three complex structures in comparison to free GdpP-C. These structures also reveal that the DHHA1 domain plays a pivotal role in substrate recognition since 5′-pApA, cyclic di-AMP, and cyclic di-GMP all reside in the DHHA1 domain while the DHH domain is separated from the DHHA1 domain when GdpP-C is in an inactive state (Fig. 5.3a).

The 3′–5′ phosphodiester bond hydrolysis by DHH–DHHA1 PDEs. (a) Structural comparison of GdpP-C (cyan), GdpP-C–cyclic di-AMP (salmon), GdpP-C–5′-pApA (yellow), and GdpP-C/cyclic di-GMP (green). The two Mn2+ ions are drawn in gray spheres and the nucleotides are shown in sticks, with the protein drawn in cartoon in the background. The Fo–Fc electron-density map for the two Mn2+ ions is contoured at 3.0σ. The residues that form H-bonds with the two Mn2+ ions and nucleotides are labeled and shown in ball-and-sticks. (b, c) Close view of the different nucleotides cyclic di-AMP (b) or 5′-pApA (c) bound to the DHHA1 domain of GdpP-C. (d) Close view of 5′-pApA bound to the active site of Rv2837c. The electron-density maps for 5′-pApA and the two Mn2+ ions of the 2Fo–Fc map are contoured at 1σ [15]. (e) Diagram showing the coordination of 5′-pApA and Mn2+ in the active site of Rv2837c and a proposed reaction mechanism of 5′-pApA hydrolysis. The arrowheads show the directions of nucleophilic attack [15]
Both adenine bases in the bound cyclic di-AMP adopt an “anti” configuration, with the adenine base of nucleotide 1 (A1) stabilized by Gln572 and Asp575, and the adenine base of nucleotide 2 (A2) stabilized by forming an H-bond with Gln628. The phosphate group facing the DHHA1 motif is stabilized by two H-bonds with Ser600 and Arg602 (Fig. 5.3b). In the structure of GdpP-C–5′-pApA, the disconnected phosphodiester bond is exposed to the solvent and is closer to the DHH motif with the remaining portion of the 5′-pApA stabilized in a way similar to the cyclic di-AMP molecule (Fig. 5.3c) [25].
Intriguingly, the cyclic di-AMP and 5′-pApA in the DHHA1 motif do not seem to interact with the two Mn2+ ions in the DHH motif, indicating that these structures possibly represent a catalytically inactive state of GdpP. The flexible linker between the DHH and DHHA1 domain makes it possible for the DHHA1 domain to draw near the active site of the DHH domain to form a catalytically active state. Mutations on most of the residues involved in cyclic di-AMP and 5′-pApA binding have decreased PDE activity, suggesting that the interactions observed in our structures potentially occurs during the catalysis. However, a ligand-bound complex structure in its active state is required to fully understand the genuine catalytic mechanism.
Fortunately, we have also obtained a crystal structure of Rv2837c in complex with the hydrolysis intermediate 5′-pApA. Since 5′-pApA is the hydrolysis intermediate from cyclic di-AMP to AMP and is also the smallest nano-RNA, the structure of 5′-pApA bound to Rv2837c can provide a solid basis for analyzing the hydrolysis reactions of both nano-RNA and 5′-pApA (the second-step of cyclic di-AMP hydrolysis). In this structure, both adenine bases of the 5′-pApA molecule are perpendicular to each other, with the adenine base of nucleotide 1 (A1) sandwiched between309GGGH312 and Arg112, and the adenine base of nucleotide 2 (A2) stabilized by a π–π interaction with His312. The phosphate group of the 3′–5′ phosphodiester bond also forms an H-bond with His312 and is coordinated with the two Mn2+ ions in the active site (Fig. 5.3d). The structure of 5′-pApA bound structure combined with available biochemical data thus allow us to propose a simplified catalytic mechanism for 3′–5′ phosphodiester bond hydrolysis by Rv2837c. The mechanism is as follows: Asp181 residue and the two Mn2+ ions together activate the water molecule W1, which then carries out a nucleophilic attack at the phosphate group of 5′-pA1pA2 to rupture the 3-phosphate-ester bond, leading to bond cleavage (Fig. 5.3e). Considering the highly conserved nature of the binuclear metal center and the residues involved in substrate binding, this catalytic mechanism might also be conserved between the two DHH–DHHA1 subfamily domains of PDEs.
6 Detailed Two-Step Hydrolysis of cyclic di-AMP
Cyclic di-AMP has two symmetric 3′–5′ phosphodiester bonds. Since there is only one binuclear center in the active site, degradation of cyclic di-AMP by Rv2837c must occur in a two-step process, in which cyclic di-AMP is first linearized to 5′-pApA, followed by hydrolysis of 5′-pApA to two AMPs. Our kinetic study has demonstrated that the two-step degradation of cyclic di-AMP by Rv2837c finishes quickly and generates the final product AMP once cyclic di-AMP enters the active site [15, 25]. Without release of the intermediate product 5′-pApA into the solvent and returning into the active site for the second hydrolysis, this mechanism may greatly improve the efficiency of catalysis.
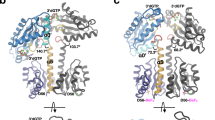
To date, how 5′-pApA is hydrolyzed to AMP is well understood, but how cyclic di-AMP is degraded to 5′-pApA remains an open question. However, structural comparison of Rv2837c and GdpP-C do provide us an unexpected discovery. Although Rv2837c and GdpP-C are quite similar in structure, these two proteins bind 5′-pApA in a quite different mode. Superposition of 5′-pApA boundRv2837c and GdpP-C shows that the 3′-adenine base of 5′-pApA in GdpP-C overlaps with the 5′-adenine in Rv2837c in the active state. In addition, the other two adenine bases reside in two separate sites. To better describe the nucleotide binding, we have further organized the substrate-binding site of the DHH/DHHA1 domain into three R, C, and G subsites. The C (common) site is occupied by a nucleoside in both Rv2837c and GdpP complexes and is surrounded by residues Leu424 and Gln628 from GdpP or Arg112 and Thr319 from Rv2837c, respectively. The position occupied by a nucleoside in Rv2837c only but not in GdpP is named the R site while the position occupied by a nucleoside in GdpP only but not in Rv2837c is named the G site. The R site is surrounded by residues Trp187 and Ala315 from Rv2837c, while the G site is surrounded by Gln572 and Asp575 from GdpP-C, respectively (Fig. 5.4a–c).

Characteristics of the three substrate binding sites of DHH–DHHA1 PDEs. (a) Structural comparison of the GdpP-C–5′-pApA complex (cyan) and the Rv2837c–5′-pApA complex (salmon) [25]. (b, c) The different locations of 5′-pApA in complex with GdpP-C and Rv2837c. The three subsites are circled in orange, and the residues involved in 5′-pApA binding of GdpP-C and Rv2837c are labeled in black and red, respectively [25]. (d) cyclic di-GMP, but not cyclic di-AMP, may exhibit steric clash with the G site of the active state model of GdpP-C. (e) GdpP-C and Rv2837c–A315N hydrolyzes 3′3′-cGAMP into a linear dinucleotide (5′-pApG and 5′-pGpA) with different yields [25], indicating that the G site of the DHH–DHHA1 domain prefers the subsite A to subsite G
Structures of GdpP-C in complex with 5′-pApA and cyclic di-AMP, as well as additional biochemical assays, together suggest that in GdpP-C, hydrolysis of cyclic di-AMP to 5′-pApA occurs in the C–G sites. In contrast, hydrolysis of 5′-pApA to AMPs occurs in the C–R sites in Rv2837c. These results then raise a question: where does the hydrolysis of cyclic di-AMP to 5′-pApA occur in Rv2837c? It is obvious that Rv2837c is structurally similar to GdpP-C. Therefore, we assume that in Rv2837c, cyclic di-AMP is also degraded to 5′-pApA in the C–G sites. This means that for the second-step hydrolysis to occur, 5′-pApA has to slide into the C–R site. A flip of 5′-pApA around its length axis is also needed for the phosphodiester bond to face the binuclear metal center after the slide. This assumption was indeed confirmed by biochemical assay with Rv2837c mutant. Compared with wildtype Rv2837c, the T180R mutant (with a blocked G site) loses half of the activity of cyclic di-AMP substrate hydrolysis whereas it still retains a full catalytic activity on the 5′-pApA intermediate. These analyses indicate that Rv2837c works as a stand-alone DHH–DHHA1 PDE adopting a sliding and flipping mechanism during the two-step hydrolysis of cyclic di-AMP.
The next question raised is: why cannot GdpP-C hydrolyze 5′-pApA to AMPs. Structural analysis shows that compared to Rv2837c, GdpP-C contains a very small R subsite that cannot accommodate an adenine base [25]. Consequently, 5′-pApA derived from the cyclic di-AMP hydrolysis in G–C sites is not able to slide into the C–R sites for further degradation but is released as the final product. We can thus speculate that as a membrane-bound DHH–DHHA1 PDE, GdpP can also adopt the similar sliding-and-flipping mechanism to hydrolyze cyclic di-AMP to AMPs if the R site is large enough. Indeed, alternation of L424R or L503A/R504W which results in a sized C or R subsite with a more suitable size will enable GdpP-C to degrade cyclic di-AMP to both 5′-pApA and AMP [25]. In conclusion, both subfamilies of DHH–DHHA1 PDEs seem to adopt a unified catalytic mechanism. The difference in function comes mainly from the surrounding architecture of the substrate binding site, which determines whether 5′-pApA can undergo sliding and flipping followed by further hydrolysis.
7 G Subsite of the DHH–DHHA1 Domain Determines the Substrate Selectivity for Cyclic Dinucleotides
One striking feature of the PDE DHH–DHHA1 domain in Rv2837c and GdpP is their wide range of substrate selectivity. This domain can hydrolyze cyclic di-AMP, cyclic di-GMP, and 3′3′-cGAMP in the order of cyclic di-AMP >> 3′3′-cGAMP > cyclic di-GMP [15, 25]. Notably, as a nano-RNAase, Rv2837c can hydrolyze 5′-pApA and 5′-pGpG at nearly the same rate; therefore, the substrate selectivity does not come from the second-step degradation of cyclic di-NMPs. In the past, while cyclic di-GMP oligomerization in aqueous solution has been shown to play a partial effect in the slow hydrolysis of cyclic di-GMP, this, however, did not seem to be the whole story.
Structural comparison between the cyclic di-AMP- and cyclic di-GMP-bound GdpP-C indicates that the two cyclic di-NMPs overlap well in the DHHA1 motif. However, in the active state model of GdpP-C, guanine of cyclic di-GMP, but not adenine of cyclic di-AMP, exhibits a steric clash with the surrounding amino acids in the G site; no such clash is observed in the C site (Fig. 5.4d). Therefore, it is reasonable to speculate that the substrate selectivity is mostly dependent on the size and shape of the G site. Consistent with this hypothesis, hydrolysis of 3′3′-cGAMP, which contains a guanine and an adenine, produces much more 5′-pApG than hydrolysis of GdpP-C. Structural analysis indicates that 5′-pApG is produced when the adenine base of 3′3′-cGAMP occupies the G site; otherwise, the product would be 5′-pGpA. From these results, we know that the G site of GdpP-C prefers an adenine base to a guanine base as a favorable substrate to play an important role in substrate selectivity.
Next, we also want to know whether a similar hydrolysis mechanism exists in Rv2837c, enabling it to prefer cyclic di-AMP to other cyclic di-NMPs. Because Rv2837c directly hydrolyzes 3′3′-cGAMP to AMP and GMP instead of producing linear dinucleotide products, the mutant A315N, which has a partially blocked R site, was selected for the experiment. Indeed, when 3′3′-cGAMP was hydrolyzed by Rv2837c–A315N, a series of products of 5′-pApG, 5′-pGpA, AMP, and GMP were produced, with higher percentage of 5′-pApG than 5′-pGpA (Fig. 5.4e). These data confirm that both Rv2837c and GdpP employ a similar mechanism for the catalysis and substrate selectivity of cyclic di-NMPs, and the architecture of the substrate binding site determines whether the second-step reaction occurs.
References
Cotter PA, Stibitz S (2007) c-di-GMP-mediated regulation of virulence and biofilm formation. Curr Opin Microbiol 10(1):17–23. https://doi.org/10.1016/j.mib.2006.12.006
Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H (2010) Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science 327(5967):866–868. https://doi.org/10.1126/science.1181185
Witte G, Hartung S, Buttner K, Hopfner KP (2008) Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol Cell 30(2):167–178. https://doi.org/10.1016/j.molcel.2008.02.020
Corrigan RM, Grundling A (2013) Cyclic di-AMP: another second messenger enters the fray. Nat Rev Microbiol 11(8):513–524. https://doi.org/10.1038/nrmicro3069
Commichau FM, Dickmanns A, Gundlach J, Ficner R, Stulke J (2015) A jack of all trades: the multiple roles of the unique essential second messenger cyclic di-AMP. Mol Microbiol 97(2):189–204. https://doi.org/10.1111/mmi.13026
Davies BW, Bogard RW, Young TS, Mekalanos JJ (2012) Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149(2):358–370. https://doi.org/10.1016/j.cell.2012.01.053
Nelson JW, Sudarsan N, Phillips GE, Stav S, Lunse CE, McCown PJ, Breaker RR (2015) Control of bacterial exoelectrogenesis by c-AMP-GMP. Proc Natl Acad Sci USA 112(17):5389–5394. https://doi.org/10.1073/pnas.1419264112
Sun LJ, Wu JX, Du FH, Chen X, Chen ZJJ (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339(6121):786–791. https://doi.org/10.1126/science.1232458
Kranzusch PJ, Wilson SC, Lee AS, Berger JM, Doudna JA, Vance RE (2015) Ancient origin of cGAS-STING reveals mechanism of universal 2′,3′ cGAMP signaling. Mol Cell 59(6):891–903. https://doi.org/10.1016/j.molcel.2015.07.022
Xiao TS, Fitzgerald KA (2013) The cGAS-STING pathway for DNA sensing. Mol Cell 51(2):135–139. https://doi.org/10.1016/j.molcel.2013.07.004
Margolis SR, Wilson SC, Vance RE (2017) Evolutionary origins of cGAS-STING signaling. Trends Immunol 38(10):733–743. https://doi.org/10.1016/j.it.2017.03.004
Jenal U, Reinders A, Lori C (2017) Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol 15(5):271–284. https://doi.org/10.1038/nrmicro.2016.190
Huynh TN, Luo S, Pensinger D, Sauer JD, Tong L, Woodward JJ (2015) An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc Natl Acad Sci USA 112(7):E747–E756. https://doi.org/10.1073/pnas.1416485112
Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, Trieu-Cuot P, Golenbock DT, Kaminski PA (2016) Group B streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe 20(1):49–59. https://doi.org/10.1016/j.chom.2016.06.003
He Q, Wang F, Liu SH, Zhu DY, Cong HJ, Gao F, Li BQ, Wang HW, Lin Z, Liao J, Gu LC (2016) Structural and biochemical insight into the mechanism of Rv2837c from Mycobacterium tuberculosis as a c-di-NMP phosphodiesterase (vol 291, pg 3668, 2016). J Biol Chem 291(27):14386–14387. https://doi.org/10.1074/jbc.A115.699801
Gao A, Serganov A (2014) Structural insights into recognition of c-di-AMP by the ydaO riboswitch. Nat Chem Biol 10(9):787–792. https://doi.org/10.1038/Nchembio.1607
Sureka K, Choi PH, Precit M, Delince M, Pensinger DA, Huynh TN, Jurado AR, Goo YA, Sadilek M, Iavarone AT, Sauer JD, Tong L, Woodward JJ (2014) The cyclic dinucleotide c-di-AMP is an allosteric regulator of metabolic enzyme function. Cell 158(6):1389–1401. https://doi.org/10.1016/j.cell.2014.07.046
Aravind L, Koonin EV (1998) A novel family of predicted phosphoesterases includes Drosophila prune protein and bacterial RecJ exonuclease. Trends Biochem Sci 23(1):17–19
Makarova KS, Koonin EV, Kelman Z (2012) The CMG (CDC45/RecJ, MCM, GINS) complex is a conserved component of the DNA replication system in all archaea and eukaryotes. Biol Direct 7:7. https://doi.org/10.1186/1745-6150-7-7
Feng L, Chang CC, Song D, Jiang C, Song Y, Wang CF, Deng W, Zou YJ, Chen HF, Xiao X, Wang FP, Liu XP (2018) The trimeric Hef-associated nuclease HAN is a 3′→5′ exonuclease and is probably involved in DNA repair. Nucleic Acids Res 46(17):9027–9043. https://doi.org/10.1093/nar/gky707
Rao F, See RY, Zhang DW, Toh DC, Ji Q, Liang ZX (2010) YybT is a signaling protein that contains a cyclic dinucleotide phosphodiesterase domain and a GGDEF domain with ATPase activity. J Biol Chem 285(1):473–482. https://doi.org/10.1074/jbc.M109.040238
Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, Metzger DW, Bai G (2013) Two DHH subfamily 1 proteins in Streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J Bacteriol 195(22):5123–5132. https://doi.org/10.1128/JB.00769-13
Corrigan RM, Abbott JC, Burhenne H, Kaever V, Grundling A (2011) c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog 7(9):e1002217. https://doi.org/10.1371/journal.ppat.1002217
Huynh TN, Woodward JJ (2016) Too much of a good thing: regulated depletion of c-di-AMP in the bacterial cytoplasm. Curr Opin Microbiol 30:22–29. https://doi.org/10.1016/j.mib.2015.12.007
Wang F, He Q, Su KX, Wei TD, Xu SJ, Gu LC (2018) Structural and biochemical characterization of the catalytic domains of GdpP reveals a unified hydrolysis mechanism for the DHH/DHHA1 phosphodiesterase. Biochem J 475:191–205. https://doi.org/10.1042/Bcj20170739
Postic G, Danchin A, Mechold U (2012) Characterization of NrnA homologs from Mycobacterium tuberculosis and Mycoplasma pneumoniae. RNA 18(1):155–165. https://doi.org/10.1261/rna.029132.111
Srivastav R, Kumar D, Grover A, Singh A, Manjasetty BA, Sharma R, Taneja B (2014) Unique subunit packing in mycobacterial nanoRNase leads to alternate substrate recognitions in DHH phosphodiesterases. Nucleic Acids Res 42(12):7894–7910. https://doi.org/10.1093/nar/gku425
Yang J, Bai Y, Zhang Y, Gabrielle VD, Jin L, Bai G (2014) Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol Microbiol 93(1):65–79. https://doi.org/10.1111/mmi.12641
Drexler DJ, Muller M, Rojas-Cordova CA, Bandera AM, Witte G (2017) Structural and biophysical analysis of the soluble DHH/DHHA1-type phosphodiesterase TM1595 from Thermotoga maritima. Structure 25(12):1887–1897. https://doi.org/10.1016/j.str.2017.10.001
Uemura Y, Nakagawa N, Wakamatsu T, Kim K, Montelione GT, Hunt JF, Kuramitsu S, Masui R (2013) Crystal structure of the ligand-binding form of nanoRNase from Bacteroides fragilis, a member of the DHH/DHHA1 phosphoesterase family of proteins. FEBS Lett 587(16):2669–2674. https://doi.org/10.1016/j.febslet.2013.06.053
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gu, L., He, Q. (2020). A Unified Catalytic Mechanism for Cyclic di-NMP Hydrolysis by DHH–DHHA1 Phosphodiesterases. In: Chou, SH., Guiliani, N., Lee, V., Römling, U. (eds) Microbial Cyclic Di-Nucleotide Signaling. Springer, Cham. https://doi.org/10.1007/978-3-030-33308-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-33308-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-33307-2
Online ISBN: 978-3-030-33308-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)