
Abstract
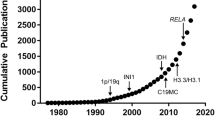
The incidence of primary malignant brain tumors has been increasing over the past 30 years, and it is estimated that in the year 2010, approximately 22,020 cases were diagnosed in the USA alone, with 13,140 deaths [1, 2]. Gliomas account for 32% of all primary brain tumors, but within the malignant subset, they account for 80% of tumors. Histological classification of tumors of the nervous system was initiated by the WHO in 1979, as a means of predicting the biological behavior of a neoplasm, and thereby determining the choice of therapies. The 2016 World Health Organization Classification of Tumors of the Central Nervous System (CNS) is both a conceptual and practical advance over its 2007 predecessor (Table 4.1). For the first time, the WHO classification of CNS tumors uses molecular parameters in addition to histology to define many tumor entities, thus formulating a concept for how CNS tumor diagnoses should be structured in the molecular era. As such, the 2016 CNS WHO presents major restructuring of the diffuse gliomas, medulloblastomas, and other embryonal tumors, and incorporates new entities that are defined by both histology and molecular features, including glioblastoma, IDH-wild-type and glioblastoma, IDH-mutant; diffuse midline glioma, H3 K27 M-mutant; RELA fusion-positive ependymoma; medulloblastoma, WNT-activated and medulloblastoma, SHH-activated; and embryonal tumor with multilayered rosettes, C19MC-altered. The 2016 edition has added newly recognized neoplasms, and has deleted some entities, variants, and patterns that no longer have diagnostic and/or biological relevance. Other notable changes include the addition of brain invasion as a criterion for atypical meningioma and the introduction of a soft tissue-type grading system for the now combined entity of solitary fibrous tumor/hemangiopericytoma a departure from the manner by which other CNS tumors are graded. Overall, it is hoped that the 2016 CNS WHO will facilitate clinical, experimental, and epidemiological studies that will lead to improvements in the lives of patients with brain tumors [3].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


4.1 General Findings
The incidence of primary malignant brain tumors has been increasing over the past 30 years, and it is estimated that in the year 2010, approximately 22,020 cases were diagnosed in the USA alone, with 13,140 deaths [1, 2]. Gliomas account for 32% of all primary brain tumors, but within the malignant subset, they account for 80% of tumors. Histological classification of tumors of the nervous system was initiated by the WHO in 1979, as a means of predicting the biological behavior of a neoplasm, and thereby determining the choice of therapies. The 2016 World Health Organization Classification of Tumors of the Central Nervous System (CNS) is both a conceptual and practical advance over its 2007 predecessor (Table 4.1). For the first time, the WHO classification of CNS tumors uses molecular parameters in addition to histology to define many tumor entities, thus formulating a concept for how CNS tumor diagnoses should be structured in the molecular era. As such, the 2016 CNS WHO presents major restructuring of the diffuse gliomas, medulloblastomas, and other embryonal tumors, and incorporates new entities that are defined by both histology and molecular features, including glioblastoma, IDH-wild-type and, IDH-mutant glioblastoma; diffuse midline glioma, H3 K27 M-mutant; RELA fusion-positive ependymoma; medulloblastoma, WNT-activated and medulloblastoma, SHH-activated; and embryonal tumor with multilayered rosettes, C19MC-altered. The 2016 edition has added newly recognized neoplasms, and has deleted some entities, variants, and patterns that no longer have diagnostic and/or biological relevance. Other notable changes include the addition of brain invasion as a criterion for atypical meningioma and the introduction of a soft tissue-type grading system for the now combined entity of solitary fibrous tumor/hemangiopericytoma a departure from the manner by which other CNS tumors are graded. Overall, it is hoped that the 2016 CNS WHO will facilitate clinical, experimental, and epidemiological studies that will lead to improvements in the lives of patients with brain tumors [3].
4.2 Molecular Biology of Gliomas
Low-grade glioma represents a spectrum of tumor types with diverse histologic features; however, recently molecular analysis of tumors has become a critical part of tumor classification and prognostication. In 2016, the WHO updated its classification of primary brain tumors to include molecular characterization, now defining tumors both on phenotype and genotype. Oligodendrogliomas on traditional hematoxylin and eosin staining have round nuclei and fine delicate branching vessels but are now also defined as having both an isocitrate dehydrogenase (IDH) gene family mutation and combined whole-arm losses of 1p and 19q (1p/19q codeletion) [4,5,6,7]. Astrocytomas are characterized by prominent glial fibrillary acidic protein processes, typically also have mutations in IDH, but have intact 1p and 19q chromosomes as well as loss of ATRX. Mutations in either IDH1 or IDH2 occur in up to 80% of grade 2 and 3 diffuse gliomas and carry a more favorable prognosis compared with IDH wild-type tumors [7].
High-grade gliomas (HGG), including glioblastoma (GBM), anaplastic astrocytoma (AA), and anaplastic oligodendroglioma (AO), originate from the supporting neuroglial cells of the CNS. GBM, the most common and most aggressive of the primary brain tumors, typically presents in late adulthood. AA and AO affect a younger age group and generally have a more protracted clinical course. High-grade gliomas can be debilitating, owing to physical disability, cognitive impairment, personality change, depression and seizure disorder, and require complex multidisciplinary care. Histologically, tumors showing anaplasia and mitotic activity are classified as grade III, while the sine qua non of grade IV tumors is microvascular proliferation and/or necrosis. Historically, all HGG have been treated in the same manner, but the treatment modality for grade III tumors is currently being investigated separate of grade IV tumors through ongoing clinical trials. The average survival time of approximately 1 year for patients with glioblastoma (GBM) has minimally improved despite decades of basic and clinical research. However, in recent years a significant survival benefit has been achieved with the addition of concurrent temozolomide (TMZ) to adjuvant RT.
There have been substantial advances in our understanding of the molecular aberrations found in malignant gliomas. Key discoveries include the isocitrate dehydrogenase (IDH) mutation, codeletion of the short arm of chromosome 1, and long arm of chromosome 19 (1p19q) and O6-methylguanine-DNA methyltransferase (MGMT) gene promoter methylation. These have emerged as being important determinants of treatment response and survival. Consequently, they are now routinely tested and have become fundamental to glioma classification.
IDH catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, and subsequently to the oncometabolite 2-hydroxyglutarate [8]. In turn, 2-hydroxyglutarate acts via a family of dioxygenases to impair epigenetic regulation and increase hypoxia-inducible factor 1-α. The prevalent IDH1 R132H mutation is detectable with immunohistochemistry in over 90% of cases [9]. IDH mutations can also be identified by sequencing IDH1 codon 132 and IDH2 codon 172. These mutations are common in low-grade gliomas and secondary GBMs, and confer significantly improved prognosis [10].
The 1p19q codeletion is an unbalanced reciprocal translocation that is a characteristic of oligodendrogliomas. Multiple studies have demonstrated the favorable prognostic and predictive utility of the 1p/19q codeletion, although the biologic basis remains unclear. Specifically, in randomized Phase III trials evaluating chemoradiotherapy with PCV for AO, patients harboring the 1p19q codeletion derived greater benefit from PCV and lived substantially longer [11, 12]. In contrast, partial 1p or 19q loss did not confer this significance.
MGMT gene promoter methylation causes epigenetic silencing of MGMT, which is necessary for DNA repair. Notably, based on the review of randomized Phase III trials evaluating temozolomide in patients with GBM, those containing the MGMT gene promoter methylation obtained meaningful survival benefit from temozolomide, whereas those without the methylation did not [13]. Initially, MGMT status was assessed with immunohistochemistry and MGMT methylation-specific PCR; however, widespread clinical use was limited by numerous technical issues including poor reliability, reproducibility, and the labor-intensive work [14, 15]. Newer methods include bisulfite sequencing, pyrosequencing, high-resolution melt analysis, and infinium methylation BeadChip, which have improved standardization and accuracy of MGMT testing [16, 17].
As mentioned above, in 2016 the WHO published the Fifth Edition Classification of Tumors of the Central Nervous System [3]. This represents a seminal update, with the introduction of integrated diagnoses combining histology and molecular parameters for many entities. This incorporates the recently established prognostic and predictive information from IDH and 1p19q.
GBM is now subdivided into IDH wild-type (predominantly primary GBM, patients over 55 years of age, poor prognosis) and IDH-mutant entities (predominantly secondary GBM, younger patients, favorable prognosis). The diagnosis of AO requires IDH-mutant and 1p19q-codeleted status, whereas AA requires IDH-mutant and noncodeleted status. Importantly, both entities are IDH mutant; a glioma that is IDH wild-type with or without 1p19q codeletion instead represents a genomically unstable GBM. In addition, 1p19q codeletion is mutually exclusive with TP53 mutation and ATRX inactivation [18]. Accordingly, a glioma that is IDH-mutant, TP53-mutant, and ATRX-inactivated is considered AA. Finally, the use of molecular parameters handles the problematic and indeterminate entity called anaplastic oligoastrocytoma, which was previously defined by a mixed histological pattern and was subject to poor interobserver agreement [19, 20]. The combination of histology and molecular parameters effectively differentiates nearly all cases as either AO or AA. To facilitate clinical decision making, the current standard is to incorporate all the tissue-based information (histology, grade, molecular findings) into an integrated diagnosis, which is then reported to clinicians.
Molecular markers have significantly contributed to diagnostic precision in high-grade glioma, and yield important therapeutic implications. The next steps will be to improve understanding of clinical and molecular heterogeneity within glioma subtypes. Ongoing efforts include assessment of additional molecular markers, methylation profiling, and a coordinated approach to histologic–molecular correlation as part of clinical trials.
4.3 Standard of Care for Malignant Low-Grade Glioma
The precise optimal management of patients with low-grade glioma after surgical resection remains to be determined. The risk–benefit ratio of treatment with radiation and chemotherapy must be weighed for each individual patient. Prior studies stratified patients into high- and low-risk low-grade glioma on the basis of clinical features of age (older or younger than 40 years) and the extent of resection. A large prospective study of observation of patients with low-risk low-grade glioma younger than 40 years who had gross total resections reported 52% of patients had recurrence within 5 years of surgery. On the basis of these data, in patients who are considered low risk, defined as age younger than 40 years with a gross total resection, it is an attractive option to forgo further treatment with radiation and chemotherapy at the time of diagnosis and instead undergo regular magnetic resonance imaging (MRI) surveillance [21]. The choice of chemotherapy is also under active investigation. PCV was originally used in early trials for low-grade glioma on the basis of efficacy in higher-grade tumors [11]. It has been largely replaced by temozolomide in later trials because of an improved adverse effect/toxicity profile and the expectation that both alkylating therapies would have similar efficacies, but a direct comparison of the two agents has yet to be completed [22].
There is no known curative therapy for low-grade gliomas. When low-grade gliomas recur, they may either be the original tumor/grade or they may also undergo malignant transformation into high-grade tumors. Oligodendrogliomas can malignantly transform into anaplastic oligodendrogliomas, and astrocytomas can transform into anaplastic astrocytomas or glioblastomas. Treatment options at the time of recurrence can include further surgery, radiation therapy and/or chemotherapy, or clinical trials. If surgical resection can safely be performed, it is again recommended. If a patient did not receive radiation at initial diagnosis or has had significant time pass before recurrence, radiation therapy may also be an option. Treatment with chemotherapy is also usually a possibility. Choices can include the original chemotherapy, if safe from a toxicity perspective, versus an alternative chemotherapeutic agent. At this time, there are few data to direct treatment decisions at recurrence, but reports do suggest that there may be at least some benefit for treatment with chemotherapy with either temozolomide or PCV [23]. Treatments after failure of alkylator-based chemotherapy vary widely, and there is no consensus opinion on the basis of current National Comprehensive Cancer Network and European Association of Neuro-Oncology guidelines [24, 25].
4.4 Standard of Care for Malignant High-Grade Glioma
Historically, glioblastoma has been treated with postoperative radiotherapy to kill remaining tumor cells. Addition of radiotherapy extends survival from 3–4 months to about 12 months [26, 27]. In the 1990s, the DNA alkylating agent temozolomide was tested and approved by the FDA as a chemotherapeutic agent for the treatment of malignant glioma [28]. Addition of temozolomide to surgical resection and radiotherapy extends median survival to 14.6 months and the 2-year survival rate to 27% compared to 10% [22]. Additional studies have shown that patients with DNA methylation in the promoter region of the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) are more likely to respond to temozolomide therapy [29]. The current standard of care for glioblastoma is GTR with concomitant temozolomide and radiotherapy followed by adjuvant temozolomide.
Carmustine is the only other FDA-approved first-line chemotherapeutic agent approved for glioblastoma. Like temozolomide, carmustine is a DNA-alkylating agent. BCNU (carmustine)-polymer wafers are positioned in the tumor bed after tumor resection. A Phase III clinical trial showed evidence of survival benefit [30]. However, the efficacy of carmustine has never been directly compared to that of temozolomide.
Nearly all patients with malignant glioma will recur [31]. Despite maximal initial resection and multimodality therapy, about 70% of GBM patients will experience disease progression within 1 year of diagnosis [32] with less than 5% of patients surviving 5 years after diagnosis [33]. Re-resection is an option for some patients, and surgical debulking can alleviate mass effect and symptoms, such as seizures, speech, and motor deficits, frequently seen at recurrence. Repeat surgery may be required to confirm a diagnosis of tumor recurrence versus pseudoprogression or radiation necrosis and may also provide tissue for molecular testing to identify potential new targeted agents [34]. Opinion varies as to whether repeat surgery enhances OS. Some evidence exists that a greater extent of resection at recurrence is associated with improved survival [35]; however, other studies have not found an absolute benefit in terms of survival [36, 37].
Additional radiation may be possible for some patients, but tolerance of healthy brain tissue to radiation is limited because of the increased risk of radiation necrosis. A wide variety of radiation techniques, including brachytherapy, gamma knife, and stereotactic radiosurgery, may be used for the treatment of recurrent disease [38].
Upon recurrence of GBM, chemotherapy and corticosteroids may be used to palliate symptoms and improve quality of life, but objective response rates are dismal, and time to progression for standard cytotoxic agents is only 3–6 months [39]. Rechallenging with TMZ may be an option, and other agents, such as carboplatin (Paraplatin®), etoposide (Toposar®), irinotecan (Camptosar®), and nitrosourea-based chemotherapy, may be tried as single agents or in regimens.
The options for the treatment of GBM are limited due to the presence of the blood–brain barrier (BBB), which prevents molecules >500 Da [40] from entering the brain. The BBB is a selective physical barrier, as the tight junctions between the adjacent endothelial cells do not allow for the normal, paracellular transport, but force molecules into a transcellular transport. Small molecules, such as O2, CO2, and ethanol may diffuse freely through the membrane [41]. The presence of specific transport systems on the membrane surface enables nutrients to enter the brain, but prevents potentially toxic substances from harming the CNS. Large molecules, such as peptides and proteins, are not able to enter the brain, unless there is a strictly regulated receptor-mediated or adsorption-mediated transcytosis [42]. The BBB has a protective role: it mediates the efflux of waste products, maintains the ionic concentrations, which may change significantly following a meal and cause a disruption of normal brain function, and it separates the pools of the neurotransmitters that act centrally and peripherally. Overall, the BBB maintains the homeostasis of the CNS. Considering the limited penetration in the brain, alternative drug-delivery strategies are required for the more effective treatment of gliomas.
4.5 Chemotherapy and More
The current standard of care for malignant glioma has limited efficacy. One limitation of radiotherapy and temozolomide chemotherapy is that the therapy is nonspecific. The therapy does not exploit specific weakness of individual tumors. As we enter a time of greater understanding of the genetic landscape and gene expression of malignant gliomas, we will have a better idea of the targets to attack [43]. The Cancer Genome Atlas (TCGA) is a project sponsored by the National Institutes of Health (NIH) to better elucidate the genetics and gene expression of multiple cancer types, including glioblastoma. The TCGA has analyzed over 500 untreated glioblastoma samples for DNA sequence and epigenetic modification, gene expression, and microRNA expression [44]. This project has led to a deeper understanding of glioblastoma enabling high-throughput pathway analysis and massive data synthesis. One of the major findings of the project was that glioblastoma is divided into four distinct subtypes: mesenchymal, proneural, classical, and neuronal [45]. Each subtype has novel mutations and expression patterns. Some of these novel pathways and targets will hopefully prove to be exploitable for effective treatments in the future.
Utilizing TCGA data and other genome-wide studies, new molecular targets for malignant gliomas have been detected. Molecular targets are common in pathways central to malignant glioma survival such as proliferation, evasion of apoptosis, invasiveness, and angiogenesis [46]. Aberrant growth factor signaling drives proliferation in many malignant gliomas. Epidermal growth factor (EGFR), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF), and fibroblast growth factor (FGF) are either highly upregulated or mutated in a large percentage of malignant gliomas [47]. Several clinical trials have tried to capitalize on blocking these pathways. EGFR is the most widely studied growth factor in malignant glioma. Several small molecular inhibitors (gefitinib, erlotinib, lapatinib, and cetuximab) have been evaluated in Phase II clinical trials for use in therapy of malignant gliomas. Unfortunately, these drugs have only shown modest efficacy for treating malignant glioma.
Personalized medicine will play a better role in identifying certain exploitable pathways or targets in an individual tumor [48, 49] . In the near future, genetic tests will determine if a patient will respond to temozolomide. Deep sequencing of tumor DNA and gene expression analysis of fresh tumor samples will eventually direct therapy for patients suffering from malignant glioma. By synthesizing ascertainable data from the tumor, therapy can be tailored and combined to select the appropriate combination of therapies to best target the tumor. As technology evolves to make medicine more personalized, new methods will be utilized to choose the proper combinatorial therapy to treat each malignant glioma.
4.6 Role of Bevacizumab
GBM is a highly vascular neoplasm, with abnormal vasculature characterized by tortuous blood vessels, vascular permeability and resulting hypoxia leading to the histological finding of pseudopalisading necrosis [50]. Tumor growth and invasion are intrinsically linked to hypoxia, which results in upregulation of hypoxia-inducible factor 1-α, and downstream upregulation of VEGF, which is associated with glioma cell stemness, mesenchymal phenotype, and an immunosuppressive cellular milieu [51]. Thus, there is a strong biologic rationale for the use of antiangiogenic agents in GBM, and these drugs have thus been extensively studied as therapeutic targets in both newly diagnosed and recurrent GBM.
Bevacizumab, a humanized monoclonal antibody which binds VEGF-A, is the most extensively studied of the antiangiogenic agents for GBM. Bevacizumab was approved by the US FDA for use in recurrent GBM in 2009 [52]. The “Bevacizumab Alone and in Combination with Irinotecan in Recurrent GBM” (BRAIN) study [53] was a randomized Phase II trial that assigned 167 patients with recurrent GBM to receive bevacizumab 10 mg/kg with or without irinotecan. This trial demonstrated objective response rates of 38 and 28% in patients treated with bevacizumab with and without irinotecan, respectively. Progression-free survival at 6 months (PFS-6) was 42% in patients treated with bevacizumab alone and 50% in the combination arm. In a single-arm study, 48 patients with recurrent GBM were treated with bevacizumab 10 mg/kg with irinotecan added upon disease progression, demonstrating an objective response rate of 35% and PFS-6 of 29% [54]. While these findings led to FDA approval for recurrent GBM in the USA, its use has not been approved in Europe due to concerns regarding the lack of a bevacizumab-free control arm, the modest improvement in OS, and difficulties with interpreting MRI-based disease progression in patients treated with bevacizumab [55].
In the USA, the widespread use of bevacizumab for recurrent GBM has limited the opportunity for further evaluation in this setting. In Europe, the randomized Phase II “Single-Agent Bevacizumab or Lomustine Versus a Combination of Bevacizumab Plus Lomustine in Patients with Recurrent GBM” (BELOB) trial [56] showed promising results for the combination of bevacizumab and lomustine versus either agent alone. Unfortunately, these findings were not borne out in the subsequent Phase III trial which compared the combination of lomustine and bevacizumab with lomustine alone [57]. This trial showed no difference in OS, although there was a significant increase in PFS from 1.5 to 4.2 months in the combination arm. Several other Phase II trials have evaluated the combination of bevacizumab with a variety of other cytotoxic and targeted agents, including temozolomide, temsirolimus, and erlotinib, but none have shown significant activity [58].
Similarly, bevacizumab has been tested in the setting of newly diagnosed GBM, with a series of Phase II trials using bevacizumab in combination with radiotherapy and temozolomide [59, 60]. As seen in the recurrent setting, PFS was prolonged in comparison to historical controls (13–14 months), while the effect on OS was modest (10–21 months). Subsequently, two randomized Phase III trials were conducted, “A Study of Avastin in Combination With Temozolomide and Radiotherapy in Patients With Newly Diagnosed GBM” (AVAGlio) [61] and RTOG-0825 [62]. These studies showed longer PFS in patients treated with bevacizumab, but failed to show OS benefit. Thus, despite encouraging preclinical results with in vivo activity and reduction of vasogenic edema, there is abundant high-quality evidence that bevacizumab is not indicated in unselected patients with newly diagnosed GBM.
4.7 Future Directions
4.7.1 Growth Factor Receptor Inhibitors
EGFRvIII, the most common variant of EGFR, is only found in GBM and other tumor cell surface without expression in normal tissue cells [63].
Biological targeted therapy targets the pathway of the EGF/EGFR ligand and involves the use of mAb against EGFR, such as cetuximab and nimotuzumab, and tyrosine kinase inhibitors (TKIs), including gefitinib, erlotinib, afatinib, canertinib, and lapatinib. A current retrospective study carried out based on clinical trials found that both gefitinib and erlotinib had good therapeutic responses in GBM patients with co-expression of EGFRvIII and PTEN [64].
Cetuximab has been used in Phase I and Phase II clinical trials and have demonstrated effective improvement in the treatment for patients with GBM.
Radiotherapy followed adjuvant therapy with the combination of ABT-414 (anti-EGFR antibody) and TMZ has shown promising results in OS among patients with newly diagnosed GBM with EGFR amplification.
4.7.2 Tumor-Treating Electric Fields for Glioblastoma
Tumor-Treating Fields (TTFields) have considered the “fourth cancer treatment modality,” after surgery, RT, and pharmacotherapy; is a locoregionally antimitotic treatment that delivers low-intensity, intermediate-frequency (200 kHz), alternating electric fields, through four transducer arrays, consisting of nine insulated electrodes applied to the shaved scalp and connected to a portable device. In vitro TTFields arrest cell division and kill tumor cells through multiple mechanisms [65].
In 2011 TTFields were approved from FDA as a therapeutic option for use in rGBM. In the EF-14 trial, an open-label Phase III study, 695 patients were treated with TTFields in combination with TMZ maintenance treatment, after chemoradiation therapy for patients with nGBM. The trial showed a significant improvement in PFS and OS. The percentage of patients alive at 2 years was 43% in the TTFields/TMZ group and 29% in the TMZ alone group (P = 0.006). In October 2015, the FDA approved TTFields for use in newly diagnosed GBM patient [66] and National Comprehensive Cancer Network has further incorporated TTFields in their updated guidelines.
4.8 Vaccine Therapy
These vaccines work by activating T cells (CD4 and CD8) against specific tumor antigens and by inducing an antitumoral cellular response by using dendritic cells (DC) and heat shock proteins [67].
4.8.1 DC Therapy
Current preparation of DC vaccines involves exposing the lysate of a patient’s tumor to the patient’s autologous DCs, which are then treated with a differentiation factor such as GM-CSF. The primed APCs are then injected back into the patient with hopes of generating a T-cell response against the tumor. The primed APCs (antigen-presenting cells) are then injected back into the patient with hopes of generating a T-cell response against the tumor.
DC vaccines have demonstrated some efficacy in improving outcomes for glioblastoma. Bregy et al. in a systematic review demonstrated that autologous DC vaccination improved median OS in patients with newly diagnosed and recurrent GBM compared to historical trends [68]. Parney and Gustafson explored the benefits of adding DC therapy with concurrent temozolomide in patients with resected newly diagnosed glioblastoma. DCs were generated from the patient’s CD14+ monocytes, pulsed with allogeneic tumor lysate from two patient-derived GBM cell cultures, and given to patients during their temozolomide therapy. After vaccination, increased circulating tumor-associated antigen-specific CD8 T cells were identified, demonstrating that allogenic tumor lysate vaccines are feasible and may generate a tumor antigen-specific immune response.
4.8.2 Heat Shock Protein (HSP) Vaccines
The HSPs intracellularly have the function to assemble and transport nascent proteins. HSPs also have a very critical role in the stress response to cellular insult and function by stabilizing proteins and preventing them from aggregating.
Tumor-derived HSPs and other proteins can be complexed together and serve as an antitumor vaccine in patients with glioblastoma. The advantage of these vaccines, respect to others, is that HSPs are not targeted to a specific predefined antigen but instead to varying types of antigenic proteins upon vaccination, which serves to broadly target the intratumoral heterogeneity that is normally seen in GBM [69].
Bloch et al. [70] reported a median overall survival of 42.6 weeks after HSP peptide complex-96 vaccination in patients with recurrent glioblastoma. Of note, 66% of patients in this study were lymphopenic prior to therapy, which is believed to have significantly impacted the antitumor immune response.
These studies demonstrate that the HSPPC-96 vaccination may be safe and deserve additional investigation.
4.9 Checkpoint Inhibitors
Immune checkpoints are very important in the balance of self-tolerance and immunogenicity. Failed immune checkpoints impede immune responses in refractory cancers that are prone to T-cell anergy and toleragenicity. Programmed cell death protein and ligand (PD-1, PDL-1), metabolic enzymes (e.g., Arginase), and inhibitory immune pathways CTLA4 (Cytotoxic T-Lymphocyte-Associated Antigen 4) have been hypothesized to play a role in immune tolerance. CTLA4, expressed on T cells, regulates the extent of the T-cell immune response by impeding the CD28 T-cell stimulatory pathway. In the clinical setting, CTLA4 blockade, through the use of monoclonal antibodies, increases CD4 T-cell activity, and inhibits regulatory T-cell immunosuppression. In glioma mouse models, systemic blockade of PD-L1 demonstrated long-term survival with concurrent inhibition of regulatory T-cell activity. In animal models, activation of co-stimulatory receptors such as OX40 and blockade of co-inhibitory receptors such as PD1 and CTLA4 induced tumor regression and increased long-term survival. Currently, several clinical trials are ongoing for assessment of monoclonal antibody checkpoint inhibitors (anti-PD-L1 and CTLA-4) for glioblastoma [71, 72].
An increasing number of clinical trials are ongoing since 2011 to evaluate the potential therapeutic efficacy of PD-1/PD-L1 inhibitors, including nivolumab, pembrolizumab, pidilizumab (anti-PD-1), and MEDI4736, MPDL3280A (anti-PD-L1) as monotherapies and combination therapies for GBMs. There are still two ongoing clinical trials to investigate the nivolumab, TMZ, and radiation therapy or their combination for newly diagnosed patients with GBM (Table 4.1). The combination of ipilimumab (anti-CTLA-4) and nivolumab was tested in a Phase III randomized trial in recurrent GBM.
4.10 Chimeric T-Cell Receptors (TCR)
Chimeric antigen receptors (CARs) are a diverse class of receptors that have been created by combining the variable region of an antibody with a T-cell-signaling molecule such as CD3. These newly created receptors are advantageous compared to the TCR-transduced T cells. CARs have the ability to mimic endogenous TCR-mediated activation without the disadvantages of classical MCH restriction as the antigen recognition site is derived from an antibody.
Brown et al. examined the bioactivity and safety of IL13Ralpha2 redirected chimeric antigen receptor CD8 T cells in the resection cavity of three patients, and noted transient immune-mediated antitumor responses in 2/3 patients with recurrent glioblastoma. Other case reports of similar IL13Ralpha2-directed CAR conducted demonstrated tumor regression and immune responses after intrathecal therapy in patients with multifocal recurrent GBM [73, 74].
4.11 Viroimmunotherapy
The use of viruses to mediate gene immunotherapy in the treatment of tumors is a promising approach and has a wide variety of applications. Treatments can include transferring genes for inflammatory proteins to tumor cells, inhibition of immunosuppressing tumor genes, or transferring proinflammatory and tumor antigen genes to professional antigen-presenting cells. Previous clinical trials have focused on conditional cytotoxicity and oncolytic viruses, which may induce a secondary immune response by generating foreign antigens and producing a proinflammatory immune beacon in tumor cells.
Several clinical trials using adenovirus, herpes simplex virus, and replicating retroviruses have been conducted with preliminary results demonstrating survival benefit [75, 76]. In a Phase I/IIa trial (ParvOryx01) the oncolytic H-1 parvovirus (H-1PV) induced markers of immune activation in patients with recurrent glioblastoma; nine patients (age 29 to 69 years) with primary (n = 2) or recurrent (n = 7) glioblastoma were treated in a compassionate use (CU) program with a combination of H-1PV followed by bevacizumab and PD-1 blockade. Seven of the patients received both intratumoral and intravenous injection of H-1PV and two patients only intravenous virus treatment. Objective tumor response was observed in seven of nine patients (78%). Two patients showed complete responses (22%), five patients had partial remissions (56%) with tumor reduction between 49% and 94%, and two patients progressive disease (22%). H-1PV-based viroimmunotherapy leads to ORR in 78% of glioblastoma patients and this is a much higher response rate than reported for treatment with either bevacizumab or checkpoint blockade.
4.12 Conclusions
The recent research in vaccine therapy, checkpoint inhibitors, chimeric antigen T-cell receptors, and viroimmunotherapy has provided an opportunity to supplement the current treatment of glioblastoma potentially, improving prognosis and overall survival for these patients. Although there are several barriers to an effective safe treatment, future larger prospective studies may help elucidate the role of immunotherapy in these patients.
Change history
05 October 2020
The original version of the chapter title “Chemotherapy Chemotherapy and Future Developments” was revised and has been corrected to “Chemotherapy and Future Developments”.
References
Thakkar JP, Dolecek TA, Horbinski C, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014;23:1985–96.
Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology. 2014;16(Suppl. 4):ivl–63.
Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–20.
Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of che- motherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473–9.
Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–61.
Griffin CA, Burger P, Morsberger L, et al. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol. 2006;65:988–94.
Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75:1560–6.
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Preusser M, Wohrer A, Stary S, et al. Value and limitations of immunohistochemistry and gene sequencing for detection of the IDH1-R132H mutation in diffuse glioma biopsy specimens. J Neuropathol Exp Neurol. 2011;70(8):715–23.
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–43.
Van Den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–50.
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;52(10):997–1003.
Preusser M, Elezi L, Hainfellner JA. Reliability and reproducibility of PCR-based testing of O6-methylguanine-DNA methyltransferase gene (MGMT) promoter methylation status in formalin-fixed and paraffin-embedded neurosurgical biopsy specimens. Clin Neuropathol. 2008;27(6):388–90.
Lalezari S, Chou AP, Tran A, et al. Combined analysis of O6-methylguanine-DNA methyltransferase protein expression and promoter methylation provides optimized prognostication of glioblastoma outcome. Neuro-Oncology. 2013;15(3):370–81.
Switzeny OJ, Christmann M, Renovanz M, et al. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clin Epigenetics. 2016;8:49.
Bady P, Sciuscio D, Diserens AC, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012;124(4):547–60.
Sahm F, Reuss D, Koelsche C, et al. Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. 2014;128(4):551–9.
Coons SW, Johnson PC, Scheithauer BW, et al. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer. 1997;79(7):1381–93.
Kros JM, Gorlia T, Kouwenhoven MC, et al. Panel review of anaplastic oligodendroglioma from European Organization for Research and Treatment of Cancer Trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss, and correlations with outcome. J Neuropathol Exp Neurol. 2007;66(6):545–51.
Shaw EG, Berkey B, Coons SW, et al. Recurrence following neurosurgeon- determined gross-total resection of adult supratentorial low-grade glioma: results of a prospective clinical trial. J Neurosurg. 2008;109:835–41.
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Nahed BV, Redjal N, Brat DJ, et al. Management of patients with recurrence of diffuse low grade glioma: a systematic review and evidence-based clinical practice guideline. J Neuro-Oncol. 2015;125:609–30.
Nabors LB, Portnow J, Ammirati M, et al. Central nervous system cancers, version 2.2014. Featured updates to the NCCN guidelines. J Natl Compr Cancer Netw. 2014;12:1517–23.
Soffietti R, Baumert BG, Bello L, et al. Guidelines on management of low-grade gliomas: report of an EFNS-EANO Task Force. Eur J Neurol. 2010;17:1124–33.
Walker MD, Alexander E, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. Acooperative clinical trial. J Neurosurg. 1978;49(3):333–43.
Walker MD, Green SB, Byar DP, et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med. 1980;303(23):1323–9.
Newlands ES, Stevens MF, Wedge SR, et al. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev. 1997;23(1):35–61.
Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003.
Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5(2):79–88.
Bloch O, Han SJ, Cha S, et al. Impact of extent of resection for recurrent glioblastoma on overall survival: clinical article. J Neurosur. 2012;117(6):1032–8.
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTCNCIC trial. Lancet Oncol. 2009;10:459–66.
Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16:896–913.
Brandes AA, Bartolotti M, Francheschi E. Second surgery for recurrent glioblastoma: advantages and pitfalls. Expert Rev Anticancer Ther. 2013;13:583–7.
Bloch O, Han SJ, Cha S, et al. Impact of extent of resection for recurrent glioblastoma on overall survival. J Neurosurg. 2012;117:1032–8.
Brandes AA, Bartolotti M, Francheschi E. Second surgery for recurrent glioblastoma: advantages and pitfalls. Expert Rev Anticancer Ther. 2013;13:583–7.
Franceschi E, Bartolotti M, Tosoni A, et al. The effect of re-operation on survival in patients with recurrent glioblastoma. Anticancer Res. 2015;35:1743–8.
Davis ME, Stoiber AM. Glioblastoma multiforme: enhancing survival and quality of life. Clin J Oncol Nurs. 2011;15:291–7.
Franceschi E, Tosoni A, Bartolini S, et al. Treatment options for recurrent glioblastoma: pitfalls and future trends. Expert Rev Anticancer Ther. 2009;9:613–9.
Pardridge WM. Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv. 2003;3:90–105.
Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–52.
Cserr HF, Bundgaard M. Blood-brain interfaces in vertebrates: a comparative approach. Am J Phys. 1984;246:R277–88.
Brennan CW, Verhaak RGW, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77.
Network TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8.
Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110.
Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013;331(2):139–46.
Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011;71(18):5945–9.
Zhu J-J, Wong ET. Personalized medicine for glioblastoma: current challenges and future opportunities. Curr Mol Med. 2013;13(3):358–67.
Weller M, Stupp R, Hegi M, et al. Individualized targeted therapy for glioblastoma: fact or fiction? Cancer J. 2012;18(1):40–4.
Wang N, Jain RK, Batchelor TT. New directions in anti-angiogenic therapy for glioblastoma. Neurotherapeutics. 2017;14(2):321–32.
Lu-Emerson C, Duda DG, Emblem KE, et al. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with glioblastoma. J Clin Oncol. 2015;33(10):1197–213.
Cohen MH, Shen YL, Keegan P, et al. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14(11):1131–8.
Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40.
Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740–5.
Wick W, Weller M, Van Den Bent M, et al. Bevacizumab and recurrent malignant gliomas: a European perspective. J Clin Oncol. 2010;28(12):e188–9.
Taal W, Oosterkamp HM, Walenkamp AM, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014;15(9):943–53.
Wick W, Brandes AA, Gorlia T, et al. EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine in patients with first progression of a glioblastoma. J Clin Oncol. 2016;34(15 Suppl):2001–10.
Lai A, Tran A, Nghiemphu PL, et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2011;29(2):142–8.
Vredenburgh JJ, Desjardins A, Kirkpatrick JP, et al. Addition of bevacizumab to standard radiation therapy and daily temozolomide is associated with minimal toxicity in newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2012;82(1):58–66.
Vredenburgh JJ, Desjardins A, Reardon DA, et al. The addition of bevacizumab to standard radiation therapy and temozolomide followed by bevacizumab, temozolomide, and irinotecan for newly diagnosed glioblastoma. Clin Cancer Res. 2011;17(12):4119–24.
Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–22.
Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708.
Johansson M, Oudin A, Tiemann K, et al. The soluble form of the tumor suppressor Lrig1 potently inhibits in vivo glioma growth irrespective of EGF receptor status. Neuro Oncol. 2013;15(9):1200–11.
Gallego O, Cuatrecasas M, Benavides M, et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J Neurooncol. 2014;116(2):413–9.
Hottinger AF, Pacheco P, Stupp R. Tumor treating fields: a novel treatment modality and its use in brain tumors. Neuro Oncol. 2016;18(10):1338–49.
Stupp R, Taillibert S, Kanner AA. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015;314(23):2535–43.
Guo C, Manjili MH, Subjeck JR. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–75.
Bregy A, Wong TM, Shah AH, et al. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat Rev. 2013;39(8):891–0.
Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel vaccines for glioblastoma: clinical update and perspective. Immunotherapy. 2016;8(11):1293–308.
Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. 2014;16:274–9.
Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–301.
Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17:1064–75.
Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–72.
O'Rourke DM, Nasrallah M, Morrissette JJ, et al. Pilot study of T cells redirected to EGFRvIII with a chimeric antigen receptor in patients with EGFRvIII+ glioblastoma. J Clin Oncol. 2016;17:110–1.
Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74.
Geletneky K, Huesing J, Rommelaere J, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. 2012;12:99.
Acknowledgement
The current chapter is a revision made by Ileana De Roma, Lucia Lombardi, and Gennaro Gadaleta-Caldarola of the original chapter written by Evaristo Maiello, Lucia Lombardi, Mario Brandi, and Tommaso Scarabino in the previous edition of the book.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
De Roma, I., Lombardi, L., Gadaleta-Caldarola, G. (2020). Chemotherapy and Future Developments. In: Scarabino, T., Pollice, S. (eds) Imaging Gliomas After Treatment. Springer, Cham. https://doi.org/10.1007/978-3-030-31210-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-31210-7_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-31209-1
Online ISBN: 978-3-030-31210-7
eBook Packages: MedicineMedicine (R0)