
Abstract
Nicotiana species, which share significant similarities with tomato, potato, eggplant, and pepper, originated in South America and subsequently spread to North America, Africa, and Australia. They are known to produce and accumulate alkaloids as part of their defense mechanism against insects and herbivores. Since their description and classification by Goodspeed in 1947, research on Nicotiana species has been ongoing, including investigations on how the genus evolved and its relationship with other Solanaceae members. The presence of diploid and polyploid species in Nicotiana and their large genome sizes resulted in the generation of multiple resources aiming at better understanding of how polyploids formed and evolved over time. The use of Nicotiana tabacum and Nicotiana benthamiana as model organisms in plant research further helped increase the interest in understanding the molecular mechanisms in these species. Here, we review the literature and summarize what is currently known about the phylogeny, cytology, and genetics of Nicotiana species. We also present the current state of the Nicotiana genomes and introduce the functional genomics insights they have provided.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1.1 Introduction
Plant species commonly referred to as “tobacco” belong to the genus Nicotiana and are members of the Solanaceae family, with at least 3,000 species described to date, the majority of which are assigned to the genus Solanum (Knapp et al. 2004a; Olmstead et al. 2008). Some Solanaceae species are important commercial crops or ornamental plants, such as potato (S. tuberosum), tomato (S. lycopersicum), tobacco (N. tabacum), eggplant (S. melongena), pepper (Capsicum annuum), and petunia (Petunia × hybrida). The evolutionary and geographical origin of Solanaceae is in South America; however, some species have been found as far away as Africa and Australia (Eich 2008). Owing to the close relationship with Solanaceae, coffee species (e.g., arabica coffee [Coffea arabica]), from the Rubiaceae family, are also often considered together with Solanaceae species, as these two families are both members of the Asterid I clade.
A large number of Solanaceae species belong to the so-called x = 12 clade (including Solanum and Nicotiana genera) (Sarkinen et al. 2013) and share the same diploid (2n = 2x = 24) or tetraploid (2n = 4x = 48) chromosome numbers and similar chromosome architecture. Similarly, coffee comprises species with either diploid (2n = 2x = 22 in C. canephora) or allotetraploid (2n = 4x = 44 in C. arabica) architectures. Thus, the Solanaceae and related Coffea genus present an appealing system to study genome evolution and polyploidization (Wu et al. 2010).
The genomes of a number of important crop plants within Solanaceae and Rubiaceae have recently become available, including S. tuberosum (potato) (Xu et al. 2011), S. lycopersicum (tomato) (Sato et al. 2012), S. pimpinellifolium (currant tomato) (Sato et al. 2012), S. pennellii (wild tomato) (Bolger et al. 2014), S. melongena (eggplant) (Hirakawa et al. 2014), C. annuum (pepper) (Kim et al. 2014; Qin et al. 2014), N. benthamiana (Australian tobacco) (Bombarely et al. 2012; Naim et al. 2012), N. sylvestris and N. tomentosiformis (Sierro et al. 2013a), N. tabacum (Edwards et al. 2017; Sierro et al. 2014) and N. otophora (Sierro et al. 2014), C. canephora (robusta coffee) (Denoeud et al. 2014), Petunia axillaris and P. inflata (Bombarely et al. 2016), N. attenuata and N. obtusifolia (Xu et al. 2017), N. glauca (Khafizova et al. 2018), and N. rustica, N. undulata, N. paniculata, and N. knightiana (Sierro et al. 2018). Genome sequencing of other species, such as Petunia x hybrida, C. arabica (Tran et al. 2018), and numerous Nicotiana species, is currently ongoing. N. attenuata has been intensively studied and established as a model of a biotic or abiotic stress (Luu et al. 2015). Sequencing data and assembled genomes for the abovementioned Solanaceae species are available at the SOL Genomics Network (http://solgenomics.net) (Fernandez-Pozo et al. 2015).
The genus Nicotiana comprises about 78 naturally occurring species (Table 1.1), of which approximately three-quarters are found in the Americas and one-quarter are found in Australia and the South Pacific, with a single species indigenous to Africa (Chase et al. 2003). The species are semi-xerophytic in nature, prefer dry rather than humid heat, and are absent from tropical zones (Goodspeed 1947). The genus was last monographed by Goodspeed (Goodspeed 1954). The most recent sectional classification of the genus, which in part reflected recent molecular phylogenetic evidence, recognized 13 sections (Table 1.1). N. tabacum is placed in the monotypic section Nicotiana and its putative progenitor species are classified in section Tomentosae (N. tomentosiformis) and in the monotypic section Sylvestres (N. sylvestris). All of the Australian, South Pacific, and African species are classified in section Suaveolentes. Five sections are believed to be of allotetraploid origin (Knapp et al. 2004b); thus, hybridization is indicated to have made an important contribution to diversification within the genus.
Tobacco (N. tabacum L.) is an allotetraploid (2n = 4x = 48) that is considered to originate from a hybridization event between ancestors of N. sylvestris (S-genome; 2n = 2x = 24) and N. tomentosiformis (T-genome; 2n = 2x = 24) (Clarkson et al. 2005; Leitch et al. 2008; Lim et al. 2004; Sierro et al. 2013a, 2014). The hybridization and polyploidization event is estimated to have occurred within the last 200,000 years (Clarkson et al. 2004, 2005). Wild plants of N. tabacum are considered to be cultivated, and truly wild populations of the species cannot be unequivocally confirmed (Lewis and Nicholson 2007). Despite the scientific significance of N. tabacum as a model plant system and its importance as an agronomic crop, until recently, the genetic resources available for genome analysis, genetic mapping, and molecular marker-assisted selection were relatively limited, at least compared with other economically important crops (Davis and Nielsen 1999). Indeed, reference genome sequences for Nicotiana species have been published only recently (Bombarely et al. 2012; Edwards et al. 2017; Sierro et al. 2013a, 2014, 2018; Xu et al. 2017). In addition to N. tabacum and N. benthamiana, certain other Nicotiana species are grown as ornamentals (N. sylvestris, N. alata, and N. langsdorffii) or for industrial purposes (N. rustica and N. glauca). N. benthamiana (section Suaveolentes) is a model plant for studies of plant–microbe interactions on account of its well-characterized transient protein expression and its amenability to virus-induced gene silencing (VIGS) (Bombarely et al. 2012; Goodin et al. 2008).
1.2 Phylogenetic Relationships Within Nicotiana
Phylogenetic relationships within Nicotiana have been explored using sequence data for four coding or noncoding chloroplast DNA (cpDNA) regions (Clarkson et al. 2004) and the internal transcribed spacer (ITS) region of nuclear ribosomal DNA (Chase et al. 2003). Relationships suggested from the two data sets for diploid taxa generally show good congruence; however, such analyses and their interpretation for elucidation of the evolution of allopolyploid taxa require extreme caution. The cpDNA trees only allow the line of maternal inheritance to be traced (Clarkson et al. 2004). The ITS region is inherited biparentally and is subject to recombination; therefore, it can provide strong evidence for the parentage of hybrid taxa, but conversely may be misleading for elucidating the evolutionary history of taxa of hybrid origin owing to concerted evolution. Interestingly, putative allopolyploid Nicotiana taxa show no evidence for “hybrid” ITS sequences, even among those taxa thought to be of recent origin, such as N. tabacum; this is believed to be a result of rapid gene conversion to one parental copy (Chase et al. 2003).
Previous phylogenetic reconstructions from cpDNA sequence data indicate that within the Solanaceae, Nicotiana is related to the Australian-endemic genus Symonanthus (tribe Anthocercideae) and diverged approximately 15 million years ago (Mya) (Clarkson et al. 2004). These authors hypothesized that Nicotiana originated in southern South America and subsequently dispersed to North America, Africa, and Australia. The divergence between Nicotiana and Petunia has been estimated at more than 23 Mya (Wikstrom et al. 2001). A recent Bayesian molecular dating analysis of the Solanaceae (Sarkinen et al. 2013) used sequence data for six plastid and nuclear DNA regions for 40% of the total number of species in the family with two fossil calibration points. The sister clades Solanoideae (containing Solanum, Capsicum, and Physalis) and Nicotianoideae (including Nicotiana, Anthocercideae, and Symonanthus) were estimated to have diverged about 24 Mya (95% highest posterior density 23–26 Mya). The two clades formed what was informally termed the “x = 12 clade” by Särkinen et al. (2013).
In both cpDNA and ITS molecular phylogenetic reconstructions, members of section Tomentosae (N. kawakamii, N. otophora, N. tomentosa, and N. tomentosiformis) were consistently members of the clade related to the remainder of the genus (Chase et al. 2003; Clarkson et al. 2004). In cpDNA phylogenies, the basal-most clades are consistently retrieved and generally well supported; thus, they are considered to be reliable groups. Although most of the major clades were well supported and generally corresponded with the sections of Knapp et al. (2004b), some internal nodes were only weakly supported, so intersectional relationships were not completely resolved.
Six independent polyploidy events and at least three homoploid hybrid events in Nicotiana are hypothesized (Kovarik et al. 2012). Polyploidization events have been estimated to occur between less than 0.2 Mya (N. tabacum, N. rustica, and N. arentsii) and more than 10 Mya (section Suaveolentes) according to the molecular clock analyses (Clarkson et al. 2004, 2005; Leitch et al. 2008). The sections containing allopolyploids are thought to originate from the ancestors of distantly related present taxa according to their placement in cpDNA trees (Knapp et al. 2004b). For example, the presumed progenitors of N. tabacum (N. sylvestris and N. tomentosiformis) are not phylogenetically close relatives. Sections Tomentosae, Undulatae, Paniculatae, Trigonophyllae, Petunioides, Alatae, Repandae, Noctiflorae, and Suaveolentes are all monophyletic, but in some cases with weak support, in the cpDNA phylogenies of Clarkson et al. (2004). The members of section Polydicliae are indicated to share the same maternal parent, but each existing allopolyploid taxon arose at different times; therefore, the section is not monophyletic in cpDNA phylogenies (Clarkson et al. 2004). Current knowledge of the origins of allopolyploid species is discussed by Kelly et al. (2013).
The monophyly of section Suaveolentes is only weakly or moderately supported, and the clade shows comparatively low genetic variation based on cpDNA sequence data compared with other clades (Clarkson et al. 2004). Evolutionary relationships in section Suaveolentes are complex, but recent studies provide more detailed insights into the genetic, morphological, and karyotypic evolution of the group (Kelly et al. 2013; Marks and Ladiges 2011; Marks et al. 2011). Nuclear and plastid DNA sequence data suggest that a species from either section Noctiflorae or section Petunioides, or their hybrid, was the maternal progenitor, while a species from the section Sylvestres was the paternal progenitor (Kelly et al. 2013).
Some of the diploid species (N. glauca, N. spegazzinii, and N. linearis) have been shown to be homoploid hybrids based on sequence data for low-copy nuclear genes (Kelly et al. 2010).
1.3 Cytology and Allopolyploidy in Nicotiana
The base haploid chromosome number in the Nicotiana genus is n = 12, which is the haploid chromosome number in seven of the 13 sections proposed by Knapp et al. (Knapp et al. 2004b), as shown in Table 1.1. Aneuploid species occur in section Alatae (n = 9 or 10) and section Suaveolentes (n = 16–22). Four sections (Nicotiana, Rusticae, Polydicliae, and Repandae) contain allopolyploid species with n = 24. These allopolyploids comprise >40% of the total number of Nicotiana species. Autoploidy is believed to have been unimportant, at least in the recent evolution of the genus, and aneuploid and/or dysploid reduction is thought to have contributed to the evolution of numerous species in sections Alatae and Suaveolentes (Reed 1991).
Cytogenetic evolution in Nicotiana is summarized by Reed (1991). The karyotype of Nicotiana species is useful to resolve species groupings. Goodspeed (1947) presented hypotheses for karyotypic evolution in the genus. A number of meiotic aberrations have been observed in some diploid and polyploid species, which may support the suggested hybrid origin of those taxa. Meiotic behavior in all of the monosomics (individuals in which one chromosome of the normal complement has been deleted) of N. tabacum, in all of the trisomics (individuals that carry three copies of a specific chromosome) of N. sylvestris, and in >200 interspecific F1 hybrids has been studied and provides evidence for chromosomal affinities among the species. Nicotiana exhibits a relatively high degree of interspecific cross-compatibility. In approximately 90% of the F1 hybrids investigated, the amount of chromosomal pairing is consistent with the taxonomic affinity of the parental taxa (Goodspeed 1947). Allopolyploidy in Nicotiana has been associated with frequent chromosomal and cytogenetic alterations, such as intergenomic translocations (currently known only in N. tabacum (Leitch et al. 2008; Skalicka et al. 2005)), changes in copy number, distribution of both simple and complex repeats, rDNA homogenization, and loss of loci (e.g., Clarkson et al. 2004, 2005; Kenton et al. 1993; Lim et al. 2000, 2004, 2007; Petit et al. 2007). The extent of chromosomal evolution may be associated with the time of the polyploidy event (Lim et al. 2007).
Goodspeed (1954) used evidence from morphology, cytology, artificial hybridization experiments, and distribution to hypothesize on the progenitors of polyploid species; for example, Goodspeed suggested that a member of section Alatae was one parent of section Suaveolentes, that N. tabacum was derived from members of sections Alatae and Tomentosae, and that N. rustica was derived from the ancestors of N. undulata and N. paniculata. Meiotic behavior of interspecific F1 hybrids may also shed light on the ancestry of allopolyploid taxa; for example, N. sylvestris and N. otophora (section Tomentosae) F1 hybrids show almost no chromosome pairing, whereas F1 progeny of N. tabacum with either N. otophora or N. sylvestris show a high frequency of multivalents (Goodspeed 1947).
Many crosses between members of section Suaveolentes and N. tabacum, which share a common progenitor (the ancestor of N. sylvestris), show hybrid lethality. Different mechanisms may be responsible in different crosses. Genes in both the S- and T-genomes of N. tabacum are indicated to be responsible for hybrid lethality in crosses with one member of section Suaveolentes, N. occidentalis (Tezuka and Marubashi 2012). Tezuka and Marubashi (2012) proposed an evolutionary model to explain the phenomenon of hybrid lethality in section Suaveolentes.
More than 300 synthetic interspecific hybrids in Nicotiana have been described (Lewis 2011). Goodspeed (1947) considered that hybridization was a major contributor to diversification and chromosomal evolution in Nicotiana and that interspecific relationships were complex and difficult to resolve, as illustrated in his intricate phyletic diagrams. Phylogenetic analyses of cpDNA sequence data have provided evidence for the maternal progenitors of the allopolyploid taxa (Clarkson et al. 2004; Knapp et al. 2004b). The ancestor of N. sylvestris is the putative maternal parent of N. tabacum and is hypothesized to have contributed to multiple allotetraploid events and the origin of three allotetraploid sections (Knapp et al. 2004b).
Genomic in situ hybridization (GISH) offered additional proof of the identity of the progenitors of N. tabacum (Kenton et al. 1993; Murad et al. 2002) to explore the origin of 15 allopolyploid Nicotiana species (Chase et al. 2003); to evaluate genome evolution in the allotetraploid species N. arentsii, N. rustica, and N. tabacum (Lim et al. 2004); and to examine the integration of geminiviral-related DNA into the Nicotiana genome (Lim et al. 2000). GISH was used to demonstrate that virus-derived sequences of N. tabacum are closer to those of N. tomentosiformis than N. tomentosa (Murad et al. 2002). On the basis of GISH with total genomic DNA of N. sylvestris and N. tomentosiformis, Lim et al. (2000) suggested that gene conversion overall had played a limited role in the evolution of N. tabacum.
Fluorescent in situ hybridization (FISH) has also been used to pinpoint the chromosomes of N. tabacum (Parokonny and Kenton 1995), for chromosomal localization of the 5S rDNA (Kitamura et al. 2005), and to examine the localization of the Tnt1 retrotransposon in 22 Nicotiana species in conjunction with GISH (Melayah et al. 2004).
1.4 Genetic Resources in Nicotiana
1.4.1 Wild and Cultivated Germplasm
Wild Nicotiana species show considerable variation in growth habit, inflorescence type, and ecophysiological adaptation. Flower variability is mainly manifested in the corolla form and color, stamen insertion, and aestivation (Goodspeed 1947). Wild Nicotiana species offer extensive phenotypic diversity, cross-compatibility, and amenity to ploidy manipulation and tissue culture (Lewis 2011) and thus represent a valuable resource for genetic improvement of cultivated tobacco.
Considerable phenotypic variation in agromorphological traits and disease resistance exists among cultivated N. tabacum (e.g., Darvishzadeh et al. 2013; Elliott et al. 2008), which demonstrates there is potential for genotypic selection to improve cultivars. Numerous studies have detected a higher level of genetic diversity among wild Nicotiana species or naturalized N. tabacum genotypes than among cultivated N. tabacum genotypes (e.g., Ren and Timko 2001; Yang et al. 2007).
Genetic variability within N. tabacum is believed to have been affected by several genetic bottlenecks during its evolution, reflecting the nature of its origin and breeding practices, but it remains unknown if more than one hybridization event occurred and whether subsequent introgression from either progenitor species occurred. Furthermore, it is speculated that only a minute portion of the genetic diversity of the diploid progenitor genomes contributed to that of N. tabacum (Lewis and Nicholson 2007).
Cultivars of some market classes of cultivated tobacco, such as cigar, dark fire-cured, and oriental, have remained largely the same. However, burley and flue-cured cultivars have been significantly improved through modern breeding practices, which considerably narrowed their germplasm diversity, resulting in limited number of available crosses between members of different classes (Lewis and Nicholson 2007).
In several studies, grouping of N. tabacum genotypes based on genetic distance is generally consistent with the main tobacco classes (e.g., Darvishzadeh et al. 2013; Davalieva et al. 2010; Fricano et al. 2012), which suggests that the gene pool of each class has largely remained isolated during subsequent selection. The degree of homogeneity of each class partially corresponds to differences in the stringency of the breeding strategy for each class. Fricano et al. (2012) resolved two heterogeneous clades that contained most wild accessions and a number of country-specific cultivars. These clades may be of particular interest for future breeding programs, as they represent the highest genetic diversity in the Nicotiana genus. Grouping of N. tabacum genotypes based on phenotypic traits or genetic distance also enables discrimination of groups of other genetically divergent genotypes.
As with many crops, breeding and selection of superior genotypes and intensification of cultivation have coincided with a decline in genetic diversity among cultivated N. tabacum genotypes. Thus, conservation of genetic resources and their use to improve cultivated genotypes is of increasing importance. For example, the U.S. Department of Agriculture Nicotiana Germplasm Collection (USDA 2016) maintains more than 1,700 tobacco accessions and seeds of more than 200 accessions representing about 60 wild Nicotiana species (Lewis and Nicholson 2007). The Tobacco Research Institute of the Chinese Academy of Agricultural Sciences (TRI 2015) maintains a collection of more than 5,300 tobacco accessions and claims to maintain the largest collection of tobacco varieties in the world.
1.4.2 Cytoplasmic Male Sterile Lines
Cytoplasmic male sterility (CMS) is frequently encountered in interspecific hybrid progeny between a wild Nicotiana species and N. tabacum (as the pollen donor) after several backcross generations. It is usually expressed as staminal sterility. Many modern N. tabacum genotypes in cultivation carry CMS (Lewis and Nicholson 2007). Lines with restored structural or functional fertility were identified within the CMS lines (Reed 1991); however, tobacco leaves rather than seeds bring the highest production value.
1.4.3 Cytogenetic Stocks
Aneuploid lines of Nicotiana have been developed and are useful for breeding and genetic investigations, but they have not been used to the same extent as in other polyploid crops, such as Triticum. As allotetraploids, both N. tabacum and N. rustica seem to be tolerant to different forms of aneuploidy. A number of genes have been localized to individual chromosomes within the monosomic lines. Trisomic lines have been developed for N. sylvestris and N. langsdorffii (Lewis 2011). Nullisomics (in which a specific chromosome pair from both genomes is deleted) have been generated, mostly in N. tabacum. The fact that most nullisomics of N. tabacum are nonviable or sterile is thought to be because of the evolution of genomic interdependence subsequent to polyploidization (Reed 1991).
In attempts to transfer foreign genes (e.g., for tobacco mosaic virus [TMV] resistance) to N. tabacum from wild species and to localize genes or molecular markers to specific loci, chromosome addition lines can be produced in tobacco (Lewis 2011). The amenability of N. tabacum to transformation and tissue culture and the recent availability of genomic resources and genetic maps may stimulate interest in genetic engineering technologies for improvement of cultivated tobacco.
1.5 Reference Genomes in Nicotiana
A methyl filtration approach (Whitelaw et al. 2003) was used to address the combined challenges of the bulky genome size and complexity of the tobacco genome within the Tobacco Genome Initiative (TGI). The project aimed to sequence more than 90% of N. tabacum hypomethylated gene-rich genomic DNA sequences. In addition, several bacterial artificial chromosome (BAC) libraries were constructed to perform BAC-by-BAC sequencing. More than 1.3 million genome survey sequences and more than 50,000 expressed sequence tags (EST) were released to GenBank. The subsequent assembly produced a highly fragmented genome with the majority of contigs containing only partial gene structures. Nevertheless, the TGI data served as a source of genomic sequences for the design of a tobacco exon array (Martin et al. 2012) and a tobacco genetic map (Bindler et al. 2011).
High-quality draft genome sequences have been published for representative genotypes of the three main cultivar classes of N. tabacum (K326, flue-cured; TN90, burley; and Basma Xanthi, oriental) (Edwards et al. 2017; Sierro et al. 2014), its progenitor species N. sylvestris and N. tomentosiformis (Sierro et al. 2013a), N. benthamiana (Bombarely et al. 2012; Naim et al. 2012), N. attenuata (Xu et al. 2017), and N. rustica and its progenitor species N. undulata, N. paniculate, and N. knightiana (Sierro et al. 2018). In addition, Sierro et al. (2013b) constructed a physical map of N. tabacum Hicks Broadleaf using whole-genome profiling technology. The physical map, which was predicted to cover the whole tobacco genome, was subsequently used for super-scaffolding of the N. tabacum genome (Sierro et al. 2014).
Bombarely et al. (2012) mapped 56.5 and 58.5% of coding sequences from the tomato and potato genomes, respectively, to the N. benthamiana genome to demonstrate syntenic relationships. The genomic ancestry of N. benthamiana is uncertain, but the progenitors of the allopolyploid section Suaveolentes might belong to the lineages represented by section Sylvestres and either section Noctiflorae or section Petunioides (Kelly et al. 2013).
Multiple insertions of cellular transferred DNA sequences (cT-DNA), derived from Agrobacterium strains and integrated into the host nuclear genome, have occurred during the evolution of Nicotiana. cT-DNAs contain genes involved in plant growth and the synthesis of opines important for bacterial growth. A number of Agrobacterium-derived genes (including mis, orf13, and rolC) have been detected by polymerase chain reaction amplification in the genomes of various Nicotiana species (see Chen et al. 2014). Four cT-DNA inserts in the genome of N. tomentosiformis have been characterized by deep sequencing. In addition, the cT-DNAs present in the genomes of N. tabacum Basma Xanthi and N. otophora have been identified, with a fifth cT-DNA detected in the latter species (Chen et al. 2014). The cT-DNA inserted exclusively in N. otophora contains intact open reading frames coding for three 6b genes derived from the original Agrobacterium 6b gene, which is known to modify plant growth. The phenotypes observed upon expression of these 6b genes in N. tabacum under the constitutive 2 × 35S promoter are however different from those previously described. Indeed, shorter plants exhibiting modified petiole wings and dark-green leaves with increased number of veins were observed upon expression of any of these three genes. Furthermore, the expression of one of them led to the additional development of outgrowths at the leaf margins and of modified flowers. In the capsules of these plants, germination of the embryos was observed at an early development stage in the capsules (Chen et al. 2018). Kovacova et al. (2014) showed that the agrobacterial mis gene has evolved independently in the N. glauca, N. tabacum, and N. tomentosiformis genomes.
1.5.1 Chloroplast Genomes
The complete chloroplast genome of N. tabacum was sequenced more than 30 years ago (Shinozaki et al. 1986) and comprises 155 kb, including two identical copies of a 25 kb inverted repeat separated by 86 kb and 18 kb unique regions.
The whole chloroplast genomes of N. sylvestris and N. tomentosiformis have also been described (Yukawa et al. 2006). The N. sylvestris chloroplast genome comprises 156 kb and, with the exception of one gene, is highly similar to the one of N. tabacum. Only seven sites were polymorphic between N. tabacum and N. sylvestris. The chloroplast genome of N. tomentosiformis is 156 kb long and also shows similar genome structure to that of N. tabacum, except for five regions with more than 1,000 single nucleotide polymorphism (SNP) differences. Overall, the chloroplast genomes of N. sylvestris and N. tomentosiformis were 99.99 and 98.54% identical to that of N. tabacum. The authors concluded that the detailed comparison clearly supported the previous finding that N. sylvestris was the source of the N. tabacum chloroplast genome (Yukawa et al. 2006).
1.5.2 Mitochondrial Genome
Bland et al. (1985) cloned fragments of known genes in the mitochondrial genome (mtDNA) of maize and unidentified genes from N. sylvestris mtDNA to use as labeled probes for Southern blot hybridization to explore N. tabacum mtDNA. The results supported the contention that N. tabacum mtDNA was inherited from N. sylvestris and not from N. tomentosiformis. Conservation in organization and sequence homology indicates that little evolutionary divergence in mtDNA between N. tabacum and N. sylvestris has occurred.
More recently, Sugiyama et al. (2005) sequenced the complete mtDNA of N. tabacum. The genome was assumed to conform to a conventional “master circle” model of mitochondrial genome structure (Sloan 2013) and comprised of 431 kb, including protein-coding, ribosomal RNA, and transfer RNA genes. The homology of repeated sequences, the gene organization, and the intergenic spacer regions differ markedly from those of other model plants (Arabidopsis thaliana or Oryza sativa), indicating that the genome structure has undergone multiple reorganizations during higher plant evolution.
1.6 Reference Genomes in Solanaceae and Coffea
The genomes of Solanaceae and Coffea species play an important role in comparative genome analysis of Nicotiana species. The knowledge accumulated over the last decades of research can be leveraged at genetics, genomics, transcriptomics, and proteomics data analysis.
The genomes of tomato (S. lycopersicum) (Sato et al. 2012) and potato (S. tuberosum) (Xu et al. 2011) are recognized as the best-quality reference genomes for the Solanaceae family, mostly due to the significant decade-long efforts by the tomato and potato communities to generate a huge collection of genomics and genetics resources. Although tomato and potato are closely related, the cultivated tomato varieties are inbred self-pollinating diploids, whereas most potato cultivars are highly heterozygous self-incompatible autotetraploids.
Due to high heterozygosity and autotetraploidy of potato cultivars, a homozygous doubled-monoploid (DM) potato was constructed to facilitate the assembly of its high-quality genome and transcriptome (Xu et al. 2011). The 727 Mb assembly of the genome with estimated size of 884 Mb was split over 443 super-scaffolds and contained at least 62% of repeats. The majority of the assembly (86%) was anchored to the potato genetic and physical maps to construct 12 pseudomolecules. Large blocks of heterochromatin were attributed to pericentromeric regions, and gene-rich euchromatin was pushed toward the distal chromosome ends. The combination of ab initio and experimental evidence (proteins and ESTs) predicted 39,031 protein-coding genes in the DM potato. Approximately 10,000 genes clustered into 1,800 paired syntenic blocks, suggesting the occurrence of at least two whole-genome duplications in the potato. Comparison between the DM and heterozygous potato (RH line) uncovered high level of structural variations comparable to the one in maize; however, no specific genes or regions were identified to be responsible for the phenomenon of inbreeding depression in the potato.
The assembled tomato genome contained 760 Mb (84% of 900 Mb) split into only 91 scaffolds linked to the BAC-based physical, genetic, and introgression line maps (Sato et al. 2012). As in the case of the potato, the tomato euchromatic gene-rich regions (71 Mb) were situated at the distal parts of the chromosome arms, while the heterochromatin was found in the pericentromeric part. The tomato genome has an unusually low proportion of retrotransposons and other complex repeats. The annotation pipeline predicted 35,000 genes in the tomato and potato, 18,320 of which were orthologous between the two species. Comparison of the tomato and potato genomes revealed ~9% nucleotide divergence in euchromatin and ~30% in heterochromatin. More than 20% of genomic regions of the grape have three orthologous regions in the tomato genome, supporting the hypothesis that triplication in the tomato occurred ~71 Mya based on Ks of paralogous genes (i.e., a long time before the divergence of the tomato and potato [~7 Mya]).
One of the interesting features of the hot pepper genome (C. annuum cv. CM334) is its large diploid size of 3.48 Gb (Kim et al. 2014), compared to less than 1 Gb for tomato or potato genomes. Kim et al. assembled 88% (3.06 Gb) of the genome into 37,989 scaffolds (with N50 = 2.47 Mb) and anchored 86% of the latter (2.63 Gb) onto 4,562 markers of the combined C. annuum high-density genetic map (Kim et al. 2014; Yarnes et al. 2013). Transposable elements occupied 76% (2.34 Gb) of the C. annuum genome, with long-terminal repeats (LTR) being the most represented (~70%) with only slight distribution preference toward the centromeric regions. Substantial expansion of the Gypsy family retrotransposons (sevenfold higher than in tomato) was put forward as the main cause of the inflated size of C. annuum. Caulimoviridae retrotransposons was another unusually abundant repeat family present ninefold higher than in tomato. The number of protein-coding genes (34,903) was very close to other Solanaceae, 17,397 of which have an ortholog in the tomato gene set. However, the expression levels among different tissues varied significantly, affecting as many as 46% of orthologous genes. Hot pepper, tomato, and potato shared 2,139 gene families, and 756 were specific to C. annuum. Genomics analysis of two other varieties of C. annuum (Qin et al. 2014) largely confirmed the observations and provided further insights into its evolution, such as evidence for the whole-genome triplication. Despite more than 600 chromosomal translocation events between the pepper and tomato genomes being uncovered, overall, the presence of a substantial number (>1,000) of large syntenic blocks indicated a close evolutionary relationship between the species.
The draft genome of Solanum melongena L. (eggplant) (Hirakawa et al. 2014) covered 833 Mb (74%) of its estimated 1.1 Gb size, with 586 Mb annotated as repetitive sequences (70% of 833 Mb). LTR of Copia and Gypsy types accounted for almost 30% of the repeats. Only 42,035 of 85,446 genes were considered protein-coding, whereas the rest were described as “transposable elements.” A total of 4,018 genes were found to be exclusive to the eggplant. Finally, Hirawaka et al. (2014) used the obtained genomics sequences to identify SNP and simple sequence repeat (SSR) polymorphic markers in the interspecific F2 population and constructed a genetic map containing on 574 SNPs and 221 SSRs.
Although the Coffea genus belongs to the Rubiaceae family, there are a few striking similarities to the Nicotiana genus, making the inclusion of Coffea species valuable. Some of the most popular cultivars of coffee belong to the allotetraploid C. arabica (2n = 4x = 44) species, which, similar to N. tabacum, emerged through an interspecific hybridization of two ancestral Coffea species still available today: C. eugenioides (2n = 2x = 22) and C. canephora (2n = 2x = 22). The latter is also known as C. robusta and is considered to be easier to produce yet more bitter coffee. While the genome of C. arabica is still in progress, the high-quality draft of C. canephora assembled at 80% of its estimated 710 Mb genome size was reported as harboring 25,574 protein-coding genes (Denoeud et al. 2014). A high-density genetic map covered 64% of the draft genome. About half of the genome was filled with repeats, 85% of which were LTR retrotransposons. Comparative genome analysis of C. canephora chromosomes provided no evidence of triplication since the origin of eudicots, thus displaying one-to-one mapping to the grape genome and very little syntenic divergence to other asterids. Denoeud et al. (2014) suggested using C. canephora as a reference species to study the evolution of asteroid angiosperms.
1.7 Genomic Evolution in Nicotiana
Goodspeed (1954) considered that genic rather than genomic evolution was of greater importance in the evolution of the allopolyploid lineages of Nicotiana. However, recent studies have provided detailed insights into the extent of genomic evolution that has occurred in N. tabacum and other allopolyploid Nicotiana species of different ages. Allopolyploidy is frequently linked to functional and structural genomic changes, and DNA loss or gain often occurs at the onset of polyploidization (Petit et al. 2007). Such genomic differentiation or “diploidization,” which decreases pairing between homoeologous chromosomes, is documented in Nicotiana allopolyploid species of different ages and synthetic hybrids.
1.7.1 Chromosomal Rearrangements
The N. tabacum genome has experienced a variety of chromosomal rearrangements subsequent to polyploidization in either wild type or synthetic species compared with the progenitor diploid genomes. The rearrangements include up to nine intergenomic translocations, retrotransposon deletion/proliferation, and transposable element mobility and loss (Bindler et al. 2011; Kenton et al. 1993; Kovarik et al. 2012; Lim et al. 2007; Petit et al. 2010; Skalicka et al. 2005; Wu et al. 2010). The T-genome of N. tabacum shows reduced genetic stability compared with the S-genome (Kovarik et al. 2012). The T-genome of N. tabacum is shown to have experienced more rapid chromosomal evolution than the S-genome, and both genomes are indicated to have evolved more rapidly than the corresponding genomes of diploid relatives. The more rapid genomic changes in N. tabacum and other polyploid species might be attributable to chromosome recombination, epigenetic variation, or higher transposable element activity (Wu et al. 2010). Matyasek et al. (2011) suggested that the lack of subtelomeric satellite repeats in the T-genome of N. tabacum may promote homoeologous pairing, which in turn might explain the frequency of intergenomic translocations in the N. tabacum genome. Wild N. tabacum plants contain reduced copy numbers of pararetroviral repeat sequences derived from N. tomentosiformis and of the Tnt1 retrotransposon compared with the number expected to be inherited from the progenitors (Gregor et al. 2004; Melayah et al. 2004). Given that most nullisomics of N. tabacum are nonviable or sterile, post-polyploidization evolution of interdependence of the progenitor genomes is also indicated (Lewis 2011). However, both dynamic and stable inheritance of different repeat sequences during N. tabacum evolution is implicated (Skalicka et al. 2005).
1.7.2 Post-polyploidization Changes
Rapid genetic changes in synthetic N. sylvestris × N. tomentosiformis hybrids have been documented, mostly targeted to the paternal T-genome donated by N. tomentosiformis. Lim et al. (2004) examined genomic rearrangements in three recent polyploid species (N. arentsii, N. rustica, and five cultivars and one wild genotype of N. tabacum; each allopolyploid originated <0.2 Mya) and three synthetic F3 progeny derived from the cross N. sylvestris × N. tomentosiformis. Intergenomic translocations were observed in all natural genotypes of N. tabacum examined but not in N. arentsii or N. rustica. Skalická et al. (2005) reported that two repeats related to endogenous viruses and two classes of N. tomentosiformis-specific noncoding tandem repeats were deleted in S4 plants generated from a synthetic allotetraploid S0 plant. All of the sequences were derived from the T-genome of N. tomentosiformis. T-genome 35S rDNA genes underwent rapid homogenization and were replaced by novel gene variants in the same S4 population (Skalicka et al. 2003, 2005). Some of the abovementioned changes are consistent with changes that are believed to have occurred during the evolution of N. tabacum. These studies also demonstrate that considerable genomic variation may arise rapidly from a single polyploid individual within only three or four generations.
1.7.3 Transposable Elements
Transposable elements are mobile DNA elements that are inserted in different positions in the genome and generate structural variations upon insertion, therefore having an important impact on genetic diversity, gene expression, and genome structure. Extensive and rapid turnover of retrotransposons and transcriptional activation and/or mobilization of transposable elements may follow interspecific hybridization and polyploidization (Petit et al. 2007, 2010). Sequence-specific amplification polymorphisms (S-SAP) have been used to examine retrotransposon polymorphism and genomic distribution in Nicotiana species (Melayah et al. 2004; Petit et al. 2007). Melayah et al. (2004) reported that Tnt1 copia-type retrotransposons are highly polymorphic and have evolved rapidly in Nicotiana, and the number of Tnt1 insertions differs widely among Nicotiana species. S-SAP profiles provide evidence for post-polyploidization changes in Tnt1 insertions in N. tabacum. Some 30% of N. tabacum S-SAP bands are shared with N. tomentosiformis, and 38% are shared with N. sylvestris. A notable proportion (28%) of Tnt1 insertions are unique to N. tabacum. A large proportion of S-SAP bands (52% for N. sylvestris and 47% for N. tomentosiformis) are present in the two progenitor species but not in N. tabacum. In N. tomentosiformis, Tnt1 insertions are distributed across all chromosomes but at different densities. N. sylvestris contains a higher number of Tnt1 insertions, which show a more homogeneous distribution in the genome. The number of insertions in N. tabacum is higher than the numbers in both progenitor species, although only 67% of the expected number based on the numbers detected in the progenitor species. These insertions are more concentrated on S-genome chromosomes than on T-genome chromosomes. Low Tnt1 polymorphism among N. tabacum lines suggests that Tnt1 content has stabilized during N. tabacum evolution. Petit et al. (2007) showed that retrotransposon turnover occurs in N. tabacum with removals counterbalanced by new insertions.
Parisod et al. (2012) examined the insertion polymorphism of seven transposable elements in the four allopolyploid species of section Repandae that show opposing evolutionary trends in N. tabacum and its progenitors. Many novel S-SAP bands were observed for two transposable elements, but considerable loss of transposable elements was detected in the four allotetraploid species of section Repandae relative to the genomes of the diploid progenitor species. In the process of diploidization of the four allotetraploid species, the transposable element genome fractions have been significantly restructured.
1.7.4 Repetitive Elements
Nicotiana genomes are notably rich in simple repeats, three superfamilies of which have been characterized in Nicotiana species (Matyasek et al. 2011). Homologous subtelomeric repeats in the allotetraploid N. arentsii, descended from progenitors of N. undulata (maternal donor) and N. wigandioides (paternal donor; both members of section Undulatae) less than 0.2 Mya, were analyzed by Matyasek et al. (2011). Intergenomic homogenization of the two homologous satellites has not occurred in N. arentsii. The authors suggested that the dissimilarity in sequence and structure of the satellite repeats protected homoeologous chromosomes from genomic intergressions.
As reported by Koukalova et al. (2010), the Nicotiana genomes of more recent allotetraploid species formed ~1 Mya (section Polydicliae) contain rearranged yet intact progenitor repeat sequences, while in older allotetraploids (section Repandae ~5 Mya), different satellite repeats partially or completely replaced the ones of the corresponding progenitors. The authors proposed a mechanism that involves removal of progenitor heterochromatic repeat-containing blocks and rolling circle replication leading to formation of new uniform blocks of satellite repeats.
Gill (1991) proposed that during the allotetraploid formation, incompatibility between maternal and paternal cytoplasms may become a source of progenitor genome instability. Lim et al. (2004) concluded that nuclear–cytoplasmic interaction was indicated to have influenced genome evolution only in N. tabacum among the taxa studied. If this hypothesis is pertinent to N. tabacum, the N. tomentosiformis-derived repeat sequences in N. tabacum would continue to be eliminated well after the polyploidization event (Lim et al. 2007). Skalická et al. (2005) raised the possibility that the N. tomentosiformis genome may simply be inherently less stable than that of N. sylvestris.
1.7.5 Other Genetic Changes
Homogenization of rDNA is often noted among allopolyploid Nicotiana species, but the extent of interlocus homogenization differs among allopolyploid species and synthetic polyploid hybrids (Dadejova et al. 2007). Furthermore, 18S rDNA coding sequences show near-complete homogenization in the diploid species N. sylvestris, N. tomentosiformis, N. otophora, and N. kawakamii, whereas the ITS1 region shows greater heterogeneity. Dadejova et al. (2007) speculated that transcriptionally active rDNA genic regions are more susceptible to homologous recombination than those that are transcriptionally silenced and thus remain unchanged. Matyasek et al. (2012) proposed that the evolution of rDNA genes is dependent on the transcriptional activity and the number of copies. The detection of putative intragenic recombination in the several low-copy nuclear genes suggests that formation of chimeric regions arising from different alleles might have been a common event during Nicotiana evolution (Kelly et al. 2010).
1.7.6 Genomic Downsizing
“Genomic downsizing” has been a frequent phenomenon found in Nicotiana ancient and more recent polyploids (Leitch and Bennett 2004). Changes in genome size in allopolyploid Nicotiana species were examined by comparing the genome sizes of nine diploid and nine polyploid species, with the extant species believed to be descended from the putative progenitors (Leitch et al. 2008). Genome size was assessed using flow cytometry and Feulgen microdensitometry. Four polyploids displayed genome downsizing, whereas five polyploids showed genome size increases. N. repanda and N. nudicaulis share the same progenitor species (ancestors of N. sylvestris and N. obtusifolia) yet show contrasting trends in genome size changes (~29% increase and ~14% decrease, respectively). The amount of genome size divergence was enhanced with increasing estimated age of the polyploid.
The progenitor sequences were still detectable in the downsized polyploids, while the signal disappears in the older upsized ones (formed ~4.5 Mya). The authors concluded that genome upsizing is likely linked with replacement of the progenitor repeat regions with the new satellite repeats.
Lim et al. (2007) analyzed genome evolution in Nicotiana allopolyploid by comparing repeat sequence distribution in selected BAC clones, in conjunction with FISH and GISH, from four allopolyploids of contrasting ages: N. nesophila (formed ~4.5 Mya), N. quadrivalvis (~1 Mya), N. tabacum (<0.2 Mya), and synthetic F1 N. sylvestris × N. tomentosiformis hybrids. The authors proposed the term “genome turnover” for the apparent genome-wide replacement of one sequence type by another, possibly through concerted evolution. The authors presented a hypothetical time-course after polyploidization of the genomic events occurring in Nicotiana allopolyploids from the initial “genomic shock,” followed by sequence mixing, to swap of progenitor DNA with new sets of genomic DNA repeats and, ultimately, a reduction in chromosome number (Lim et al. 2007).
1.8 Functional Genomics and Gene Mining
1.8.1 Alkaloid Metabolism
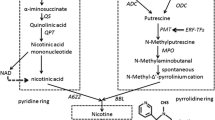
The biosynthesis and transport of major tobacco alkaloids (e.g., nicotine, nornicotine, anabasine, and anatabine) have been extensively reviewed elsewhere (Dewey and Xie 2013; Eich 2008). Nicotine is synthesized in the roots of tobacco plants and is transported to the leaves. The majority of genes involved in the nicotine biosynthesis pathway are sequenced and well characterized. Nicotine is the principal alkaloid that accumulates in N. sylvestris, whereas nornicotine is predominant in N. tomentosiformis, typical for members of section Tomentosae. Unlike most Nicotiana species, N. sylvestris accumulates higher quantities of nicotine in the roots than in the leaves. Transcriptome data indicate that the marked differences in alkaloid metabolism in N. sylvestris and N. tomentosiformis are due to strong upregulation of a set of key enzymes in the nicotine biosynthesis pathway in the roots of N. sylvestris (e.g., aspartate oxidase, putrescine N-methyltransferase [PMT], quinolinate synthase, and quinolinic acid phosphoribosyl transferase) compared with N. tomentosiformis (Sierro et al. 2013a).
In the senescing leaf of N. tabacum, nicotine is accumulated as the main alkaloid, whereas in N. sylvestris, N. tomentosiformis, and synthetic N. sylvestris × N. tomentosiformis and N. sylvestris × N. otophora amphidiploids, nicotine is converted to nornicotine. This pathway is mediated by nicotine N-demethylase (CYP82E) genes, copies of which are present in the genomes of the progenitor species. Transcriptome data indicate that the higher accumulation of nornicotine in the leaf of N. tomentosiformis reflects expression of three CYP82E genes that enhance nornicotine production (Sierro et al. 2013a). Mutation of CYP82E in N. tabacum decreases conversion of nicotine to nornicotine compared with the wild type (Chakrabarti et al. 2007).
Gene expression analysis of the tobacco BY-2 cell cultures has identified a set of genes responsible for alkaloid biosynthesis (Goossens et al. 2003). Transcription factors (TF) were screened using a transient expression assay, leading to identification of two AP-2 domain TFs that enhance the expression of PMT, a key enzyme in nicotine biosynthesis (De Sutter et al. 2005). Häkkinen et al. (2007) isolated full-length clones for several genes putatively involved in pyridine alkaloid metabolism. Upon their transformation into tobacco BY-2 cell and hairy root cultures, overexpression of a GH3-like enzyme (designated NtNEG1) was shown to increase nicotine accumulation.
In general, the number of isoforms of genes implicated in alkaloid biosynthesis present in the N. tabacum reference genomes corresponds roughly to the sum of genes inherited from the progenitor species, N. tomentosiformis and N. sylvestris (Sierro et al. 2014).
1.8.2 Cadmium Accumulation
An unusual property of Nicotiana species is the ability to accumulate relatively high concentrations of cadmium and other heavy metals compared with other plants. N. sylvestris and N. tomentosiformis genomic sequences and transcriptome data for the root, leaf, and flower have been analyzed to identify genes associated with heavy metal accumulation and transport (Sierro et al. 2013a). Certain differences in transcription were detected in the different organs and between the two Nicotiana species, and between A. thaliana and the Nicotiana species. Expression of osmotin- and thaumatin-like proteins in both leaf tissue and trichomes is induced by cadmium treatment (Harada et al. 2010).
1.8.3 Disease Resistance and Viral Infection
Many of the disease resistance genes currently present in N. tabacum germplasm were introgressed from wild Nicotiana species (Battey and Ivanov 2014). These genes often consist of a single locus (e.g., TMV resistance derived from N. glutinosa (Lewis et al. 2005) and tomato spotted wilt virus resistance inherited from N. alata (Moon and Nicholson 2007)), but often additional genetic material from the donor Nicotiana species is also introgressed with the resistance gene (Battey and Ivanov 2014).
TMV resistance was introduced into N. tabacum in the 1930s as a single dominant locus through interspecific hybridization with N. glutinosa. The locus harbors the N gene, which encodes a TIR-NBS-LRR protein, triggering a hypersensitive response involving formation of a necrotic lesion (Holmes 1938). It was cloned (Whitham et al. 1994) and subsequently successfully transformed (Lewis et al. 2007). Adoption of flue-cured cultivars carrying the N gene was low because of unfavorable “linkage drag effects” (Lewis et al. 2005). Among the three N. tabacum cultivars with genome sequences, only TN90 is TMV resistant and includes a putative N. glutinosa-derived genomic segment containing the N gene (Sierro et al. 2014).
N. tabacum are vulnerable to infections by potyviruses, such as potato virus Y (PVY), tobacco vein mottling virus, and tobacco etch virus. Unlike N. tabacum and N. sylvestris, N. tomentosiformis is resistant to PVY. Next-generation sequencing has been used to characterize the recessive PVY resistance gene in a recombinant inbred line population of N. tabacum (Julio et al. 2014). A correlation between susceptibility and expression of a eukaryotic translation initiation factor 4E (eIF4E) was confirmed. The eIF4E gene was mapped on linkage group 21 and was inherited from the progenitor of N. sylvestris. Missense mutations in the eIF4E gene conferred resistance of N. tabacum lines to PVY infection. PVY resistance is associated with deletion of a chromosomal segment containing the eIF4E gene, which is corroborated by the genome sequence of the PVY-resistant N. tabacum TN90 (Sierro et al. 2014), although identification of other PVY-resistant accessions with a functional eIF4E gene indicates that additional sources of resistance may exist (Julio et al. 2014).
N. benthamiana serves as a model system to study plant–pathogen interactions, in large part because of its susceptibility to a wide spectrum of bacteria and viruses (Goodin et al. 2008). The unique virus susceptibility of N. benthamiana is at least partly attributed to an SNP in an RNA-dependent RNA polymerase gene (NbRdRP1m) (Yang et al. 2004). Senthil et al. (2005) used microarrays derived from S. tuberosum ESTs to monitor gene expression to shed light on the mechanism of N. benthamiana infection with a variety of viruses. In addition, progress is being made on the development of VIGS cDNA libraries for N. benthamiana (Goodin et al. 2008).
Bombarely et al. (2012) detected orthologs for more than 20 immunity-associated genes putatively transferred into the N. benthamiana genome from other species. Due to the allotetraploid nature of N. benthamiana, with a couple of exceptions, each gene was represented by at least two homoeologous sequences in its genome sequence.
1.9 Conclusion
Among Nicotiana species, N. tabacum and N. benthamiana have been extensively used as model systems for plant biology. The availability of a growing number of genomic resources for the genus contributed significantly to understanding the Nicotiana biology and evolution. Tobacco plants thus continue to play a significant role in plant research. Furthermore, N. tabacum remains an important agricultural crop for which these resources are opening new avenues (e.g., in numerous biotechnological applications for the production of valuable compounds for the pharma or cosmetic industries). With the release of additional resources and, consequently, the increased understanding of the specificities of each clade and species, other members of the Nicotiana genus will eventually also develop into efficient biological systems for the production of these compounds.
References
Battey JND, Ivanov NV (2014) Data mining of plant metabolic pathways. In: Hoffmann RD, Gohier A, Pospisil P (eds) Data mining in drug discovery. Wiley, pp 101–130
Bindler G, Plieske J, Bakaher N, Gunduz I, Ivanov N, Van der Hoeven R, Ganal M, Donini P (2011) A high density genetic map of tobacco (Nicotiana tabacum L.) obtained from large scale microsatellite marker development. Theor Appl Genet 123:219–230
Bland MM, Matzinger DF, Levings CS (1985) Comparison of the mitochondrial genome of Nicotiana tabacum with its progenitor species. Theor Appl Genet 69:535–541
Bolger A, Scossa F, Bolger ME, Lanz C, Maumus F, Tohge T, Quesneville H, Alseekh S, Sorensen I, Lichtenstein G et al (2014) The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat Genet 46:1034–1038
Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller L, Martin G (2012) A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol. Plant Microbe Interact 25:1523–1530
Bombarely A, Moser M, Amrad A, Bao M, Bapaume L, Barry CS, Bliek M, Boersma MR, Borghi L, Bruggmann R et al (2016) Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat Plants 2:16074
Chakrabarti M, Meekins KM, Gavilano LB, Siminszky B (2007) Inactivation of the cytochrome P450 gene CYP82E2 by degenerative mutations was a key event in the evolution of the alkaloid profile of modern tobacco. New Phytol 175:565–574
Chase MW, Knapp S, Cox AV, Clarkson JJ, Butsko Y, Joseph J, Savolainen V, Parokonny AS (2003) Molecular systematics, GISH and the origin of hybrid taxa in Nicotiana (Solanaceae). Ann Bot 92:107–127
Chen K, Dorlhac de Borne F, Szegedi E, Otten L (2014) Deep sequencing of the ancestral tobacco species Nicotiana tomentosiformis reveals multiple T-DNA inserts and a complex evolutionary history of natural transformation in the genus Nicotiana. Plant J 80:669–682
Chen K, Dorlhac de Borne F, Sierro N, Ivanov NV, Alouia M, Koechler S, Otten L (2018) Organization of the TC and TE cellular T-DNA regions in Nicotiana otophora and functional analysis of three diverged TE-6b genes. Plant J Cell Mol Biol 94:274–287
Clarkson JJ, Knapp S, Garcia VF, Olmstead RG, Leitch AR, Chase MW (2004) Phylogenetic relationships in Nicotiana (Solanaceae) inferred from multiple plastid DNA regions. Mol Phylogenet Evol 33:75–90
Clarkson JJ, Lim KY, Kovarik A, Chase MW, Knapp S, Leitch AR (2005) Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytol 168:241–252
Dadejova M, Lim KY, Souckova-Skalicka K, Matyasek R, Grandbastien MA, Leitch A, Kovarik A (2007) Transcription activity of rRNA genes correlates with a tendency towards intergenomic homogenization in Nicotiana allotetraploids. New Phytol 174:658–668
Darvishzadeh R, Mirzaei L, Maleki HH, Laurentin H, Alavi SR (2013) Genetic variation in oriental tobacco (Nicotiana tabacum L.) by agro-morphological traits and simple sequence repeat markers. Rev Ciênc Agronômica 44:347–355
Davalieva K, Maleva I, Filiposki K, Spiroski O, Efremov GD (2010) Genetic Variability of Macedonian tobacco varieties determined by microsatellite marker analysis. Diversity 2:439–449
Davis DL, Nielsen MT (1999) Tobacco : production, chemistry and technology. Blackwell Science Ltd
De Sutter V, Vanderhaeghen R, Tilleman S, Lammertyn F, Vanhoutte I, Karimi M, Inze D, Goossens A, Hilson P (2005) Exploration of jasmonate signalling via automated and standardized transient expression assays in tobacco cells. Plant J 44:1065–1076
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G et al (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184
Dewey RE, Xie J (2013) Molecular genetics of alkaloid biosynthesis in Nicotiana tabacum. Phytochemistry 94:10–27
Edwards KD, Fernandez-Pozo N, Drake-Stowe K, Humphry M, Evans AD, Bombarely A, Allen F, Hurst R, White B, Kernodle SP et al (2017) A reference genome for Nicotiana tabacum enables map-based cloning of homeologous loci implicated in nitrogen utilization efficiency. BMC Genom 18:448
Eich E (2008) Solanaceae and convolvulaceae: secondary metabolites: biosynthesis, chemotaxonomy, biological and economic significance (a handbook). Springer
Elliott PE, Lewis RS, Shew HD, Gutierrez WA, Nicholson JS (2008) Evaluation of tobacco germplasm for seedling resistance to stem rot and target spot caused by Thanatephorus cucumeris. Plant Dis 92:425–430
Fernandez-Pozo N, Menda N, Edwards JD, Saha S, Tecle IY, Strickler SR, Bombarely A, Fisher-York T, Pujar A, Foerster H et al (2015) The Sol Genomics Network (SGN)-from genotype to phenotype to breeding. Nucleic Acids Res 43:D1036–D1041
Fricano A, Bakaher N, Del Corvo M, Piffanelli P, Donini P, Stella A, Ivanov NV, Pozzi C (2012) Molecular diversity, population structure, and linkage disequilibrium in a worldwide collection of tobacco (Nicotiana tabacum L.) germplasm. BMC Genet 13:18
Gill BS (1991) Nucleocytoplasmic interaction (NCI) hypothesis of genome evolution and speciation in polyploid plants. In: Sasakuma T, Kinoshita T (eds) Proceedings of the Kihara Memorial International Symposium on Cytoplasmic Engineering in Wheat, pp 48–53
Goodin MM, Zaitlin D, Naidu RA, Lommel SA (2008) Nicotiana benthamiana: its history and future as a model for plant-pathogen interactions. Mol Plant Microbe Interact 21:1015–1026
Goodspeed TH (1947) On the evolution of the genus Nicotiana. Proc Natl Acad Sci USA 33:158–171
Goodspeed TH (1954) The genus Nicotiana. Chron Bot 16:1–536
Goossens A, Hakkinen ST, Laakso I, Seppanen-Laakso T, Biondi S, De Sutter V, Lammertyn F, Nuutila AM, Soderlund H, Zabeau M et al (2003) A functional genomics approach toward the understanding of secondary metabolism in plant cells. Proc Natl Acad Sci USA 100:8595–8600
Gregor W, Mette MF, Staginnus C, Matzke MA, Matzke AJM (2004) A distinct endogenous pararetrovirus family in Nicotiana tomentosiformis, a diploid progenitor of polyploid tobacco. Plant Physiol 134:1191–1199
Hakkinen ST, Tilleman S, Swiatek A, De Sutter V, Rischer H, Vanhoutte I, Van Onckelen H, Hilson P, Inze D, Oksman-Caldentey KM et al (2007) Functional characterisation of genes involved in pyridine alkaloid biosynthesis in tobacco. Phytochemistry 68:2773–2785
Harada E, Kim JA, Meyer AJ, Hell R, Clemens S, Choi YE (2010) Expression profiling of tobacco leaf trichomes identifies genes for biotic and abiotic stresses. Plant Cell Physiol 51:1627–1637
Hirakawa H, Shirasawa K, Miyatake K, Nunome T, Negoro S, Ohyama A, Yamaguchi H, Sato S, Isobe S, Tabata S et al (2014) Draft genome sequence of eggplant (Solanum melongena L.): the representative solanum species indigenous to the old world. DNA Res 21:649–660
Holmes FO (1938) Inheritance of resistance to tobacco-mosaic disease in tobacco. Phytopathology 28:553–561
Julio E, Cotucheau J, Decorps C, Volpatti R, Sentenac C, Candresse T, Dorlhac de Borne F (2014) A Eukaryotic translation initiation factor 4E (eIF4E) is responsible for the “va” tobacco recessive resistance to Potyviruses. Plant Mol Biol Report 1–15
Kelly LJ, Leitch AR, Clarkson JJ, Hunter RB, Knapp S, Chase MW (2010) Intragenic recombination events and evidence for hybrid speciation in Nicotiana (Solanaceae). Mol Biol Evol 27:781–799
Kelly LJ, Leitch AR, Clarkson JJ, Knapp S, Chase MW (2013) Reconstructing the complex evolutionary origin of wild allopolyploid tobaccos (Nicotiana section suaveolentes). Evolution 67:80–94
Kenton A, Parokonny AS, Gleba YY, Bennett MD (1993) Characterization of the Nicotiana tabacum L. genome by molecular cytogenetics. Mol Gen Genet 240:159–169
Khafizova G, Dobrynin P, Polev D, Matveeva T (2018) Nicotiana glauca whole-genome investigation for cT-DNA study. BMC Res Notes 11:18
Kim S, Park M, Yeom SI, Kim YM, Lee JM, Lee HA, Seo E, Choi J, Cheong K, Kim KT et al (2014) Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet 46:270–278
Kitamura S, Tanaka A, Inoue M (2005) Genomic relationships among Nicotiana species with different ploidy levels revealed by 5S rDNA spacer sequences and FISH/GISH. Genes Genet Syst 80:251–260
Knapp S, Bohs L, Nee M, Spooner DM (2004a) Solanaceae—a model for linking genomics with biodiversity. Comp Funct Genomics 5:285–291
Knapp S, Chase MW, Clarkson JJ (2004b) Nomenclatural changes and a new sectional classification in Nicotiana (Solanaceae). Taxon 53:73–82
Koukalova B, Moraes AP, Renny-Byfield S, Matyasek R, Leitch AR, Kovarik A (2010) Fall and rise of satellite repeats in allopolyploids of Nicotiana over c. 5 million years. New Phytol 186:148–160
Kovacova V, Zluvova J, Janousek B, Talianova M, Vyskot B (2014) The evolutionary fate of the horizontally transferred agrobacterial mikimopine synthase gene in the genera Nicotiana and Linaria. PLoS ONE 9:e113872
Kovarik A, Renny-Byfield S, Grandbastien M-A, Leitch A (2012) Evolutionary implications of genome and karyotype restructuring in Nicotiana tabacum L. In: Soltis PS, Soltis DE (eds) Polyploidy and genome evolution. Springer, Berlin Heidelberg, pp 209–224
Leitch IJ, Bennett MD (2004) Genome downsizing in polyploid plants. Biol J Linn Soc 82:651–663
Leitch IJ, Hanson L, Lim KY, Kovarik A, Chase MW, Clarkson JJ, Leitch AR (2008) The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae). Ann Bot 101:805–814
Lewis RS (2011) Nicotiana. Wild Crop Relat. Genomic Breed Resour 185–208
Lewis RS, Nicholson JS (2007) Aspects of the evolution of Nicotiana tabacum L. and the status of the United States Nicotiana germplasm collection. Genet Resour Crop Evol 54:727–740
Lewis RS, Milla SR, Levin JS (2005) Molecular and genetic characterization of Nicotiana glutinosa L. chromosome segments in tobacco mosaic virus-resistant tobacco accessions. Crop Sci 45:2355–2362
Lewis RS, Linger LR, Wolff MF, Wernsman EA (2007) The negative influence of N-mediated TMV resistance on yield in tobacco: linkage drag versus pleiotropy. Theor Appl Genet 115:169–178
Lim KY, Matyášek R, Lichtenstein CP, Leitch AR (2000) Molecular cytogenetic analyses and phylogenetic studies in the Nicotiana section Tomentosae. Chromosoma 109:245–258
Lim KY, Matyasek R, Kovarik A, Leitch AR (2004) Genome evolution in allotetraploid Nicotiana. Biol J Linn Soc 82:599–606
Lim KY, Kovarik A, Matyasek R, Chase MW, Clarkson JJ, Grandbastien MA, Leitch AR (2007) Sequence of events leading to near-complete genome turnover in allopolyploid Nicotiana within five million years. New Phytol 175:756–763
Luu VT, Schuck S, Kim SG, Weinhold A, Baldwin IT (2015) Jasmonic acid signalling mediates resistance of the wild tobacco Nicotiana attenuata to its native Fusarium, but not Alternaria, fungal pathogens. Plant Cell Env. 38:572–584
Marks CE (2010) Definition of South Pacific taxa of Nicotiana section Suaveolentes. Muelleria 28:74–84
Marks CE, Ladiges PY (2011) Comparative morphology and phylogeny of Nicotiana section Suaveolentes (Solanaceae) in Australia and the South Pacific. Aust Syst Bot 24:61–86
Marks CE, Ladiges PY, Newbigin E (2011) Karyotypic variation in Nicotiana section Suaveolentes. Genet Resour Crop Evol 58:797–803
Martin F, Bovet L, Cordier A, Stanke M, Gunduz I, Peitsch MC, Ivanov NV (2012) Design of a Tobacco Exon Array with application to investigate the differential cadmium accumulation property in two tobacco varieties. BMC Genom 13:674
Matyasek R, Fulnecek J, Leitch AR, Kovarik A (2011) Analysis of two abundant, highly related satellites in the allotetraploid Nicotiana arentsii using double-strand conformation polymorphism analysis and sequencing. New Phytol 192:747–759
Matyasek R, Renny-Byfield S, Fulneček J, Macas J, Grandbastien M-A, Nichols R, Leitch A, Kovařík A (2012) Next generation sequencing analysis reveals a relationship between rDNA unit diversity and locus number in Nicotiana diploids. BMC Genom 13:722
Melayah, D, Lim, KY, Bonnivard, E, Chalhoub, B, Dorlhac De Borne, F, Mhiri, C, Leitch, AR, Grandbastien, MA (2004). Distribution of the Tnt1 retrotransposon family in the amphidiploid tobacco (Nicotiana tabacum) and its wild Nicotiana relatives. Biol. J. Linn. Soc. 82, 639–649
Moon H, Nicholson JS (2007) AFLP and SCAR markers linked to tomato spotted wilt virus resistance in tobacco. Crop Sci 47:1887–1894
Murad L, Lim KY, Christopodulou V, Matyasek R, Lichtenstein CP, Kovarik A, Leitch AR (2002) The origin of tobacco’s T genome is traced to a particular lineage within Nicotiana tomentosiformis (Solanaceae). Am J Bot 89:921–928
Naim F, Nakasugi K, Crowhurst RN, Hilario E, Zwart AB, Hellens RP, Taylor JM, Waterhouse PM, Wood CC (2012) Advanced engineering of lipid metabolism in Nicotiana benthamiana using a draft genome and the V2 viral silencing-suppressor protein. PLoS ONE 7:e52717
Olmstead RG, Bohs L, Migid HA, Santiago-Valentin E, Garcia VF, Collier SM (2008) A molecular phylogeny of the Solanaceae. Taxon 57:1159–1181
Parisod C, Mhiri C, Lim KY, Clarkson JJ, Chase MW, Leitch AR, Grandbastien M-A (2012) Differential dynamics of transposable elements during long-term diploidization of Nicotiana section Repandae (Solanaceae) allopolyploid genomes. PLoS ONE 7:e50352
Parokonny, A.S., and Kenton, A.Y. (1995). Comparative physical mapping and evolution of the Nicotiana tabacum L. karyotype. pp. 301–320
Petit M, Lim KY, Julio E, Poncet C, Dorlhac de Borne F, Kovarik A, Leitch AR, Grandbastien MA, Mhiri C (2007) Differential impact of retrotransposon populations on the genome of allotetraploid tobacco (Nicotiana tabacum). Mol Genet Genomics 278:1–15
Petit M, Guidat C, Daniel J, Denis E, Montoriol E, Bui QT, Lim KY, Kovarik A, Leitch AR, Grandbastien M-A (2010) Mobilization of retrotransposons in synthetic allotetraploid tobacco. New Phytol 186:135–147
Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y et al (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 111:5135–5140
Reed, S.M. (1991). Cytogenetic evolution and aneuploidy in Nicotiana. Chromosome Eng Plants Genet Breed Evol Part B Elsevier Dordr Neth 483–505
Ren N, Timko MP (2001) AFLP analysis of genetic polymorphism and evolutionary relationships among cultivated and wild Nicotiana species. Genome 44:559–571
Sarkinen T, Bohs L, Olmstead RG, Knapp S (2013) A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol Biol 13:214
Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Egholm M (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641
Senthil G, Liu H, Puram VG, Clark A, Stromberg A, Goodin MM (2005) Specific and common changes in Nicotiana benthamiana gene expression in response to infection by enveloped viruses. J Gen Virol 86:2615–2625
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5:2043
Sierro N, Battey JND, Ouadi S, Bovet L, Goepfert S, Bakaher N, Peitsch MC, Ivanov NV (2013a) Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol 14:R60
Sierro N, van Oeveren J, van Eijk MJ, Martin F, Stormo KE, Peitsch MC, Ivanov NV (2013b) Whole genome profiling physical map and ancestral annotation of tobacco hicks broadleaf. Plant J 75:880–889
Sierro N, Battey JND, Ouadi S, Bakaher N, Bovet L, Willig A, Goepfert S, Peitsch MC, Ivanov NV (2014) The tobacco genome sequence and its comparison with those of tomato and potato. Nat Commun 5:3833
Sierro N, Battey JND, Bovet L, Liedschulte V, Ouadi S, Thomas J, Broye H, Laparra H, Vuarnoz A, Lang G et al (2018) The impact of genome evolution on the allotetraploid Nicotiana rustica—an intriguing story of enhanced alkaloid production. BMC Genom 19:855
Skalicka K, Lim KY, Matyášek R, Koukalová B, Leitch AR, Kovařík A (2003) Rapid evolution of parental rDNA in a synthetic tobacco allotetraploid line. Am J Bot 90:988–996
Skalicka K, Lim KY, Matyasek R, Matzke M, Leitch AR, Kovarik A (2005) Preferential elimination of repeated DNA sequences from the paternal, Nicotiana tomentosiformis genome donor of a synthetic, allotetraploid tobacco. New Phytol 166:291–303
Sloan DB (2013) One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol 200:978–985
Sugiyama Y, Watase Y, Nagase M, Makita N, Yagura S, Hirai A, Sugiura M (2005) The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: comparative analysis of mitochondrial genomes in higher plants. Mol Genet Genomics 272:603–615
Symon DE, Lepschi BJ (2007) A new status in Nicotiana (Solanaceae): N. monoschizocarpa (P. Horton) Symon & Lepschi. J. Adelaide Bot. Gard. 21:92
Tezuka T, Marubashi W (2012) Genes in S and T subgenomes are responsible for hybrid lethality in interspecific hybrids between Nicotiana tabacum and Nicotiana occidentalis. PLoS ONE 7:e36204
Tran HTM, Ramaraj T, Furtado A, Lee LS, Henry RJ (2018) Use of a draft genome of coffee (Coffea arabica) to identify SNPs associated with caffeine content. Plant Biotechnol J 16:1756–1766
TRI (2015). Tobacco Research Institute of the Chinese Academy of Agricultural Sciences
USDA (2016). National Plant Germplasm System
Whitelaw CA, Barbazuk WB, Pertea G, Chan AP, Cheung F, Lee Y, Zheng L, van Heeringen S, Karamycheva S, Bennetzen JL et al (2003) Enrichment of gene-coding sequences in maize by genome filtration. Science 302:2118–2120
Whitham S, Dinesh-Kumar SP, Choi D, Hehl R, Corr C, Baker B (1994) The product of the tobacco mosaic virus resistance gene N: Similarity to toll and the interleukin-1 receptor. Cell 78:1101–1115
Wikstrom N, Savolainen V, Chase MW (2001) Evolution of the angiosperms: calibrating the family tree. Proc Biol Sci 268:2211–2220
Wu F, Eannetta NT, Xu Y, Plieske J, Ganal M, Pozzi C, Bakaher N, Tanksley SD (2010) COSII genetic maps of two diploid Nicotiana species provide a detailed picture of synteny with tomato and insights into chromosome evolution in tetraploid N. tabacum. Theor Appl Genet 120:809–827
Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, Zhang G, Yang S, Li R, Wang J (2011) Genome sequence and analysis of the tuber crop potato. Nature 475:189
Xu S, Brockmoller T, Navarro-Quezada A, Kuhl H, Gase K, Ling Z, Zhou W, Kreitzer C, Stanke M, Tang H et al (2017) Wild tobacco genomes reveal the evolution of nicotine biosynthesis. Proc Natl Acad Sci USA 114:6133–6138
Yang S-J, Carter SA, Cole AB, Cheng N-H, Nelson RS (2004) A natural variant of a host RNA-dependent RNA polymerase is associated with increased susceptibility to viruses by Nicotiana benthamiana. Proc Natl Acad Sci USA 101:6297–6302
Yang BC, Xiao BG, Chen XJ, Shi CH (2007) Assessing the genetic diversity of tobacco germplasm using intersimple sequence repeat and inter-retrotransposon amplification polymorphism markers. Ann Appl Biol 150:393–401
Yarnes SC, Ashrafi H, Reyes-Chin-Wo S, Hill TA, Stoffel KM, Van Deynze A (2013) Identification of QTLs for capsaicinoids, fruit quality, and plant architecture-related traits in an interspecific Capsicum RIL population. Genome 56:61–74
Yukawa M, Tsudzuki T, Sugiura M (2006) The chloroplast genome of Nicotiana sylvestris and Nicotiana tomentosiformis: complete sequencing confirms that the Nicotiana sylvestris progenitor is the maternal genome donor of Nicotiana tabacum. Mol Genet Genomics 275:367–373
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sierro, N., Ivanov, N.V. (2020). Background and History of Tobacco Genome Resources. In: Ivanov, N.V., Sierro, N., Peitsch, M.C. (eds) The Tobacco Plant Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-030-29493-9_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-29493-9_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-29492-2
Online ISBN: 978-3-030-29493-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)