
Abstract
Genomic study of the endometrium may shed light on the regulation of embryo implantation and how the process is disrupted in women with reproductive failure. There are several essential requirements in planning a genomic study of the endometrium, including precise timing of endometrial biopsy specimens and the recruitment of not only subjects with a well-defined category of reproductive failure but also a separate population of fertile control subjects. Genomic regulation of implantation may be achieved at various levels, and the measurement of each requires a different analytical method. Current literature data will be summarized.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Understanding Endometrium and Reproductive Disorder
The primary function of the endometrium is to support embryo implantation, a process whereby the embryo attaches itself to the endometrium, followed by migration across the luminal epithelium and invasion into the endometrial stroma layer to become embedded. The process involves a complex sequence of cellular and molecular changes [1]. The deranged endometrial function may result in a spectrum of reproductive disorders, ranging from implantation failure, miscarriage, abnormal placentation, fetal growth restriction, and possibly preeclampsia.
Many methods have been used to study the endometrium and understand if the observed reproductive disorder in a particular individual is due to an underlying endometrial problem. Histological examination of the endometrial biopsy is often utilized to assess if the histological transformation of the endometrium, especially secretory transformation, is adequate. Immunohistochemical techniques offer to examine the expression of a specific protein or putative marker of implantation. Cell counting techniques are used to determine the density of certain immune cell types such as uterine natural killer cell. Imaging techniques such as ultrasonography or hysteroscopy are utilized to examine the uterine cavity, and the sampling of fluid in the endometrial cavity provides an opportunity to measure proteins or cytokine concentrations in the endometrial fluid.
Depending on the outcome of the initial investigation, one of the frequently asked questions is why and how the observed abnormality is brought up and if there is an effective treatment available to correct the underlying abnormality. In this regard, the genomic study of the endometrium is gaining increasing popularity as a modern approach to the study of endometrial receptivity and its regulation. A well-known earlier example is microarray analysis of endometrial biopsy using endometrial samples obtained at the putative time of embryo transfer to determine if the endometrial development at the time of implantation is in-phase, ahead, or behind schedule.
Prerequisites for the Genomic Study of the Endometrium
An important prerequisite for conducting a genomic study of the endometrium is to collect the specimen precisely at a well-defined time point in the menstrual cycle, because the endometrium undergoes profound, rapid changes in the secretory phase, especially around the time of implantation. Endometrial genes, including LIF, HOXA10, MUC1, EMX2, IGFBP-1, CSF-1, and IL-1, have been shown to fluctuate during the menstrual cycle [2,3,4] profoundly. The examination of RNA transcripts in precisely timed endometrial specimens and the comparison of results between women with recurrent miscarriage and recurrent implantation failure have provided new insights into how these two types of reproductive failure differ from each other [2].
A second prerequisite of genomic study of the endometrium is to collect an endometrial sample from a homogenous, well-defined population. The specific type of reproductive failure to be studied must be clearly defined. For example, it is necessary to understand that subjects with “infertility” of “recurrent implantation failure” or “recurrent miscarriage” are all rather heterogeneous with several underlying etiologies. Every effort must be made to define the inclusion criteria and exclusion criteria carefully to rule out known contributory or confounding factors such as the presence of any congenital or acquired uterine anomalies such as fibroid, congenital uterine anomaly, or intrauterine adhesions.
A third prerequisite is the need to recruit a suitable group of fertile, control subjects with whom the result of the studied population can be compared. Without a proper control group, it is difficult to draw meaningful conclusions. The control groups ought to have proven fertility in the recent past and similarly have undergone investigations to verify the absence of uterine anomalies.
Transcriptomic Study
Transcriptomic studies by using microarray analysis or RNA sequencing have been applied to examine the endometrium during the peri-implantation period (before or during the window of implantation) [3, 4] using various patient populations such as PCOS [5], RIF [6,7,8], or RM [9, 10], with different hormonal treatments [11, 12], and data were compared between unexplained RM and RIF [2]. It is of interest that the endometrial gene expression in unexplained RM and recurrent RIF shared something in common. For example, IL-15 has been reported to be increased in both conditions [13, 14]. On the other hand, certain markers are deranged in one condition but not the other. A notable example is beta3 integrin, which is downregulated in RM [15] but not in RIF [16], whereas leukemia inhibitory factor (LIF) is downregulated in RIF [17], but not in RM [18, 19]. Recently, Brosens et al. [20,21,22] hypothesized that RM is associated with an over-receptive endometrium which would allow defective or abnormal embryos to implant and in turn leads to super-fertility, but followed by an increased risk of miscarriage of an abnormal embryo. In contrast, in women with RIF, implantation often fails to take place despite the replacement of many good-quality embryos, implying an underlying etiology of a different nature, possibly a presence of endometrial pathology.
Genes in complement and coagulation cascade pathways of women with RIF are upregulated in the endometrium when compared to those of women with RM. The complement system, represented by complement component 3 (C3), is a proteolytic cascade in plasma, which also mediates innate immunity. One of the major immune functions of this pathway is to form a membrane attack complex, which leads to cell lysis [23]. Genetic association study also found that the loss of functional mutation of some genes in this pathway was associated with RM [24] and other adverse pregnancy outcomes, such as preeclampsia [25]. It seems like that the upregulation of the complement pathway is associated with impairment of endometrial receptivity leading to implantation failure. The downregulation of this pathway observed in the endometrium of women with RM may explain the apparent increase in endometrial receptivity observed in this group of women.
On the contrary, the calcium signaling pathway in the endometrium of women with RIF is downregulated when compared with RM. It has long been known that Ca2+ channels are involved in a variety of implantation processes, and increased Ca2+ mobilization can assist blastocyst-endometrium adhesion [20, 26, 27]. It is suggested that in women with RM, the endometrium is more favorable for implantation when compared with RIF, consistent with the in vitro study carried out by Brosens and co-workers [20].
Single-Cell Gene Expression Study
One of the limitations of endometrial gene expression study is a lack of information about gene expression patterns in various cell types. Given the fact that the endometrium consists of various cell populations including luminal epithelium, glandular epithelium, stroma, blood vessels, fibroblasts, and immune cells, it would be more informative if gene expression changes are investigated in various cell types. For example, C3 gene expression is low in both epithelial and stromal cells in the proliferative phase endometrium. However, it gradually increases during the cycle, reaching a significantly higher level in the mid-secretory phase [2]. When stimulated by human chorionic gonadotropin (hCG), C3 gene expression is further increased in stromal cells but not in glandular cells. Similarly, after endometrial scratch, CCL19 and ITGB1 gene expressions were altered in the stromal but not in glandular cells.
On the other hand, single-cell sequencing (Sc-seq) may be performed in a single cell and amplified by whole genome or transcriptome for high-throughput sequencing, which reveals the genetic structure and gene expression status of the particular individual cell [28, 29]. Unlike a conventional sequencing technique, Sc-seq is the sequencing of the genome of a single cell, which avoids the heterogeneity of multiple cells by ordinary sequencing [29]. As a result, it helps to better understand the difference in genomic expression between different cell types [30]. The single-cell sequencing technology includes single-cell separation, cytolysis and genomic DNA acquisition, whole genome amplification, sequencing, and data analysis. The difficulty often lies in the isolation of single-cell samples and whole genome amplification [31].
Earlier, the single-cell isolation was made by the manual selection under the microscope and serial dilution method, which were problematic but did not require special instruments [32]. Later on, laser capture microdissection (LCM) and flow cytometry emerged and utilized for the single-cell isolation [33]. However, these techniques have a certain impact on cell viability, and throughput is not high. Recently, updated single-cell sorting methods, including fluorescence activated cell sorting (FACS), magnetic activated cell sorting (MACS), microdroplet, microfluidics, and nanopore chip technology, are available [34]. These new technologies cannot only achieve high throughput but have the advantage of sorting thousands of the same cells at a time [35, 36].
A special challenge of single-cell genomic study relates to whole genome amplification (WGA). High-throughput sequencing requires at least 200 ng of DNA sample, but the total DNA in a single cell is only a few picograms, so even with the use of third-generation sequencing technology, it may not be able to meet the minimal amount of DNA required for the analysis [29]. Therefore, DNA needs to be amplified for the single-cell sequencing [37]. Early whole genome amplification technology was performed by PCR, including ligation-mediated PCR (LM-PCR), primer extension pre-amplification PCR (PEP-PCR), and degenerate oligonucleotide-primed PCR (DOP-PCR). At present, multi-displacement amplification (MDA) and primase-based whole genome amplification (pWGA) are the most commonly used whole genome amplification methods based on constant temperature conditions [38]. Recently, a new method called multiple annealing and looping-based amplification cycles (MALBAC), which combines the former two amplification methods, has been introduced [39]. These genome amplification methods can be selected according to different experimental requirements (Table 7.1).
In the endometrium, Sc-seq has the potential to detect cell-to-cell variability, map possible subpopulations, discover possible rare cell types, and study clinically relevant but rare endometrial adult stem cells [40]. The first and so far the only study on endometrial Sc-seq was performed in 2016. Frozen-thawed endometrial biopsy samples were used for ScRNA-seq. Endometrial stromal cells were cultured from two patients obtained in the mid-secretory (Day 21, LH + 8) and late-secretory phase (Day 25). However, due to a small number of the endometrial epithelial cell sorted by FACS and low reads mapping, this research only focused on endometrial stromal cells. The results showed that cultured cells had more high-quality mRNA than frozen-thawed biopsy sample. Besides, of the 8622 detected genes, 2661 were more active in cultured stromal cells than those in the biopsy sample [41]. It suggests that ScRNA-seq technique may be utilized to study the function of different compartments and cell types of human endometrium. The single-cell preparation method for endometrial epithelial cells needs to be optimized due to the minimal amount of transcriptome data per individual epithelial cell, which compromised further analysis.
Epigenetics
Classical genetics refers to genes as the molecular basis of inheritance in terms of nucleic acids as the structural and functional unit of an organism. A gene determines a protein required for activities of life [42]. However, it is now known that many genetic phenomena cannot be entirely explained by classical genetics. Change of the genome without affecting the nucleic acids not only can affect the function of genes in an individual but also can be inherited to the offspring. This phenomenon is known as epigenetics, which is first introduced by Waddington in 1942 [43]. The current definition of epigenetics is the science studying the inheritance of gene function that cannot be explained by nucleic acids changes during mitosis and meiosis [44]. In general, epigenetics has three characteristics, including being heritable, reversible, and regulatory, without DNA sequences change [45]. They are influenced by genetic variability and other factors, such as environmental and nutritional status. Currently, epigenetics includes DNA methylation, histone modification, non-coding RNA, genomic imprinting, and chromosomal inactivation, and each affects gene transcription in various ways. DNA methylation adds methyl to cytosine in a gene, particularly promotor regions, resulting in gene silencing. Histone modification alters coiling of genes for replication and functioning. Non-coding RNAs target complementary sequence interrupting transcription or post-transcription of genes. Genomic imprinting activates allele, while chromosomal inactivation changes chromatin location and structure.
Several studies have shown that epigenetics plays an important role in embryo implantation, placental formation, organ formation, and fetal growth [46]. Epigenetic modification may result in aberrant endometrial receptivity, leading to a reproductive failure [47].
DNA Methylation and Endometrium Receptivity
DNA methylation is the epigenetic regulatory mechanism which was discovered earliest and still one of the hotspots in current epigenetic research. It refers to the selective addition of methyl to cytosine in CpG dinucleotides to form 5-methylcytosine (5-mc) catalyzed by DNA methyltransferases (DNMTs), including DNMT1, DNMT3a, DNMT3b, and DNMT3l. DNA methylation is generally associated with gene silencing, and DNA demethylation is associated with gene activation [48].
DNA methylation in the endometrium is involved in the regulation of endometrial receptivity-related cytokines, gene expression, cell adhesion mechanism, and sex hormones. The endometrium undergoes periodic morphological and functional changes during the menstrual cycle to prepare for the implantation of the embryo. These changes include many gene modifications under the regulation of estrogen and progesterone, and DNA methylation is also involved in the regulation [49].
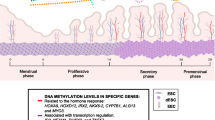
On the one hand, DNA methylation regulates the cyclical changes of the endometrium during the menstrual cycle. The expression of DNMTs in the endometrium was significantly changed during the menstrual cycle. At the mRNA expression level, the expression of DNMT1, DNMT3a, and DNMT3b in the secretory phase was significantly lower than that in the proliferative phase, with the lowest expression in the mid-secretory phase [50]. However, another research used whole genome-wide methylation sequencing to investigate the changes in DNA methylation during the menstrual cycle and found methylome remained relatively stable during the menstrual cycle with small changes affecting only 5% of the CpG sites [51]. These findings suggest that DNA methylation probably plays a significant but a specific role in the endometrium.
Several studies examined how DNA methylation regulates the expression of genes involved in endometrial receptivity. E-cadherin and homeobox genes 10 (HOXA10) are important for endometrial receptivity and implantation. Rahnama et al. used an in vitro embryo implantation model and demonstrated that inhibition of DNA methylation promoted E-cadherin expression which was regulated by DNMT-1, 3a, and 3b. Also, hypermethylation of HOXA10 promoter led to reduced expression of HOXA10 and then resulted in poor endometrial receptivity [52]. 5-Aza-2′-deoxycytidine (AZA) inhibits DNA methyltransferase and in turn increases implantation rate with increased HOXA10 gene expression in Jeg-3 spheroid-endometrial cells [53]. It is now known that high progesterone (P) on the day of hCG trigger in fresh ART cycle was associated with poor endometrium receptivity. The expression of 5-mC in the high P group was significantly higher than that in the normal P group in human endometrial glandular epithelium. It indicated that DNA methylation modification associated with high P level might lead to poor endometrium receptivity [54]. However, the effects of high progesterone on the whole genome methylation changes in the endometrium are still lacking. While DNA methylation is considered an important factor in the regulation of endometrial receptivity, the specific role of DNA methylation in this important process requires further research.
Histone Modification and Endometrial Receptivity
Histone modifications refer to the basal amino-terminal tail of histones undergoing posttranscriptional modifications (PTMs), such as methylation, acetylation, phosphorylation, and ubiquitination [55]. Modifications constitute a rich “histone code” that affects the degree of compression tightness of chromatin and therefore plays an important regulatory role in gene expression [56].
The most studied histone modifications in endometrial receptivity and embryo implantation are histone acetylation and deacetylation. Histone acetylation is mainly regulated by two opposite enzymes: histone acetylase (HATs) and histone deacetylases (HDACs) [57, 58]. HATs and HDACs are expressed in the endometrium in the different menstrual cycles, which regulate the cyclical changes of the endometrium. The histone deacetylase inhibitors (HDACI) induced the expression, morphology, and function of endometrial stromal and epithelial cell differentiation markers and cell differentiation [59]. During the menstrual cycle, HDAC-1, HDAC-2, and HDAC-3 mRNAs were expressed periodically in the endometrium. HDAC-1 expression was significantly different among individuals, while HDAC-2 expression was significantly increased in the secretory phase, and HDAC-3 expression constituted throughout the menstrual cycle [60]. Estella et al. showed that histone acetylation affected the balance of extracellular matrix regulators, which in turn inhibited trophoblast invasion into the endometrium [61].
Histone modification and DNA methylation can also be regulated by each other. For example, DNMTs can recruit complexes containing HDACs [50]. Inhibition of HDAC can lead to DNA methylation modification [62]. Further study on the interaction of histone modification and DNA methylation in endometrial receptivity may help better understand the embryo implantation process in synchronized endometrium.
ncRNAs and Endometrial Receptivity
Non-coding RNAs (ncRNAs) are RNA molecules that do not translate into a protein. ncRNAs contain microRNAs (miRNAs), transcribed ultraconserved region (T-UCR), small nucleolar RNAs (snoRNAs), PIWI-interacting RNAs (piRNAs), long non-coding RNAs (lncRNAs), and many other RNA molecules, accounting for more than 97% of the human genome [63]. Although ncRNAs rarely encode or do not have the function to encode proteins, they are involved in many biological processes such as stem cell maintenance, embryo development, cell proliferation, apoptosis, and differentiation [64]. Among them, miRNAs are the most studied ncRNAs in human endometrium receptivity. The research on other ncRNAs in human endometrium receptivity is still in its infancy, with only limited reports on related functions and mechanisms.
MicroRNA is a small ncRNA molecule of about 21–23 nucleotides widely distributed in eukaryotes [65]. It specifically inhibits posttranscriptional gene expression by binding to target messenger RNA (mRNA). It plays an important role in regulating gene expression and involves in cells cycles, developmental timing, and many other biological aspects [66]. More than 700 miRNAs have been discovered in humans, many of which are mutated or downregulated during disease development, leading to abnormal gene expression and related diseases [67].
Similar to DNA methylation and histone modifications, the expression of miRNAs is also cycle-dependent. Lam showed that expressions of 12 kinds of miRNAs (miR-29b, miR-29c, miR-30b, miR-30d, miR-31, miR-193a-3p, miR-203, miR-204, miR-200c, miR-210, miR-582-5p, and miR-345) in the secretory phase were significantly higher than those in the proliferative phase [68]. Some miRNAs (miR-15b, miR-20a, and miR-21) have higher expression in the proliferative phase, while others (e.g., miR-135a) have lower expression in the proliferative phase [69].
Revel et al. compared the expression of different miRNAs in the secretory phase between women with recurrent implantation failures (RIF) and normal fertile women. The result showed that 13 miRNAs were abnormally expressed, and these 13 miRNAs potentially regulated the expression of 3800 genes. Through miRNA-mRNA parallel analysis, molecular signaling pathways regulated by miRNAs were observed, including cell adhesion, Wnt, P53, and cell cycle. These molecular pathways are closely related to endometrial receptivity [70]. Petracco et al. observed that miRNA-135a and miRNA-135b inhibited the expression of HOXA10, and overexpression of the miRNA may inhibit the expression of genes involved in embryo implantation, such as HOXA10 and cyclooxygenase-2 (COX-2) [71]. It showed miRNAs might affect endometrial receptivity by regulating genes and signaling pathways involved in embryo implantation and implantation window.
At present, miRNAs are one of the hotspots in the field of epigenetics research, and its role in endometrial receptivity is far from clear. Further work will be required to better understand how miRNA regulates endometrial receptivity.
Genomic Imprinting and Endometrial Receptivity
In 1984, scientists proposed that mammalian parental genomes have asymmetric functions. In these genes, one of the alleles is expressed in a family-dependent way, and the other is not expressed or expressed very weakly. This is called genomic imprinting [72]. The expression of these imprinted genes depends on whether they are from the father or the mother. The paternal and maternal genes have the opposite effect. The paternal genes direct the development of extraembryonic tissues, while the maternal genes direct the embryonic development. Therefore, genomic imprinting is a self-regulation and monitoring mechanism, which can effectively prevent parthenogenesis, ensure the simultaneous existence of both embryonic parental genomes, and maintain genetic diversity. However, it increases the disease risk caused by the mutation as well [73, 74]. At present, there are 73 imprinted genes in mice, and 243 are already discovered in humans [75].
It has been shown that the expression of imprinted genes may be involved in the establishment of endometrial receptivity [8]. Specifically, the expression of imprinted gene H19 in the endometrium appears to be related to the development of endometrial cancer [76]. Expression of H19 gene in sheep embryos can only be detected in the embryonic tissue after implantation, and no expression of H19 is observed in the embryonic ectoderm at any time before the implantation [77]. It seems, therefore, that normal expression of imprinted genes may play an important role in endometrial receptivity. Abnormal expression of imprinted genes may lead to structural and functional changes of endometrial microenvironment and thus reduce the implantation rate.
Chromosomal Inactivation and Endometrium Receptivity
X-chromosomal inactivation (XCI ) refers to the silencing of one of the X chromosome pairs in the mammalian female embryo at the transcriptional level. Up to 1000 silencing genes on the X chromosome cause a balanced expression of sex-linked genes between male and female. The balance of gene expression is a mechanism to maintain dose-compensation effects in mammalian individuals and an important part of epigenetic research [78]. However, there is so far no reported study on the association between XCI and endometrium receptivity.
Sequencing Techniques for Epigenetic Study
Given that there are various types of epigenetic study, the laboratory methods should be chosen depending on the specific question to be addressed. First, there are two categories of methods to study DNA methylation: the one category is the methylation analysis at the whole genome level, including high-performance liquid chromatography, methylation-sensitive amplification polymorphism (MSAP) detection, DNA methylation immunoprecipitation (MeDIP), and DNA methylation chip technology [79,80,81,82]. The other category is a DNA methylation analysis for target sites, including bisulfite sequencing, sodium bisulfite treatment combined with enzymatic analysis, methylation-sensitive single-nucleotide amplification, methylation-sensitive high-resolution amplification, and methylation fluorescence PCR [83, 84]. These methods are generally based on the principle that cytosine becomes uracil under the treatment of bisulfite, while methylated cytosine does not change. This principle is the gold standard for DNA methylation analysis.
As for histone modifications, there are many types, including methylation, acetylation, phosphorylation, ubiquitination, and ADP ribosylation. To study histone modification, the most commonly used method is chromatin immunoprecipitation (ChIP) technique, including CHIP-chip and ChIP-seq [85, 86].
For non-coding RNA detection, the methods used include Northern blot, real-time PCR and expression library cloning, as well as ChIP technology, surface-enhanced Raman spectroscopy, and next-generation sequencing. ChIP technology and next-generation sequencing are the most commonly used technology for miRNA [87, 88].
Conclusions
Genomic study of the endometrium holds a great promise to increase our knowledge of the biology of implantation and its regulation, especially in women suffering from various forms of reproductive failure. In planning a study to examine genomic regulation of implantation, it is necessary to obtain precisely timed endometrial biopsy specimens and to recruit not only subjects with a well-defined category of reproductive failure but also a separate population of fertile control subjects.
References
Cha J, Sun X, Dey SK. Mechanisms of implantation: strategies for successful pregnancy. Nat Med. 2012;18(12):1754–67.
Huang J, Qin H, Yang Y, Chen X, Zhang J, Laird S, et al. A comparison of transcriptomic profiles in endometrium during window of implantation between women with unexplained recurrent implantation failure and recurrent miscarriage. Reproduction. 2017;153(6):749–58.
Diaz-Gimeno P, Horcajadas JA, Martinez-Conejero JA, Esteban FJ, Alama P, Pellicer A, et al. A genomic diagnostic tool for human endometrial receptivity based on the transcriptomic signature. Fertil Steril. 2011;95(1):50–60, e1–15.
Hu S, Yao G, Wang Y, Xu H, Ji X, He Y, et al. Transcriptomic changes during the pre-receptive to receptive transition in human endometrium detected by RNA-Seq. J Clin Endocrinol Metab. 2014;99(12):E2744–53.
Qiao J, Wang L, Li R, Zhang X. Microarray evaluation of endometrial receptivity in Chinese women with polycystic ovary syndrome. Reprod Biomed Online. 2008;17(3):425–35.
Koot YE, van Hooff SR, Boomsma CM, van Leenen D, Groot Koerkamp MJ, Goddijn M, et al. An endometrial gene expression signature accurately predicts recurrent implantation failure after IVF. Sci Rep. 2016;6:19411.
Ruiz-Alonso M, Blesa D, Diaz-Gimeno P, Gomez E, Fernandez-Sanchez M, Carranza F, et al. The endometrial receptivity array for diagnosis and personalized embryo transfer as a treatment for patients with repeated implantation failure. Fertil Steril. 2013;100(3):818–24.
Ledee N, Munaut C, Aubert J, Serazin V, Rahmati M, Chaouat G, et al. Specific and extensive endometrial deregulation is present before conception in IVF/ICSI repeated implantation failures (IF) or recurrent miscarriages. J Pathol. 2011;225(4):554–64.
Kosova G, Stephenson MD, Lynch VJ, Ober C. Evolutionary forward genomics reveals novel insights into the genes and pathways dysregulated in recurrent early pregnancy loss. Hum Reprod. 2015;30(3):519–29.
Othman R, Omar MH, Shan LP, Shafiee MN, Jamal R, Mokhtar NM. Microarray profiling of secretory-phase endometrium from patients with recurrent miscarriage. Reprod Biol. 2012;12(2):183–99.
Mirkin S, Nikas G, Hsiu JG, Diaz J, Oehninger S. Gene expression profiles and structural/functional features of the peri-implantation endometrium in natural and gonadotropin-stimulated cycles. J Clin Endocrinol Metab. 2004;89(11):5742–52.
Haouzi D, Assou S, Mahmoud K, Tondeur S, Reme T, Hedon B, et al. Gene expression profile of human endometrial receptivity: comparison between natural and stimulated cycles for the same patients. Hum Reprod. 2009;24(6):1436–45.
Tuckerman E, Mariee N, Prakash A, Li TC, Laird S. Uterine natural killer cells in peri-implantation endometrium from women with repeated implantation failure after IVF. J Reprod Immunol. 2010;87(1–2):60–6.
Tuckerman E, Laird SM, Prakash A, Li TC. Prognostic value of the measurement of uterine natural killer cells in the endometrium of women with recurrent miscarriage. Hum Reprod. 2007;22(8):2208–13.
Germeyer A, Savaris RF, Jauckus J, Lessey B. Endometrial beta3 integrin profile reflects endometrial receptivity defects in women with unexplained recurrent pregnancy loss. Reprod Biol Endocrinol. 2014;12:53.
Coughlan C, Sinagra M, Ledger W, Li TC, Laird S. Endometrial integrin expression in women with recurrent implantation failure after in vitro fertilization and its relationship to pregnancy outcome. Fertil Steril. 2013;100(3):825–30.
Mariee N, Li TC, Laird SM. Expression of leukaemia inhibitory factor and interleukin 15 in endometrium of women with recurrent implantation failure after IVF; correlation with the number of endometrial natural killer cells. Hum Reprod. 2012;27(7):1946–54.
Xu B, Sun X, Li L, Wu L, Zhang A, Feng Y. Pinopodes, leukemia inhibitory factor, integrin-beta3, and mucin-1 expression in the peri-implantation endometrium of women with unexplained recurrent pregnancy loss. Fertil Steril. 2012;98(2):389–95.
Karaer A, Cigremis Y, Celik E, Urhan GR. Prokineticin 1 and leukemia inhibitory factor mRNA expression in the endometrium of women with idiopathic recurrent pregnancy loss. Fertil Steril. 2014;102(4):1091–5 e1.
Brosens JJ, Salker MS, Teklenburg G, Nautiyal J, Salter S, Lucas ES, et al. Uterine selection of human embryos at implantation. Sci Rep. 2014;4:3894.
Teklenburg G, Salker M, Heijnen C, Macklon NS, Brosens JJ. The molecular basis of recurrent pregnancy loss: impaired natural embryo selection. Mol Hum Reprod. 2010;16(12):886–95.
Macklon NS, Brosens JJ. The human endometrium as a sensor of embryo quality. Biol Reprod. 2014;91(4):98.
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–97.
Mohlin FC, Mercier E, Fremeaux-Bacchi V, Liszewski MK, Atkinson JP, Gris JC, et al. Analysis of genes coding for CD46, CD55, and C4b-binding protein in patients with idiopathic, recurrent, spontaneous pregnancy loss. Eur J Immunol. 2013;43(6):1617–29.
Salmon JE, Heuser C, Triebwasser M, Liszewski MK, Kavanagh D, Roumenina L, et al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med. 2011;8(3):e1001013.
Ruan YC, Chen H, Chan HC. Ion channels in the endometrium: regulation of endometrial receptivity and embryo implantation. Hum Reprod Update. 2014;20(4):517–29.
Thie M, Denker H-W. In vitro studies on endometrial adhesiveness for trophoblast: cellular dynamics in uterine epithelial cells. Cells Tissues Organs. 2002;172(3):237–52.
Nawy T. Single-cell sequencing. Nat Methods. 2014;11(1):18.
Lasken RS. Single-cell genomic sequencing using multiple displacement amplification. Curr Opin Microbiol. 2007;10(5):510–6.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82.
Shintaku H, Nishikii H, Marshall LA, Kotera H, Santiago JG. On-chip separation and analysis of RNA and DNA from single cells. Anal Chem. 2014;86(4):1953–7.
Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW, et al. Sequencing genomes from single cells by polymerase cloning. Nat Biotechnol. 2006;24(6):680–6.
Chiou PY, Ohta AT, Wu MC. Massively parallel manipulation of single cells and microparticles using optical images. Nature. 2005;436(7049):370–2.
Hosic S, Murthy SK, Koppes AN. Microfluidic sample preparation for single cell analysis. Anal Chem. 2016;88(1):354–80.
Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2018;18(1):35–45.
Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17(9):557–69.
Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13(3):718–25.
Nakamura T, Yabuta Y, Okamoto I, Aramaki S, Yokobayashi S, Kurimoto K, et al. SC3-seq: a method for highly parallel and quantitative measurement of single-cell gene expression. Nucleic Acids Res. 2015;43(9):e60.
Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–6.
Vassena R, Eguizabal C, Heindryckx B, Sermon K, Simon C, van Pelt AM, et al. Stem cells in reproductive medicine: ready for the patient? Hum Reprod. 2015;30(9):2014–21.
Krjutskov K, Katayama S, Saare M, Vera-Rodriguez M, Lubenets D, Samuel K, et al. Single-cell transcriptome analysis of endometrial tissue. Hum Reprod. 2016;31(4):844–53.
Weiling F. Historical study: Johann Gregor Mendel 1822-1884. Am J Med Genet. 1991;40(1):1–25; discussion 6.
Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41(1):10–3.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–3.
Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol. 2014;2:49.
Horsthemke B, Ludwig M. Assisted reproduction: the epigenetic perspective. Hum Reprod Update. 2005;11(5):473–82.
Das L, Parbin S, Pradhan N, Kausar C, Patra SK. Epigenetics of reproductive infertility. Front Biosci (Schol Ed). 2017;9:509–35.
Koukoura O, Sifakis S, Spandidos DA. DNA methylation in the human placenta and fetal growth (review). Mol Med Rep. 2012;5(4):883–9.
Houshdaran S, Zelenko Z, Irwin JC, Giudice LC. Human endometrial DNA methylome is cycle-dependent and is associated with gene expression regulation. Mol Endocrinol. 2014;28(7):1118–35.
Yamagata Y, Asada H, Tamura I, Lee L, Maekawa R, Taniguchi K, et al. DNA methyltransferase expression in the human endometrium: down-regulation by progesterone and estrogen. Hum Reprod. 2009;24(5):1126–32.
Kukushkina V, Modhukur V, Suhorutsenko M, Peters M, Magi R, Rahmioglu N, et al. DNA methylation changes in endometrium and correlation with gene expression during the transition from pre-receptive to receptive phase. Sci Rep. 2017;7(1):3916.
Rahnama F, Thompson B, Steiner M, Shafiei F, Lobie PE, Mitchell MD. Epigenetic regulation of E-cadherin controls endometrial receptivity. Endocrinology. 2009;150(3):1466–72.
Wang L, Tan YJ, Wang M, Chen YF, Li XY. DNA methylation inhibitor 5-Aza-2’-deoxycytidine modulates endometrial receptivity through upregulating HOXA10 expression. Reprod Sci. 2019;26(6):839–46.
Xiong Y, Wang J, Liu L, Chen X, Xu H, Li TC, et al. Effects of high progesterone level on the day of human chorionic gonadotrophin administration in in vitro fertilization cycles on epigenetic modification of endometrium in the peri-implantation period. Fertil Steril. 2017;108(2):269–76 e1.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80.
Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100.
Uchida H, Maruyama T, Arase T, Ono M, Nagashima T, Masuda H, et al. Histone acetylation in reproductive organs: significance of histone deacetylase inhibitors in gene transcription. Reprod Med Biol. 2005;4(2):115–22.
Uchida H, Maruyama T, Nagashima T, Ono M, Masuda H, Arase T, et al. Human endometrial cytodifferentiation by histone deacetylase inhibitors. Hum Cell. 2006;19(1):38–42.
Krusche CA, Vloet AJ, Classen-Linke I, von Rango U, Beier HM, Alfer J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum Reprod. 2007;22(11):2956–66.
Estella C, Herrer I, Atkinson SP, Quinonero A, Martinez S, Pellicer A, et al. Inhibition of histone deacetylase activity in human endometrial stromal cells promotes extracellular matrix remodelling and limits embryo invasion. PLoS One. 2012;7(1):e30508.
Maass N, Biallek M, Rosel F, Schem C, Ohike N, Zhang M, et al. Hypermethylation and histone deacetylation lead to silencing of the maspin gene in human breast cancer. Biochem Biophys Res Commun. 2002;297(1):125–8.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74.
Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nat Rev Genet. 2010;11(8):559–71.
Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Creighton CJ, Benham AL, Zhu H, Khan MF, Reid JG, Nagaraja AK, et al. Discovery of novel microRNAs in female reproductive tract using next generation sequencing. PLoS One. 2010;5(3):e9637.
Lam EW, Shah K, Brosens JJ. The diversity of sex steroid action: the role of micro-RNAs and FOXO transcription factors in cycling endometrium and cancer. J Endocrinol. 2012;212(1):13–25.
Ramon LA, Braza-Boils A, Gilabert-Estelles J, Gilabert J, Espana F, Chirivella M, et al. microRNAs expression in endometriosis and their relation to angiogenic factors. Hum Reprod. 2011;26(5):1082–90.
Revel A, Achache H, Stevens J, Smith Y, Reich R. MicroRNAs are associated with human embryo implantation defects. Hum Reprod. 2011;26(10):2830–40.
Petracco R, Grechukhina O, Popkhadze S, Massasa E, Zhou Y, Taylor HS. MicroRNA 135 regulates HOXA10 expression in endometriosis. J Clin Endocrinol Metab. 2011;96(12):E1925–33.
McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37(1):179–83.
Haig D, Graham C. Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell. 1991;64(6):1045–6.
Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7(2):45–9.
Regha K, Latos PA, Spahn L. The imprinted mouse Igf2r/Air cluster--a model maternal imprinting system. Cytogenet Genome Res. 2006;113(1–4):165–77.
Tanos V, Ariel I, Prus D, De-Groot N, Hochberg A. H19 and IGF2 gene expression in human normal, hyperplastic, and malignant endometrium. Int J Gynecol Cancer. 2004;14(3):521–5.
Lee RS, Depree KM, Davey HW. The sheep (Ovis aries) H19 gene: genomic structure and expression patterns, from the preimplantation embryo to adulthood. Gene. 2002;301(1–2):67–77.
Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–3.
Yan PS, Chen CM, Shi H, Rahmatpanah F, Wei SH, Huang TH. Applications of CpG island microarrays for high-throughput analysis of DNA methylation. J Nutr. 2002;132(8 Suppl):2430S–4S.
Schilling E, Rehli M. Global, comparative analysis of tissue-specific promoter CpG methylation. Genomics. 2007;90(3):314–23.
Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26(7):779–85.
Butcher LM, Beck S. AutoMeDIP-seq: a high-throughput, whole genome, DNA methylation assay. Methods. 2010;52(3):223–31.
Begemann M, Leisten I, Soellner L, Zerres K, Eggermann T, Spengler S. Use of multilocus methylation-specific single nucleotide primer extension (MS-SNuPE) technology in diagnostic testing for human imprinted loci. Epigenetics. 2012;7(5):473–81.
Konishi Y, Hayashi H, Suzuki H, Yamamoto E, Sugisaki H, Higashimoto H. Comparative analysis of methods to determine DNA methylation levels of a tumor-related microRNA gene. Anal Biochem. 2015;484:66–71.
Sandmann T, Jakobsen JS, Furlong EE. ChIP-on-chip protocol for genome-wide analysis of transcription factor binding in Drosophila melanogaster embryos. Nat Protoc. 2006;1(6):2839–55.
O’Neill LP, Turner BM. Immunoprecipitation of chromatin. Methods Enzymol. 1996;274:189–97.
Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci China Life Sci. 2013;56(10):876–85.
Liu CG, Spizzo R, Calin GA, Croce CM. Expression profiling of microRNA using oligo DNA arrays. Methods. 2008;44(1):22–30.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Huang, J., Zhang, R., Wang, C.C., Li, T.C. (2020). Endometrium Gene Expression and Epigenetic Regulation in Reproductive Failure. In: Kwak-Kim, J. (eds) Endometrial Gene Expression. Springer, Cham. https://doi.org/10.1007/978-3-030-28584-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-28584-5_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28583-8
Online ISBN: 978-3-030-28584-5
eBook Packages: MedicineMedicine (R0)