
Abstract
Hodgkin lymphoma (HL) comprises two distinct disease entities, classical HL (cHL) and nodular lymphocyte-predominant HL (LPHL). They have in common that the tumor cells—Hodgkin and Reed-Sternberg (HRS) cells in cHL and lymphocyte-predominant (LP) cells in LPHL—are typically large, either mononucleated or multinucleated cells that represent at most a few percent of cells in the lymphoma tissue. The two forms of HL differ in numerous aspects of their histological picture, and the morphology of the lymphoma cells, their phenotype, and their genetic features. HRS and LP cells both derive from germinal center B cells, albeit likely from distinct subsets of these mature B cells. HRS cells have lost most of the B cell phenotype and gene expression pattern of their cellular origin. HRS cells show an activation of numerous signaling pathways, and particularly the NF-κB and JAK/STAT pathways are often constitutively activated through genetic lesions. Other genetic lesions contribute to immune evasion of the HRS cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Germinal center
- Hodgkin lymphoma
- Hodgkin and Reed-Sternberg cells
- JAK/STAT
- LP cells
- NF-κB
- Nodular lymphocyte-predominant Hodgkin lymphoma
1 Subclassification and Pathology
The history of Hodgkin lymphoma (HL) dates back to the first half of the nineteenth century (see Chap. 1), and it has also been an established view for quite some time that HL comprises two different disease entities, namely, classical Hodgkin lymphoma (cHL) and nodular lymphocyte-predominant Hodgkin lymphoma (LPHL) [1]. Both entities have in common that the neoplastic cell population, which can be mononucleated or multinucleated, makes up only a small percentage of all cells present in an affected lymph node. However, morphological, clinical, epidemiologic, and molecular evidence strongly support the belief that the pathogenesis of these lymphomas is distinct enough to be considered separate entities. From a diagnostic point of view, morphological details and immunohistochemistry for a selected set of markers almost always allow for a proper classification of a given lymphoma into the group of LPHL or cHL, the latter of which can be further subdivided into nodular sclerosis cHL (NSCHL), mixed cellularity cHL (MCCHL), lymphocyte-depleted cHL (LDCHL), and lymphocyte-rich cHL (LRCHL) [1].
The following sections summarize the key morphological aspects and important immunohistochemical features of HL, as well as key biological and genetic features of the HL tumor cells. For microenvironmental, clinical, and epidemiologic parameters, please refer to the respective other chapters of this book.
1.1 Nodular Lymphocyte-Predominant Hodgkin Lymphoma
Although the morphology of the tumor cell population of LPHL can occasionally mimic Hodgkin and Reed-Sternberg (HRS) cells of cHL, in most instances the tumor cells in LPHL, which are termed lymphocyte-predominant (LP) cells according to the current WHO classification (previously called L&H cells, for lymphocytic and/or histiocytic Reed-Sternberg (RS) cell variants), carry one large nucleus that is often multilobated (“popcorn cell”) (Fig. 3.1a). In contrast to classic HRS cells, the number of nucleoli is increased, but they are usually less prominent and less eosinophilic. LP cells are found in a nodular or follicular background that is dominated by small B lymphocytes that usually express IgD, but a more diffuse growth pattern can also be encountered, especially during progression. The follicular infiltration pattern is highlighted by the presence of CD21-positive follicular dendritic cells that tend to form a well-developed meshwork in the nodules. Immunohistochemically, LP cells demonstrate a complete B cell phenotype with expression of CD20, CD75, and, frequently, CD79a (Fig. 3.1b; Table 3.1). Moreover, the essential B cell transcription factors BOB.1 and OCT-2 are usually positive, and the expression of BCL6 and activation-induced cytidine deaminase (AID) is well in line with a germinal center (GC) derivation of the tumor cells, although CD10 is generally negative [1,2,3]. The negativity of the tumor cells for CD30, CD15, and Epstein-Barr virus (EBV) helps to distinguish LP cells from HRS cells in cHL, although occasionally a weak positivity for CD30 can be present in LP cells (Table 3.1). Whereas in initial lesions small B cells dominate the background, histiocytes and T cells may become more prominent during the evolution of LPHL, to an extent that LPHL may be hardly distinguishable from T cell/histiocyte-rich large B cell lymphoma (THRLBCL). “Variant histology” (e.g., depletion of small B cells in the background or unusual localization of the LP cells) appears to be associated with an inferior prognosis [4]. A prominent feature of LPHL is the often impressive rosetting of LP cells by T cells that belong to the subset of follicular T helper cells and therefore express CD57 and PD-1 [5,6,7].

Nodular lymphocyte-predominant Hodgkin lymphoma (LPHL). (a) HE-stained lymph node infiltrate showing multiple characteristic, multilobated tumor cells—termed lymphocyte-predominant (LP) cells—in a background of small lymphocytes and histiocytes (×400). (b) Strong CD20 expression in LP cells, but also in reactive, small B cells in the background (×400). Note that some of the tumor cells show rosetting by a CD20-negative lymphocyte population. These cells are T cells that often express the follicular T helper cell marker PD-1
1.2 Classical Hodgkin Lymphoma: The HRS Cells
The characteristic tumor cell of cHL, the RS cell, is large and contains at least two nuclear lobes or nuclei, usually with a prominent nuclear membrane (Fig. 3.2a). In contrast to LP cells in LPHL, the nucleoli of RS cells are often eosinophilic. The mononuclear variant of RS cells is termed the Hodgkin cell. However, the morphological spectrum of the tumor cell population in cHL can be broad and includes variants such as lacunar cells and mummified cells. In general, the tumor cells in cHL are called Hodgkin and Reed-Sternberg cells. Immunohistochemically, the HRS cells stain positive for CD30 (Fig. 3.2c), and CD15 is coexpressed in the majority of cases, occasionally with prominent staining of the Golgi area of the tumor cell. However, CD15 is negative in a significant proportion of cHL (20–25%) and therefore not required to establish the diagnosis of cHL [1]. CD45 is usually negative, as are the B cell transcription factors BOB.1 and OCT-2. In the vast majority of cases, the derivation of the tumor cells from the B cell lineage is indicated by a nuclear positivity for the B cell-specific activator protein PAX5/BSAP, but the staining is usually weaker compared to the staining intensity in the small reactive B cell population in the background of the infiltrate [11]. CD20 expression can be observed in HRS cells in 30–40% of cases, but the expression is frequently restricted to a subset of the tumor cell population, and even within one HRS cell, it is of varying intensity in different parts of the cell membrane. In comparison to CD20 expression, CD79a expression is observed less frequently [12, 13]. An EBV association, either demonstrated by immunohistochemical staining for LMP1 (latent membrane protein 1; Fig. 3.2d) or by EBER in situ hybridization, is found in a significant proportion of cHL, but the frequency varies considerably between different histological subtypes and across geographical areas [1]. Whether cHL cases exist with a bona fide derivation from the T cell lineage is currently a matter of debate. Single cases have been reported, in which a T cell receptor rearrangement could be proven in the HRS cells [14, 15], but others argue that such cases might represent only mimics of cHL which are not to be included in a disease entity that—based on fundamental principles of current lymphoma classification schemes—is of B cell derivation [16]. HRS cells reside in a cellular background that varies among the different histological subtypes of cHL which will be discussed in the following sections.

Classical Hodgkin lymphoma (cHL). (a) Characteristic Hodgkin and Reed-Sternberg (HRS) cells in a mixed background of small lymphocytes, histiocytes, and eosinophils in a mixed cellularity cHL (MCCHL) (HE, ×400). (b) Nodular sclerosis subtype of cHL that demonstrates thick collagen bands surrounding the nodular infiltrates (PAS, ×20). (c) CD30 expression in HRS cells (×400). (d) Immunohistochemical staining for latent membrane protein 1 (LMP1) shows Epstein-Barr virus (EBV) association of HRS cells (×400)
1.2.1 Nodular Sclerosis Classical Hodgkin Lymphoma
In NSCHL, affected lymph nodes frequently show a markedly thickened capsule and a nodular infiltrate whereby individual nodules are surrounded by broad collagen bands (Fig. 3.2b). HRS cells are present in a background of small lymphocytes and other nonneoplastic cells such as histiocytes and eosinophils. The number of HRS cells can vary significantly between NSCHL cases and also within a single infiltrated lymph node. Occasionally, HRS cells can form sheets that can be associated with necrosis and an intense fibrohistiocytic reaction. Morphologically, HRS cells in NSCHL often show a retraction artifact of the cytoplasmic membrane that appears to be a consequence of formalin fixation, which has led to the term “lacunar cell variant” of HRS cells. The immunohistochemical phenotype of HRS cells in NSCHL as described above is the classic phenotype; however, association with EBV is less common as compared to other cHL subtypes, especially MCCHL.
1.2.2 Mixed Cellularity Classical Hodgkin Lymphoma
HRS cells in MCCHL usually have a classic morphological appearance and are scattered in a background that can contain small lymphocytes, eosinophils, neutrophils, plasma cells, and histiocytes. The infiltration pattern can be diffuse or vaguely nodular; sometimes, the lymph node architecture and especially some B cell areas are partially preserved leading to an interfollicular infiltration pattern. The characteristic features of other histologic cHL subtypes (e.g., the formation of nodular collagen bands) are absent, and, thus, MCCHL is sometimes considered as the “wastebasket” of cHL. The EBV association of HRS cells is the highest among all cHL subtypes and can reach 75% [1].
1.2.3 Lymphocyte-Depleted Classical Hodgkin Lymphoma
LDCHL is the rarest histological subtype of cHL (<1% of cases) and probably the most problematic one to define. It is characterized by an increased number of HRS cells present in the infiltrate and/or depletion of small lymphocytes in the nonneoplastic background population. In some cases, HRS cells are of anaplastic appearance, and in other cases, the background is composed of extensive diffuse fibrosis. However, if the pattern of fibrosis is nodular and therefore characteristic of NSCHL, a given case should be classified as NSCHL, regardless of whether there is a high number of HRS cells. Since the definition of LDCHL has changed over the past decades, some of the established clinical and biological features appear outdated in the context of the current definition. Moreover, with the increase in knowledge and the development of additional immunohistochemical markers, some of the cHL cases that were previously assigned to the LDCHL category would nowadays be included into borderline categories or even different entities [1].
1.2.4 Lymphocyte-Rich Classical Hodgkin Lymphoma
In LRCHL, the HRS cells are present in a lymphocyte-rich background that can be nodular or, rarely, diffuse. Often, B cell follicles are partially preserved with recognizable GC, and HRS cells can be found in expanded mantle and marginal zones, thus providing a B cell-rich background. HRS cells in LRCHL may resemble LP cells in LPHL morphologically to such an extent that they are indistinguishable from each other without additional immunohistochemical characterization. It is of significance that eosinophils and neutrophils should be absent from the nodular infiltrates and may only be found in low numbers in interfollicular zones and close to vascular structures. The immunophenotype of the HRS cells is classic, and an EBV association is occasionally observed, though at a lower frequency compared to MCCHL [1].
2 Differential Diagnosis
In most instances, the diagnosis of LPHL and cHL is unambiguous on the basis of morphological, clinical, and, especially, immunohistochemical features (Table 3.1). However, a gray area between cHL and diffuse large B cell lymphoma (DLBCL), specifically with primary mediastinal large B cell lymphoma (PMBL), has long been known, and the most recent WHO classification introduced the category of “B cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin lymphoma” [1]. It is important to note that lymphomas falling into this category are not considered a separate disease entity; rather, it was felt that lymphomas in which there is a discordance between morphological aspects of the infiltrate and the expected immunophenotype should be labeled as “intermediate” to allow a more precise definition of biological and clinical features of these lymphomas in the future. Frequently, these borderline lymphomas present with large mediastinal masses. Morphologically, they consist of large, pleomorphic B cells that grow in a sheetlike pattern in a background of a fibrotic stroma. A subset of the tumor cells may resemble HRS cells, specifically the lacunar variant, and parts of the infiltrate may correspond to the growth pattern of cHL, particularly the nodular sclerosis subtype. Immunophenotypically, there is often a preserved expression program of cHL including expression of CD30 and CD15, while markers of the B cell lineage that are often downregulated in cHL, such as CD20 and CD79a, are equally expressed in the tumor cells [1]. It is important to note that these gray zone lymphomas appear to be more common in male patients, in contrast to NSCHL and PMBL that are more frequent in females [17]. Clinically, these tumors may behave more aggressively than NSCHL and PMBL; it has to be determined in the future whether treatment regimens for aggressive B cell lymphomas or for cHL are more beneficial.
The differential diagnosis between cHL and ALK-negative anaplastic large cell lymphoma (ALCL) of T cell lineage can usually be resolved using an appropriate panel of immunohistochemical markers including T cell, cytotoxic, and other markers. Problems arise when morphological features favor cHL, but tumor cells lack PAX5/BSAP expression while cytotoxic markers are expressed. As discussed above, it is a matter of current debate whether such cases should be grouped into the cHL category or diagnosed as ALCL. Remarkably, a global gene expression study revealed surprisingly few consistent differences in the gene expression of HRS cells and ALK-negative ALCL cells [18].
Finally, EBV-associated lymphoproliferations, e.g., in the context of a coexisting T cell non-HL as well as EBV-associated DLBCL of the elderly, a subgroup of DLBCL introduced in the new WHO classification [1], can harbor HRS or HRS-like cells and therefore mimic cHL [19]. Besides other morphological and immunohistochemical features and information on the clinical setting, the pattern of EBV infection, determined by LMP1 staining or EBER in situ hybridization, might help to distinguish between these tumors.
3 Histogenesis of HRS and LP Cells
3.1 Cellular Origin of HRS and LP Cells
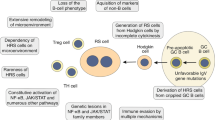
The unusual immunophenotype of HRS cells, which does not resemble any normal hematopoietic cell, has hampered the identification of the cellular origin of these cells considerably. Moreover, only few cell lines were available for detailed genetic studies, and the rarity of the HRS cells in the tissue posed a problem for their molecular analysis. Finally, by microdissection of HRS cells from tissue sections and single-cell polymerase chain reaction analysis of these cells, it was clarified that HRS cells derive from B cells in nearly all cases [20, 21]. This is because rearranged immunoglobulin (Ig) heavy (IgH) and light (IgL) chain gene rearrangements were detected in these cells. The detection of identical IgV gene rearrangements in the HRS cells of a given HL case also established the monoclonal nature of these cells, a hallmark of malignant cancer cells. With a few exceptions, somatic mutations were detected in the rearranged V genes of HRS cells [20,21,22,23]. As the process of somatic hypermutation, which generates such mutations, is specifically active in antigen-activated mature B cells proliferating in the GC microenvironment in the course of T-dependent immune responses [24], the presence of mutated IgV genes in the HRS cells established their derivation from GC-experienced B cells. A surprising finding was that about 25% of cases of cHL showed destructive IgV gene mutations, such as nonsense mutations or deletions causing frameshifts that rendered originally functional V region genes nonfunctional [20]. When such mutations happen in normal GC B cells, these cells quickly undergo apoptosis. On this basis, it was proposed that HRS cells in these cases derive from pre-apoptotic GC B cells that were rescued from apoptosis because they harbored or acquired some transforming events [20, 25]. It is important to note that crippling mutations, such as those generating premature stop codons, represent only a small fraction of disadvantageous IgV gene mutations that cause apoptotic death of GC B cells, and it is therefore likely that also most or even all other cases of cHL are derived from pre-apoptotic GC B cells. Even a few HL with unmutated IgV genes may derive from these precursors, because GC founder cells proliferating in GC become prone to apoptosis before the onset of somatic hypermutation activity [26]. The GC B cell origin of HRS cells was further supported by the molecular analysis of composite lymphomas, composed of a cHL and a B cell non-HL. Such cases are often clonally related and show an intriguing pattern of shared as well as distinct somatic V gene mutations [27,28,29,30]. This pattern supports the assumption that both lymphomas were derived from distinct members of a proliferating GC B cell clone.
A comparison of the transcriptomes of HRS cells and normal GC and extrafollicular CD30+ B cells revealed that HRS cells are in their global gene expression pattern more similar to the normal CD30+ B cells than to bulk GC B cells [31]. However, a direct derivation of HRS cells from CD30+ GC B cells seems unlikely, as CD30+ GC B cells are positively selected GC B cells with functional BCR that are preparing to return to the dark zone of the GC for a further round of proliferation and IgV gene mutation. Perhaps, in the course of their malignant transformation, the HRS cell precursors that managed to escape from apoptosis acquired the gene expression program of the positively selected and proliferation prepared CD30+ GC B cells.
A few cases of cHL appear to originate from T cells, because T cell receptor gene rearrangements were detected in some cases diagnosed as HL and expressing some typical T cell molecules [14, 15]. However, it is debated whether these are true HL (see above). Remarkably, among HL cases with expression of one or more T cell markers, the majority nevertheless derives from B cells [14, 15].
The expression of multiple B cell markers by LP cells of LPHL already indicated a B cell derivation of these cells. Moreover, LP cells express several markers typically expressed by GC B cells, such as BCL6, AID, centerin, and hGAL, and the cells grow in a follicular pattern in close association with typical constituents of normal GC, i.e., follicular dendritic cells and GC-type T helper cells [2, 3, 5, 6, 32, 33]. This pointed to a close relationship between LP cells and GC B cells. This is indeed supported by the detection of clonally related and somatically mutated IgV genes in these cells [21, 34,35,36]. As opposed to cHL, the V genes are selected for functionality, and a fraction of cases shows ongoing somatic hypermutation during clonal expansion, a hallmark of GC B cells [21, 34, 35]. Thus, these findings altogether indicate a GC B cell origin of LP cells. A large-scale gene expression profiling of isolated LP cells in comparison to the main subsets of mature B cells has led to a further specification of the derivation of LP cells by showing that the gene expression pattern of LP cells resembles that of GC B cells that have already acquired some features of post-GC memory B cells [37].
3.2 Relationship of Hodgkin Cells and Reed-Sternberg Cells and Putative HRS Cell Precursors
The relationship of the mononucleated Hodgkin cells to the multinuclear RS cells and the potential existence of HRS precursor cells has been a matter of debate. Based on the “mixed” phenotype of HRS cells and many numerical chromosomal aberrations in these cells, it has been speculated that HRS cells as such or, specifically, the RS cells may derive from cell fusions of different cells (e.g., a B cell and a non-B cell). However, a detailed study of antigen receptor loci revealed that HRS cells do not carry more than two different alleles of these loci, which strongly supports the assumption that these cells do not derive from cell fusions [38]. Several studies of HL cell lines showed that the mononuclear Hodgkin cells give rise to the RS cells and that the latter have little proliferative activity [39,40,41]. Long-term time lapse-microscopy analyses revealed that mononucleated Hodgkin cells undergo incomplete cytokinesis and refusion to give rise to the multinucleated RS cells [42, 43].
Two studies reported the existence of a small subpopulation of side population cells among the mononuclear Hodgkin cells. Side population cells extrude the Hoechst dye, because they express multidrug transporters, such as MDR1 and/or ABCG2. In several types of cancers, there is an overlap between side population cells and cancer stem cells. Side population cells of cHL cell lines were CD30+CD20- and showed increased resistance against chemotherapeutic drugs [44, 45]. However, it has not yet been determined whether they have a higher capacity to sustain the HRS cell clone in long term than other mononuclear Hodgkin cells, and the fact that side population cells were not identified in all cHL cell lines analyzed argues against an essential role of these cells for the survival of the HRS cell clone.
Another debated issue relates to the question whether the CD30+ typical HRS cells represent the entire tumor clone in HL or whether members of the HRS cell clones exist among small CD30– cells. An initial study for numerical chromosomal abnormalities indeed suggested that such CD30– clone members might exist [46]. However, trisomies of chromosomes as studied in that work are not a stringent clonal marker. Moreover, a molecular analysis of EBV-positive HL cases for members of the malignant clones among small, CD30– EBV+ B cells in the HL lymph nodes suggested that the small EBV+ B cells rarely, if at all, belong to the HRS cell clones [47]. Two HL cell lines were reported to contain small subpopulations of CD20+CD30–Ig+ B cells coexpressing the stem cell marker aldehyde dehydrogenase (ALDH) [48]. These cells had clonogenic potential and gave rise to the typical HRS cells of these lines. It is important to note that ALDHhigh cells were also detectable in the peripheral blood of most HL patients, and it was reported that these cells were often clonally related to the HRS cells [48]. However, the clonal relationship between the HRS cells and ALDHhigh peripheral blood B cells was not clearly shown [49], so it remains to be clarified whether ALDHhigh B cells indeed represent precursors of the HRS cell clones. A previous study using a highly sensitive PCR for HRS cell-specific Ig gene rearrangements failed to detect members of the HRS cell clone in the peripheral blood or bone marrow of two HL patients [50].
4 Genetic Lesions
HRS cells have a much higher number of chromosomal aberrations, including multiple numerical as well as structural abnormalities, than most other lymphomas [51]. However, it is still unclear whether this is mostly a side effect of some type of genetic instability and whether the expression of specific oncogenes or tumor suppressor genes is recurrently affected by these lesions. When the B cell origin of HRS cells became clear, HRS cells were studied for the presence of chromosomal translocations involving the Ig loci, as such translocations are a hallmark of many B cell lymphomas. Fluorescence in situ hybridization (FISH) studies indeed provided evidence for such translocations in about 20% of cases, but most of the translocation partners involved remain to be identified [52, 53]. In a few cases, the translocation partners were BCL2, BCL3, REL, BCL6, or MYC [52,53,54,55]. Recurrent translocations affecting the major histocompatibility complex (MHC) class II transactivator (MHC2TA) were detected in about 15% of cHL cases [56]. These translocations appear to cause downregulation of MHC class II expression by HRS cells. In LPHL, translocations of the BCL6 gene have been found in about 30% of cases [57, 58]. These translocations can involve the Ig loci, but also multiple other partners [59].
Due to the difficulty to analyze the few HRS and LP cells for mutations in oncogenes and tumor suppressor genes, only relatively few of such genes have been analyzed so far in these cells. There was a major interest to understand the apoptosis resistance of HRS cells, but it turned out that mutations in the CD95 gene, an important death receptor, as well as in members of the CD95 signaling pathway (FADD, caspase 8, caspase 10) were rare or not found at all [60,61,62]. Likewise, no mutations were found in the BCL2 family member BAD, and also ATM lesions are very rare [63,64,65]. The TP53 tumor suppressor gene was mutated in less than 10% of cases where the exons of TP53 usually carrying mutations were studied in isolated HRS cells [66, 67]. However, studies of HL cell lines indicate that HRS cells may additionally carry untypical TP53 mutations and that the frequency of TP53 mutations may therefore be higher than previously thought [68]. MDM2, a negative regulator of TP53, frequently shows gains in HRS cells, which might contribute to impaired functions of TP53 in these cells [69].
Further candidate gene mutation studies revealed frequent mutations in the exportin 1 gene (XPO1) [70], which encodes a nuclear expert receptor for numerous RNAs and proteins, and inactivating mutations in and deletions of CD58 [71, 72]. CD58 is important for targeting of cells by cytotoxic T cells and NK cells, so that CD58 inactivation may contribute to immune escape of HRS cells from an attack by these cells.
HRS cells show constitutive activity of the NF-κB transcription factor (see below), which is essential for the survival of these cells. The mechanisms of this activation were originally not understood. Consequently, members and regulators of this signaling pathway were studied for genetic lesions (Table 3.1). Inactivating mutations in the main NF-κB inhibitor NFKBIA (IκBα) were found in about 10–20% of HL cases and also in several HL cell lines (Fig. 3.3) [73,74,75,76]. Recurrent mutations were also detected in another NF-κB inhibitor, NFKBIE (IκBε) [77, 78]. Inactivating mutations or deletions in two further negative regulators of NF-κB signaling, CYLD and TRAF3, have also been detected in HL cell lines and a few primary cases, but overall these events are rare [79, 80]. Moreover, HRS cells frequently harbor genomic gains or amplifications of the REL gene [81,82,83], encoding an NF-κB family member, and a correlation between such gains and strong REL protein expression was found [84]. The MAP3K14 gene, which encodes the NIK kinase, a major activating component of the alternative NF-κB pathway, shows gains or amplifications in about 15% of cHL [79, 85]. Also the IκB family member BCL3, which acts as a positive regulator of NF-κB activity, is affected by chromosomal gains or translocations in a small fraction of cHL [86, 87]. Somatic and clonal inactivating mutations were found in the TNFAIP3 gene in about 40% of cHL [88, 89]. TNFAIP3 encodes for the A20 protein, which is a dual ubiquitinase and deubiquitinase that functions as a negative regulator of NF-κB. It inhibits signaling from the receptor-interacting protein (RIP) and TNF receptor-associated factors (TRAFs) to the IKK kinases, which are essential mediators of NF-κB signaling. TNFAIP3 mutations were mainly found in EBV-negative cases. Nearly 70% of EBV– cases carried TNFAIP3 mutations, indicating that EBV infection and A20 inactivation are alternative pathogenetic mechanisms in HL [88, 89]. As LMP1 of EBV, which is expressed in EBV-positive HRS cells, mimics an active CD40 receptor and signals through NF-κB [90, 91], LMP1 may replace the role of A20 inactivation in EBV+ HL.

NF-κB and JAK/STAT activity in HRS cells. In the classical NF-κB signaling pathway, stimulation of numerous receptors leads via TNF receptor-associated factors (TRAFs), which are often associated with the receptor-interacting protein (RIP), to activation of the IKK complex, which is composed of IKKα, IKKβ, and NEMO. The IKK complex subsequently phosphorylates the NF-κB inhibitors IκBα and IκBε. This marks them for ubiquitination and subsequent proteasomal degradation. Thereby the NF-κB transcription factors (p50/p65 or p50/REL heterodimers) are no longer retained in the cytoplasm and translocate into the nucleus, where they activate multiple genes. The signal transduction from TRAFs/RIP to the IKK complex can be inhibited by TNFAIP3, which removes activating ubiquitins from RIP and TRAFs and additionally links ubiquitins to these molecules to mark them for proteasomal degradation. In the alternative NF-κB pathway, activation of receptors such as CD40, BCMA, and TACI causes stimulation of the kinase NIK, which then activates an IKKα complex. Activated IKKα processes p100 precursors to p52 molecules, which translocate as active p52/RELB NF-κB heterodimers into the nucleus. HRS cells show constitutive activity of the classical and alternative NF-κB signaling pathway. This activity is probably mediated by diverse mechanisms, including receptor signaling through CD40, RANK, BCMA, and TACI; genomic REL and MAP3K14 (NIK) amplification; destructive mutations in the TNFAIP3, NFKBIA, and NFKBIE genes; and signaling through the EBV-encoded LMP1. The role of CD30 signaling in HRS cells is controversially discussed. HRS cells may also harbor nuclear BCL3/(p50)2 complexes, and in a few cases, the strong BCL3 expression appears to be mediated by genomic gains or chromosomal translocations. The JAK/STAT pathway is the main signaling pathway for cytokines. Upon binding of cytokines to their receptors, members of the JAK kinase family become activated by phosphorylation. The activated JAKs then phosphorylate and thereby activate STAT transcription factors. These phosphorylated factors homo- or heterodimerize and translocate into the nucleus where they activate target genes. Main inhibitors of the JAK/STAT pathway are the phosphatase PTPN1 and SOCS (suppressor of cytokine signaling) factors, which function by binding to JAK molecules and inhibiting their enzymatic activity and additionally by inducing proteasomal JAK degradation. In HRS cells, STAT3, 5, and 6 are constitutively active. Besides activation of cytokine receptors (e.g., IL13 receptor and IL21 receptor) through cytokines, activation of this pathway is mediated by genomic gains or rare translocations of the JAK2 gene, activating mutations in the STAT6 gene, and frequent inactivating mutations in the SOCS1 and PTPN1 gene. The frequency of genetic lesions and viral infections affecting NF-κB or STAT activity in cHL cases is indicated
As it was recently revealed that also the LP cells of LPHL show strong constitutive NF-κB activity [37], also these cells were studied for mutations in NFKBIA and TNFAIP3, but clonal destructive mutations were not found (Table 3.1) [92].
Genetic lesions were also found in members of the JAK/STAT pathway, which is constitutively activated in HRS and LP cells. In about 40% of cases analyzed, both HRS and LP cells showed somatic mutations in the SOCS1 gene, which encodes a main inhibitor of STAT signaling (Fig. 3.3) [93, 94]. In HRS cells, recurrent mutations were additionally found in the gene of another negative regulator of JAK/STAT signaling, namely, the PTPN1 gene, which encodes a phosphatase [95]. Furthermore, a fraction of cHL cases show genomic gains or amplifications of the JAK2 locus, which encodes one of the kinases activating the STAT factors (Table 3.1) [82, 96]. Importantly, the genomic gains at 9p24 do not only affect the JAK2 locus, but additionally the PD-L1, PD-L2, and JMJD2C genes [97, 98]. PD-L1 and PD-L2 are inhibitory receptors for PD1-positive T cells and may hence inhibit a cytotoxic T cell attack on HRS cells. JMJD2C encodes a histone demethylase and plays a role in the epigenetic remodeling of HRS cells. Finally, the JAK2 gene is in rare instances also deregulated by chromosomal translocations [99]. Activating point mutations in the STAT6 gene and genomic gains involving this gene were also detected in HRS cells [10, 100]. Thus, multiple types of genetic lesions cause a constitutive JAK/STAT signaling, suggesting an essential role of its deregulated activity for cHL pathogenesis.
With the availability of high-throughput sequencing methods, tumor cells can now be studied for genetic lesions at a genome-wide level. An exome sequencing analysis of six cHL lines and the only LPHL cell line (DEV) revealed over 400 genes mutated in at least two of the lines [8]. This is a valuable database that should be considered when performing functional studies with these cell lines. A first whole exome sequencing study of primary HRS cells used flow-cytometry isolated lymphoma cell from ten cases of cHL [9]. Between 100 and 500 somatic mutations were found per case. A main finding of this analysis was recurrent inactivating mutations in the B2M gene. B2M is essential for MHC class I expression, so that the loss of its expression presumably leads to immune evasion from CD8+ cytotoxic T cells. Other novel recurrent mutations identified in that work affect several histone genes, the inositol-trisphosphate 3-kinase B (ITPKB), the B cell transcription factor EBF1, and the G protein subunit GNA13 [9]. Tiacci and colleagues performed a whole exome sequencing analysis of pools of HRS cells microdissected from 34 cases of cHL [10]. A median of 47 non-silent somatic mutations in the exomes was found. This study confirmed recurrent mutations in ITPKB and GNA13, and newly revealed recurrent point mutations in STAT6, further adding to the complexity of JAK/STAT deregulation in cHL. Although only four EBV+ cases were included in the study by Tiacci et al., it seems that such cases carry considerably fewer somatic mutations than the EBV-negative cases. A mutation study of LP cells of LPHL was based on a whole genome analysis of DLBCL clonally related to LPHL in the same patient, followed by targeted sequencing analysis of microdissected LP cells. In this work, three genes were found to be each mutated in about half of the cases of LPHL (also in cases without co-occurring DLBCL), namely, the genes encoding the kinase SGK1, the AP-1 family member JUNB, and the phosphatase DUSP2 [101].
5 Deregulated Transcription Factor Networks and Signaling Pathways
5.1 The Lost B Cell Phenotype
Early immunohistochemical studies already revealed that HRS cells usually do not express typical B cell markers, such as CD20, CD79b, or the BCR [13, 102,103,104]. This lack of expression of B cell markers was indeed one of the reasons why the B cell origin of HRS cells was not revealed until genetic studies for Ig gene rearrangements unequivocally demonstrated a B cell identity of these cells (see above). Gene expression profiling studies of HRS cells in comparison to normal B cells then showed that there is a global loss of the B cell typical gene expression in HRS cells [105]. This downregulation involved all types of genes with important functions in these cells, for example, cell surface receptors (CD37, CD53), components of signaling pathways (SYK, BLK, SLP-65), and transcription factors (PU.B, A-MYB, SPI-B). As plasma cells also show a downregulation of many B cell-typical genes, it had been speculated that HRS cells lost their B cell gene expression and acquired a partial plasma cell differentiation program [2, 106]. However, a gene expression profiling study of microdissected HRS cells revealed that HRS cells have not acquired a plasma cell phenotype [107].
Remarkably, HRS cells have retained expression of molecules that are involved in antigen-presenting functions and the interaction with CD4+ T helper cells. HRS cells usually express CD40, CD80, and CD86 and often MHC class II [105, 108]. This indicates that an interaction with T helper cells is important for HRS cell survival. In line with this view, HRS cells are typically surrounded by CD40L expressing CD4+ T cells [109].
We are now beginning to understand which factors contribute to the lost B cell phenotype of HRS cells. First, several transcription factors that positively regulate the expression of multiple genes in B cells are downregulated, including OCT-2, PU.1, EBF1, ETS1, and BOB.1 [102, 103, 110,111,112]. The downregulation of ETS1 may often be due to heterozygous deletions of the gene, which have been observed in over 60% of cHL analyzed [112]. Second, although E2A, a master regulator of the B cell transcription program, is still expressed, HRS cells also show deregulated expression of ID2 and ABF1 [113,114,115], which bind to E2A and inhibit its function [114]. The physiological role of ABF1 is poorly understood, but ID2 is normally expressed in dendritic cells and natural killer cells, and supports the generation of these cells concomitant with suppression of B cell development [116, 117]. Third, HRS cells express activated NOTCH1, which normally induces T cell differentiation in lymphocyte precursors and suppresses a B lineage differentiation of such cells [118, 119]. Activation of NOTCH1 is probably caused by interaction with its ligand Jagged-1, which is expressed by other cells in the HL microenvironment [119], and by high-level expression of the NOTCH coactivator mastermind-like 2 (MAML2) [120]. Moreover, HRS cells have downregulated the NOTCH1 inhibitor Deltex1 [118]. Fourth, STAT5A and STAT5B are activated in HRS cells and have been reported to induce an HRS cell-like phenotype in normal B cells [121]. Constitutive active STAT5 induced expression of CD30 and of the T cell transcription factor GATA3 in the B cells and led to downregulation of BCR expression. Aberrant GATA3 expression in HRS cells is furthermore mediated by NOTCH1 and NF-κB activity in HRS cells [122]. Fifth, the downregulation of multiple B cell genes in HRS cells is further caused by epigenetic mechanisms, as DNA methylation has been detected for numerous such genes [123,124,125]. Sixth, HRS cells express several transcription factors that have important roles in hematopoietic stem cells and early lymphoid precursors, including GATA2, BMI1, RING1, and RYBP [126,127,128,129]. The expression of these factors may contribute to a “dedifferentiated” phenotype of HRS cells.
Surprisingly, PAX5, the main B lineage commitment and maintenance factor, is still expressed in HRS cells, albeit at reduced levels [11]. As many of its direct target genes are not expressed, it is likely that PAX5 activity is inhibited. NOTCH1 is a candidate for this inhibition [118]. It may also be that PAX5 target genes are not expressed because other transcription factors needed for the efficient expression of these genes are missing.
Expression of the myeloid specific colony-stimulating factor 1 receptor (CSF1R) by HRS cells is a further important example of aberrant expression of a non-B cell gene in HRS cells [130]. CSF1R expression promotes HRS cell survival. The mechanism of its deregulated expression is remarkable, because this is mediated by derepression of an endogenous long terminal repeat upstream of the CSF1R gene that replaces the function of the normal CSF1R promoter [130].
The downregulation of many B cell transcription factors that also suppress the expression of non-B lineage genes, combined with the upregulated expression of genes promoting expression of genes of other hematopoietic cell types (e.g., NOTCH1, ID2), not only explains the lost B cell phenotype of HRS cells but also the heterogeneous expression of genes specifically expressed by dendritic cells, T cells, or other cell types. It is an intriguing question whether the lost B cell phenotype of HRS cells is related to their origin from crippled GC B cells. Perhaps, due to the stringent selection of B cells for expression of a functional BCR (a high-affinity one in the GC), there is a selection in HRS cell pathogenesis downregulating the B cell gene expression program to escape the selection forces that induce apoptosis in GC B cells with unfavorable IgV gene mutations. The observation that enforced re-expression of the B cell transcription factors PU.1, FOXO1, or E2A or the pharmacological restoration of the B cell phenotype in HL cell lines induces apoptosis is in line with this view [131,132,133,134]. However, the lost B cell phenotype could also be a side effect of so far unknown transforming events.
5.2 Constitutive Activation of Multiple Signaling Pathways
It is obvious that tumor cells need to activate and deregulate signaling pathways and transcription factors that promote their survival and proliferation. Nevertheless, it is striking how many of such pathways are constitutively activated in HRS cells, and cHL appears to be rather unique among lymphoid malignancies in the extent to which multiple signaling pathways contribute to the survival and expansion of HRS cells. It has already been mentioned above that HRS cells show constitutive NF-κB activity. This activity is essential for HRS cell survival [135] and is most likely not only mediated by genetic lesions (see above) but also by signaling through receptors. NF-κB factors of both the canonical pathway (p50/p65) and the noncanonical NF-κB pathway (p52/RelB) are activated (Fig. 3.3). HRS cells express the TNF receptor family members CD30, CD40, RANK, TACI, and BCMA, which activate NF-κB, and cells expressing the respective ligands are found in the HL microenvironment [109, 136,137,138,139,140]. There are, however, conflicting data about the role of CD30 in NF-κB activation [141, 142]. In EBV-positive cases of cHL, the virally encoded LMP1 mimics an active CD40 receptor and hence also contributes to NF-κB activation [143].
Another central signaling pathway, which is like NF-κB activated both by genetic lesions and by ligand-mediated receptor triggering, is the JAK/STAT pathway (Fig. 3.3). This is the main signaling pathway for cytokines. Activation of cytokine receptors causes activation of JAK kinases which in turn phosphorylate and thereby activate STAT transcription factors. The phosphorylated STAT factors dimerize and then translocate into the nucleus where they activate transcription of target genes. HRS cells show activation of STAT3, STAT5, and STAT6 [121, 144,145,146]. The activation of STAT6 is at least partly mediated by signaling through IL13. As HRS cells express IL13 and its receptor, STAT6 activation can be mediated through an autocrine stimulation loop [147, 148]. Signaling through the IL21 receptor contributes to STAT3 and STAT5 activation in HRS cells, which is also enhanced by the NF-κB activity in the cells [121, 149, 150]. As mentioned above, STAT5 activity may contribute to the lost B cell phenotype of HRS cells. Inhibition of STAT activity in HL cell lines resulted in reduced proliferation of the cells, further supporting an important pathogenetic role of this signaling pathway [144, 145, 147].
Receptor tyrosine kinases (RTKs) are important regulators of cell growth, survival, and proliferation. In multiple cancers, specific RTKs are activated, often by somatic mutations [151]. In contrast, HRS cells show multiple activated RTKs, and their activation does not appear to be due to activating mutations but at least partly to ligand-mediated stimulation [152]. RTKs that are often expressed in varying combinations in HRS cells include PDGFRA, DDR2, EPHB1, RON, TRKA, TRKB, CSF1R, and MET [130, 152, 153]. The expression of most of these is aberrant, as they are not expressed by normal GC B cells [130, 152]. They are also usually not expressed by other B cell non-HL, showing that this is a specific feature of HL among B cell lymphomas [152, 154]. Expression of multiple RTKs is most pronounced in EBV-negative cases of cHL, suggesting that EBV activates pathways in HRS cells replacing the function of RTKs [155]. For PDGFRA, TRKA, and CSF1R, a growth-inhibitory effect has been shown upon their inhibition in HL cell lines, giving a first indication that the activity of RTKs is important for HRS cell proliferation [130, 152, 156].
Signaling through various receptors is mediated by the mitogen-activated protein kinase (MAPK)/ERK pathway. In HRS cells, the serine/threonine kinases ERK1, ERK2, and ERK5 are activated [157, 158]. Inhibition of their activity has antiproliferative effects on HL cell lines [158]. Signaling through CD30, CD40, and RANK may contribute to the stimulation of this pathway [158].
The transcription factor AP-1 acts as homo- or heterodimers of JUN, FOS, and ATF components. In HRS cells, JUN and JUNB are overexpressed and constitutively active [159]. The overexpression of JUNB is mediated by NF-κB [159]. AP-1 induces many target genes and promotes proliferation of HRS cells. Target genes of AP-1 include CD30 and galectin-1, the latter of which has immunomodulatory functions [160, 161]. HRS cells also show strong expression of BATF3, another member of the AP-1 transcription factor family [162, 163]. BATF3 expression is induced by STAT3 and STAT6 in HRS cells. It forms heterodimers with JUN and JUNB, and the proto-oncogene MYC was identified as one of the direct BATF3 target genes [162]. Importantly, downregulation of BATF3 in HL cell lines is toxic for these cells, revealing an essential role of this factor in cHL pathophysiology [162].
Finally, also the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which is a main promoter of cell survival, shows activity in HRS cells [164, 165]. AKT is a serine/threonine kinase that is activated in HRS cells, as evident from its phosphorylated state and phosphorylation of known target proteins [164, 165]. Inhibition of AKT in HL cell lines causes cell death, suggesting an important role of active AKT in HRS cell survival [164, 165]. PI3K may be activated in HRS cells by signaling through CD30, CD40, RANK, and RTK. Moreover, downregulation of the AKT inhibitor INPP5D in HRS cells may further contribute to strong AKT activity in these cells [107].
While we have a relatively detailed insight into signaling pathways active in HRS cells, less is known about signaling pathways constitutively active in LP cells of LPHL. However, LP cells also show a high constitutive activity of NF-κB [37]. RTKs are partly also aberrantly expressed by these cells [152], and activation of the JAK/STAT pathway has been observed [93].
In conclusion, HRS cells are characterized by the deregulated and constitutive activation of multiple signaling pathways and transcription factors that contribute to the survival and proliferation of these cells. The multitude of different stimulated pathways appears to be rather unique among human B cell lymphomas. Often, these pathways are activated by common mechanisms, and they may interact in numerous ways.
6 Anti-apoptotic Mechanisms
With a presumed origin from pre-apoptotic GC B cells, it is critical to understand through which mechanisms HRS cells escape from apoptosis. A number of factors contributing to HRS cell survival have already been discussed in the previous section: constitutive activity of NF-κB, STAT, PI3K, NOTCH1, AP-1, RTK, and ERK. Several specific inhibitors of the two main apoptosis pathways deserve specific mentioning. Although HRS cells express the CD95 death receptor of the extrinsic apoptosis pathway as well as its activating ligand, HL cell lines are resistant to CD95-mediated death induction, suggesting a specific inhibition of this pathway [166,167,168]. As mentioned above, this resistance is neither due to mutations in the CD95 receptor itself nor in its interaction partners FADD, caspase 8, or caspase 10. However, HRS cells show strong expression of the CD95 inhibitor CFLAR (previously known as cFLIP, cellular FADD-like interleukin 1b-converting enzyme-inhibitory protein), and this factor impairs CD95 signaling in HRS cells [166, 167]. Inhibition of the intrinsic (mitochondrial) apoptosis pathway is probably mediated through strong expression of the anti-apoptotic factors BCLXL and XIAP (X-linked inhibitor of apoptosis) and downregulation of the pro-apoptotic factor BIK [107, 169, 170]. BCLXL inhibits apoptosis at the level of the mitochondrial apoptosis induction, whereas XIAP inhibits activity of caspases 3 and 9, which are downstream executioners of the mitochondrial apoptosis program. Although HRS cells also express pro-apoptotic SMAC, which can inhibit XIAP, the cells show an impaired release of SMAC from the mitochondria into the cytoplasm [171]. As mentioned above, HRS cells express high levels of the pro-apoptotic TP53 factor, but resistance to TP53-mediated apoptosis appears to be rarely due to inactivating mutations in the TP53 gene. An important factor for the inhibition of TP53 activity is MDM2, which is expressed at high levels in HRS cells [172]. The functional role of MDM2 as a TP53 inhibitor in HRS cells is supported by the fact that HL cell lines expressing wild-type TP53 are rendered apoptosis-sensitive toward pharmacological apoptosis inducers upon inhibition of MDM2 by its antagonist nutlin 3 [173, 174].
References
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H et al (2008) Classification of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC Press, Lyon
Carbone A, Gloghini A, Gaidano G, Franceschi S, Capello D, Drexler HG et al (1998) Expression status of BCL-6 and syndecan-1 identifies distinct histogenetic subtypes of Hodgkin’s disease. Blood 92:2220–2228
Greiner A, Tobollik S, Buettner M, Jungnickel B, Herrmann K, Kremmer E et al (2005) Differential expression of activation-induced cytidine deaminase (AID) in nodular lymphocyte-predominant and classical Hodgkin lymphoma. J Pathol 205:541–547
Hartmann S, Eichenauer DA, Plutschow A, Mottok A, Bob R, Koch K et al (2013) The prognostic impact of variant histology in nodular lymphocyte-predominant Hodgkin lymphoma: a report from the German Hodgkin Study Group (GHSG). Blood 122:4246–4252
Hansmann ML, Fellbaum C, Hui PK, Zwingers T (1988) Correlation of content of B cells and Leu7-positive cells with subtype and stage in lymphocyte predominance type Hodgkin's disease. J Cancer Res Clin Oncol 114:405–410
Kamel OW, Gelb AB, Shibuya RB, Warnke RA (1993) Leu 7 (CD57) reactivity distinguishes nodular lymphocyte predominance Hodgkin's disease from nodular sclerosing Hodgkin's disease, T-cell-rich B-cell lymphoma and follicular lymphoma. Am J Pathol 142:541–546
Nam-Cha SH, Roncador G, Sanchez-Verde L, Montes-Moreno S, Acevedo A, Dominguez-Franjo P et al (2008) PD-1, a follicular T-cell marker useful for recognizing nodular lymphocyte-predominant Hodgkin lymphoma. Am J Surg Pathol 32:1252–1257
Liu Y, Abdul Razak FR, Terpstra M, Chan FC, Saber A, Nijland M et al (2014) The mutational landscape of Hodgkin lymphoma cell lines determined by whole-exome sequencing. Leukemia 28:2248–2251
Reichel J, Chadburn A, Rubinstein PG, Giulino-Roth L, Tam W, Liu Y et al (2015) Flow-sorting and exome sequencing reveals the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 125:1061–1072
Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V et al (2018) Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 131:2454–2465
Foss HD, Reusch R, Demel G, Lenz G, Anagnostopoulos I, Hummel M et al (1999) Frequent expression of the B-cell-specific activator protein in Reed-Sternberg cells of classical Hodgkin’s disease provides further evidence for its B-cell origin. Blood 94:3108–3113
Korkolopoulou P, Cordell J, Jones M, Kaklamanis L, Tsenga A, Gatter KC et al (1994) The expression of the B-cell marker mb-1 (CD79a) in Hodgkin’s disease. Histopathology 24:511–515
Kuzu I, Delsol G, Jones M, Gatter KC, Mason DY (1993) Expression of the Ig-associated heterodimer (mb-1 and B29) in Hodgkin’s disease. Histopathology 22:141–144
Müschen M, Rajewsky K, Bräuninger A, Baur AS, Oudejans JJ, Roers A et al (2000) Rare occurrence of classical Hodgkin’s disease as a T cell lymphoma. J Exp Med 191:387–394
Seitz V, Hummel M, Marafioti T, Anagnostopoulos I, Assaf C, Stein H (2000) Detection of clonal T-cell receptor gamma-chain gene rearrangements in Reed-Sternberg cells of classic Hodgkin disease. Blood 95:3020–3024
Mani H, Jaffe ES (2009) Hodgkin lymphoma: an update on its biology with new insights into classification. Clin Lymphoma Myeloma 9:206–216
Traverse-Glehen A, Pittaluga S, Gaulard P, Sorbara L, Alonso MA, Raffeld M et al (2005) Mediastinal gray zone lymphoma: the missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am J Surg Pathol 29:1411–1421
Eckerle S, Brune V, Döring C, Tiacci E, Bohle V, Sundstrom C et al (2009) Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia 23(11):2129–2138
Asano N, Yamamoto K, Tamaru J, Oyama T, Ishida F, Ohshima K et al (2009) Age-related Epstein-Barr virus (EBV)-associated B-cell lymphoproliferative disorders: comparison with EBV-positive classic Hodgkin lymphoma in elderly patients. Blood 113:2629–2636
Kanzler H, Küppers R, Hansmann ML, Rajewsky K (1996) Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 184:1495–1505
Küppers R, Rajewsky K, Zhao M, Simons G, Laumann R, Fischer R et al (1994) Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 91:10962–10966
Marafioti T, Hummel M, Foss H-D, Laumen H, Korbjuhn P, Anagnostopoulos I et al (2000) Hodgkin and Reed-Sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 95:1443–1450
Müschen M, Küppers R, Spieker T, Bräuninger A, Rajewsky K, Hansmann ML (2001) Molecular single-cell analysis of Hodgkin- and Reed-Sternberg cells harboring unmutated immunoglobulin variable region genes. Lab Invest 81:289–295
Küppers R, Zhao M, Hansmann ML, Rajewsky K (1993) Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO J 12:4955–4967
Küppers R, Rajewsky K (1998) The origin of Hodgkin and Reed/Sternberg cells in Hodgkin’s disease. Annu Rev Immunol 16:471–493
Lebecque S, de Bouteiller O, Arpin C, Banchereau J, Liu YJ (1997) Germinal center founder cells display propensity for apoptosis before onset of somatic mutation. J Exp Med 185:563–571
Bräuninger A, Hansmann ML, Strickler JG, Dummer R, Burg G, Rajewsky K et al (1999) Identification of common germinal-center B-cell precursors in two patients with both Hodgkin’s disease and Non-Hodgkin’s lymphoma. N Engl J Med 340:1239–1247
Küppers R, Sousa AB, Baur AS, Strickler JG, Rajewsky K, Hansmann ML (2001) Common germinal-center B-cell origin of the malignant cells in two composite lymphomas, involving classical Hodgkin's disease and either follicular lymphoma or B-CLL. Mol Med 7:285–292
Marafioti T, Hummel M, Anagnostopoulos I, Foss HD, Huhn D, Stein H (1999) Classical Hodgkin's disease and follicular lymphoma originating from the same germinal center B cell. J Clin Oncol 17:3804–3809
Küppers R, Dührsen U, Hansmann ML (2014) Pathogenesis, diagnosis, and treatment of composite lymphomas. Lancet Oncol 15:e435–e446
Weniger MA, Tiacci E, Schneider S, Arnolds J, Rüschenbaum S, Duppach J et al (2018) Human CD30+ B cells represent a unique subset related to Hodgkin lymphoma cells. J Clin Invest 128:2996–3007
Montes-Moreno S, Roncador G, Maestre L, Martinez N, Sanchez-Verde L, Camacho FI et al (2008) Gcet1 (centerin), a highly restricted marker for a subset of germinal center-derived lymphomas. Blood 111:351–358
Natkunam Y, Lossos IS, Taidi B, Zhao S, Lu X, Ding F et al (2005) Expression of the human germinal center-associated lymphoma (HGAL) protein, a new marker of germinal center B-cell derivation. Blood 105:3979–3986
Braeuninger A, Küppers R, Strickler JG, Wacker HH, Rajewsky K, Hansmann ML (1997) Hodgkin and Reed-Sternberg cells in lymphocyte predominant Hodgkin disease represent clonal populations of germinal center-derived tumor B cells. Proc Natl Acad Sci U S A 94:9337–9342
Marafioti T, Hummel M, Anagnostopoulos I, Foss HD, Falini B, Delsol G et al (1997) Origin of nodular lymphocyte-predominant Hodgkin's disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 337:453–458
Ohno T, Stribley JA, Wu G, Hinrichs SH, Weisenburger DD, Chan WC (1997) Clonality in nodular lymphocyte-predominant Hodgkin’s disease. N Engl J Med 337:459–465
Brune V, Tiacci E, Pfeil I, Döring C, Eckerle S, van Noesel CJM et al (2008) Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J Exp Med 205:2251–2268
Küppers R, Bräuninger A, Müschen M, Distler V, Hansmann ML, Rajewsky K (2001) Evidence that Hodgkin and Reed-Sternberg cells in Hodgkin disease do not represent cell fusions. Blood 97:818–821
Drexler HG, Gignac SM, Hoffbrand AV, Minowada J (1989) Formation of multinucleated cells in a Hodgkin’s-disease-derived cell line. Int J Cancer 43:1083–1090
Newcom SR, Kadin ME, Phillips C (1988) L-428 Reed-Sternberg cells and mononuclear Hodgkin's cells arise from a single cloned mononuclear cell. Int J Cell Cloning 6:417–431
Ikeda J, Mamat S, Tian T, Wang Y, Rahadiani N, Aozasa K et al (2010) Tumorigenic potential of mononucleated small cells of Hodgkin lymphoma cell lines. Am J Pathol 177:3081–3088
Rengstl B, Newrzela S, Heinrich T, Weiser C, Thalheimer FB, Schmid F et al (2013) Incomplete cytokinesis and re-fusion of small mononucleated Hodgkin cells lead to giant multinucleated Reed-Sternberg cells. Proc Natl Acad Sci U S A 110:20729–20734
Xavier de Carvalho A, Maiato H, Maia AF, Ribeiro SA, Pontes P, Bickmore W et al (2015) Reed-Sternberg cells form by abscission failure in the presence of functional Aurora B kinase. PLoS One 10:e0124629
Nakashima M, Ishii Y, Watanabe M, Togano T, Umezawa K, Higashihara M et al (2010) The side population, as a precursor of Hodgkin and Reed-Sternberg cells and a target for nuclear factor-kappaB inhibitors in Hodgkin’s lymphoma. Cancer Sci 101:2490–2496
Shafer JA, Cruz CR, Leen AM, Ku S, Lu A, Rousseau A et al (2010) Antigen-specific cytotoxic T lymphocytes can target chemoresistant side-population tumor cells in Hodgkin lymphoma. Leuk Lymphoma 51:870–880
Jansen MP, Hopman AH, Bot FJ, Haesevoets A, Stevens-Kroef MJ, Arends JW et al (1999) Morphologically normal, CD30-negative B-lymphocytes with chromosome aberrations in classical Hodgkin’s disease: the progenitor cell of the malignant clone? J Pathol 189:527–532
Spieker T, Kurth J, Küppers R, Rajewsky K, Bräuninger A, Hansmann ML (2000) Molecular single-cell analysis of the clonal relationship of small Epstein-Barr virus-infected cells and Epstein-Barr virus-harboring Hodgkin and Reed/Sternberg cells in Hodgkin disease. Blood 96:3133–3138
Jones RJ, Gocke CD, Kasamon YL, Miller CB, Perkins B, Barber JP et al (2009) Circulating clonotypic B cells in classic Hodgkin lymphoma. Blood 113:5920–5926
Küppers R (2009) Clonogenic B cells in classic Hodgkin lymphoma. Blood 114:3970–3971
Vockerodt M, Soares M, Kanzler H, Küppers R, Kube D, Hansmann ML et al (1998) Detection of clonal Hodgkin and Reed-Sternberg cells with identical somatically mutated and rearranged VH genes in different biopsies in relapsed Hodgkin’s disease. Blood 92:2899–2907
Weber-Matthiesen K, Deerberg J, Poetsch M, Grote W, Schlegelberger B (1995) Numerical chromosome aberrations are present within the CD30+ Hodgkin and Reed-Sternberg cells in 100% of analyzed cases of Hodgkin’s disease. Blood 86:1464–1468
Martin-Subero JI, Klapper W, Sotnikova A, Callet-Bauchu E, Harder L, Bastard C et al (2006) Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Cancer Res 66:10332–10338
Szymanowska N, Klapper W, Gesk S, Küppers R, Martin-Subero JI, Siebert R (2008) BCL2 and BCL3 are recurrent translocation partners of the IGH locus. Cancer Genet Cytogenet 186:110–114
Gravel S, Delsol G, Al Saati T (1998) Single-cell analysis of the t(14;18)(q32;p21) chromosomal translocation in Hodgkin's disease demonstrates the absence of this transformation in neoplastic Hodgkin and Reed-Sternberg cells. Blood 91:2866–2874
Poppema S, Kaleta J, Hepperle B (1992) Chromosomal abnormalities in patients with Hodgkin's disease: evidence for frequent involvement of the 14q chromosomal region but infrequent bcl-2 gene rearrangement in Reed-Sternberg cells. J Natl Cancer Inst 84:1789–1793
Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P et al (2011) MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 471:377–381
Renné C, Martin-Subero JI, Hansmann ML, Siebert R (2005) Molecular cytogenetic analyses of immunoglobulin loci in nodular lymphocyte predominant Hodgkin’s lymphoma reveal a recurrent IGH-BCL6 juxtaposition. J Mol Diagn 7:352–356
Wlodarska I, Nooyen P, Maes B, Martin-Subero JI, Siebert R, Pauwels P et al (2003) Frequent occurrence of BCL6 rearrangements in nodular lymphocyte predominance Hodgkin lymphoma but not in classical Hodgkin lymphoma. Blood 101:706–710
Wlodarska I, Stul M, De Wolf-Peeters C, Hagemeijer A (2004) Heterogeneity of BCL6 rearrangements in nodular lymphocyte predominant Hodgkin’s lymphoma. Haematologica 89:965–972
Maggio EM, van den Berg A, de Jong D, Diepstra A, Poppema S (2003) Low frequency of FAS mutations in Reed-Sternberg cells of Hodgkin’s lymphoma. Am J Pathol 162:29–35
Müschen M, Re D, Bräuninger A, Wolf J, Hansmann ML, Diehl V et al (2000) Somatic mutations of the CD95 gene in Hodgkin and Reed-Sternberg cells. Cancer Res 60:5640–5643
Thomas RK, Schmitz R, Harttrampf AC, Abdil-Hadi A, Wickenhauser C, Distler V et al (2005) Apoptosis-resistant phenotype of classical Hodgkin’s lymphoma is not mediated by somatic mutations within genes encoding members of the death-inducing signaling complex (DISC). Leukemia 19:1079–1082
Bose S, Starczynski J, Chukwuma M, Baumforth K, Wei W, Morgan S et al (2007) Down-regulation of ATM protein in HRS cells of nodular sclerosis Hodgkin’s lymphoma in children occurs in the absence of ATM gene inactivation. J Pathol 213:329–336
Lespinet V, Terraz F, Recher C, Campo E, Hall J, Delsol G et al (2005) Single-cell analysis of loss of heterozygosity at the ATM gene locus in Hodgkin and Reed-Sternberg cells of Hodgkin’s lymphoma: ATM loss of heterozygosity is a rare event. Int J Cancer 114:909–916
Schmitz R, Thomas RK, Harttrampf AC, Wickenhauser C, Schultze JL, Hansmann ML et al (2006) The major subtypes of human B-cell lymphomas lack mutations in BCL-2 family member BAD. Int J Cancer 119:1738–1740
Maggio EM, Stekelenburg E, Van den Berg A, Poppema S (2001) TP53 gene mutations in Hodgkin lymphoma are infrequent and not associated with absence of Epstein-Barr virus. Int J Cancer 94:60–66
Montesinos-Rongen M, Roers A, Küppers R, Rajewsky K, Hansmann M-L (1999) Mutation of the p53 gene is not a typical feature of Hodgkin and Reed-Sternberg cells in Hodgkin’s disease. Blood 94:1755–1760
Feuerborn A, Moritz C, Von Bonin F, Dobbelstein M, Trümper L, Sturzenhofecker B et al (2006) Dysfunctional p53 deletion mutants in cell lines derived from Hodgkin’s lymphoma. Leuk Lymphoma 47:1932–1940
Küpper M, Joos S, Von Bonin F, Daus H, Pfreundschuh M, Lichter P et al (2001) MDM2 gene amplification and lack of p53 point mutations in Hodgkin and Reed-Sternberg cells: results from single-cell polymerase chain reaction and molecular cytogenetic studies. Br J Haematol 112:768–775
Jardin F, Pujals A, Pelletier L, Bohers E, Camus V, Mareschal S et al (2016) Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am J Hematol 91:923–930
Abdul Razak FR, Diepstra A, Visser L, van den Berg A (2016) CD58 mutations are common in Hodgkin lymphoma cell lines and loss of CD58 expression in tumor cells occurs in Hodgkin lymphoma patients who relapse. Genes Immun 17:363–366
Schneider M, Schneider S, Zühlke-Jenisch R, Klapper W, Sundström C, Hartmann S et al (2015) Alterations of the CD58 gene in classical Hodgkin lymphoma. Genes Chromosomes Cancer 54:638–645
Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT (1999) Mutations in the IκΒα gene in Hodgkin’s disease suggest a tumour suppressor role for IκΒα. Oncogene 18:3063–3070
Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F et al (1999) Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood 94:3129–3134
Jungnickel B, Staratschek-Jox A, Bräuninger A, Spieker T, Wolf J, Diehl V et al (2000) Clonal deleterious mutations in the IkBa gene in the malignant cells in Hodgkin’s disease. J Exp Med 191:395–401
Lake A, Shield LA, Cordano P, Chui DT, Osborne J, Crae S et al (2009) Mutations of NFKBIA, encoding IkappaBalpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int J Cancer 125:1334–1342
Emmerich F, Theurich S, Hummel M, Haeffker A, Vry MS, Döhner K et al (2003) Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol 201:413–420
Mansouri L, Noerenberg D, Young E, Mylonas E, Abdulla M, Frick M et al (2016) Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood 128:2666–2670
Otto C, Giefing M, Massow A, Vater I, Gesk S, Schlesner M et al (2012) Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br J Haematol 157:702–708
Schmidt A, Schmitz R, Giefing M, Martin-Subero JI, Gesk S, Vater I et al (2010) Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer 49:803–809
Joos S, Granzow M, Holtgreve-Grez H, Siebert R, Harder L, Martin-Subero JI et al (2003) Hodgkin's lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer 103:489–495
Joos S, Menz CK, Wrobel G, Siebert R, Gesk S, Ohl S et al (2002) Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 99:1381–1387
Martin-Subero JI, Gesk S, Harder L, Sonoki T, Tucker PW, Schlegelberger B et al (2002) Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 99:1474–1477
Barth TF, Martin-Subero JI, Joos S, Menz CK, Hasel C, Mechtersheimer G et al (2003) Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 101:3681–3686
Steidl C, Telenius A, Shah SP, Farinha P, Barclay L, Boyle M et al (2010) Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 116:418–427
Martin-Subero JI, Wlodarska I, Bastard C, Picquenot JM, Höppner J, Giefing M et al (2006) Chromosomal rearrangements involving the BCL3 locus are recurrent in classical Hodgkin and peripheral T-cell lymphoma. Blood 108:401–402
Mathas S, Jöhrens K, Joos S, Lietz A, Hummel F, Janz M et al (2005) Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 106:4287–4293
Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K et al (2009) Frequent inactivation of A20 in B-cell lymphomas. Nature 459:712–716
Schmitz R, Hartmann S, Giefing M, Mechtersheimer G, Zuhlke-Jenisch R, Martin-Subero JI et al (2007) Inactivating mutations of TNFAIP3 (A20) indicate a tumor suppressor role for A20 in Hodgkin's lymphoma and primary mediastinal B cell lymphoma. Haeamtologica. Hematol J 92(Suppl. 5):41
Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E (1995) The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80:389–399
Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N et al (1999) Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286:300–303
Schumacher MA, Schmitz R, Brune V, Tiacci E, Döring C, Hansmann ML et al (2010) Mutations in the genes coding for the NF-kappaB regulating factors IkappaBalpha and A20 are uncommon in nodular lymphocyte-predominant Hodgkin’s lymphoma. Haematologica 95:153–157
Mottok A, Renné C, Willenbrock K, Hansmann ML, Bräuninger A (2007) Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood 110:3387–3390
Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K et al (2006) Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 25:2679–2684
Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL et al (2014) Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet 46:329–335
Joos S, Küpper M, Ohl S, von Bonin F, Mechtersheimer G, Bentz M et al (2000) Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res 60:549–552
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E et al (2010) Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116:3268–3277
Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G et al (2010) Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 18:590–605
Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B et al (2011) JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood 117:4056–4064
Hartmann S, Martin-Subero JI, Gesk S, Husken J, Giefing M, Nagel I et al (2008) Detection of genomic imbalances in microdissected Hodgkin and Reed-Sternberg cells of classical Hodgkin's lymphoma by array-based comparative genomic hybridization. Haematologica 93:1318–1326
Hartmann S, Schuhmacher B, Rausch T, Fuller L, Döring C, Weniger M et al (2016) Highly recurrent mutations of SGK1, DUSP2 and JUNB in nodular lymphocyte predominant Hodgkin lymphoma. Leukemia 30:844–853
Re D, Müschen M, Ahmadi T, Wickenhauser C, Staratschek-Jox A, Holtick U et al (2001) Oct-2 and Bob-1 deficiency in Hodgkin and Reed Sternberg cells. Cancer Res 61:2080–2084
Stein H, Marafioti T, Foss HD, Laumen H, Hummel M, Anagnostopoulos I et al (2001) Down-regulation of BOB.1/OBF.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant Hodgkin disease correlates with immunoglobulin transcription. Blood 97:496–501
Watanabe K, Yamashita Y, Nakayama A, Hasegawa Y, Kojima H, Nagasawa T et al (2000) Varied B-cell immunophenotypes of Hodgkin/Reed-Sternberg cells in classic Hodgkin’s disease. Histopathology 36:353–361
Schwering I, Bräuninger A, Klein U, Jungnickel B, Tinguely M, Diehl V et al (2003) Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 101:1505–1512
Carbone A, Gloghini A, Larocca LM, Antinori A, Falini B, Tirelli U et al (1999) Human immunodeficiency virus-associated Hodgkin’s disease derives from post-germinal center B cells. Blood 93:2319–2326
Tiacci E, Döring C, Brune V, van Noesel CJ, Klapper W, Mechtersheimer G et al (2012) Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood 120:4609–4620
Poppema S (1996) Immunology of Hodgkin’s disease. Baillieres Clin Haematol 9:447–457
Carbone A, Gloghini A, Gruss HJ, Pinto A (1995) CD40 ligand is constitutively expressed in a subset of T cell lymphomas and on the microenvironmental reactive T cells of follicular lymphomas and Hodgkin’s disease. Am J Pathol 147:912–922
Torlakovic E, Tierens A, Dang HD, Delabie J (2001) The transcription factor PU.1, necessary for B-cell development is expressed in lymphocyte predominance, but not classical Hodgkin’s disease. Am J Pathol 159:1807–1814
Bohle V, Döring C, Hansmann ML, Küppers R (2013) Role of early B-cell factor 1 (EBF1) in Hodgkin lymphoma. Leukemia 27:671–679
Overbeck BM, Martin-Subero JI, Ammerpohl O, Klapper W, Siebert R, Giefing M (2012) ETS1 encoding a transcription factor involved in B-cell differentiation is recurrently deleted and down-regulated in classical Hodgkin’s lymphoma. Haematologica 97:1612–1614
Küppers R, Klein U, Schwering I, Distler V, Bräuninger A, Cattoretti G et al (2003) Identification of Hodgkin and Reed-Sternberg cell-specific genes by gene expression profiling. J Clin Invest 111:529–537
Mathas S, Janz M, Hummel F, Hummel M, Wollert-Wulf B, Lusatis S et al (2006) Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol 7:207–215
Renné C, Martin-Subero JI, Eickernjager M, Hansmann ML, Küppers R, Siebert R et al (2006) Aberrant expression of ID2, a suppressor of B-cell-specific gene expression, in Hodgkin’s lymphoma. Am J Pathol 169:655–664
Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C et al (2003) Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 4:380–386
Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S et al (1999) Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 397:702–706
Jundt F, Acikgoz O, Kwon SH, Schwarzer R, Anagnostopoulos I, Wiesner B et al (2008) Aberrant expression of Notch1 interferes with the B-lymphoid phenotype of neoplastic B cells in classical Hodgkin lymphoma. Leukemia 22:1587–1594
Jundt F, Anagnostopoulos I, Förster R, Mathas S, Stein H, Dörken B (2002) Activated Notch 1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood 99:3398–3403
Köchert K, Ullrich K, Kreher S, Aster JC, Kitagawa M, Johrens K et al (2011) High-level expression of Mastermind-like 2 contributes to aberrant activation of the NOTCH signaling pathway in human lymphomas. Oncogene 30(15):1831–1840
Scheeren FA, Diehl SA, Smit LA, Beaumont T, Naspetti M, Bende RJ et al (2008) IL-21 is expressed in Hodgkin lymphoma and activates STAT5; evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood 111:4706–4715
Stanelle J, Döring C, Hansmann ML, Küppers R (2010) Mechanisms of aberrant GATA3 expression in classical Hodgkin lymphoma and its consequences for the cytokine profile of Hodgkin and Reed/Sternberg cells. Blood 116:4202–4211
Doerr JR, Malone CS, Fike FM, Gordon MS, Soghomonian SV, Thomas RK et al (2005) Patterned CpG methylation of silenced B cell gene promoters in classical Hodgkin lymphoma-derived and primary effusion lymphoma cell lines. J Mol Biol 350:631–640
Ushmorov A, Leithäuser F, Sakk O, Weinhausel A, Popov SW, Möller P et al (2006) Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood 107:2493–2500
Ammerpohl O, Haake A, Pellissery S, Giefing M, Richter J, Balint B et al (2012) Array-based DNA methylation analysis in classical Hodgkin lymphoma reveals new insights into the mechanisms underlying silencing of B cell-specific genes. Leukemia 26:185–188
Dukers DF, van Galen JC, Giroth C, Jansen P, Sewalt RG, Otte AP et al (2004) Unique polycomb gene expression pattern in Hodgkin's lymphoma and Hodgkin’s lymphoma-derived cell lines. Am J Pathol 164:873–881
Raaphorst FM, van Kemenade FJ, Blokzijl T, Fieret E, Hamer KM, Satijn DP et al (2000) Coexpression of BMI-1 and EZH2 polycomb group genes in Reed-Sternberg cells of Hodgkin’s disease. Am J Pathol 157:709–715
Sanchez-Beato M, Sanchez E, Garcia JF, Perez-Rosado A, Montoya MC, Fraga M et al (2004) Abnormal PcG protein expression in Hodgkin’s lymphoma. Relation with E2F6 and NFkappaB transcription factors. J Pathol 204:528–537
Schneider EM, Torlakovic E, Stuhler A, Diehl V, Tesch H, Giebel B (2004) The early transcription factor GATA-2 is expressed in classical Hodgkin’s lymphoma. J Pathol 204:538–545
Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D et al (2010) Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med 16:571–579
Yuki H, Ueno S, Tatetsu H, Niiro H, Iino T, Endo S et al (2013) PU.1 is a potent tumor suppressor in classical Hodgkin lymphoma cells. Blood 121:962–970
Guan H, Xie L, Wirth T, Ushmorov A (2016) Repression of TCF3/E2A contributes to Hodgkin lymphomagenesis. Oncotarget 7:36854–36864
Xie L, Ushmorov A, Leithäuser F, Guan H, Steidl C, Farbinger J et al (2012) FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 119:3503–3511
Du J, Neuenschwander M, Yu Y, Dabritz JH, Neuendorff NR, Schleich K et al (2017) Pharmacological restoration and therapeutic targeting of the B-cell phenotype in classical Hodgkin lymphoma. Blood 129:71–81
Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W et al (1997) Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest 100:2961–2969
Carbone A, Gloghini A, Gattei V, Aldinucci D, Degan M, De Paoli P et al (1995) Expression of functional CD40 antigen on Reed-Sternberg cells and Hodgkin’s disease cell lines. Blood 85:780–789
Chiu A, Xu W, He B, Dillon SR, Gross JA, Sievers E et al (2007) Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 109:729–739
Fiumara P, Snell V, Li Y, Mukhopadhyay A, Younes M, Gillenwater AM et al (2001) Functional expression of receptor activator of nuclear factor kappaB in Hodgkin disease cell lines. Blood 98:2784–2790
Molin D, Fischer M, Xiang Z, Larsson U, Harvima I, Venge P et al (2001) Mast cells express functional CD30 ligand and are the predominant CD30L-positive cells in Hodgkin’s disease. Br J Haematol 114:616–623
Schwab U, Stein H, Gerdes J, Lemke H, Kirchner H, Schaadt M et al (1982) Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 299:65–67
Hirsch B, Hummel M, Bentink S, Fouladi F, Spang R, Zollinger R et al (2008) CD30-induced signaling is absent in Hodgkin’s cells but present in anaplastic large cell lymphoma cells. Am J Pathol 172:510–520
Horie R, Watanabe T, Morishita Y, Ito K, Ishida T, Kanegae Y et al (2002) Ligand-independent signaling by overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene 21:2493–2503
Kilger E, Kieser A, Baumann M, Hammerschmidt W (1998) Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J 17:1700–1709
Baus D, Pfitzner E (2006) Specific function of STAT3, SOCS1, and SOCS3 in the regulation of proliferation and survival of classical Hodgkin lymphoma cells. Int J Cancer 118:1404–1413
Kube D, Holtick U, Vockerodt M, Ahmadi T, Behrmann I, Heinrich PC et al (2001) STAT3 is constitutively activated in Hodgkin cell lines. Blood 98:762–770
Skinnider BF, Elia AJ, Gascoyne RD, Patterson B, Trümper L, Kapp U et al (2002) Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 99:618–626
Kapp U, Yeh WC, Patterson B, Elia AJ, Kagi D, Ho A et al (1999) Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J Exp Med 189:1939–1946
Skinnider BF, Elia AJ, Gascoyne RD, Trumper LH, von Bonin F, Kapp U et al (2001) Interleukin 13 and interleukin 13 receptor are frequently expressed by Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 97:250–255
Hinz M, Lemke P, Anagnostopoulos I, Hacker C, Krappmann D, Mathas S et al (2002) Nuclear factor kappaB-dependent gene expression profiling of Hodgkin's disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J Exp Med 196:605–617
Lamprecht B, Kreher S, Anagnostopoulos I, Johrens K, Monteleone G, Jundt F et al (2008) Aberrant expression of the Th2 cytokine IL-21 in Hodgkin lymphoma cells regulates STAT3 signaling and attracts Treg cells via regulation of MIP-3{alpha}. Blood 112:3339–3347
Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411:355–365
Renné C, Willenbrock K, Küppers R, Hansmann M-L, Bräuninger A (2005) Autocrine and paracrine activated receptor tyrosine kinases in classical Hodgkin lymphoma. Blood 105:4051–4059
Teofili L, Di Febo AL, Pierconti F, Maggiano N, Bendandi M, Rutella S et al (2001) Expression of the c-met proto-oncogene and its ligand, hepatocyte growth factor, in Hodgkin disease. Blood 97:1063–1069
Renné C, Willenbrock K, Martin-Subero JI, Hinsch N, Döring C, Tiacci E et al (2007) High expression of several tyrosine kinases and activation of the PI3K/AKT pathway in mediastinal large B cell lymphoma reveals further similarities to Hodgkin lymphoma. Leukemia 21:780–787
Renné C, Hinsch N, Willenbrock K, Fuchs M, Klapper W, Engert A et al (2007) The aberrant coexpression of several receptor tyrosine kinases is largely restricted to EBV-negative cases of classical Hodgkin’s lymphoma. Int J Cancer 120:2504–2509
Renne C, Minner S, Küppers R, Hansmann ML, Bräuninger A (2008) Autocrine NGFbeta/TRKA signalling is an important survival factor for Hodgkin lymphoma derived cell lines. Leuk Res 32:163–167
Nagel S, Burek C, Venturini L, Scherr M, Quentmeier H, Meyer C et al (2007) Comprehensive analysis of homeobox genes in Hodgkin lymphoma cell lines identifies dysregulated expression of HOXB9 mediated via ERK5 signaling and BMI1. Blood 109:3015–3023
Zheng B, Fiumara P, Li YV, Georgakis G, Snell V, Younes M et al (2003) MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood 102:1019–1027
Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F et al (2002) Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J 21:4104–4113
Juszczynski P, Ouyang J, Monti S, Rodig SJ, Takeyama K, Abramson J et al (2007) The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Natl Acad Sci U S A 104:13134–13139
Watanabe M, Ogawa Y, Ito K, Higashihara M, Kadin ME, Abraham LJ et al (2003) AP-1 mediated relief of repressive activity of the CD30 promoter microsatellite in Hodgkin and Reed-Sternberg cells. Am J Pathol 163:633–641
Lollies A, Hartmann S, Schneider M, Bracht T, Weiss AL, Arnolds J et al (2018) An oncogenic axis of STAT-mediated BATF3 upregulation causing MYC activity in classical Hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia 32:92–101
Vrzalikova K, Ibrahim M, Vockerodt M, Perry T, Margielewska S, Lupino L et al (2018) S1PR1 drives a feedforward signalling loop to regulate BATF3 and the transcriptional programme of Hodgkin lymphoma cells. Leukemia 32:214–223
Dutton A, Reynolds GM, Dawson CW, Young LS, Murray PG (2005) Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin's lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol 205:498–506
Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Mills GB, Younes A (2006) Inhibition of the phosphatidylinositol-3 kinase/Akt promotes G1 cell cycle arrest and apoptosis in Hodgkin lymphoma. Br J Haematol 132:503–511
Dutton A, O'Neil JD, Milner AE, Reynolds GM, Starczynski J, Crocker J et al (2004) Expression of the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin’s lymphoma cells from autonomous Fas-mediated death. Proc Natl Acad Sci U S A 101:6611–6616
Mathas S, Lietz A, Anagnostopoulos I, Hummel F, Wiesner B, Janz M et al (2004) c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor-induced apoptosis. J Exp Med 199:1041–1052
Re D, Hofmann A, Wolf J, Diehl V, Staratschek-Jox A (2000) Cultivated H-RS cells are resistant to CD95L-mediated apoptosis despite expression of wild-type CD95. Exp Hematol 28:31–35
Chu WS, Aguilera NS, Wei MQ, Abbondanzo SL (1999) Antiapoptotic marker Bcl-X(L), expression on Reed-Sternberg cells of Hodgkin’s disease using a novel monoclonal marker, YTH-2H12. Hum Pathol 30:1065–1070
Kashkar H, Haefs C, Shin H, Hamilton-Dutoit SJ, Salvesen GS, Krönke M et al (2003) XIAP-mediated caspase inhibition in Hodgkin’s lymphoma-derived B cells. J Exp Med 198:341–347
Kashkar H, Seeger JM, Hombach A, Deggerich A, Yazdanpanah B, Utermohlen O et al (2006) XIAP targeting sensitizes Hodgkin lymphoma cells for cytolytic T-cell attack. Blood 108:3434–3440
Sanchez-Beato M, Piris MA, Martinez-Montero JC, Garcia JF, Villuendas R, Garcia FJ et al (1996) MDM2 and p21WAF1/CIP1, wild-type p53-induced proteins, are regularly expressed by Sternberg-Reed cells in Hodgkin's disease. J Pathol 180:58–64
Drakos E, Thomaides A, Medeiros LJ, Li J, Leventaki V, Konopleva M et al (2007) Inhibition of p53-murine double minute 2 interaction by nutlin-3A stabilizes p53 and induces cell cycle arrest and apoptosis in Hodgkin lymphoma. Clin Cancer Res 13:3380–3387
Janz M, Stuhmer T, Vassilev LT, Bargou RC (2007) Pharmacologic activation of p53-dependent and p53-independent apoptotic pathways in Hodgkin/Reed-Sternberg cells. Leukemia 21:772–779
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rosenwald, A., Küppers, R. (2020). Pathology and Molecular Pathology of Hodgkin Lymphoma. In: Engert, A., Younes, A. (eds) Hodgkin Lymphoma. Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-32482-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-32482-7_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-32481-0
Online ISBN: 978-3-030-32482-7
eBook Packages: MedicineMedicine (R0)