
Abstract
Green tea polyphenols, especially catechins, have shown great promise for use as anticancer agents. One such catechin, epigallocatechin-3 gallate (EGCG), is of particular interest due to mounting evidence of its ability to interrupt a variety of essential signaling pathways in cancer cells and target multiple tumor-specific proteins, and its synergistic effects when paired with commonly used anticancer therapies. However, EGCG has a number of disadvantages, e.g., low stability and poor bioavailability, as well as rapid metabolic transformations in vivo. We previously developed a prodrug of EGCG (Pro-EGCG or 1) which shows increased levels of stability, bioavailability, and biological activity in human cancer cells and xenografts as compared to EGCG. In order to potentially reduce the susceptibility of EGCG analogs to be methylated and thus inhibited by catechol-O-methyltransferase (COMT), we synthesized novel analogs and prodrugs where one or two hydroxyl or acetoxy groups, respectively, have been removed from the gallate ester moiety. Such prodrugs (2a and 4a) were reported recently to have potent antiproliferative, antiangiogenic, and antifibrotic properties. We previously also reported that synthetic EGCG analogs 4 and 6 were more potent AMPK activators than metformin and EGCG. Here we review the activity of 4 and 6 in inhibiting growth of uterine fibroid cells. We also review the potential of these novel EGCG analogs and prodrugs as anticancer drugs and discuss the potential mechanisms of action involved.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
14.1 Introduction
Cancer is an umbrella term for a group of diseases in which dysregulated cells proliferate. As of 2018, cancer was the world’s second most prevalent and fatal disease (World Health Organization 2018). There was an estimated 1,735,350 new cases of cancer in the United States in 2018, and it was estimated that 609,640 deaths will occur due to cancer in the United States in 2018 (Siegel et al. 2018). Using global data, experts have estimated that there will be 21.7 million new cancer cases and 13 million cancer fatalities in 2030 (National Cancer Institute 2018). Conventional cancer therapies such as surgery, chemotherapy, and radiotherapy have significant limitations, including a high-cost development of resistance, and adverse side effects. Therefore, there is a pressing need for new drugs and new therapeutic strategies that are safe, selective, and effective for cancer treatment. In recent years, many physiological compounds and natural products, including steroid hormones, vitamin analogs, cytokines, and plant phytochemicals, have demonstrated the ability to induce differentiation, cellular death via apoptosis, and growth inhibition in human cancer cells (Wang et al. 2012).
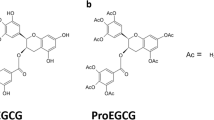
In particular, the naturally occurring flavanols—polyphenols—which are found in green tea have been researched for such anticancer properties (Ahmed et al. 2016; Zhang et al. 2010a; Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009). A subgroup of polyphenols, catechins, is known for their powerful antioxidant properties. Studies by many groups have demonstrated that green tea catechins can act as cancer-preventative compounds by modulating multiple cell signaling pathways that are involved in carcinogenesis (Ahmed et al. 2016; Zhang et al. 2010a; Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009). The most abundant and potent of these green tea polyphenols is epigallocatechin-3-gallate (EGCG) (Fig. 14.1a). Several reports show that EGCG can reduce proliferation and growth of human tumors including breast, prostate, and leiomyoma (Ahmed et al. 2016; Zhang et al. 2010a). However, there are some identified limits to EGCG’s therapeutic efficacy. EGCG has poor bioavailability especially when ingested. Not only will most of the EGCG be excreted, but it also has poor stability and is susceptible to biotransformation within the human body during digestion and absorption. Many researchers (including ourselves) have developed novel EGCG analogs and prodrugs to address these limitations (Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009).

Chemical structures of EGCG (a), methylated EGCG metabolites (b), EGCG-like prodrugs 1 (c), 2a, and 4a (d), as well as EGCG analogs 4 (e) and 6 (f)
14.2 Green Tea, EGCG, and Cancer
The origin of the tea plant, Camellia sinensis, is believed to be located somewhere in Asia; however experts cannot be entirely sure where, especially considering that the “wild” plant is extinct. Tea is thought to have originated as a medicine in ancient China. From sometime about 3000 BC to 2100 BC, tea leaves were chewed for stimulation and refreshment (Du et al. 2012). One ancient Chinese tells the story of how a stray leaf fell from a wild bush and landed into a pot of water that was being boiled by the Chinese emperor Shen-Nung in 2737 BC. According to the legend, this led to the discovery of the beverage known as tea.
Historically, tea has been used in brews and meal preparation since 722 BC–221 BC. For the subsequent years there is evidence of a variety of teas’ widespread consumption. In actuality, all “real” tea varieties originate from the herb Camellia sinensis; it is simply a matter of when the leaves are picked and how they are preserved. Any teas made from a different plant are called herbal teas. From China, tea migrated to Japan, and then along the trade routes to Tibet, the Arabic nations, the Turks, the nomadic tribes of the Indian Himalayas, and India. Finally, in the sixteenth century, tea crossed the continents to Europe. It is at this point that Europeans developed the process for black tea to increase the durability and thus the profit margin on tea. The increased hardiness allowed the beverage to become one of the world’s most popular drinks after water.
Most research focuses on green tea’s potential as a cancer preventative and cure, rather than black tea. This is because although they both come from the same plant they are not exactly the same due to the curing process. For example, green tea contains 30–40% catechins (like EGCG) whereas black tea contains only 3–10% (Hu et al. 2009). This high percentage of catechins largely explains why green tea has remained a focus of nutraceutical research. In recent years, green tea has become the focus of many research studies due to its potential for human health benefits, as well as in epidemiological and clinical settings as an anti-disease agent.
In one example, an investigation into the frequency of cancer across racial groups has shown that Asian and Pacific Islander populations have lower occurrences and mortality rates due to cancer when compared with other races. There may be a correlation between these low cancer statistics and the dietary intake of green tea, a popular beverage which is often consumed daily in Asian and islander societies (Dou et al. 2008). Green tea leaves may contain roughly 35–42% of catechins and flavonols upon extraction. Additionally, it is estimated that approximately one cup of green tea contains 300–400 mg of polyphenols (Chen et al. 2011). Catechins, a subgroup of polyphenols, are found in relatively high concentrations in green tea (Dou 2009), including epicatechin (EC), epigallocatechin (EGC), epicatechin-3-gallate (ECG), and EGCG. EGCG has been identified as the most common polyphenol in green tea (Du et al. 2012). It has been well documented that EGCG has anticancer activity associated with its ability to inhibit key cancer-related targets including tumor necrosis factor alpha, the proteasome, and others (Yang and Wang 1993; Dou et al. 2008; Dou 2009). This suggests that EGCG could be used as an agent for treatment and prevention of cancer.
14.2.1 EGCG as a Chemopreventative
The diseases known as cancer occur where cellular homeostasis has been interrupted by hyperplastic, dysplastic, or regenerative changes. These changes are brought about by chemical, physical, biological, and/or genetic assaults on cells inducing changes in the genome (Park and Dong 2003). Cancer essentially has two “phases,” carcinogenesis and oncogenesis. Oncogenesis is the maintenance and evolution of the existing transformed cells and tumors. Carcinogenesis is the creation of those transformed cells and tumors. It causes the tumor initiation and growth within the host. While there are multiple theories of carcinogenesis, the multistage theory is the most widely held and accepted. This theory allows for three stages of carcinogenesis. The first two stages induce transformations of the cells, while the third stage is more involved in final transformations to malignancy.
The first stage of carcinogenesis is initiation. This stage occurs when DNA is targeted. These changes to genetic material are irreversible. They are subtle, and fast; initiation can occur from within minutes to within hours (Park and Dong 2003; Afaq et al. 2003). Initiation can be caused by mutations, transitions, deletions, and even changes in the gene function such as histone modification, transcription activity, and DNA methylation (Park and Dong 2003; Afaq et al. 2003). This first stage aligns with both the genetic mutation and viral theories of carcinogenesis. Cells can remain in the initiation stage for an unlimited amount of time and most initiated cells will never move to the next two stages remaining quiescent for the duration of their life cycle (Park and Dong 2003; Afaq et al. 2003). Unfortunately, cells at the initiation stages have no phenotype expression making it very hard to detect and target for treatment.
The second stage of carcinogenesis—promotion—is easy to identify. It acts on genetic programs of terminal proliferation and differentiation much like the aberrant differentiation theory of carcinogenesis suggests (Afaq et al. 2003). At this stage cells no longer recognize the differentiation signals that would usually remove them. Unlike initiation, promotion targets the cell membrane and is reversible by remediation and apoptosis (Park and Dong 2003; Afaq et al. 2003). It leads to the expression of the genome and is not an essential stage in carcinogenesis; promotion may be bypassed. Initiation can go directly to stage three, progression (Park and Dong 2003; Afaq et al. 2003).
Progression, the final stage of carcinogenesis, is when evident malignant neoplasms appear (Park and Dong 2003; Afaq et al. 2003). It is characterized by unstable karyotypes caused by major genetic alterations (Park and Dong 2003; Afaq et al. 2003). There are multiple types of evolving karyotypic instabilities caused by increasing chromosomal irregularities and cellular proliferation (Park and Dong 2003). These include (1) invasion, (2) metastatic growth, (3) anaplasia, and (4) increased proliferation and growth to ever-increasing malignancy, potentially leading to the death of the host (Park and Dong 2003; Afaq et al. 2003). Once cells have reached progression they must be treated.
Green tea polyphenols are of interest not only for their anti-oncogenesis capabilities, but also as possible chemopreventatives. Research has shown that green tea polyphenols can prolong the stages of carcinogenesis either individually or in tandem (Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009). While EGCG’s mechanisms as a chemopreventative are not 100% understood they are assumed to include protecting the DNA from damage and methylation, which will stop carcinogenesis at its initiation stage. Similarly, it may inhibit cell proliferation and tumor promotion (Chen et al. 2011). EGCG inhibits oncogene expression and uses its pro-apoptotic properties to induce apoptosis to reverse the promotion and progression stages of carcinogenesis (Park and Dong 2003). In the latter stages of carcinogenesis EGCG interferes with cell cycle regulation by disrupting signaling pathways. These affected pathways include (1) the ubiquitin proteasome system (UPS), (2) kinase signaling and activation (i.e., MAPKs), (3) EGFR-mediated pathways, and (4) insulin growth factor-I (IGF-I)-mediated signal transduction pathways (Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009). Studies have proven that decreased IGF-1 levels correlate to a decrease of the incidence of cancer (Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009). This supports the use of green tea polyphenols—EGCG in particular—as chemopreventatives.
Animal studies conducted using EGCG showed that green tea inhibits tumor occurrence, interfering with some stage of carcinogenesis, possibly initiation, through its antioxidant properties. A mouse study, using fluid ingestion of green tea, done on NDEA-involved lung tumors, found a 36–44% decrease of tumor incidence (Chen et al. 2011). The same study observed a decrease of 44–60% in tumor multiplicity (stage three). A different animal study showed reduced UVB-induced skin cancer incidence by 35%; the multiplicity was reduced by 63% and growth by 55% using oral green tea polyphenols (Chen et al. 2011). Even topical application demonstrated decreases in incidence, growth, and multiplicity in animal studies. These effects were demonstrated on numerous cancer sites including skin, lung, stomach, colon, liver, and mammary gland. All the while, they proved to be relatively safe (Chen et al. 2011; Yang and Wang 1993; Dou et al. 2008; Dou 2009).
14.2.2 EGCG Can Target Multiple Molecular Signaling Pathways Required for Cancer Cell Survival
It has been shown that EGCG exhibits antioxidant, anti-inflammatory, and antitumor activity in various types of cancer (Hu et al. 2009). These potent effects are related to EGCG’s ability to regulate various signaling pathways involved in cancer cell growth and proliferation (Zaveri 2006). Studies have shown that EGCG is able to halt tumor cell growth at the G1 phase (or Gap 1 phase, the first of four phases of the cell cycle that takes place in eukaryotic cell division) by inhibiting key enzymes known as cyclin-dependent kinases (CDKs), which prevents cells from continuing to mitosis and therefore inhibits tumor growth. EGCGs can also act by inducing other CDK inhibitors in breast, prostate, and other cancer cells (Park and Dong 2003). Additionally, EGCG has been shown to inhibit the DNA transcription factor NF-κB, a protein complex that controls cytokine production and cell survival (Afaq et al. 2003). Reactive oxygen species (ROS) can activate NF-κB, which subsequently activates the transcription of survival genes that may also be anti-apoptotic, thus creating hardier cells. NF-κB also cross talk to another cancer survival signaling pathway, the PI3-kinase/AKT pathway, ensuring cancer cell survival (Hussain et al. 2012). EGCG possesses ROS-scavenging properties, which prevent NF-κB from initializing (Singh et al. 2011), and also inhibits PI-3K/AKT pathway (Van Aller et al. 2011).
When used with conventional cancer therapies, EGCG can increase their efficacy. In one study, breast cancer patients undergoing radiotherapy were split into two groups—one that was given 400 mg oral EGCG capsules thrice daily in addition to their therapy, and a control group that was not. The success of the EGCG treatment was determined by measuring cell proliferation factors in blood samples collected at various time points. The group who consumed the EGCG capsules followed by radiotherapy demonstrated lower serum levels of vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and reduced activation of metalloproteinase-9 and metalloproteinase-2 (MMP9/MMP2) as compared with the group who only underwent radiotherapy (Zhang et al. 2012). In a different study that used pancreatic cancer cells, EGCG was shown to increase the effect of gemcitabine, a nucleotide analog, and CP690550, a small-molecule immunosuppressant. This study also showed that EGCG decreased expression and activation of STAT3, an oncogene transcription factor, while also inducing apoptotic pathways in the pancreatic cancer cells (Tang et al. 2012).
EGCG demonstrates specific discernment in its effect on tumors, or transformed cells. One study showed that when tested on normal and transformed NIH-pATM Ras fibroblasts, EGCG showed an inhibitory and pro-apoptotic effect on the tumor cells without any effect on normal cells (Wang and Bachrach 2002). The study analyzed various growth factors in response to EGCG treatment of normal and transformed fibroblasts. The cellular proliferation signal molecule, ornithine decarboxylase (ODC), displayed decreased levels in transformed cells, but not in normal cells. ECGC also caused a reduction in numerous kinase activities, including those involved in the MAPK pathway, and a decrease in the oncogenes Ras and Jun. These effects were also specific to transformed cells and were not in evidence in normal cells (Wang and Bachrach 2002).
14.2.3 EGCG as a Tumor 20S Proteasome Inhibitor
It has been reported that EGCG can act as a 20S proteasome inhibitor, and EGCG treatment of human cancer cells results in the accumulation of pro-apoptotic proteins, such as IκB and Bax, both of which are NF-κB inhibitors (Nam et al. 2001). Cells regulate their proteasome pathway as a survival strategy to respond to environmental changes and fluctuations in its growth needs. It is estimated that ~90% of proteins in the cell are degraded by the ubiquitin-proteasome system (UPS) (Fig. 14.2) (Shen et al. 2012). Many of the proteins involved in carcinogenesis and tumor growth are degraded through the UPS. The UPS consists of enzymatic ubiquitination followed by proteolysis. A cascade of enzymes—E1, E2, and E3—tag misfolded old or unneeded proteins with ubiquitin (Ub). The 26S proteasome is the executive end of the UPS. It is comprised of two 19S proteasome caps and a 20S proteolytic core (Fig. 14.3) (Shen et al. 2012; Lin et al. 2002). The 19S subunit recognizes Ub-tagged protein substrates; it can then release the protein or its deubiquitinating (DUB) enzymes remove the Ub tags and then the proteins are linearized in preparation for their cleavage by the 20S catalytic core particle. The 20S subunit is a cylindrical structure comprised of two identical outer α-rings and two identical inner β-rings. The α-rings have seven subunits and act as “gates,” while the β-rings contain the active proteolytic sites. The β-subunits contain chymotrypsin-like (CT), trypsin-like, and post-glutamyl peptidyl hydrolytic-like (PGHP) activities (Shen et al. 2012; Lin et al. 2002; Adams 2004).

The ubiquitin-proteasome system or UPS. Substrate proteins are tagged for degradation with ubiquitin molecules via an ATP-dependent pathway, facilitated by three enzymes (E1, E2, and E3)

The 26S proteasome, consisting of the 19S “lid” and 20S hollow core with α- and β-subunits
The first FDA-approved proteasome inhibitor was bortezomib (BTZ) also known by its trade name Velcade (Adams 2004; Chen et al. 2008; Dou and Zonder 2014). BTZ is administered to patients with multiple myeloma and mantle cell lymphoma intravenously. Bortezomib targets the β5 and β1 but not β2 active sites of the catalytic subunits of the 20S proteasome, leading to cancer cell growth inhibition (Adams 2004; Chen et al. 2008; Dou and Zonder 2014). Specifically, the boron atom in bortezomib can interact with the amino-terminus of β5 and β1 subunits, resulting in proteasome inhibition by preventing cleavage. Proteasome inhibition has been demonstrated as an effective cancer treatment strategy, and BTZ and future-generation proteasome inhibitors are considered to be important cancer therapies. Like all cancer therapies, BTZ has several limitations, including the following: (1) not all patients respond to treatment, and relapse occurs in many patients who initially responded; (2) solid tumors, in particular, are often resistant; and (3) treatment causes the induction of dose-limiting peripheral neuropathy. Several second-generation proteasome inhibitors have been developed to address these disadvantages. Carfilzomib (CFZ) can induce responses in a minority of multiple myeloma patients relapsed from or refractory to BTZ with less peripheral neuropathy (Dou and Zonder 2014). While the mechanism of BTZ resistance in human cancers remains unclear, targeting the immunoproteasome, ubiquitin E3 ligases, 19S proteasome, and deubiquitinases (DUBs) in preclinical studies represents possible directions for future-generation inhibitors of the UPS in the treatment of not only multiple myeloma but other cancers as well. Along this line, EGCG-based proteasome inhibitors may play an important role in this field whether individually or in conjunction with current proteasome inhibitors.
14.2.4 Development of Pro-EGCG to Improve EGCG’s Stability, Bioavailability, and Activity
In our effort to increase the stability of EGCG, we converted its eight reactive hydroxyl groups into the corresponding acetates, named Pro-EGCG or 1 (Fig. 14.1c). In a previous study it was demonstrated that Pro-EGCG could be converted into EGCG, thus functioning as a prodrug of EGCG under in vitro and in vivo conditions (Lam et al. 2004; Landis-Piwowar et al. 2007a). It was also found that when breast cancer MDA-MB-231 cells were treated with Pro-EGCG, the EGCG metabolite was produced and accumulated intracellularly, accompanied by increased levels of proteasome inhibition, growth suppression, and apoptosis induction, as compared to tumor cells treated with EGCG (Lam et al. 2004). To investigate whether Pro-EGCG can work as a novel prodrug that converts to a proteasome inhibitor and antitumor agent in vivo, MDA-MB-231 xenografts were induced in nude mice, followed by daily treatment with Pro-EGCG or EGCG for 1 month. From this in vivo study a significant inhibition of tumor growth by Pro-EGCG was observed, as compared to EGCG, associated with increased proteasome inhibition and apoptosis induction in tumor tissues (Landis-Piwowar et al. 2007a). It should be noted that independent research groups using various models have confirmed the increased stability, bioavailability, and biological activity of Pro-EGCG in vitro and in vivo as compared to EGCG (Lambert et al. 2006; Vyas et al. 2007; Lee et al. 2008; Chiou et al. 2012, 2013; Wang et al. 2013).
14.2.5 Novel EGCG Analogs Resistant to COMT-Mediated Methylation and Inhibition
One drawback to EGCG is its susceptibility to methylation by catechol-O-methyltransferase (COMT) (Fig. 14.1b) (Landis-Piwowar et al. 2007b). COMT is an enzyme found in many mammalian tissues. It carries an important function in catechol metabolism, especially amine hormones, and eliminates biologically active or toxic catechols. COMT’s substrates include catecholamines and catecholestrogens. It catalyzes the transfer of a methyl group from S-adenosylmethionine to one of the hydroxy groups of the catechol ring (Axelrod and Tomchick 1958). With regard to EGCG, COMT seems to have a higher affinity for methylation of the 4′-hydroxy group of the D-ring rather than the hydroxyl groups of the B-ring (Lu et al. 2003). Methylation of EGCG results in decreased potency in terms of anticancer effects, possibly due to decreased proteasomal inhibition (Landis-Piwowar et al. 2007b).
The COMT protein is expressed in two forms: S-COMT, found in the cytoplasm, and MB-COMT, which is membrane bound. Both proteins are expressed by the same gene and are nearly identical in amino acid sequence, the only difference being an additional 50 amino acids on the NH2-terminal of the MB-COMT protein, which allows it to anchor to the membrane. A single-nucleotide polymorphism has been found in the COMT gene, involving a methionine substitution in the place of valine. The addition of this allele (alternative forms of a gene that arise by mutation and are found at the same place on a chromosome) results in three genotypes: Val/Val, Val/Met, and Met/Met (Dawling et al. 2001). Each genotype results in a different level of COMT activity: the Val/Val genotype results in high COMT activity, Val/Met results in intermediate activity, and Met/Met results in low activity. Since EGCG is a substrate of COMT, EGCG potency is likely affected by COMT status. One study specifically examined the relationship between tea consumption and breast cancer. The study found that the risk of developing breast cancer was significantly lower in women who drank green tea, but only in those with one or more low-activity COMT alleles. Those with the high-activity COMT allele (Val/Val genotype) had the same risk of breast cancer regardless of whether or not they consumed green tea (Wu et al. 2003).
There are several COMT inhibitors that are commercially available. Studies have shown that COMT inhibition can increase EGCG potency and thus allow for greater proteasome inhibition (Forester and Lambert 2014)]. Another approach to overcoming COMT methylation is to develop novel EGCG analogs and their corresponding prodrugs (2a and 4a) (Fig. 14.1d), which are less susceptible to COMT methylation. Design and synthesis of EGCG analogs and prodrugs may be an effective way to overcome COMT methylation and therefore provide increased potency of anticancer effects (Chen et al. 2011). We hypothesized that EGCG analogs missing one hydroxyl at the 4′-position or two hydroxyl groups on the gallate ester moiety could result in compounds not having a catechol structure at the D-ring and would be less susceptible to COMT methylation than EGCG. With this in mind, two prodrugs (2a and 4a) of such EGCG analogs were synthesized. Following this, we conducted a study using human leiomyoma cell lines and found that 2a and 4a have potent antiproliferative, antiangiogenic, and antifibrotic properties ((Ahmed et al. 2016); see below Sect. 3).
14.3 Activities of EGCG Prodrugs and Analogs in Uterine Fibroids
Uterine leiomyomas, or uterine fibroids, are the most common type of benign tumors seen in reproductive-age women (Styer and Rueda 2015). Women of African-American descent are at greater risk for developing uterine fibroids, with incidence rates in this population being 2–3 times higher than in white women of the same age (Wise et al. 2016). Other risk factors include obesity, age, early menarche, and increasing age up to menopause (Sparic et al. 2016). Though benign, uterine fibroids can cause pain, excessive bleeding, bladder issues, and complications during pregnancy (Downes et al. 2010; Sparic 2014). These tumors are the most common cause for hysterectomy in the United States, causing a negative impact on the lives of patients and resulting in billions of dollars per year in healthcare costs (Yang et al. 2016). Alternatives to hysterectomy, such as uterine artery embolization and magnetic resonance-guided focused ultrasound surgery, have many limitations, and new fibroids often recur after the procedure (Taylor and Leppert 2012). Currently, there are few noninvasive long-term treatment options for women with uterine fibroids, creating a need for development of other approaches.
It has been reported that the green tea polyphenol EGCG inhibits the growth of uterine leiomyoma cells in vitro and in vivo (Zhang et al. 2010a, b; Ozercan et al. 2008) and the use of a green tea extract (containing 45% EGCG) suggests potential clinical activity as a safe therapeutic agent for women with uterine fibroids (Roshdy et al. 2013). However, EGCG has a number of shortcomings, as stated above, including low stability, poor bioavailability, and rapid metabolic transformations under physiological conditions, presenting challenges for its development as a therapeutic agent (Johnson et al. 2010; Sang et al. 2011; Kanwar et al. 2012).
Most recently we have performed a study to expand our understanding of the mechanism of action of Pro-EGCG and Pro-EGCG analogs and assess whether Pro-EGCG and its analogs 2a and 4a display antiproliferative, antiangiogenic, and antifibrotic properties using human leiomyoma cell lines (Ahmed et al. 2016), as a prelude to advancing one or more compounds to human clinical trials.
14.3.1 Pro-EGCG and Its Analogs
Proliferation, fibrosis, and angiogenesis each plays a role in uterine fibroid growth and formation (Islam et al. 2014). Studies have shown that all of these processes can be inhibited by EGCG (Xu et al. 2009; Larsen and Dashwood 2010; Matsuzaki and Darcha 2014). We first studied EGCG in comparison to pro-EGCG and analogs 2a and 4a with regard to their effect on proliferation of human uterine leiomyoma cell lines (Ahmed et al. 2016).
UtLM-ht or UtLM cell lines were treated with the solvent DMSO, EGCG, Pro-EGCG (1), analog 2a, or analog 4a, followed by the MTT assay. It was found that growth of both cell lines was inhibited by each of the compounds, with 2a and 4a having the greatest antiproliferative effect (Ahmed et al. 2016).
The study also aimed to determine whether UtLM and UtLM-ht cell lines could form 3D spheres and if so whether EGCG, 1, 2a, and 4a could inhibit proliferation of the spheres. 3D spheres were successfully grown for both cell lines and it was found that 4a was able to significantly inhibit growth of the spheres, as well as 2a, 1, and EGCG to a lesser effect (Ahmed et al. 2016).
Additionally, western blots were performed to measure levels of S-phase markers PCNA and cyclin A as well as apoptosis-related proteins PARP and caspase-3 (Ahmed et al. 2016). Compounds 2a and 4a were able to significantly reduce S-phase markers, and they were also able to significantly reduce the apoptosis-related proteins. Western blots were also performed to look for biomarkers related to fibrosis and angiogenesis, both of which are critical to uterine fibroid development and growth (Islam et al. 2014). Compounds 2a and 4a were able to significantly reduce expression of fibrosis biomarkers α-smooth muscle actin (αSMA) and collagen type I (Col-I) in UtLM-ht cells, as well as expression levels of angiogenesis biomarkers vascular endothelial growth factor receptor 2 (VEGFR-2) and vascular endothelial growth factor C (VEGF-C). A scratch-wound-healing assay was performed next on both cell lines to measure migration of cells. While untreated control cells were able to migrate into the gap produced by the scratch after a 24-h recovery period, migration of cells treated with EGCG, 1, 2a, or 4a was inhibited in both cell lines. Again, 2a and 4a were the most effective (Ahmed et al. 2016).
Next, proteasomal activity was measured in UtLM-ht cells. It was found that 4a was able to most potently inhibit proteasomal activity, followed by 2a, 1, and EGCG, respectively (Ahmed et al. 2016). As EGCG has also been shown to inhibit tumor cell growth via Akt kinase activity inhibition (Tang et al. 2003; Zhang et al. 2006; Qin et al. 2007), a western blot was performed to measure pAkt and Akt levels in UtLM and UtLM-ht cells. pAkt expression was inhibited most successfully by 2a and 4a in both cell lines, followed by 1 and then EGCG. This indicated that 2a and 4a are capable of inhibiting the Akt pathway in this cell line (Ahmed et al. 2016).
Finally, it was hypothesized that 2a and 4a are more potent than 1 and EGCG due to the analogs’ resistance to methylation by COMT. UtLM-ht cells were treated with dinitrocatechol (DNC), an inhibitor of COMT (Pérez et al. 1993), or a control solvent to test this. The cells were then treated with EGCG, 1, 2a, or 4a. An MTT assay was then performed, showing that antiproliferative activity in cells treated with EGCG or 1 was significantly higher, while the DNC treatment had a much lesser effect on 2a- or 4a-treated cells. This indicated that EGCG and 1 are more susceptible to COMT methylation than the analogs (Ahmed et al. 2016).
14.3.2 Compounds 4 and 6
Two EGCG analogs (compounds 4 and 6) (Fig. 14.1e, f) (Chen et al. 2012) were studied alongside EGCG and 1 as a comparison to examine their effects on two human uterine leiomyoma cell lines, UtLM and UtLM-ht (Fig. 14.4). Previously, we reported that these synthetic EGCG analogs were more potent AMPK activators than metformin and EGCG in breast cancer cells (Chen et al. 2012).

Inhibition of human uterine leiomyoma cell growth. UtLM-ht (a) or UtLM (b) cells, grown in a 96-well plate, were treated with either the control solvent DMSO, EGCG, or Pro-EGCG 4 or 6 (at indicated concentration, μM) daily for up to 3 days (with each drug repeatedly added every 24 h), followed by the MTT assay
Human UtLM-ht (Fig. 14.4a) or UtLM cells (Fig. 14.4b) were treated with DMSO, EGCG, 1, 4, and 6 at indicated concentrations (10, 25, 35, 50, or 75 μM) for 3 days (with a repeat dose of each drug given every 24 h), followed by an MTT assay to determine cell viability. We found that 4 and 6 inhibited growth of these two uterine leiomyoma cell lines in a dose-dependent manner, similar to 1 and more or equally potent to EGCG (Fig. 14.4).
Spheroid formation assay of UtLM-ht cells was then performed and the effects of 4 and 6 vs. EGCG and 1 were determined. As we found in our previous study (Ahmed et al. 2016), both uterine leiomyoma cell lines successfully formed spheroid masses (Fig. 14.5). When treated daily with 15 or 30 μM of EGCG, 1, 4, or 6 for up to 6 days, decreased number and size of uterine leiomyoma spheroids were observed, in the order of 1, 4 > 6 > EGCG.

Inhibition of spheroid formation and morphology in 3-D culture of human uterine leiomyoma cells. UtLM-ht cells were grown in a 6-well plate (in triplicates), followed by either no treatment (NT) or treatment with the control solvent DMSO or EGCG, 1, 4, or 6 (at 15 or 30 μM) daily for up to 6 days (with each drug repeatedly added every 24 h). The primary spheres were photographed at 200× magnification
These results suggested that 4 and 6 can inhibit proliferation of uterine leiomyoma cells. Further studies were then conducted on the molecular level in order to validate the above results. The effects of 4 and 6 on the expression of cell cycle-specific proteins were analyzed using western blot analysis (Fig. 14.6). UtLM-ht cells were treated with DMSO or EGCG, 1, 4, or 6 at the indicated concentrations (25, 35, or 45 μM) for 24 h. A western blot was then performed, using antibodies to cell-specific proteins of interest. We found that in UtLM-ht line, the order of the inhibition of PCNA expression, an S-phase marker, by these compounds was 6 > 4 > 1 > EGCG (Fig. 14.6).

Reduction of protein biomarkers of cell proliferation, apoptosis, and angiogenesis by Pro-EGCG analogs. UtLM-ht cells were either untreated (NT) or treated with the control solvent DMSO, EGCG, or Pro-EGCG 1, 4, or 6 (at 25, 35, or 45 μM) for 24 h, followed by western blotting using specific antibodies to PCNA (MW 37 kDa), caspase-3 (MW 32 kDa), VEGF-R2 (MW 152 kDa; the 200 kDa band might be a tetra-ubiquitinated form), VEGF-C (MW 46 kDa), and actin (MW 46 kDa)
We also found that Pro-EGCG potently reduced full-length caspase-3 protein levels, indicating caspase activation, in uterine leiomyoma cells and was more effective than EGCG, 4 and 6 (Fig. 14.6).
Angiogenesis is an essential component of uterine fibroid growth (Tal and Segars 2014). The effect of 4 and 6 on vascular endothelial growth factor-receptor 2 (VEGF-R2; MW 150 kDa), which is important for the process of angiogenesis, was investigated. UtLM-ht cell lines were either untreated (NT) or treated with the control solvent DMSO or 25, 35, or 45 μM of EGCG, 1, 4, or 6 for 24 h, followed by western blot analysis using a specific antibody to VEGF-R2 (polyclonal antibody 2). The order of inhibition of expression of VEGF-R2/p150 was 6 > 4 > 1 > EGCG (Fig. 14.6; we had no clear explanation for the effect of 4 at 35 μM in this experiment).
The effect of 4 and 6 on vascular endothelial growth factor-C (VEGF-C), which is also essential for angiogenesis, was then studied (Fig. 14.6). Human UtLM-ht cells were either untreated (NT) or treated with the control solvent DMSO or 25, 35, or 45 μM of EGCG, 1, 4, or 6 for 24 h, followed by western blot analysis using antibodies specific to VEGF-C. Partial inhibition on VEGF-C was observed by 1 but not by others (Fig. 14.6).
The effect of 4 and 6 on migration of uterine leiomyoma cells was examined next by performing a scratch wound-healing assay (Fig. 14.7). A scratch was made in several dishes of UtLM-ht cells. The cells were either untreated or treated with DMSO or 4 or 6 at 25, 35, or 45 μM. The cells were then allowed to recover for 24 h. It was found that the untreated UtLM-ht cells could migrate into the gap left by the scratch. Cells treated with DMSO were able to migrate into the gap with 90% recovery. However, migration of UtLM-ht cells treated with 4 or 6 was inhibited. The order of inhibition potency was 6, 1 > 4 > EGCG (Fig. 14.7).

Inhibition of human uterine leiomyoma UtLM-ht cell migration (wound-healing assay). Cells were grown in a 12-well Matrigel-coated plate until they reached ~80% confluence. A scratch was then generated by using a yellow tip in each dish, and cells were either untreated (NT) or treated with the control solvent DMSO or EGCG 1, 4, or 6 at indicated concentration for 24 h. The percentage of the gap remaining under each experimental condition was measured and the values were represented as the average of triplicate samples (±SD)
14.4 Conclusions
Cancer is a prevalent and growing concern. Conventional treatments are not sufficient to address the problem. Research shows that green tea polyphenols have the potential as anticancer agents. The catechin EGCG is an especially effective anticancer compound. EGCG has demonstrated the potential to inhibit cancer cell growth and induce tumor cell death through a variety of different mechanisms. EGCG can disrupt cancer cell survival pathways such as CDK, or it can induce tumor cell death by proteasome inhibition and controlling the UPS. Despite its promise, EGCG has certain drawbacks that have hindered its use as a viable cancer therapy. There is no established delivery system that overcomes EGCG’s poor bioavailability; and susceptibility to biotransformation via COMT methylation reduces its efficacy, which can vary based on genotype. Researchers are working on overcoming these hurdles through the design of new synthetic analogs and prodrugs. These analogs are designed to make the compound less susceptible to methylation by COMT. Studies have shown that prodrugs and synthetic analogs have performed better than natural EGCG in terms of effectiveness as well as stability demonstrating that they may be an effective way to circumvent EGCG’s above problems.
Of all the green tea polyphenols EGCG and its analogs have shown the most promise as compounds for the treatment of cancer. Continued research on EGCG, particularly in design and synthesis of more stable analogs as well as their prodrugs, is strongly encouraged in hopes of developing new drug candidates for clinical research. In the future, EGCG analogs may be developed into effective new drugs for the treatment of many cancers.
References
Adams J (2004) The proteasome: a suitable antineoplastic target. Nat Rev Cancer 4(5):349–360
Afaq F et al (2003) Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea constituent (−)-epigallocatechin-3-gallate. Oncogene 22(7):1035–1044
Ahmed RS et al (2016) Biological and mechanistic characterization of novel prodrugs of green tea polyphenol epigallocatechin gallate analogs in human leiomyoma cell lines. J Cell Biochem 117(10):2357–2369
Axelrod J, Tomchick R (1958) Enzymatic O-methylation of epinephrine and other catechols. J Biol Chem 233(3):702–705
Chen D et al (2008) Tea polyphenols, their biological effects and potential molecular targets. Histol Histopathol 23(4):487–496
Chen D et al (2011) EGCG, green tea polyphenols and their synthetic analogs and prodrugs for human cancer prevention and treatment. Adv Clin Chem 53:155–177
Chen D et al (2012) Novel epigallocatechin gallate (EGCG) analogs activate AMP-activated protein kinase pathway and target cancer stem cells. Bioorg Med Chem 20(9):3031–3037
Chiou YS et al (2012) Peracetylated (−)-epigallocatechin-3-gallate (AcEGCG) potently suppresses dextran sulfate sodium-induced colitis and colon tumorigenesis in mice. J Agric Food Chem 60(13):3441–3451
Chiou YS et al (2013) Peracetylated (−)-epigallocatechin-3-gallate (AcEGCG) potently prevents skin carcinogenesis by suppressing the PKD1-dependent signaling pathway in CD34+ skin stem cells and skin tumors. Carcinogenesis 34(6):1315–1322
Dawling S et al (2001) Catechol-O-methyltransferase (COMT)-mediated metabolism of catechol estrogens. Cancer Res 61(18):6716–6722
Dou QP (2009) Molecular mechanisms of green tea polyphenols. Nutr Cancer 61(6):827–835
Dou QP, Zonder JA (2014) Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets 14(6):517–536
Dou QP et al (2008) Green tea polyphenols as a natural tumour cell proteasome inhibitor. Inflammopharmacology 16(5):208–212
Downes E et al (2010) The burden of uterine fibroids in five European countries. Eur J Obstet Gynecol Reprod Biol 152(1):96–102
Du G-J et al (2012) Epigallocatechin gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 4(11):1679–1691
Forester SC, Lambert JD (2014) Synergistic inhibition of lung cancer cell lines by (−)-epigallocatechin-3-gallate in combination with clinically used nitrocatechol inhibitors of catechol-O-methyltransferase. Carcinogenesis 35(2):365–372
Hu J, Zhou D, Chen Y (2009) Preparation and antioxidant activity of green tea extract enriched in epigallocatechin (EGC) and epigallocatechin gallate (EGCG). J Agric Food Chem 57(4):1349–1353
Hussain AR et al (2012) Cross-talk between NFkB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS One 7(6):e39945
Islam MS et al (2014) Use of dietary phytochemicals to target inflammation, fibrosis, proliferation, and angiogenesis in uterine tissues: promising options for prevention and treatment of uterine fibroids? Mol Nutr Food Res 58(8):1667–1684
Johnson JJ, Bailey HH, Mukhtar H (2010) Green tea polyphenols for prostate cancer chemoprevention: a translational perspective. Phytomedicine 17(1):3–13
Kanwar J et al (2012) Recent advances on tea polyphenols. Front Biosci (Elite Ed) 4:111–131
Lam WH et al (2004) A potential prodrug for a green tea polyphenol proteasome inhibitor: evaluation of the peracetate ester of (−)-epigallocatechin gallate [(−)-EGCG]. Bioorg Med Chem 12(21):5587–5593
Lambert JD et al (2006) Peracetylation as a means of enhancing in vitro bioactivity and bioavailability of epigallocatechin-3-gallate. Drug Metab Dispos 34(12):2111–2116
Landis-Piwowar KR et al (2007a) A novel prodrug of the green tea polyphenol (−)-epigallocatechin-3-gallate as a potential anticancer agent. Cancer Res 67(9):4303–4310
Landis-Piwowar KR et al (2007b) Methylation suppresses the proteasome-inhibitory function of green tea polyphenols. J Cell Physiol 213(1):252–260
Larsen CA, Dashwood RH (2010) (−)-Epigallocatechin-3-gallate inhibits Met signaling, proliferation, and invasiveness in human colon cancer cells. Arch Biochem Biophys 501(1):52–57
Lee SC et al (2008) Effect of a prodrug of the green tea polyphenol (−)-epigallocatechin-3-gallate on the growth of androgen-independent prostate cancer in vivo. Nutr Cancer 60(4):483–491
Lin H-K et al (2002) Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem 277(39):36570–36576
Lu H, Meng X, Yang CS (2003) Enzymology of methylation of tea catechins and inhibition of catechol-O-methyltransferase by (−)-epigallocatechin gallate. Drug Metab Dispos 31(5):572–579
Matsuzaki S, Darcha C (2014) Antifibrotic properties of epigallocatechin-3-gallate in endometriosis. Hum Reprod 29(8):1677–1687
Nam S, Smith DM, Dou QP (2001) Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem 276(16):13322–13330
National Cancer Institute (2018) Cancer statistics. Understanding cancer. Available from: https://www.cancer.gov/about-cancer/understanding/statistics
Ozercan IH et al (2008) Chemoprevention of fibroid tumors by [−]-epigallocatechin-3-gallate in quail. Nutr Res 28(2):92–97
Park AM, Dong Z (2003) Signal transduction pathways: targets for green and black tea polyphenols. J Biochem Mol Biol 36(1):66–77
Pérez RA et al (1993) Inhibition of catechol-O-methyltransferase by 1-vinyl derivatives of nitrocatechols and nitroguaiacols. Biochem Pharmacol 45(10):1973–1981
Qin J et al (2007) A component of green tea, (−)-epigallocatechin-3-gallate, promotes apoptosis in T24 human bladder cancer cells via modulation of the PI3K/Akt pathway and Bcl-2 family proteins. Biochem Biophys Res Commun 354(4):852–857
Roshdy E et al (2013) Treatment of symptomatic uterine fibroids with green tea extract: a pilot randomized controlled clinical study. Int J Womens Health 5:477–486
Sang S et al (2011) The chemistry and biotransformation of tea constituents. Pharmacol Res 64(2):87–99
Shen M, Chan TH, Dou QP (2012) Targeting tumor ubiquitin-proteasome pathway with polyphenols for chemosensitization. Anti Cancer Agents Med Chem 12(8):891–901
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68(1):7–30
Singh BN, Shankar S, Srivastava RK (2011) Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol 82(12):1807–1821
Sparic R (2014) Uterine myomas in pregnancy, childbirth and puerperium. Srp Arh Celok Lek 142(1–2):118–124
Sparic R et al (2016) Epidemiology of uterine myomas: a review. Int J Fertil Steril 9(4):424–435
Styer AK, Rueda BR (2015) The epidemiology and genetics of uterine leiomyoma. Best Pract Res Clin Obstet Gynaecol 34:3–12
Tal R, Segars JH (2014) The role of angiogenic factors in fibroid pathogenesis: potential implications for future therapy. Hum Reprod Update 20(2):194–216
Tang F-Y, Nguyen N, Meydani M (2003) Green tea catechins inhibit VEGF-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt molecule. Int J Cancer 106(6):871–878
Tang S-N et al (2012) EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PLoS One 7(2):e31067
Taylor DK, Leppert PC (2012) Treatment for uterine fibroids: searching for effective drug therapies. Drug Discov Today Ther Strateg 9(1):e41–e49
Van Aller GS et al (2011) Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res Commun 406(2):194–199
Vyas S et al (2007) Design, semisynthesis, and evaluation of O-acyl derivatives of (−)-epigallocatechin-3-gallate as antitumor agents. J Agric Food Chem 55(15):6319–6324
Wang YC, Bachrach U (2002) The specific anti-cancer activity of green tea (−)-epigallocatechin-3-gallate (EGCG). Amino Acids 22(2):131–143
Wang H et al (2012) Plants against cancer: a review on natural phytochemicals in preventing and treating cancers and their druggability. Anti Cancer Agents Med Chem 12(10):1281–1305
Wang CC et al (2013) Prodrug of green tea epigallocatechin-3-gallate (pro-EGCG) as a potent anti-angiogenesis agent for endometriosis in mice. Angiogenesis 16(1):59–69
Wise LA et al (2016) History of uterine leiomyoma and risk of endometrial cancer in black women. Cancer Causes Control 27(4):545–552
World Health Organization (2018) Cancer. Available from: http://www.who.int/news-room/fact-sheets/detail/cancer
Wu AH et al (2003) Tea intake, COMT genotype, and breast cancer in Asian-American women. Cancer Res 63(21):7526–7529
Xu H et al (2009) Anti-angiogenic effects of green tea catechin on an experimental endometriosis mouse model. Hum Reprod 24(3):608–618
Yang CS, Wang ZY (1993) Tea and cancer. J Natl Cancer Inst 85(13):1038–1049
Yang Q, Diamond MP, Al-Hendy A (2016) Early life adverse environmental exposures increase the risk of uterine fibroid development: role of epigenetic regulation. Front Pharmacol 7:40
Zaveri NT (2006) Green tea and its polyphenolic catechins: medicinal uses in cancer and noncancer applications. Life Sci 78(18):2073–2080
Zhang Q et al (2006) Green tea extract and (−)-epigallocatechin-3-gallate inhibit hypoxia- and serum-induced HIF-1α protein accumulation and VEGF expression in human cervical carcinoma and hepatoma cells. Am Assoc Cancer Res 5(5):1227–1238
Zhang D et al (2010a) Antiproliferative and proapoptotic effects of epigallocatechin gallate on human leiomyoma cells. Fertil Steril 94(5):1887–1893
Zhang D et al (2010b) Green tea extract inhibits proliferation of uterine leiomyoma cells in vitro and in nude mice. Am J Obstet Gynecol 202(3):289.e1–289.e9
Zhang G et al (2012) Anti-cancer activities of tea epigallocatechin-3-gallate in breast cancer patients under radiotherapy. Curr Mol Med 12(2):163–176
Acknowledgments
Partially supported by Canadian Institutes of Health Research (CIHR).
Conflicts of interest: T.H.C., Q.P.D., and R.F. are the named inventors of patents and patent applications; other authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ahmed, R.S.I. et al. (2019). Discovery of Green Tea Polyphenol-Based Antitumor Drugs: Mechanisms of Action and Clinical Implications. In: Joshee, N., Dhekney, S., Parajuli, P. (eds) Medicinal Plants. Springer, Cham. https://doi.org/10.1007/978-3-030-31269-5_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-31269-5_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-31268-8
Online ISBN: 978-3-030-31269-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)