
Abstract
Oral carcinogenesis is a consequence of multiple alterations in important pathways of a cell’s life. These altered pathways provide capabilities to tumorigenesis that include sustained proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprograming cellular energetics, and evading immune destruction. Several of the genes and proteins involved in these acquired capacities for oral carcinogenesis will be developed in this chapter. Many of them could be useful as diagnostic biomarkers and also as predictors of malignant transformation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Oral carcinogenesis is a consequence of multiple alterations in important pathways of a cell’s life. These altered pathways provide capabilities to tumorigenesis that include sustained proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprograming cellular energetics, and evading immune destruction. Several of the genes and proteins involved in these acquired capacities for oral carcinogenesis will be developed in this chapter. Many of them could be useful as diagnostic biomarkers and also as predictors of malignant transformation .
14.1 Introduction
Oral cancer arises from the accumulation of successive genetic and epigenetic alterations in a multistep process over many years, transforming a single cell or a clone of cells of the oral mucosa into a malignant tumor. Although many genes and proteins can be affected by carcinogenic agents, including tobacco or alcohol , a group of them when deregulated are decisive for driving a cell or a clone of cells to transform and form a malignant tumor. These key genes and proteins that are affected belong to several signaling pathways related to cell proliferation, regulation, and differentiation that, when deregulated, predispose to tumor growth and progression. They include some normal genes (termed proto-oncogenes) that when modified (e.g. by mutation, gene amplification, or chromosome rearrangements ) become oncogenes, leading to persistent cell proliferation. Contrarily, tumor suppressor genes that normally prevent deregulated cell growth but when altered by mutations, deletions, or epigenetic modifications may allow clones of cells to acquire insensitivity to growth inhibition and avoidance of apoptosis [1, 2].
Most oral cancers are squamous cell carcinomas, which means they represent a homogeneous group of tumors arising from lining epithelia comparable with squamous cancers in other locations. This suggests a carcinogenesis model with common characteristics such as Califano’s model based on serial and sequential amounts of genetic errors occurring in key oncogenes. Thus tumor suppressor genes would be the basis for transformation of a normal mucosal cell into a cancer [3]. As Hahn and Weinberg had proposed around the beginning of this century for cancer in general, several pathways would be typically affected along the landscape for oral cancer [4, 5]. These altered pathways provide capabilities to tumorigenesis that include sustained proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis. Additionally, to these features of cancer, more emerging capacities include reprograming cellular energetics and evading immune destruction [6].
As with other cancers, molecular and genomics studies have thrown new insights into several new aspects of oral carcinogenesis and we can no longer define a linear and unidirectional oral carcinogenesis model driving cells, but instead what has emerged is a dynamic, personalized, or multidirectional model with heterogeneity [7, 8]. These genetic studies suggested that some subclasses of head and neck cancers can be defined and identified through genetic profiles that have been recently mapped out. One such group of head and neck tumors (especially in the oropharynx ) could be clearly related with the presence of HPV infection (HPV + ve) [9, 10]. Comparative genomic hybridization and ploidy status studies have revealed subgroups of HPV -ve tumors with few copy number alterations or with diploid status with a relatively good survival when compared with the rest of HPV -ve tumors [8, 10, 11]. Moreover, other subgroups include patients typically presenting with tobacco-induced tumors with alterations in, for example, the epidermal growth factor receptor (EGFR) pathway that indicate a poor prognosis [7, 10].
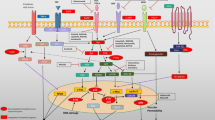
This means that the model of carcinogenesis, such as presented in ◘ Fig. 14.1, is not similar or uniform for all oral cancers and the identification of the molecular signatures could have important and useful implications for diagnosis, prognosis, and treatment planning of oral cancers. Moreover, each tumor will have its own molecular signature and novel capacity to explore new molecular escapes. The identification of key genes or proteins involved in this tumor escape could be important for the individual characterization of the tumors and could function as biomarkers of the disease not only for early diagnosis but also for predicting prognosis [12].

An integrative oral carcinogenesis model depicting changes from a normal cell to a transformed cell indicating possible alterations in clinical, histological, and molecular changes. Several genes/proteins could be important for the oral carcinogenesis development
In the following sections, we will discuss several genes, proteins, and their related pathways most often associated with oral cancer.
Definition
A cancer biomarker refers to a substance that is indicative of the presence of cancer in the body. It could have a diagnostic, predictive or prognostic value.
14.2 Altered Pathways Involved in Oral Carcinogenesis
In physiological states, the cell cycle is controlled by several proteins and genes with activating or suppressing activity which drive cells through the cell cycle, allowing for cell division when needed. Important genes and proteins have a strong capacity to stop and cause inhibition of cell proliferation, especially when DNA errors are detected. Several tumor suppressor genes operate on this principle, such as RB and TP53, but when affected by mutations or inhibitions such as HPV proteins, they lead to pathways to oral cancer [4].
We present here some commonly altered genes/proteins in oral cancers that have been reported.
Important
A biomarker may be a molecule secreted by a tumor or a specific response of the body to the presence of cancer.
14.2.1 Evading Growth Suppressors
-
P53
P53, a phosphoprotein of 53 kD, is a transcription factor present in most cells with several significant functions: the regulation of gene transcription, regulation of DNA synthesis, and repair and apoptosis. It is encoded by TP53, an oncosuppressor gene located on chromosome 17 (17p13.1) [13].
In the presence of cellular stress (including DNA errors, hypoxia, oxidative damage, or exposure to radiation), P53 levels increase dramatically, initiating a cell protective response. Briefly, an increase in P53 induces transcription of CDKN1A gene encoding the P21 protein, responsible for cell cycle arrest at the G1 phase checkpoint, by inhibition of the complex CDK4/6 – cyclin D1. At this time, non-phosphorylated pRB binds and inhibits E2F family preventing the transcription of factors required for the cell cycle and for other cyclins such as cyclin E and cyclin A to act in consort [14]. This cell cycle arrest allows DNA repair mainly by the activation of the GADD45 family of genes in association with PCNA, p48/DDB2, ERCC2, and ERC3. Nevertheless, if the DNA injury is too severe, or if p53 DNA repair is not possible, apoptosis occurs. P53 is capable of inducing pro-apoptotic genes such as BAX, PUMA, PIG3, NOXA, TRAIL, or PTEN and can inactivate anti-apoptotic genes such as BCL2 or SURVIVIN. In the case of successful DNA repair, the now active MDM2 promotes P53 degradation and the break of the cell cycle [14]. Hence, this protein could be considered as a guardian of the genome [13].
Alterations in P53 protein occurs in several types of tumors and in half of them by mutations in TP53. Most of the mutations are located in the DNA binding domain, impairing the P53 binding to target genes [15]. Interestingly, many of the TP53 mutations observed in head and neck carcinomas affect guanine nucleotide (G) and are caused by tobacco carcinogens [14].
Loss of P53 function can occur by other mechanisms even without any mutations of TP53 and in the presence of the normal protein, such as its upregulation by amplification or due to polymorphisms of the MDM2 , leading to P53 degradation by the ubiquitin proteasome system. Infection of cells with high risk types of human papillomavirus (HPV) can also lead to P53 degradation, brought about by viral E6 protein which binds to P53 leading to ubiquitin proteasome degradation.
Somatic mutations in TP53 gene with consequent and frequent P53 overexpression represent one of the most reported changes in squamous cell carcinomas of the oral cavity and found in more than 50% of cases and correspond to an early event already present in potentially malignant disorders [16,17,18,19]. Overexpression of P53 is associated with poor survival of oral squamous cell carcinomas [15, 19,20,21].
In addition to TP53 mutations, other early events in oral carcinogenesis include the loss of chromosome 9p, which is the locus for this gene [22]. CDKN2A is a gene located on chromosome 9p21 and encodes P16, a tumor suppressor protein that promotes cycle arrest in G1/S check-point, by binding to the complex cyclin D1/CDK4, which inactivates pRb. Loss or inactivation of this gene is frequent (by mutations, methylation, chromosome loss, or homozygous deletion) in early oral carcinogenesis. Many oral carcinomas have reduced expression of P16 and this has been correlated with a poor prognosis [22].
-
pRb
pRb protein is encoded by the RB1 gene (3q14.1-q14.2) and the loss of both alleles of this gene leads to retinoblastoma. In the normal cell, pRb is in a hypo-phosphorylated state. When a mitogenic stimulus is transmitted, the transcription of cyclins A, D, E increase dramatically leading to the phosphorylation of pRb. Now, the phosphorylated form of pRb becomes permissive with the transcription of genes involved in DNA replication and cell cycle progression [1]. RB1mutations in oral cancer are rare, but pRB protein could be inactivated by other forms such as the action by E7 from HPV [22].
14.2.2 Enabling Replicative Immortality
Telomerase and TERT Proteins
Other mechanisms of cell cycle persistency would involve other genes, such as telomerase and TERT proteins. Although the data on these proteins for oral cancer are not obvious yet, the altered malignant cells could undergo alternative lengthening of telomeres (ALT), a related TERT process of telomere lengthening [22].
Tumor cells could gain the capacity to sustain proliferative signaling using mitogenic signaling pathways by producing growth factors themselves in an autocrine proliferative manner, to induce stromal tumor cells to produce mitogenic factors for tumor cells, or simply by constitutive activation of components of mitogenic pathways.
In oral cancer, the most commonly affected pathways includes EGFR pathway, MAP kinases or PI3K /AKT/mTOR pathways, or even the endpoint of mitotic pathways in the nucleus as cyclins and kinases.
14.2.3 Sustaining Proliferative Signaling
-
EGFR
The epidermal growth factor receptor (EGFR) is a transmembrane receptor encoded by the c-erbB proto-oncogene (located at 7p12). This glycoprotein is composed of an extracellular ligand-binding part, an intermediate transmembrane region, and an intracellular domain with tyrosine kinase capacity (◘ Fig. 14.2).

A schematic resume of EGFR pathway is shown including the dimerization and phosphorylation of the tyrosine kinase receptor EGFR and subsequent activation of several pathways resulting in cell growth, proliferation, and survival
When a ligand such as epidermal growth factor (EGF), transforming growth factor alpha (TGF-α), epiregulin (ER), or amphiregulin (RA) binds to EGFR, a homo- or heterodimer is formed with one or more members of the ErbB family (such as ErbB2/HER-2, ErbB3/ HER-3, or ErbB4/HER-4) leading to the phosphorylation of cytoplasmic tyrosine residues mainly at positions 992, 1068, 1086, 1148, or 1173 [23]. The activated tyrosine kinase domain induces the transduction of mitogenic and survival signals by mitogen-activated protein kinase (MAPK) pathway, phosphatidylinositol 3-kinase (PI3K)/Akt pathway, or phospholipase Cγ (PLCγ1) [24].
EGFR is involved in cell proliferation and survival not only in a normal cell but also promotes tumor growth and resistance to apoptosis, promotion of cell motility, alteration and reduction of adhesion molecules such as E-cadherin , stimulating metalloproteinases (MMP-9), or even in the process of angiogenesis by regulating VEGF [24].
EGFR overexpression has been reported in several cancers and interestingly in more than 90% of head and neck squamous cell carcinomas (HNSCC) and has been associated with aggressive disease and poor prognosis [7, 25,26,27]. Some studies have shown that both membranous and cytoplasmic expression of EGFR could have an adverse influence in the overall survival of patients with oral squamous cell carcinoma [28].
EGFR overexpression can be caused by several mechanisms. EGFR gene amplification has been reported in 10–30% of head and neck cancers [24]. EGFR mutations have been described with a fewer frequency (1–7%) and could include point mutations (e.g., exon 21 (L858R)), deletions in exons 2–7 that result in the EGFRv III variant, lacking the extracellular binding domain but with active constituent [29,30,31,32,33] and by other mechanisms such as an autocrine expression with EGF and TGF-α, albeit unusual in head and neck cancers [34].
EGFR is one of the popular molecular targets for therapeutic agents against head and neck cancers due to the high overexpression rate of this receptor found in these cancers. The inhibition of this receptor can be achieved using monoclonal antibodies, tyrosine kinase inhibitors (TKIs), ligand-toxin conjugates, or immunoconjugates [35]. Monoclonal antibodies, such as Cetuximab , block the binding of the growth factor to the external domain of EGFR impairing the activation of the receptor. This anti-EGFR drug has shown to increase the overall survival of HNSCC patients presenting in advanced stages as part of combination therapies with radiation and chemotherapy [36]. EGFR can be inactivated also using tyrosine kinase inhibitors, small molecules which inhibit the tyrosine kinase activity of this receptor. TKIs include gefitinib, erlotinib, and lapatinib [37] (see for details ► Chapter 27).
-
c-MET
Another tyrosine-protein kinase receptor that has been shown to be involved in oral cancer is the mesenchymal-epithelial transition factor (c-MET), a receptor for the hepatocyte growth factor (HGF) encoded by the proto-oncogene MET [38]. Mutations and gene amplifications of MET have been found in several cancers, including oral cancers [22]. c-MET has been found overexpressed in tumor cells and also in carcinoma-associated fibroblasts (CAFs) promoting cell growth, motility, and lymphangiogenesis in oral squamous cell carcinomas (OSCC) via PI3K/AKT, ERK1/2, and NF-κB pathways [38, 39].
-
TGFβ/SMAD Pathway
Signaling through TGFβ/SMAD pathway can be involved in oral tumorigenesis [40]. Transforming growth factor-β (TGF-β) is a receptor with inhibitory growth control regulation function [39]. This receptor can phosphorylate SMAD2 and SMAD3 proteins, and then together they can activate SMAD4 protein that will regulate the expression of target genes such as p15, p21, or p57 [22]. Interestingly, mutations have been found on SMAD2, SMAD3, and SMAD4 proteins in OSCC [22, 41]. Numerous studies showed that TGF-β/SMAD signaling pathway is associated with tumor progression and worsening of prognosis of OSCC [41, 42].
-
PI3K /AKT and mTOR Pathways
PI3K/AKT/PTEN /mTOR has become a recognized important dysregulated signaling pathway in head and neck cancer. This multiple role pathway can influence proliferation and cellular survival, as well as cell motility, migration, and glucose metabolism [43]. Several genes and proteins are involved in this pathway, such as PI3K proteins, mTOR complex, AKT protein, and PTEN .
The phosphatidyl inositol 3-Kinase (PI3K) is composed of the subunits p100 and p87 which works as a heterodimer coupled to tyrosine kinase receptors such as EGFR. When the receptor is activated, p100 promotes the phosphorylation of PIP2 into PIP3. This attracts the PDK1 and phosphorylates the AKT protein a serine/threonine kinase, which in turn stimulates several proto-oncogenes and suppresses other tumor oncosuppressor genes, resulting in cell proliferation and inhibition of apoptosis [22]. The p110 subunit is encoded by PIK3CA, on locus 3q26. Interestingly, this locus or the gene have been found to be amplified or to have activating mutations, in some head and neck cancers. AKT can also be phosphorylated by activation of the mTOR complex [22].
One of the main targets of the PI3K/AKT signaling pathway is the mammalian target of rapamycin (mTOR ), a serine-threonine protein kinase that makes part of two different protein complexes – mTOR complex 1 (rapamycin sensitive) and mTOR complex 2 with multiple actions, including cell proliferation and survival, cell motility, protein synthesis, or insulin receptors regulation (◘ Fig. 14.3). In the presence of nutrient or oxygen stimuli or other factors such as insulin, growth factors, ATP, or toxins of tobacco, mTOR becomes phosphorylated and activates the eukaryotic translation factor 4E (eIF4E), the p70 ribosomal S6 kinase (p70S6 kinase), and elongation factor 2 (eEF2) that will modulate protein biosynthesis. pS6 protein is one of the targets of mTORC1 and is inhibited by rapamycin and is used as a marker for the mTORC1 pathway [44].

Representation of the AKT/mTOR pathway from a membrane receptor activation to cell growth and proliferation. The inhibition effect of rapamycin is illustrated
mTOR has been found to be overexpressed and is related with poor overall survival in several cancers including oral cancer [45, 46]. Importantly, mTOR has been used as a molecular target for anticancer therapy including everolimus, temsirolimus, and ridaforolimus [47]. Interestingly, some authors have reported on the anti-cancer effect of metformin by inhibition of mTOR activity [48].
Another protein involved in this pathway is PTEN. PTEN gene (phosphatase and tensin homolog deleted on chromosome TEN) is a tumor suppressor gene, located at 10q23.3, that encodes a protein phosphatase with lipid and protein phosphatase activity. The AKT activation is normally turned off by PTEN gene that promotes the switch of phosphatidylinositol (3,4,5)-triphosphate (PIP-3) to PIP2. Loss of the PTEN function is reported in ~10% of head and neck cancers. This may lead to increasing levels of PIP-3, resulting in a hyperactivation of AKT and unrestricted activity of mTOR [46].
-
RAS and MAPK Pathway
The classical cellular signal transducers include a family of proteins with ~21 kda protein with guanosine triphosphate (GTP) activity known as RAS proteins. They were named because they were discovered from the genome of murine leukemia virus (rat sarcoma virus) in rodent sarcomas. Currently, three genes characterize the RAS gene family: HRAS (Harvey sarcoma virus–associated oncogene), KRAS (Kirsten sarcoma virus), and NRAS (neuroblastoma-derived sarcoma virus) [49].
RAS becomes active after the phosphorylation of a tyrosine kinases receptor (rtks) thought to result in Grb2–SOS complexes and G proteins (guanine nucleotide-binding proteins). Active RAS leads to the stimulation of several pathways, including mitogen-activated protein kinase (MAPK) by RAF1 kinase, of both MEK1/2 or the phosphatidylinositol-3-kinase (RAS)/AKT, resulting in cell growth and differentiation [49].
RAS is commonly mutated in several cancers including oral cancer, especially as point mutations. Some mutations occur in codons 12, 13, or 61, and the RAS protein becomes permanently activated with a continuous cell growth. Although mutations can appear in all three isoforms of the RAS gene, most of them appear in HRAS (0–55%) especially in South Asian populations [49, 50]. Other mechanisms of RAS overexpression can be related to gene amplification .
-
Cyclins and Mitotic Checkpoint
Cyclins correspond to several forms of proteins divided into two groups based on their function: the G1 cyclins (C, D, E), regulating the passage of cells through the G1 phase and their entry into the S phase, and the mitotic cyclins (A, B) [2, 51]. Cyclins have no phosphorylation capacity, so they work together with several kinases (CDK) during the passage of the cell cycle. The cyclin D1, encoded by CCND1 proto-oncogene (11q13), is the great opener or activator of the cell cycle. After activation, by second messengers such as proteins from MAPK the pathway, this cyclin binds and activates proteins CDK4 and CDK6, leading to phosphorylation of pRb, driving the cell cycle from the G1 to the S-phase. Other cyclins conduct the completion of the rest of the phases of cell cycle. Cyclin A (CCNA2 gene 4q25-q31) is required for DNA synthesis during the S phase and progression through the G2/M transition. Cyclin E (CCNE1 gene in 19q12) is expressed in the middle of G1 and ends at the beginning of the S phase, and cyclin B1 (CCNB1 gene in 5q12) is crucial to drive cells into mitosis phase.
CCND1 amplification and Cyclin D1 overexpression have been reported to be a frequent event in several tumors, including head and neck cancers, and are related to poor survival [20, 52,53,54,55]. Cyclin E and cyclin B1 overexpression may lead to accelerated G1/S transition or even to premature entry into mitosis, contributing to increased chromosomal instability [56] and abnormal cell proliferation. Cyclin A, E, or B1 overexpression have been found to be adverse prognostic factors in oral potentially malignant disorders and oral malignancies [51, 56,57,58,59,60].
Genetic instability is one of the hallmarks of cancer and is a known process for tumor development. Spindle assembly checkpoint (SAC) is one of the most important checkpoints that controls cell division and prevents genetic instability. During this phase, when errors in the attachment of chromosomes are detected, there is formation of an inhibitory complex, called mitotic checkpoint complex (MCC), composed by mitotic checkpoint proteins such as Mad2, Bub3, or BubR1 [61]. This complex prevents the function of other proteins such as CDC20 that normally lead to activation of the anaphase-promoting complex/cyclosome (APC/C), and the 26S proteasome degradation of securin and cyclin B, preventing premature anaphase and aneuploidy due to deregulation of chromosomal alignment and separation (◘ Fig. 14.4, adapted from Teixeira et al. 2014) [61]. After normal sister chromatid separation and anaphase onset, the destruction of securin and cyclin B promotes the exit from mitosis and the beginning of interphase [61,62,63].

Signaling pathway of the spindle assembly checkpoint (SAC). An inappropriate attached kinetochore activates the spindle assembly checkpoint via an association of Mad2, Bub3, BubR1, and Cdc20 [61]
Among the proteins mentioned, CDC20 and BubR1 have been found overexpressed and related to reduced survival rates in OSCC [64, 65].
-
Wingless-Related Integration Site (WNT)
WNT signaling pathway is an important signaling pathway composed of several proteins including WNT ligands, the protein AXIN, and APC. During an non-activated phase a complex including AXIN, APC, and glycogen synthase kinase 3b sequesters β-catenin leading to their proteasomal degradation. By contrast, in the presence of a WNT ligand this complex is attached to the cell membrane leaving β-catenin free into cytoplasm and nucleus with the activation of several Wnt target genes, resulting in cell proliferation, tumor growth, and a stem cell phenotype [66].
Deregulation of the Wnt/β-catenin pathway lead to the carcinogenesis of many types of human cancers. Recently, other proteins were associated with this pathway including the Fat1 protein, a cadherin-like protein, and the NOCTH , a tumor suppressor gene, in oral cancer. FAT1 mutations in multiple cancer types suggests that FAT1 is a major cause of Wnt pathway activation in several human cancers. The inactivation of FAT1 by mutation has been reported to increase the Wnt signaling and tumor progression carrying an adverse prognosis in patients with head and neck cancers) [67].
-
NOTCH
NOTCH signaling is an evolutionarily conserved pathway already present in unicellular eukaryotic cells and in multicellular organisms, regulating cell proliferation, apoptosis, and differentiation. In humans, the NOTCH family is composed of four receptors (NOTCH1–4) and five ligands (JAGGED1 and 2, and DLL1, 3, and 4) [68]. The binding of the ligand to NOTCH receptor leads to the proteolytic release of the NOTCH intracellular domain (NIC) by secretases and its translocation to the nucleus, starting the transcription of the NOTCH target genes [69].
Dysregulation of NOTCH pathway has been reported in several cancers, including non-small cell lung cancer, ovarian carcinomas, colon cancer, pancreatic cancer, osteosarcoma, T-cell acute lymphoblastic leukemia, and head and neck carcinoma [70, 71]. NOTCH-1 has been reported as the second most frequently mutated gene in head and neck carcinoma after TP53 [72]. This was reported especially in a Chinese population and was related to the use of high alcohol -containing beverages in China [70]. NOTCH-1 mutations were also found in oral potentially malignant disorders, suggesting a role for NOTCH receptor in early stages of oral carcinogenesis and OSCC progression [73, 74]. Molecular therapies directed to NOTCH pathway could be interesting, such as the γ-secretase inhibitor (GSI), a pharmacological agent, which is capable of blocking NOTCH activation, preventing the in vitro growth of OSCC cells and resulting in the delay of tumorigenesis [75].
14.2.4 Invasion and Angiogenesis
Many oral cancers show an invasive phenotype and are capable of metastasis, especially to the regional lymph nodes. Regional spread occurs in more than one-third of the cases [76]. Invasion and metastatic dissemination are sequential processes in which, with acquired capacities, tumor cells escape from their tissue of origin, enter the stroma, and travel to distant sites. As part of such acquired capabilities, tumor cells must lose their surface adhesion molecules, which bind them to their own tissue, must be capable of migrating into and through the connective tissue and must be capable of entering lymphatic or vascular channels to escape to other locations.
A group of molecules play a key role in intercellular adhesion of keratinocytes in the oral epithelium.
-
Cadherins, Claudins, and Occludins
Cadherins are a family of junctional cell-surface glycoproteins commonly represented by E-cadherin , a 120-kDa transmembrane glycoprotein encoded by the CDH1 gene located on chromosome 16q22.1. E-cadherin is also involved in the transduction of signals controlling various cellular events, including polarity, differentiation, cell growth, and cell migration [77]. Reduced expression of E-Cadherin has been found in oral cancers and was related to tumor progression, dissemination, and poor prognosis [77]. Another group of adhesion molecules belongs to tight junctions . These form intercellular junctional complexes located at the apical side of the lateral membranous surface cells and are important in maintaining cell polarity. This group of proteins includes claudins and occludins. Their deregulation has been reported in a variety of cancers, including oral squamous cell carcinomas, and is related with poor survival rates [78, 79].
Tumor cells and specially tumor microenvironment (TME) cells, such as tumor-associated fibroblasts, can produce factors that stimulate the production of collagenases such as matrix metalloproteinase (MMPs) . There are several types of MMPs and related proteins such as extracellular matrix metalloproteinase inducer (EMMPRIN) , which increase their expression and function or decreases such as TIMPs. EMMPRIN and MMP-9 have been related to tumor progression and invasion in oral cancers [80, 81].
-
EMMPRIN and MMP-9
One of these molecules is the EMMPRIN , also known as CD147. It is a highly glycosylated transmembrane protein that has shown a strong capacity to induce the expression of matrix metalloproteinases . EMMPRIN also contributes to cell adhesion modulation, tumor growth, invasion, and angiogenesis. Overexpression of EMMPRIN was found in OSCC, with an autocrine and paracrine positive effect for MMP production enhancing tumor invasion and dissemination [80] (◘ Fig. 14.5).

The stimulation effect of EMMPRIN on fibroblast and endothelial cells resulting in MMP or VEGF production promoting tumor cell invasion
Matrix metalloproteinase 9 (MMP-9), also known as gelatinase-B or type IV collagenase, is a 92-kDa zinc-dependent endopeptidase, involved in the degradation of the extracellular matrix. Overexpression of MMP-9 could promote the degradation of the basement membrane and the extracellular matrix, in particular collagen IV, contributing to tumor invasion. Overexpression of MMP-9 has been associated with the lymph node and distant metastasis in oral squamous cell carcinomas and is related to adverse overall survival [81].
Neoangiogenesis has been considered an important hallmark of tumorigenesis and tumor dissemination. Since the work of Folkman, it is well known that solid tumors cannot exceed 1–2 mm3 without the existence of a new blood supply formed from the adjacent connective tissue vessels [80, 82]. Several molecules participate in this angiogenic process, including vascular endothelial growth factor (VEGF) and its receptors VEGFRs [83].
-
VEGF and VEGFRs
VEGF is a heparin glycoprotein produced by tumor cells, and also by peritumoral endothelial cells and inflammatory cells such as macrophages in the presence of hypoxic-inducing factors (HIF). VEGF can increase vascular permeability, stimulate production of proteases, migration, proliferation, and differentiation of endothelial cells and capillary tube formation, increasing vascular support within tumor cells [83, 84]. The majority of solid cancers overexpress this factor and this is associated with a higher risk of recurrence, metastasis, and poor survival [83].
VEGF effects are mediated by vascular endothelial growth factor receptors (VEGFR) composed of 3 tyrosine kinase receptors including VEGFR-1 (Flt-1), VEGFR-2 (KDR / flk-1), and VEGFR-3 (flt-4) [83,84,85].
VEGFR-1 and VEGFR-2 are located on the vascular endothelial cells and macrophages, while VEGFR-3 is found mostly in the endothelium of lymphatic vessels. Interestingly, all can be found in the cells of several tumors, including head and neck cancers. Each receptor could contribute differently to tumorigenesis. VEGFR-1 has been related to the infiltration of macrophages and increases in MMP-9 in lung tissues before the appearance of lung metastasis. VEGFR-2 has been related to the recruitment of hematopoietic and endothelial precursor cells from bone marrow, while VEGFR-3 is mostly involved in tumoral lymphangiogenesis, contributing to its metastatic effect [83,84,85,86].
Targeting tumoral angiogenesis is an attractive therapeutic approach. Several antibodies and selective inhibitors have been studied and validated and some are already in clinical use. Bevacizumab is a VEGF antibody approved for anti-angiogenic therapy for several types of cancers, while vandetanib, sorafenib, and sunitinib are tyrosine kinase inhibitors [85, 87] (see ► Chapter 27).
-
Podoplanin
Podoplanin is a transmembrane glycoprotein encoded by the PDPN gene and was named after its discovery in kidney podocytes [88, 89]. It is a classic marker of lymphatic endothelium but not blood vessel endothelium. Some recent reports gave visibility to this protein as a predictive marker of malignant transformation in oral leukoplakia. Podoplanin is normally not expressed in normal oral epithelium and when expressed, sometimes in cluster points, it indicates that it could be a stem cell marker, or even in a diffuse pattern it may represent a sign of an increased risk of malignant transformation . Moreover, podoplanin could promote cell motility and migration of tumor cells to the invasive front of the tumors, many times working along with metalloproteins such as MMP-9 [90,91,92]. As a lymphatic endothelium marker, podoplanin has been related with lymph node metastasis [93, 94]. Overexpression of podoplanin has been reported in cancers of the lung, breast, skin, larynx, uterine cervix, esophagus, germ cell tumors, as well as head and neck cancers including oral cancers and is related with poor prognosis [81, 95,96,97,98].
Interestingly, molecular therapies against PDPN have been evaluated, including antibodies against PDPN with promising results in preclinical studies [99].
14.2.5 Reprograming Cellular Energetics and Evading Immune Destruction
Malignant cells can recruit and corrupt adjacent non-transformed cells and become involved in interactions that create the tumor microenvironment (TME) . Such interactions modify the tumor stroma and, ultimately, promote regulation of energy availability, angiogenesis, and tumor metastasis [6, 100, 101]. Moreover, in addition to the fibroblasts, cells of tumor vasculature and lymphatics, the non-transformed cells include cells of the immune system, suggesting a relation between tumor cells and the immune system [6].
Proliferative tumors have also developed energy pathways responsible for sustained tumor growth and survival in adverse conditions. This is obtained essentially by glycolysis, even in an aerobic environment, a condition known as “Warburg effect ” that results in lactic acid secretion in the stroma [101, 102]. To compensate the missing energetic efficiency of aerobic energy, tumor cells use glucose transporters such as GLUT1, which increase glucose transportation to the cytoplasm. In the presence of hypoxia, HIF1 and HIF2 also upregulate glycolysis. As a consequence, high concentration of lactic acid is produced, which is exported out of the cells by monocarboxylate transporters (MCTs). Deregulated expression of MCT1 and MCT4 has been reported in many cancers including oral cancers and has been correlated with poor prognosis [6, 103].
For many years there have been reports of infiltration of immune cells in the tumor microenvironment but without any known significance. Nowadays, it is believed that cancers, such oral cancer, can avoid their identification by immune cells, escaping any host defense mechanisms. A hypothesis for this could be the related remodulation of tumor cells in order to eliminate some high immunogenic clones, a process called immunoediting. Deficiencies in the CD8+ and CD4+ T lymphocytes and NK cells had been related with increased tumor incidence [6]. Other explanations could include the capacity of tumor cells to produce immunosuppressive factors. In particular, special attention has been put on some molecules that can control the function of T-cells, programmed death protein one (PD-1) and its ligands, programmed death ligand one and two (PD-L1, PD-L2 ). PD-1 and PD-L1 are immune-checkpoint proteins that primarily function to limit the effector function of T-cells in peripheral tissues during inflammatory responses and limit autoimmunity. These are considered as one of the immune evasion mechanisms for cancer. Recently, strategies to help improve the efficacy of the immune system against cancer represent an important breakthrough in cancer treatment. In humans, clinical trials with anti-programmed death (PD)-1/PD-ligand 1 (L1) monoclonal antibodies have shown objective clinical activity of these agents (e.g., nivolumab , pembrolizumab ) in several malignancies, including melanoma, non-small-cell lung cancer, bladder cancer, and squamous cell head and neck cancer [104,105,106].
Other immune cells that could be of importance in tumor microenvironment (TME) are the tumor-associated macrophages (TAMs) . In this context, macrophages contribute to tumor progression through wound-healing and tissue-repair mechanisms that allow cancerous tissues to repair damages caused by low oxygen tension and acidic pH that result from metabolic reprogramming of tumor cells and vascular abnormalities of the tumor. TAMs belong to the monocyte-macrophage lineage and, according to the stimulus, there are two main phenotypes of macrophages: the pro-inflammatory (anti-tumoral) M1 and the immunosuppressive (pro-tumoral) M2 macrophages [107]. Soluble tumor-derived factors initiate the polarization of macrophages into M2 macrophages, leading to the expression of molecules that support angiogenesis, immunosuppression , tumor growth, and metastasis. M2 macrophage phenotypes have been identified in oral cancer and were related with more aggressive tumors [108].
14.3 HPV+ve Pathways for Oral Cancer
A new group of head and neck cancers, referred to as HPV + ve cancers, are now identified and related with the identification of human papillomavirus in tumor cells. They are present in cancers located mainly in the oropharynx , especially in the tonsillar crypt epithelium (47%). By contrast, HPV-related SCC in oral cavity correspond only to 3.9% of all tumors [109]. There are more than 200 genotypes of HPV, and some are related with tumor carcinogenesis – these are the high-risk HPVs. HPV-16 type is the most common genotype found in these tumors. The virus contains a circular double-stranded DNA with several areas such E6 and E7 oncogenes . These proteins, E6 and E7 can bind and inhibit two proteins p53 and pRb. In these HPV + ve tumors no mutations are found on these tumor suppressor genes, but their function is inhibited by E6 and E7 viral proteins which represent an early event in oral carcinogenesis [6]. The same is observed in the CDKN2A, the gene encoding p16, without mutations or deletions on HPV + ve cancer but overexpressed in these tumors. In the view of this, p16 overexpression in a surrogate marker of HPV-16 infection of tumor cells in oral cancers. Interestingly, genetic studies have shown that PI3K pathway genes are commonly altered in HPV + ve cancers with mutations or amplifications of the gene PIK3CA [10, 109]. Knowledge of these molecular characteristics is important in the selection of a treatment plan for patients with HPV + ve tumors as they have a more favorable prognosis than tumors that harbor TP53 mutations or p16 loss, generally HPV -ve tumors. Recent genomic analysis has identified two subgroups of HPV + ve tumors – one having a mesenchymal and immunological signature (HPV-IMU), and the other having a keratinocyte differentiation and oxidative stress genes signature (HPV-KRT) [110].
Warning
Some of the biomarkers published in the literature are neither necessary nor sufficient for the evolution of a cancer.
14.4 Prognostic Biomarkers
The genes and proteins involved in the multiple pathways most often disrupted in oral cancer permit the possible use of such alterations as biomarkers of prognosis. We present in ◘ Table 14.1 several genes or proteins that have been reported as usefulness prognostic biomarkers in oral cancers [108, 109, 111,112,113,114,115].
14.5 Conclusion
Oral carcinogenesis is a multistep process where many biological pathways could be affected. These pathways are not always similar or common to all patients. Subgroups of head and neck cancers have been identified, including patients with HPV infection (HPV + ve), tumors with aneuploidy status, tumors with few copy number alterations, and some pathways have been linked to some subclasses of tumors. This highlights the importance of the molecular knowledge of the biological tumoral phenotype for each patient and they could function as biomarkers of disease, not only for early diagnosis but also for predicting prognosis as presented in this chapter. The significant markers include p53, EGFR, p16, cyclin A, or Akt/mTOR pathways.
References
Khan Z, Bisen PS. Oncoapoptoticsignaling and deregulated target genes in cancers: special reference to oral cancer. Biochim Biophys Acta. 2013;1836(1):123–45.
Hahn WC, Weinberg RA. Rules for making human tumors cells. N Engl J Med. 2002;347:1593–603.
Ha PK, Benoit NE, Yochem R, Sciubba J, Zahurak M, Sidransky D, Pevsner J, Westra WH, Califano J. A transcriptional progression model for head and neck cancer. Clin Cancer Res. 2003;9:3058–64.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Bernstein JM, Bernstein CR, West CM, Homer JJ. Molecular and celular processes underlying the hallmarks of head and neck cancer. Eur Arch Otorhinolaryngol. 2013;270(10):2585–93.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, Butterfoss D, Xiang D, Zanation A, Yin X, Shockley WW, Weissler MC, Dressler LG, Shores CG, Yarbrough WG, Perou CM. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5(5):489–500.
Smeets SJ, Brakenhoff RH, Ylstra B, van Wieringen WN, van de Wiel MA, Leemans CR, Braakhuis BJ. Genetic classification of oral and oropharyngeal carcinomas identifies subgroups with a different prognosis. Cell Oncol. 2009;31(4):291–300.
Slebos RJ, Jehmlich N, Brown B, Yin Z, Chung CH, Yarbrough WG, Liebler DC. Proteomic analysis of oropharyngeal carcinomas reveals novel HPV-associated biological pathways. Int J Cancer. 2013;132(3):568–79.
Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82.
Hermsen M, Guervós MA, Meijer G, Baak J, van Diest P, Marcos CA, Sampedro A. New chromosomal regions with high-level amplifications in squamous cell carcinomas of the larynx and pharynx, identified by comparative genomic hybridization. J Pathol. 2001;194(2):177–82.
Sinevici N, O'sullivan J. Oral cancer: deregulated molecular events and their use as biomarkers. Oral Oncol. 2016;61:12–8.
Lane DP. Cancer. P53, guardian of the genome. Nature. 1992;358:15–6.
Partridge M, Costea DE, Huang X. The changing face of p53 in head and neck cancer. Int J Oral Maxillofac Surg. 2007;36(12):1123–38.
Li VD, Li KH, Li JT. TP53 mutations as potential prognostic markers for specific cancers: analysis of data from The Cancer Genome Atlas and the International Agency for Research on Cancer TP53 database. J Cancer Res Clin Oncol. 2019;145(3):625–36.
Oliveira LR, Ribeiro-Silva A, Costa JP, Simões AL, Matteo MA, Zucoloto S. Prognostic factors and survival analysis in a sample of oral squamous cell carcinoma patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106(5):685–95.
Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. Tp53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–61.
van Houten VM, Tabor MP, van den Brekel MW, Kummer JA, Denkers F, Dijkstra J, Leemans R, van der Waal I, Snow GB, Brakenhoff RH. Mutated p53 as a molecular marker for the diagnosis of head and neck cancer. J Pathol. 2002;198:476–86.
Warnakulasuriya S. Lack of molecular markers to predict malignant potential of oral precancer. J Pathol. 2000;190(4):407–9.
Lu XJD, Liu KYP, Soares RC, Thomson T, Prisman E, Wu J, Poh CF. Potential clinical implications of HPV status and expressions of p53 and cyclin D1 among oropharyngeal cancer patients. J Oral Pathol Med. 2018;47(10):945–53.
Monteiro LS, Diniz-Freitas M, Warnakulasuriya S, Garcia-Caballero T, Forteza J, Fraga M. An immunohistochemical score to predict the outcome for oral squamous cell carcinoma. J Oral Pathol Med. 2018;47(4):375–81.
Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9–22.
Monteiro L, Ricardo S, Delgado M, Garcez F, do Amaral B, Lopes C. Phosphorylated EGFR at tyrosine 1173 correlates with poor prognosis in oral squamous cell carcinomas. Oral Dis. 2014;20(2):178–85.
O-charoenrat P, Rhys-Evans PH, Archer DJ, Eccles SA. C-erbB receptors in squamous cell carcinomas of the head and neck: clinical significance and correlation with matrix metalloproteinases and vascular endothelial growth factors. Oral Oncol. 2002;38:73–80.
Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5(4):311–6.
Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345(26):1890–900.
Grandis JR, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90(11):824–32.
Monteiro LS, Diniz-Freitas M, Garcia-Caballero T, Warnakulasuriya S, Forteza J, Fraga M. Combined cytoplasmic and membranous EGFR and p53 overexpression is a poor prognostic marker in early stage oral squamous cell carcinoma. J Oral Pathol Med. 2012;41(7):559–67.
Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–84.
Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, Hilbe W, Schwentner I, Sprinzl GM, Utermann G, Zwierzina H. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;42:109–11.
Temam S, Kawaguchi H, El-Naggar AK, Jelinek J, Tang H, Liu DD, Lang W, Issa JP, Lee JJ, Mao L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25:2164–70.
Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD, Gooding WE, Furnari FB, Grandis JR. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12:5064–73.
Sauer T, Guren MG, Noren T, Dueland S. Demonstration of EGFR gene copy loss in colorectal carcinomas by fluorescence in situ hybridization (FISH): a surrogate marker for sensitivity to specific anti-EGFR therapy? Histopathology. 2005;47:560–4.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37.
Sacco AG, Worden FP. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. Onco Targets Ther. 2016;9:1927–43.
Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78.
Venook AP. Epidermal growth factor receptor-targeted treatment for advanced colorectal carcinoma. Cancer. 2005;103:2435–46.
Gao P, Li C, Chang Z, Wang X, Xuan M. Carcinoma associated fibroblasts derived from oral squamous cell carcinoma promote lymphangiogenesis via c-Met/PI3K/AKT in vitro. Oncol Lett. 2018;15(1):331–7.
Sun Z, Liu Q, Ye D, Ye K, Yang Z, Li D. Role of c-Met in the progression of human oral squamous cell carcinoma and its potential as a therapeutic target. Oncol Rep. 2018;39(1):209–16.
Cheng CM, Shiah SG, Huang CC, Hsiao JR, Chang JY. Up-regulation of miR-455-5p by the TGF-β-SMAD signalling axis promotes the proliferation of oral squamous cancer cells by targeting UBE2B. J Pathol. 2016;240(1):38–49.
Sivadas VP, George NA, Kattoor J, Kannan S. Novel mutations and expression alterations in SMAD3/TGFBR2 genes in oral carcinoma correlate with poor prognosis. Genes Chromosomes Cancer. 2013;52(11):1042–52.
Hwang YS, Park KK, Chung WY. Stromal transforming growth factor-beta 1 is crucial for reinforcing the invasive potential of low invasive cancer. Arch Oral Biol. 2014;59(7):687–94.
Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45(4–5):324–34.
Xu K, Liu P, Wei W. mTORsignaling in tumorigenesis. Biochim Biophys Acta. 2014;1846(2):638–54.
Marques AE, Elias ST, Porporatti AL, Castilho RM, Squarize CH, De Luca Canto G, Guerra EN. mTOR pathway protein immunoexpression as a prognostic factor for survival in head and neck cancer patients: a systematic review and meta-analysis. J Oral Pathol Med. 2016;45(5):319–28.
Monteiro LS, Delgado ML, Ricardo S, Garcez F, do Amaral B, Warnakulasuriya S, Lopes C. Phosphorylated mammalian target of rapamycin is associated with na adverse outcome in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115(5):638–45.
Simpson DR, Mell LK, Cohen EE. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015;51(4):291–8.
Vitale-Cross L, Molinolo AA, Martin D, Younis RH, Maruyama T, Patel V, Chen W, Schneider A, Gutkind JS. Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prev Res (Phila). 2012;5(4):562–73.
Murugan AK, Munirajan AK, Tsuchida N. Ras oncogenes in oral cancer: the past 20 years. Oral Oncol. 2012;48(5):383–92.
Saranath D, Chang SE, Bhoite LT, Panchal RG, Kerr IB, Mehta AR, Johnson NW, Deo MG. High frequency mutation in codons 12 and 61 of H-ras oncogene in chewingtobacco-related human oral carcinoma in India. Br J Cancer. 1991;63(4):573–8.
Saarilahti K, Kajanti M, Kouri M, Aaltonen LM, Franssila K, Joensuu H. Cyclin A and Ki-67 expression as predictors for locoregional recurrence and outcome in laryngeal cancer patients treated with surgery and postoperative radiotherapy. Int J Radiat Oncol Biol Phys. 2003;57(4):986–95.
Hanken H, Gröbe A, Cachovan G, Smeets R, Simon R, Sauter G, Heiland M, Blessmann M. CCND1 amplification and cyclin D1 immunohistochemical expression in head and neck squamous cell carcinomas. Clin Oral Investig. 2014;18(1):269–76.
Huang SF, Cheng SD, Chuang WY, Chen IH, Liao CT, Wang HM, Hsieh LL. Cyclin D1 overexpression and poor clinical outcomes in Taiwanese oral cavity squamous cell carcinoma. World J Surg Oncol. 2012;10:40.
Noorlag R, van Kempen PM, Stegeman I, Koole R, van Es RJ, Willems SM. The diagnostic value of 11q13 amplification and protein expression in the detection of nodal metastasis from oral squamous cell carcinoma: a systematic review and meta-analysis. Virchows Arch. 2015;466(4):363–73.
Zhao Y, Yu D, Li H, Nie P, Zhu Y, Liu S, Zhu M, Fang B. Cyclin D1 overexpression is associated with poor clinicopathological outcome and survival in oral squamous cell carcinoma in Asian populations: insights from a meta-analysis. PLoS One. 2014;9(3):e93210.
Fraczek M, Wozniak Z, Ramsey D, Zatonski T, Krecicki T. Clinicopathologic significance and prognostic role of cyclin E and cyclin A expression in laryngeal epithelial lesions. Acta Otolaryngol. 2008;128(3):329–34.
Chen HM, Yen-Ping Kuo M, Lin KH, Lin CY, Chiang CP. Expression of cyclin A is related to progression of oral squamous cell carcinoma in Taiwan. Oral Oncol. 2003;39(5):476–82.
Tandon R, Cunningham LL, White DK, Herford AS, Cicciu M. Overexpression of cyclin A in oral dysplasia: an international comparison and literature review. Indian J Cancer. 2014;51(4):502–5.
Ko MT, Su CY, Huang SC, Chen CH, Hwang CF. Overexpression of cyclin E messenger ribonucleic acid in nasopharyngeal carcinoma correlates with poor prognosis. J Laryngol Otol. 2009;123(9):1021–6.
Hoffmann TK, Trellakis S, Okulicz K, Schuler P, Greve J, Arnolds J, Bergmann C, Bas M, Lang S, Lehnerdt G, Brandau S, Mattheis S, Scheckenbach K, Finn OJ, Whiteside TL, Sonkoly E. Cyclin B1 expression and p53 status in squamous cell carcinomas of the head and neck. Anticancer Res. 2011;31(10):3151–7.
Teixeira JH, Silva PM, Reis RM, Moura IM, Marques S, Fonseca J, Monteiro LS, Bousbaa H. An overview of the spindle assembly checkpoint status in oral cancer. Biomed Res Int. 2014;2014:145289.
Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93.
Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–85.
Moura IM, Delgado ML, Silva PM, Lopes CA, do Amaral JB, Monteiro LS, Bousbaa H. High CDC20 expression is associated with poor prognosis in oral squamous cell carcinoma. J Oral Pathol Med. 2014;43(3):225–31.
Teixeira JH, Silva P, Faria J, Ferreira I, Duarte P, Delgado ML, Queirós O, Moreira R, Barbosa J, Lopes CA, do Amaral JB, Monteiro LS, Bousbaa H. Clinicopathologicsignificanceof BubR1 and Mad2 overexpression in oral cancer. Oral Dis. 2015;21(6):713–20.
Shiah SG, Shieh YS, Chang JY. The role of WntSignaling in squamous cell carcinoma. J Dent Res. 2016;95(2):129–34.
Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan Ş, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, Vakiani E, Solit D, Singh B, Ganly I, Liau L, Cloughesy TC, Mischel PS, Mellinghoff IK, Chan TA. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45(3):253–61.
Kayamori K, Katsube K, Sakamoto K, Ohyama Y, Hirai H, Yukimori A, Ohata Y, Akashi T, Saitoh M, Harada K, Harada H, Yamaguchi A. NOTCH3 is induced in cancer-associated fibroblasts and promotes angiogenesis in oral squamous cell carcinoma. PLoS One. 2016;11(4):e0154112.
Hijioka H, Setoguchi T, Miyawaki A, Gao H, Ishida T, Komiya S, Nakamura N. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int J Oncol. 2010;36(4):817–22.
Yap LF, Lee D, Khairuddin A, Pairan MF, Puspita B, Siar CH, Paterson IC. The opposing roles of NOTCH signalling in head and neck cancer: a mini review. Oral Dis. 2015;21(7):850–7.
Song X, Xia R, Li J, Long Z, Ren H, Chen W, Mao L. Common and complex Notch1 mutations in Chinese oral squamous cell carcinoma. Clin Cancer Res. 2014;20:701–10.
Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Treviño L, Drummond JA, Muzny DM, Wu Y, Wood LD, Hruban RH, Westra WH, Koch WM, Califano JA, Gibbs RA, Sidransky D, Vogelstein B, Velculescu VE, Papadopoulos N, Wheeler DA, Kinzler KW, Myers JN. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–7.
Izumchenko E, Sun K, Jones S, Brait M, Agrawal N, Koch W, McCord CL, Riley DR, Angiuoli SV, Velculescu VE, Jiang WW, Sidransky D. Notch1 mutations are drivers of oral tumorigenesis. Cancer Prev Res (Phila). 2015;8:277–86.
Yoshida R, Nagata M, Nakayama H, Niimori-Kita K, Hassan W, Tanaka T, Shinohara M, Ito T. The pathological significance of Notch1 in oral squamous cell carcinoma. Lab Investig. 2013;93(10):1068–81.
Zhang JP, Qin HY, Wang L, Liang L, Zhao XC, Cai WX, Wei YN, Wang CM, Han H. Overexpression of Notch ligand Dll1 in B16 melanoma cells leads to reduced tumor growth due to attenuated vascularization. Cancer Lett. 2011;309:220–7.
Monteiro LS, Amaral JB, Vizcaíno JR, Lopes CA, Torres FO. A clinical-pathological and survival study of oral squamous cell carcinomas from a population of the North of Portugal. Med Oral Patol Oral Cir Bucal. 2014;19(2):e120–6.
Diniz-Freitas M, García-Caballero T, Antúnez-López J, Gándara-Rey JM, García-García A. Reduced E-cadherin expression is an indicator of unfavourable prognosis in oral squamous cell carcinoma. Oral Oncol. 2006;42(2):190–200.
DE Vicente JC, Fernández-Valle Á, Vivanco-Allende B, Santamarta TR, Lequerica-Fernández P, Hernández-Vallejo G, Allonca-Campa E. The prognostic role of claudins −1 and −4 in oral squamous cell carcinoma. Anticancer Res. 2015;35(5):2949–59.
Lourenço SV, Coutinho-Camillo CM, Buim ME, Pereira CM, Carvalho AL, Kowalski LP, Soares FA. Oral squamous cell carcinoma: status of tight junction claudins in the different histopathological patterns and relationship with clinical parameters. A tissue-microarray-based study of 136 cases. J Clin Pathol. 2010;63(7):609–14.
Monteiro LS, Delgado ML, Ricardo S, Garcez F, do Amaral B, Pacheco JJ, Lopes C, Bousbaa H. EMMPRIN expression in oral squamous cell carcinomas: correlation with tumor proliferation and patient survival. Biomed Res Int. 2014;2014:905680.
Monteiro LS, Delgado ML, Ricardo S, do Amaral B, Salazar F, Pacheco JJ, Lopes CA, Bousbaa H, Warnakulasuryia S. Prognostic significance of CD44v6, p63, podoplanin and MMP-9 in oral squamous cell carcinomas. Oral Dis. 2016;22(4):303–12.
Folkman J. Tumor angiogenesis. Therapeutic implications. N Engl J Med. 1971;285:1182–6.
Zang J, Li C, Zhao LN, Shi M, Zhou YC, Wang JH, Li X. Prognostic value of vascular endothelial growth factor in patients with head and neck cancer: a meta-analysis. Head Neck. 2013;35(10):1507–14.
Neuchrist C, Erovic BM, Handisurya A, Steiner GE, Rockwell P, Gedlicka C, Burian M. Vascular endothelial growth factor receptor 2 (VEGFR2) expression in squamous cell carcinomas of the head and neck. Laryngoscope. 2001;111(10):1834–41.
Vassilakopoulou M, Psyrri A, Argiris A. Targeting angiogenesis in head and neck cancer. Oral Oncol. 2015;51(5):409–15.
Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, Shipley JM, Senior RM, Shibuya M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2(4):289–300.
Hsu HW, Wall NR, Hsueh CT, Kim S, Ferris RL, Chen CS, Mirshahidi S. Combination antiangiogenic therapy and radiation in head and neck cancers. Oral Oncol. 2014;50(1):19–26.
Preuss SF, Anagiotos A, Seuthe IM, Drebber U, Wedemeyer I, Kreppel M, Semrau R, Eslick GD, Klussmann JP, Huebbers CU. Expression of podoplanin and prognosis in oropharyngeal cancer. Eur Arch Otorhinolaryngol. 2015;272(7):1749–54.
Kahn HJ, Marks A. A new monoclonal antibody, D2-40, for detection of lymphatic invasion in primary tumors. Lab Investig. 2002;82(9):1255–7.
Ochoa-Alvarez JA, Krishnan H, Pastorino JG, Nevel E, Kephart D, Lee JJ, Retzbach EP, Shen Y, Fatahzadeh M, Baredes S, Kalyoussef E, Honma M, Adelson ME, Kaneko MK, Kato Y, Young MA, Deluca-Rapone L, Shienbaum AJ, Yin K, Jensen LD, Goldberg GS. Antibody and lectin target podoplanin to inhibit oral squamous carcinoma cell migration and viability by distinct mechanisms. Oncotarget. 2015;6(11):9045–60.
Li YY, Zhou CX, Gao Y. Podoplanin promotes the invasion of oral squamous cell carcinoma in coordination with MT1-MMP and Rho GTPases. Am J Cancer Res. 2015;5(2):514–29.
Inoue H, Miyazaki Y, Kikuchi K, Yoshida N, Ide F, Ohmori Y, Tomomura A, Sakashita H, Kusama K. Podoplanin promotes cell migration via the EGF-Src-Cas pathway in oral squamous cell carcinoma cell lines. J Oral Sci. 2012;54(3):241–50.
Huber GF, Fritzsche FR, Züllig L, Storz M, Graf N, Haerle SK, Jochum W, Stoeckli SJ, Moch H. Podoplanin expression correlates with sentinel lymph node metastasis in early squamous cell carcinomas of the oral cavity and oropharynx. Int J Cancer. 2011;129(6):1404–9.
Cueni LN, Hegyi I, Shin JW, Albinger-Hegyi A, Gruber S, Kunstfeld R, Moch H, Detmar M. Tumorlymphangiogenesis and metastasis to lymph nodes induced by cancer cell expression of podoplanin. Am J Pathol. 2010;177(2):1004–16.
Swain N, Kumar SV, Routray S, Pathak J, Patel S. Podoplanin-a novel marker in oral carcinogenesis. Tumour Biol. 2014;35(9):8407–13.
Yuan P, Temam S, El-Naggar A, Zhou X, Liu DD, Lee JJ, Mao L. Overexpression of podoplanin in oral cancer and its association with poor clinical outcome. Cancer. 2006;107(3):563–9.
Vormittag L, Thurnher D, Geleff S, Pammer J, Heiduschka G, Brunner M, Grasl MC, Erovic BM. Co-expression of Bmi-1 and podoplanin predicts overall survival in patients with squamous cell carcinoma of the head and neck treated with radio(chemo)therapy. Int J Radiat Oncol Biol Phys. 2009;73(3):913–8.
Kreppel M, Drebber U, Wedemeyer I, Eich HT, Backhaus T, Zöller JE, Scheer M. Podoplanin expression predicts prognosis in patients with oral squamous cell carcinoma treated with neoadjuvantradiochemotherapy. Oral Oncol. 2011;47(9):873–8.
Retzbach EP, Sheehan SA, Nevel EM, Batra A, Phi T, Nguyen ATP, Kato Y, Baredes S, Fatahzadeh M, Shienbaum AJ, Goldberg GS. Podoplanin emerges as a functionally relevant oral cancer biomarker and therapeutic target. Oral Oncol. 2018;78:126–36.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52.
Yu M, Chen S, Hong W, Gu Y, Huang B, Lin Y, Zhou Y, Jin H, Deng Y, Tu L, Hou B, Jian Z. Prognostic role of glycolysis for cancer outcome: evidence from 86 studies. J Cancer Res Clin Oncol. 2019;145(4):967–99.
Vander Heiden M, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Romero-Garcia S, Moreno-Altamirano M, Prado-Garcia H, Sánchez-García F. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52.
Festino L, Botti G, Lorigan P, Masucci GV, Hipp JD, Horak CE, Ascierto PA. Cancer treatment with anti-PD-1/PD-L1 agents: is PD-L1 expression a biomarker for patient selection? Drugs. 2016;76(9):925–45.
Pai SI, Zandberg DP, Strome SE. The role of antagonists of the PD-1:PD-L1/PD-L2 axis in head and neck cancer treatment. Oral Oncol. 2016;61:152–8.
Zandberg DP, Strome SE. The role of the PD-L1:PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2014;50(7):627–32.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86.
Mori K, Hiroi M, Shimada J, Ohmori Y. Infiltration of m2 tumor-associated macrophages in oral squamous cell carcinoma correlates with tumor malignancy. Cancers (Basel). 2011;3(4):3726–39.
Castellsagué X, Alemany L, Quer M, Halec G, Quirós B, Tous S, Clavero O, Alòs L, Biegner T, Szafarowski T, Alejo M, Holzinger D, Cadena E, Claros E, Hall G, Laco J, Poljak M, Benevolo M, Kasamatsu E, Mehanna H, Ndiaye C, Guimerà N, Lloveras B, León X, Ruiz-Cabezas JC, Alvarado-Cabrero I, Kang CS, Oh JK, Garcia-Rojo M, Iljazovic E, Ajayi OF, Duarte F, Nessa A, Tinoco L, Duran-Padilla MA, Pirog EC, Viarheichyk H, Morales H, Costes V, Félix A, Germar MJ, Mena M, Ruacan A, Jain A, Mehrotra R, Goodman MT, Lombardi LE, Ferrera A, Malami S, Albanesi EI, Dabed P, Molina C, López-Revilla R, Mandys V, González ME, Velasco J, Bravo IG, Quint W, Pawlita M, Muñoz N, de Sanjosé S, Xavier Bosch F, ICO International HPV in Head and Neck Cancer Study Group. HPV involvement in head and neck cancers: comprehensive assessment of biomarkers in 3680 patients. J Natl Cancer Inst. 2016;108(6):djv403.
Zhang Y, Koneva LA, Virani S, Arthur AE, Virani A, Hall PB, Warden CD, Carey TE, Chepeha DB, Prince ME, McHugh JB, Wolf GT, Rozek LS, Sartor MA. Subtypes of HPV-positive head and neck cancers are associated with HPV characteristics, copy number alterations, PIK3CA mutation, and pathway signatures. Clin Cancer Res. 2016;22(18):4735–45.
Rivera C, Oliveira AK, Costa RAP, De Rossi T, PaesLeme AF. Prognostic biomarkers in oral squamous cell carcinoma: a systematic review. Oral Oncol. 2017;72:38–47.
Almangush A, Heikkinen I, Mäkitie AA, Coletta RD, Läärä E, Leivo I, Salo T. Prognostic biomarkers for oral tongue squamous cell carcinoma: a systematic review and meta-analysis. Br J Cancer. 2017;117(6):856–66.
Lakshminarayana S, Augustine D, Rao RS, Patil S, Awan KH, Venkatesiah SS, Haragannavar VC, Nambiar S, Prasad K. Molecular pathways of oral cancer that predict prognosis and survival: a systematic review. J Carcinog. 2018;17:7.
Peterle GT, Maia LL, Trivilin LO, de Oliveira MM, Dos Santos JG, Mendes SO, Stur E, Agostini LP, Rocha LA, Moysés RA, Cury PM, Nunes FD, Louro ID, Dos Santos M, da Silva AMÁ. PAI-1, CAIX, and VEGFA expressions as prognosis markers in oral squamous cell carcinoma. J Oral Pathol Med. 2018;47(6):566–74.
Götz C, Bissinger O, Nobis C, Wolff KD, Drecoll E, Kolk A. ALDH1 as a prognostic marker for lymph node metastasis in OSCC. Biomed Rep. 2018;9(4):284–90.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Monteiro, L., Warnakulasuriya, S. (2020). Molecular and Signaling Pathways During Oral Carcinogenesis. In: Warnakulasuriya, S., Greenspan, J. (eds) Textbook of Oral Cancer. Textbooks in Contemporary Dentistry. Springer, Cham. https://doi.org/10.1007/978-3-030-32316-5_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-32316-5_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-32315-8
Online ISBN: 978-3-030-32316-5
eBook Packages: MedicineMedicine (R0)