
Abstract
Porphyromonas gingivalis is an oral pathogen with the ability to induce oral dysbiosis and periodontal disease. Nevertheless, the mechanisms by which P. gingivalis could abrogate the host–microbe symbiotic relationship leading to oral dysbiosis remain unclear. We have recently demonstrated that P. gingivalis specifically increased the antimicrobial properties of oral epithelial cells, through a strong induction of the expression of PLA2-IIA in a mechanism that involves activation of the Notch-1 receptor. Moreover, gingival expression of PLA2-IIA was significantly increased during initiation and progression of periodontal disease in non-human primates and interestingly, those PLA2-IIA expression changes were concurrent with oral dysbiosis. In this chapter, we present an innovative hypothesis of a potential mechanism involved in P. gingivalis-induced oral dysbiosis and inflammation based on our previous observations and a robust body of literature that supports the antimicrobial and proinflammatory properties of PLA2-IIA as well as its role in other chronic inflammatory diseases.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
Introduction
Variation in the abundance of bacterial species that normally colonize epithelial surfaces and favors overgrowth of pathobionts (i.e., microorganisms that are part of the indigenous oral flora but transition to pathogenic after disruption of homeostasis) with a concomitant decrease in bacterial species normally associated with health (also named symbionts) is known as dysbiosis. Growing evidence indicates that dysbiosis is associated with several diseases such as inflammatory bowel disease, obesity, diabetes mellitus, and periodontal disease (DeGruttola et al. 2016; Maruvada et al. 2017; Carding et al. 2015; Hajishengallis et al. 2011; Dutzan et al. 2018). Enhancers of dysbiosis include environmental (e.g., smoking, diet, antibiotics), host factors (e.g., immunosuppression), and some pathogens such as Salmonella typhimurium and Helicobacter pylori (Kau et al. 2011; Baumler and Sperandio 2016; Brawner et al. 2017).
Porphyromonas gingivalis is an oral gram-negative pathobiont with the ability to enhance oral dysbiosis and periodontal disease (Hajishengallis et al. 2011). The mechanisms by which P. gingivalis enhances oral dysbiosis remain not fully elucidated; nevertheless, evidence indicates that P. gingivalis could be manipulating the host immune response for its own benefit. For example, it has been demonstrated in humans and mice that P. gingivalis manipulates complement C5a receptor 1 (C5aR1) and Toll-like receptor 2 (TLR2) cross talk signaling to block phagocytosis through a suppression of phagolysosomal maturation and simultaneously promotion of inflammation (Maekawa et al. 2014; Wang et al. 2010). Another described mechanism involved in P. gingivalis-induced oral dysbiosis is a localized chemokine paralysis. This refers to the ability of P. gingivalis to suppress the expression of chemokines in oral epithelial cells (OECs) (e.g., CXCL8, CXCL9, CXCL10, and CXCL11) through a mechanism that involves negative regulation of NFκB by a serine phosphatase B secreted by P. gingivalis. As a consequence, decreased chemokine expression leads to a reduction in the number of immune cells normally recruited to the subgingival environment that are critical for immune surveillance and maintaining homeostatic host–bacteria interactions (Darveau et al. 1998; Jauregui et al. 2013; Takeuchi et al. 2013).
Epithelial cells, in addition to orchestrating the recruitment of immune cells through the production of chemokines in response to the microbiota, also produce antimicrobial factors such as human beta defensins (hBDs) that are critical to maintain host–bacteria homeostatic interactions (Dale 2002; Chung et al. 2007). It has been shown that OECs express several antimicrobial factors (e.g., hBD-2 and hBD-3, CCL20) in response to oral bacterial species, and these responses seem to be differentially induced by both oral pathogenic and commensal bacterial species (Dommisch et al. 2012; Hosokawa et al. 2006; Greer et al. 2013; Chung and Dale 2008; Ji et al. 2007).
Specific modulation of OEC antimicrobial responses by oral commensal and pathogenic bacteria could be a plausible mechanism contributing to the maintenance or disruption of the homeostatic host–microbe interactions during health and disease, respectively (Lamont et al. 2018). In fact, some oral bacterial species specifically induce the production of classical antimicrobial peptides (AMPs) such as hBDs by OECs, while exhibiting antimicrobial resistance to the AMPs. For example, Fusobacterium nucleatum has been shown to induce significant hBD2 expression in OECs and be resistant to hBD2 killing; however, P. gingivalis seems to be a weak hBD2 inducer and is more susceptible to killing by hBD2 (Ghosh et al. 2018). In contrast, P. gingivalis enhances a remarkable expression of the antimicrobial protein phospholipase A2-group IIA (PLA2-IIA) by OECs, and is resistant, whereas F. nucleatum appears susceptible to PLA2-IIA–induced killing (Al-Attar et al. 2018).
Whether P. gingivalis’ ability to specifically modulate antimicrobial responses in OECs is a strategy to enhance oral dysbiosis remains unknown; however, the potentiation of the antimicrobial properties of OECs through up-regulation of PLA2-IIA seems to be a plausible mechanistic pathway as we have recently reported (Al-Attar et al. 2018). In this chapter, we will review the general properties and functions of PLA2-IIA and postulate a hypothesis concerning its potential contribution to the pathogenesis of P. gingivalis-induced oral dysbiosis and periodontal disease.
PLA2-IIA, General Properties
PLA2-IIA (also known as non-pancreatic secretory- or synovial secretory-PLA2) belongs to a family of ten secreted PLA2s (IB, IIA, IIC, IID, IIE, IIF, III, V, X, and XIIA) identified in mammals and is the only isoform detectable in the blood. PLA2-IIA is a highly cationic lipolytic enzyme, with catalytic activity (as for all secreted PLA2s) that is calcium (Ca2+) dependent (Murakami et al. 2011a, b). PLA2-IIA mRNA has been detected in placenta, tonsil, skin keratinocytes, kidney, rheumatoid synovial cells, cartilage, colon, ileum, stomach, spleen, and lung tissues (Ishizaki et al. 1989; Kramer et al. 1989; Nevalainen and Haapanen 1993; Ilic et al. 2014). Among the cell types that have been shown to produce PLA2-IIA in response to different stimuli such as pathogenic bacteria (e.g., Pseudomonas aeruginosa) or proinflammatory cytokines are hepatocytes, platelets, macrophages, and intestinal epithelial cells (i.e., Paneth cells) (Crowl et al. 1991; Okita et al. 2016; Mueller et al. 1993; Piris-Gimenez et al. 2005; Ouellette 2011; Pernet et al. 2014).
Given that PLA2-IIA is secreted, it has been suggested that its primary targets should be extracellular non-cellular phospholipids including microvesicles, microbial membranes, and dietary phospholipids (Murakami et al. 2011a). Nevertheless, cell-associated functions have also been attributed to PLA2-IIA in concert with other PLA2s such as cytoplasmic PLA2 isoforms that release arachidonic acid (AA) from cell membranes with the consequent production of inflammatory lipids (e.g., prostaglandins) (Murakami et al. 1996, 1997a). These observations remain controversial since PLA2-IIA has low affinity for phosphatidylcholine (PC), a major phospholipid in the outer leaflet of the plasma membrane. In contrast, due to its high cationic nature, PLA2-IIA exhibits high affinity for phosphatidylethanolamine (PE), phosphatidylglycerol (PG), or phosphatidylserine (PS), which are enriched in bacterial membranes and apoptotic cells making them suitable targets for the enzyme (Atsumi et al. 1997; Bayburt et al. 1993; Nevalainen et al. 2008).
In addition to its enzymatic activities, it has been suggested that PLA2-IIA could also modulate biological responses in an enzymatic-independent manner through interaction with cellular receptors. Particularly, the muscular-type receptor (M-type-R) or sPLA2 mouse receptor (PLA2R1) are type I transmembrane glycoproteins composed of multiple C-type lectin domains that can be activated by PLA2-IIA (and other PLA2s such as group IB and group X) leading to internalization into phagolysosomes, in which the enzyme is rapidly degraded. Thus, it could be a mechanism by which PLA2-IIA can be physiologically cleared from fluids (Zvaritch et al. 1996; Cupillard et al. 1999; Murakami et al. 2014). Nevertheless, the lack of an intracellular tail of PLA2R1 with cell signaling molecular features suggests that cellular responses mediated by this receptor could be linked to other receptors such as Toll-like receptors (TLRs) (Murakami et al. 2011b). PLA2R1 expression has been shown in skeletal muscle, lung, liver, heart, and kidney as well as in myeloid cells such as neutrophils, macrophages, and eosinophils (Lambeau and Lazdunski 1999; Silliman et al. 2002; Granata et al. 2005; Triggiani et al. 2003). Interestingly, in vitro studies demonstrated that PLA2R1 expression is increased during cellular senescence and regulates senescence in human fibroblasts through the p53 pathway (Augert et al. 2009). These findings were recently validated in vivo showing that PLA2R1 mediates premature aging (Griveau et al. 2018).
In general, important regulatory roles of PLA2-IIA in inflammation and infection have been demonstrated. Details about the potential mechanisms involved in these critical biological processes will be discussed in the following sections.
Proinflammatory Properties of PLA2-IIA
PLA2-IIA was identified in humans in 1989 as an inducible enzyme during inflammatory responses isolated from rheumatoid arthritis synovial fluid (Kramer et al. 1989; Seilhamer et al. 1989). Since then, several studies have demonstrated that expression of PLA2-IIA can be induced by proinflammatory cytokines including interleukin-1alpha (IL-1α), IL-1β, tumor necrosis factor-alpha (TNFα), as well as lipopolysaccharide (LPS) in macrophages. Consistently with these observations, elevated PLA2-IIA levels in sera or inflammatory exudates are correlated with the severity of inflammation and infection (Gronroos et al. 2002; Tan and Goh 2017). On the other hand, glucocorticoids, anti-inflammatory cytokines, and growth factors (e.g., dexamethasone, transforming growth factor β, platelet-derived growth factor, and insulin-like growth factors I and II) are potent suppressors for the induction of PLA2-IIA (Murakami et al. 1997b; Jacques et al. 1997).
It has been suggested that the proinflammatory properties of PLA2-IIA would be related to its enzymatic activity participating in the release of AA and contributing to the production of eicosanoids such as prostaglandins, or independently of its enzymatic activity, modulating immune cell responses through the PLA2R1. In general, several studies suggest that the role of PLA2-IIA in AA release from cell membranes is minor and seems to be cell-type dependent when compared with other PLA2 enzymes such as cytosolic PLA2, whose role in AA production has been clearly demonstrated (Murakami et al. 2017; Fonteh et al. 1994; Kuwata et al. 1999; Marshall et al. 2000; Mounier et al. 1993).
Another proinflammatory mechanism is the activation of immune cells through the PLA2R1. For example, macrophages stimulated with PLA2-IIA showed activation of PI3K, AKT, ERK1/2, JNK, NFκB pathways resulting in up-regulation of inducible nitric oxide synthase (iNOS) and NO generation, as well as increased expression of cytokines/chemokines (Baek et al. 2000, 2001; Hernandez et al. 2002; Triggiani et al. 2002). Similarly, in neutrophils, the activation of PLA2R1 led to the release of elastase and changes in cell adhesion through activation of the p38-MAPK pathway (Silliman et al. 2002). Most recently, the ability of PLA2-IIA to bind integrins (e.g., αvβ3 and α4β1) was demonstrated, which suggested that immune cell activation by PLA2-IIA could also be mediated by this pathway (Fujita et al. 2015; Takada and Fujita 2017).
A most recent and plausible mechanism by which PLA2-IIA could induce inflammation is through generation of inflammatory mediators such as lysophospholipids, fatty acids, and mitochondrial DNA as a product of the hydrolysis of the mitochondrial membrane. This is interesting given that platelets during inflammatory processes can release mitochondria that serve as a PLA2-IIA substrate (Boudreau et al. 2014). Notably, neutrophils can internalize platelet microparticles (which contain transcription factors, nucleic acids, and mitochondria) in a mechanism mediated by PLA2-IIA and platelet-type 12-lipoxygenase, which could also be contributing to pathologic inflammation in arthritis (Duchez et al. 2015).
A better understanding of the mechanisms by which PLA2-IIA could be contributing to enhance inflammation is an evolving field. Future studies will be needed to clarify the role of PLA2-IIA in inflammatory pathologies in which higher levels have been found locally and systemically.
Antibacterial Properties of PLA2-IIA
As indicated earlier in this chapter, the antibacterial properties of secreted PLA2s have been broadly documented (Nevalainen et al. 2008; Koduri et al. 2002). Among all sPLA2s, the antibacterial effect of PLA2-IIA is the most potent (ng) among all secreted isoforms (Koduri et al. 2002).
It has been shown that PLA2-IIA has a greater affinity for gram-positive than gram-negative bacteria. This seems to be due to the structural characteristics of the cell wall of these bacteria phyla, whereby multiple layers of peptidoglycan cross-linked with peptide interbridges also contain higher levels of teichoic and lipoteichoic acids in gram-positive bacteria. These components proscribe a negative charge to the bacterial wall that creates an ideal/complementary substrate for the cationic PLA2-IIA (Foreman-Wykert et al. 1999; Weiss 2015; Dore and Boilard 2019). PLA2-IIA is therefore able to penetrate and hydrolyze the plasma membrane of gram-positive bacteria (Beers et al. 2002). In contrast, gram-negative bacteria normally have a thinner peptidoglycan layer that is covered by lipopolysaccharide, which prevent the entry of PLA2-IIA and subsequent bacterial killing (Beers et al. 2002). It is likely that for PLA2-IIA to kill gram-negative bacteria, other host factors working in concert with the enzyme, such as complement, would be necessary (Madsen et al. 1996; Gronroos et al. 2005).
PLA2-IIA seems to be a critical innate factor to protect against infections. For example, PLA2-IIA sera levels are increased during group B streptococcus (GBS) infections and clinically relevant GBS strains are rapidly killed by PLA2-IIA (Movert et al. 2013). Similarly, PLA2-IIA has been shown to be an important innate factor that plays a protective role against Bacillus anthracis infections (Piris-Gimenez et al. 2005). Moreover, normal serum PLA2-IIA concentrations effectively kill the commensal Enterococcus faecium (Paganelli et al. 2018). Interestingly, some pathogenic strains of Staphylococcus aureus appear to modulate PLA2-IIA expression at epithelial surfaces to avoid being killed and enabling disease processes (Pernet et al. 2015).
The potent antimicrobial properties of PLA2-IIA at low concentrations are of particular clinical interest, given that PLA2-IIA concentrations in body fluids under normal conditions have been shown to prevent some gram-positive bacterial infections. In addition, the bactericidal effect of acute phase serum seems to be determined mostly by the presence of elevated levels of PLA2-IIA. For example, PLA2-IIA serum levels from healthy individuals (1.5 ng/mL) or during sepsis (300 ng/mL) can kill Listeria monocytogenes or S. aureus, respectively, in 2 h (Gronroos et al. 2002). Similarly, PLA2-IIA concentrations (50 ng/mL) in expectorations from adult cystic fibrosis patients can efficiently kill S. aureus (Pernet et al. 2014). Likewise, higher levels of PLA2-IIA have been detected in tears exceeding about 400 times those found in sera from healthy subjects, which suggests that this enzyme could play a significant role in the innate protective immunity of the ocular surfaces (Nevalainen et al. 1994).
This evidence suggests that PLA2-IIA is an important innate antimicrobial factor that plays a significant role at mucosal surfaces maintaining homeostatic host–bacteria interactions as well as contributing to the control and eradication of different pathogens during the acute phase. It is tempting to hypothesize that increased PLA2-IIA levels at mucosal surfaces may contribute to modify the normal abundance of bacterial species, especially reducing gram-positive bacterial numbers that could enhance a dysbiotic microbial ecology and disease.
PLA2-IIA, Role in Disease
PLA2-IIA has been associated with several diseases such as atherosclerosis, rheumatoid arthritis, Alzheimer’s disease, cancer, and inflammatory bowel disease. Interestingly, all these diseases have as a common denominator chronic inflammation, as well as microbial components that could be involved in the pathologic progression. Some of the main findings relating PLA2-IIA with some of these diseases will be presented in this section.
Atherosclerosis
The association between PLA2-IIA and increased risk for atherosclerotic cardiovascular disease has been established (Sun et al. 2016). Clinical studies have indicated that elevated blood levels of PLA2-IIA are an independent risk factor for coronary artery disease and predict clinical coronary events independent of other risk factors (Kugiyama et al. 1999). Similarly, higher circulation PLA2-IIA levels and enzymatic activity were associated with an increased risk of incident and recurrent major vascular events such as cardiovascular death, myocardial infarction, and stroke. This association was similar to that observed between C-reactive protein and coronary artery disease (Boekholdt et al. 2005; Mallat et al. 2007; O’Donoghue et al. 2011).
It is well described that augmented levels of low-density lipoprotein (LDL) cholesterol particles increase the risk of coronary heart disease through stimulation of foam cell formation. A potential pro-atherogenic mechanism of PLA2-IIA could involve induction of foam cell formation and LDL lipoprotein modification, both events associated with higher levels of PLA2-IIA expressed in vascular smooth muscle cells from both medial and intimal layers and in macrophage-rich regions of atherosclerotic plaques (Menschikowski et al. 1995; Jaross et al. 2002). Consistent with these findings, it was shown that PLA2-IIA is able to increase the adhesion and migration of monocytic cells, which could increase macrophage recruitment to endothelium during the atherogenic process (Ibeas et al. 2009). Evidence supporting the association between PLA2-IIA with atherosclerotic disease has generated great interest in developing inhibitors that may help to reduce atherosclerosis (Magrioti and Kokotos 2010; Garcia-Garcia and Serruys 2009).
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is an autoimmune and inflammatory disease that causes inflammation and tissue damage of the joints leading to chronic pain and deformity (www.cdc.gov). The initial observations linking PLA2-IIA and inflammation were performed in patients with RA and osteoarthritis, where characterization of PLA2-IIA and detection of its enzymatic activity in sera and synovial fluids were determined (Vadas et al. 1985; Pruzanski et al. 1985). Further studies have documented that serum PLA2-IIA concentrations are elevated in patients with RA and seem to correlate with severity of disease (Pruzanski et al. 1988, 1994; Kortekangas et al. 1994; Lin et al. 1996; Jamal et al. 1998). PLA2-IIA has also been found in articular and extra-articular cartilage from RA patients, which is consistent with the ability of human chondrocytes to synthesize and release PLA2-IIA (Nevalainen et al. 1993; Pruzanski et al. 1990). Encouraged by these observations, several groups have investigated the potential role of PLA2-IIA in the pathogenesis of RA. For example, injection of pure recombinant PLA2-IIA elicited a dramatic inflammatory and arthritogenic response in rabbit joints characterized by hyperplasia of the synovial lining cells and prostaglandin production in comparison to pancreatic PLA2, which had little activity in this model (Bomalaski et al. 1991). The authors suggested that given that proinflammatory cytokines can induce PLA2-IIA synthesis and secretion, synovial PLA2-IIA could play a role in acute episodes in chronic inflammatory conditions such as RA. Most recently, it was elegantly shown by Boilard’s group that PLA2-IIA could specifically potentiate inflammatory responses in vivo by acting with a type 12 lipoxygenase (12-LO) expressed by platelet extracellular vesicles (also called platelet microparticles-MP) to enhance internalization of those MP by neutrophils and promoting autoimmune inflammatory arthritis (Duchez et al. 2015).
Cancer
Among the secreted PLA2s, group IIA, III, and X enzymes are significantly dysregulated in various types of cancer such as lung, gastric, and colon. Several biological mechanisms involved in PLA2s-induced pro-tumorigenic responses have been described such as stimulation of cell proliferation and cell survival (mediated by antiapoptotic effects), inflammation, and angiogenesis. Nevertheless, the role of PLA2-IIA in cancer seems to exert either a pro- or anti-tumorigenic role, depending on the tissue and cancer type (Brglez et al. 2014). For example, patients with prostate (Jiang et al. 2002; Dong et al. 2010), esophageal (Menschikowski et al. 2013), and lung (Kupert et al. 2011) cancer exhibit elevated PLA2-IIA levels compared with healthy controls. Of note, prolonged survival time was seen in patients with lower PLA2-IIA levels (Menschikowski et al. 2013).
In contrast, the increased expression of PLA2-IIA in gastric cancer cell lines and tumors appears to be associated with an anti-tumorigenic role of the enzyme, as it reduces cell migration and invasiveness in vitro, its expression correlates with longer survival and is a predictor of a favorable outcome for patients with gastric cancer (Minami et al. 1997; Ganesan et al. 2008; Wang et al. 2013). Interestingly, the expression of PLA2-IIA mRNA was found to be elevated in primary, early-stage gastric tumors but decreased in late-stage metastatic tumors, suggesting that the loss of PLA2-IIA activity may contribute to the development of more aggressive tumors (Ganesan et al. 2008).
The expression of PLA2-IIA is also up-regulated in colon cancer; however, its role in pathogenesis remains controversial with studies showing either pro- or anti-tumorigenic effects. This seems to be partly due to the difficulty of extrapolating results from mouse to human malignancies and inconsistent results in both mouse models and human tissues (Brglez et al. 2014; Mounier et al. 2008). A recent report demonstrated that intracellular PLA2-IIA and PLA2-X produced by Paneth cells are stem cell niche factors that, during homeostasis, inhibit Wnt signaling (pathway that has an anti-tumorigenic role); however, with inflammation PLA2-IIA and -X can be secreted into the intestinal lumen, where the enzymes promote prostaglandin synthesis and Wnt signaling. Thus, these PLA2 enzymes seem to be important genetic modifiers of inflammation and colon cancer (Schewe et al. 2016).
Alzheimer’s Disease
Expression of major groups of PLA2 enzymes has been described in the central nervous system including PLA2-IIA, which seems to be particularly associated with neurons and glial cells (Sun et al. 2004, 2010). Specifically, PLA2-IIA and PLA2-IVA seems to be contributors of neurodegeneration (Schaeffer et al. 2010; Sanchez-Mejia et al. 2008). As such, increased expression of PLA2-IIA mainly by astrocytes was found in Alzheimer’s disease (AD) brains as compared to non-demented elderly brains. Likewise, PLA2-IIA–positive astrocytes were associated with amyloid beta-containing plaques, which seem to play a major role in neuronal cell death and neuro-inflammation, two critical factors leading to neurodegeneration as the principal underlying cause of the cognitive decline seen in AD (Moses et al. 2006; Culmsee and Landshamer 2006). Supporting these observations, it has been demonstrated that PLA2-IIA is highly cytotoxic for neurons and astrocytes, with proinflammatory mediators such as IL-1β and TNFα being major drivers for PLA2-IIA expression in neurons and microglia (Schaeffer et al. 2010; Villanueva et al. 2012; Jensen et al. 2009; Thomas et al. 2000; Yagami et al. 2002). Most recently, a significant increase of secreted PLA2 activity was observed in cerebrospinal fluid of AD patients, which presented the possibility of using secreted PLA2 activity as a biomarker for neuroinflammation. Nevertheless, the specific contribution of PLA2-IIA to these findings was not addressed (Chalbot et al. 2009).
Inflammatory Bowel Disease (IBD)
Inflammatory bowel disease is a group of conditions [i.e., Crohn’s disease (CD) and ulcerative colitis (UC)] that are characterized by chronic inflammation of the gastrointestinal (GI) tract and leads to damage of the GI mucosal surfaces and the onset of clinical symptoms such as abdominal pain, persistent diarrhea, bloody stool, weight loss, and fatigue (www.cdc.gov). Although less evidence exists about a potential role of PLA2-IIA in the pathogenesis of IBD, some studies have shown that increased PLA2-IIA serum levels and activity are associated with CD and UC (Minami et al. 1994, 1997) and PLA2-IIA activity decreased in parallel with clinical improvement and correlated with serum C-reactive protein and erythrocyte sedimentation rate (Minami et al. 1992). It seems like most attention has been placed on the inflammatory properties of PLA2-IIA within the context of IBD, with little research exploring its role in intestinal microbial dysbiosis associated with its potent antimicrobial effects.
In general, the above evidence suggests that PLA2-IIA is associated with several systemic diseases and seems to play an important role in their pathogenesis. Modulation of PLA2-IIA expression could be an innovative strategy for preventing/treating them. For instance, the molecular structure of PLA2-IIA was resolved, which has contributed to improve the understanding of its functions as well as the possibility to explore different strategies for identification and testing candidate chemical/drug inhibitors (Scott et al. 1991; Wery et al. 1991; Church et al. 2001; Thwin et al. 2007; Magrioti and Kokotos 2013). Interestingly, it has been shown that lipophilic tetracyclines such as minocycline and doxycycline inhibit PLA2-IIA, probably by interaction with the substrate (Pruzanski et al. 1992).
Periodontal disease has also been associated as a risk factor for some of the chronic inflammatory diseases discussed above in which oral pathogenic bacteria such as P. gingivalis (a strong inducer of PLA2-IIA) seem to contribute to their pathogenesis (de Pablo et al. 2009; Gibson 3rd et al. 2006; Velsko et al. 2015; Hayashi et al. 2010). It is tempting to hypothesize that P. gingivalis virulence factors (e.g., gingipains) may also be contributing to enhance atherosclerosis, joint inflammation, and central nervous system inflammation through stimulation of PLA2-IIA production by tissue-specific cells (i.e., endothelial cells, articular fibroblasts and chondrocytes, as well astrocytes and microglia).
Potential Role of PLA2-IIA in P. gingivalis-Induced Oral Dysbiosis and Periodontitis
Although PLA2-IIA has been associated with several chronic inflammatory diseases as described in the previous section, its role in the pathogenesis of periodontal disease remains surprisingly unexplored. Only two small clinical studies demonstrated that increased PLA2-IIA levels and activity in gingival crevicular fluid (GCF) and gingival tissue, respectively, were significantly elevated in periodontitis sites when compared with healthy sites; the levels returned to those comparable with health after periodontal treatment (Shinohara et al. 1995; Ishida et al. 1994). Consistent with these clinical findings, we have recently reported that gingival mRNA expression of PLA2-IIA was significantly increased during initiation and progression of periodontal disease in non-human primates (NHPs), and interestingly, the PLA2-IIA expression changes were concurrent with oral dysbiosis (Al-Attar et al. 2018). This is of interest given the clinically demonstrated proinflammatory and potent antimicrobial effects of PLA2-IIA. Recent immunohistochemistry and immunofluorescence analysis of gingival tissues from NHPs and mice suggests that PLA2-IIA is constitutively expressed by oral epithelium and some infiltrating inflammatory cells are also positive (Fig. 1a, b). Epithelial cells from the stratum basale and spinosum in NHP gingival tissue show the highest expression intensity for PLA2-IIA, which seems to increase early after disease induction at 2 weeks and 1 month, with cells from outer layers (stratum granulosum and lucidum) showing also positive staining during disease progression (Fig. 1b).


Patterns of PLA2-IIA expression in gingival tissues from (a) mice (BALB/c) by immunofluorescence and (b) non-human primates (rhesus) by immunohistochemistry before ligature-induced periodontitis (baseline), 2 weeks (2 W) and 1 month (1 M) after disease induction. CT Connective Tissue; E Epithelium
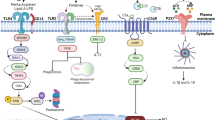
The proposed hypothesis is that elevated oral local PLA2-IIA levels could be contributing to early variations in the abundance of susceptible oral bacterial communities (i.e., oral dysbiosis) and gingival inflammation (Fig. 2). Supporting this hypothesis, we have recently demonstrated that P. gingivalis specifically increased the antimicrobial properties of oral epithelial cells through a remarkable induction of PLA2-IIA expression via a mechanism that involved activation of the Notch-1 receptor, and some oral bacterial species (but not P. gingivalis) were differentially susceptible to the antimicrobial effects of recombinant PLA2-IIA (Al-Attar et al. 2018). This is relevant given the key role that P. gingivalis seems to play in oral dysbiosis and periodontal disease (Hajishengallis et al. 2011; Maekawa et al. 2014).


Hypothesis for the role of P. gingivalis-induced PLA2-IIA through Notch-1 activation in oral epithelial cells as contributor to oral dysbiosis, inflammation, and tissue destruction
While the role of PLA2-IIA in the pathogenesis of periodontal disease in vivo remains to be determined, previous evidence supports the hypothesis that this enzyme may indeed play a significant role. For example, mouse strains (e.g., C57BL/6, 129/J) that have a natural mutation in the PLA2G2A gene (leading to the production of an inactive enzyme) are more resistant to P. gingivalis-induced periodontitis than mouse strains (e.g., BALB/c, DBA/2J) that express an active PLA2-IIA enzyme (Baker et al. 2000; Kennedy et al. 1995). Therefore, a non-functional PLA2-IIA in periodontitis-resistant mice strains may prevent potential P. gingivalis-induced antimicrobial and proinflammatory responses mediated by PLA2-IIA that could lead to oral dysbiosis and periodontitis.
Ongoing preliminary mouse studies in our laboratory suggest that the oral microbiome of C57BL/6 transgenic mice (more resistant strain to P. gingivalis-induced periodontitis) expressing the human functional PLA2-IIA (hPLA2-IIA-Tg) has significant differences in the abundance of several bacterial species that belong to the Firmicutes and Proteobacteria phyla compared with their corresponding wild-type (WT) co-caged littermates (unpublished data). Notably, the oral microbiome of hPLA2-IIA-Tg mice showed a significant decrease in the abundance of Firmicutes species in contrast to Proteobacteria species that were enriched. In particular, Lactobacillus plantarum and clostridia species were reduced in hPLA2-IIA-Tg mice. L. plantarum is a gram-positive lactic acid bacterium currently used as a probiotic due to its anti-inflammatory and antioxidant activities, its positive effects in maintaining epithelial barrier integrity, and its ability to inhibit the growth of pathogens, which enhances the maintenance of intestinal microbial symbiosis (Jang et al. 2018; Wu et al. 2017; Yin et al. 2018; Wang et al. 2018). It has been shown that the distribution of oral lactobacilli species including L. plantarum seems to change with periodontal disease and could play an important role in the maintenance of the microbiologic balance in the oral cavity associated with their antimicrobial properties against periodontopathogenic bacteria such as P. gingivalis, Aggregatibacter actinomycetemcomitans, and Prevotella intermedia, as well as their anti-inflammatory effects (Schmitter et al. 2018; Koll-Klais et al. 2005; Teanpaisan et al. 2011; Khalaf et al. 2016).
In contrast, hPLA2-IIA-Tg mice showed a significant enrichment in Proteobacteria species. Bacteria from this phylum are all gram-negative and have been broadly associated with mucosal inflammation and disease (Eom et al. 2018; Rizzatti et al. 2017). Likewise, some oral Proteobacteria species such as A. actinomycetemcomitans, Campylobacter rectus, Eikenella corrodens, Desulfobulbus sp., Acinetobacter baumannii, and Escherichia coli among others have been systematically isolated and associated with periodontal lesions (Kumar et al. 2005; Camelo-Castillo et al. 2015; Oliveira et al. 2016; Richards et al. 2015; Miller et al. 2018; da Silva-Boghossian et al. 2011; Perez-Chaparro et al. 2014). Based on this evidence, it is reasonable to hypothesize that elevation in the gingival PLA2-IIA levels could impact the oral microbiome, decreasing the numbers of bacterial species critical for health and enhancing the growth of more pathogenic bacteria.
The above changes in the oral microbiome of hPLA2-IIA-Tg mice are concurrent with variations in the expression of gingival innate genes. For example, down-regulation of genes associated with antimicrobial responses (e.g., S100a8, Defb1, B2m, Cxcl13) and recruitment of immune cells (e.g., MCP-1, MCP-3, MCP-5 and RANTES) have been identified in hPLA2-IIA-Tg in comparison to wild-type (WT) co-caged mice. In contrast, increased expression of bacterial sensing genes such as nucleotide-binding oligomerization domain-containing protein 2 (NOD2) was seen in PLA2-IIA Tg vs. WT mice (unpublished data). Of note, activation of NOD2-RIP2-NFkB pathway by peptidoglycan internalized via phagocytosis by alveolar macrophages also led to PLA2-IIA expression (Pernet et al. 2015). Thus, up-regulation of NOD2 associated with a dysbiotic oral microbiome could also amplify PLA2-IIA responses (Fig. 2). Whether these host innate immune changes are a consequence of a direct effect of PLA2-IIA on specific cells or an indirect effect that is driven by a PLA2-IIA–induced dysbiotic oral microbiome warrants further investigation.
Finally, our ongoing pilot clinical study suggests that PLA2-IIA is detectable in unstimulated saliva, within a range from 7 to 100 ng/mL (unpublished data). These findings suggest that salivary PLA2-IIA concentrations seem to be about 5–7 times higher than normal/healthy serum levels (1.5 ng/mL), which have been shown to kill pathogens like L. monocytogenes in about 2 h after exposure (Gronroos et al. 2002). This is relevant and suggests that elevations of local (salivary and gingival) PLA2-IIA levels could contribute to modify the abundance of susceptible bacterial species in the oral microbiome leading to oral dysbiosis.
The early mechanisms associated with the breakdown of homeostasis between the resident microbiota and the periodontal tissues that results in progressing disease remain unclear. Elucidation of these processes will require novel approaches, likely evaluating unique cellular mechanisms that have evolved by the host to actively discriminate “friend from foe.” Additionally, novel mechanisms employed by pathogenic bacteria to enhance their capacity to emerge and survive in the commensal autochthonous microbiota by “short circuiting” host cellular responses likely contribute to the observations of the capability of P. gingivalis to act as a keystone species and functionally hijack the microbial ecology to enhance chronic inflammation and destructive disease processes. This review presents an innovative hypothesis for a potential mechanism through intersecting functional pathways, by which P. gingivalis could accomplish this goal. Thus, the biological active molecule PLA2-IIA with its targeted antimicrobial activity and ability to modulate immunoinflammatory responses, combined with the pathogen specifically activating the Notch-1 receptor pathway, makes this a new strategy, likely among others, used by P. gingivalis to modify the local ecology that starts disease.
References
Al-Attar, A., Alimova, Y., Kirakodu, S., Kozal, A., Novak, M. J., Stromberg, A. J., et al. (2018). Activation of Notch-1 in oral epithelial cells by P. gingivalis triggers the expression of the antimicrobial protein PLA2-IIA. Mucosal Immunology, 11(4), 1047–1059.
Atsumi, G., Murakami, M., Tajima, M., Shimbara, S., Hara, N., & Kudo, I. (1997). The perturbed membrane of cells undergoing apoptosis is susceptible to type II secretory phospholipase A2 to liberate arachidonic acid. Biochimica et Biophysica Acta, 1349(1), 43–54.
Augert, A., Payre, C., de Launoit, Y., Gil, J., Lambeau, G., & Bernard, D. (2009). The M-type receptor PLA2R regulates senescence through the p53 pathway. EMBO Reports, 10(3), 271–277.
Baek, S. H., Kwon, T. K., Lim, J. H., Lee, Y. J., Chang, H. W., Lee, S. J., et al. (2000). Secretory phospholipase A2-potentiated inducible nitric oxide synthase expression by macrophages requires NF-kappa B activation. Journal of Immunology, 164(12), 6359–6365.
Baek, S. H., Lim, J. H., Park, D. W., Kim, S. Y., Lee, Y. H., Kim, J. R., et al. (2001). Group IIA secretory phospholipase A(2) stimulates inducible nitric oxide synthase expression via ERK and NF-kappaB in macrophages. European Journal of Immunology, 31(9), 2709–2717.
Baker, P. J., Dixon, M., & Roopenian, D. C. (2000). Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infection and Immunity, 68(10), 5864–5868.
Baumler, A. J., & Sperandio, V. (2016). Interactions between the microbiota and pathogenic bacteria in the gut. Nature, 535(7610), 85–93.
Bayburt, T., Yu, B. Z., Lin, H. K., Browning, J., Jain, M. K., & Gelb, M. H. (1993). Human nonpancreatic secreted phospholipase A2: Interfacial parameters, substrate specificities, and competitive inhibitors. Biochemistry, 32(2), 573–582.
Beers, S. A., Buckland, A. G., Koduri, R. S., Cho, W., Gelb, M. H., & Wilton, D. C. (2002). The antibacterial properties of secreted phospholipases A2: A major physiological role for the group IIA enzyme that depends on the very high pI of the enzyme to allow penetration of the bacterial cell wall. The Journal of Biological Chemistry, 277(3), 1788–1793.
Boekholdt, S. M., Keller, T. T., Wareham, N. J., Luben, R., Bingham, S. A., Day, N. E., et al. (2005). Serum levels of type II secretory phospholipase A2 and the risk of future coronary artery disease in apparently healthy men and women: The EPIC-Norfolk Prospective Population Study. Arteriosclerosis, Thrombosis, and Vascular Biology, 25(4), 839–846.
Bomalaski, J. S., Lawton, P., & Browning, J. L. (1991). Human extracellular recombinant phospholipase A2 induces an inflammatory response in rabbit joints. Journal of Immunology, 146(11), 3904–3910.
Boudreau, L. H., Duchez, A. C., Cloutier, N., Soulet, D., Martin, N., Bollinger, J., et al. (2014). Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood, 124(14), 2173–2183.
Brawner, K. M., Kumar, R., Serrano, C. A., Ptacek, T., Lefkowitz, E., Morrow, C. D., et al. (2017). Helicobacter pylori infection is associated with an altered gastric microbiota in children. Mucosal Immunology, 10(5), 1169–1177.
Brglez, V., Lambeau, G., & Petan, T. (2014). Secreted phospholipases A2 in cancer: Diverse mechanisms of action. Biochimie, 107(Pt A), 114–123.
Camelo-Castillo, A., Novoa, L., Balsa-Castro, C., Blanco, J., Mira, A., & Tomas, I. (2015). Relationship between periodontitis-associated subgingival microbiota and clinical inflammation by 16S pyrosequencing. Journal of Clinical Periodontology, 42(12), 1074–1082.
Carding, S., Verbeke, K., Vipond, D. T., Corfe, B. M., & Owen, L. J. (2015). Dysbiosis of the gut microbiota in disease. Microbial Ecology in Health and Disease, 26, 26191.
Chalbot, S., Zetterberg, H., Blennow, K., Fladby, T., Grundke-Iqbal, I., & Iqbal, K. (2009). Cerebrospinal fluid secretory Ca2+-dependent phospholipase A2 activity is increased in Alzheimer disease. Clinical Chemistry, 55(12), 2171–2179.
Chung, W. O., & Dale, B. A. (2008). Differential utilization of nuclear factor-kappaB signaling pathways for gingival epithelial cell responses to oral commensal and pathogenic bacteria. Oral Microbiology and Immunology, 23(2), 119–126.
Chung, W. O., Dommisch, H., Yin, L., & Dale, B. A. (2007). Expression of defensins in gingiva and their role in periodontal health and disease. Current Pharmaceutical Design, 13(30), 3073–3083.
Church, W. B., Inglis, A. S., Tseng, A., Duell, R., Lei, P. W., Bryant, K. J., et al. (2001). A novel approach to the design of inhibitors of human secreted phospholipase A2 based on native peptide inhibition. The Journal of Biological Chemistry, 276(35), 33156–33164.
Crowl, R. M., Stoller, T. J., Conroy, R. R., & Stoner, C. R. (1991). Induction of phospholipase A2 gene expression in human hepatoma cells by mediators of the acute phase response. The Journal of Biological Chemistry, 266(4), 2647–2651.
Culmsee, C., & Landshamer, S. (2006). Molecular insights into mechanisms of the cell death program: Role in the progression of neurodegenerative disorders. Current Alzheimer Research, 3(4), 269–283.
Cupillard, L., Mulherkar, R., Gomez, N., Kadam, S., Valentin, E., Lazdunski, M., et al. (1999). Both group IB and group IIA secreted phospholipases A2 are natural ligands of the mouse 180-kDa M-type receptor. The Journal of Biological Chemistry, 274(11), 7043–7051.
da Silva-Boghossian, C. M., do Souto, R. M., Luiz, R. R., & Colombo, A. P. (2011). Association of red complex, A. actinomycetemcomitans and non-oral bacteria with periodontal diseases. Archives of Oral Biology, 56(9), 899–906.
Dale, B. A. (2002). Periodontal epithelium: A newly recognized role in health and disease. Periodontology 2000, 30, 70–78.
Darveau, R. P., Belton, C. M., Reife, R. A., & Lamont, R. J. (1998). Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infection and Immunity, 66(4), 1660–1665.
de Pablo, P., Chapple, I. L., Buckley, C. D., & Dietrich, T. (2009). Periodontitis in systemic rheumatic diseases. Nature Reviews Rheumatology, 5(4), 218–224.
DeGruttola, A. K., Low, D., Mizoguchi, A., & Mizoguchi, E. (2016). Current understanding of dysbiosis in disease in human and animal models. Inflammatory Bowel Diseases, 22(5), 1137–1150.
Dommisch, H., Reinartz, M., Backhaus, T., Deschner, J., Chung, W., & Jepsen, S. (2012). Antimicrobial responses of primary gingival cells to Porphyromonas gingivalis. Journal of Clinical Periodontology, 39(10), 913–922.
Dong, Z., Liu, Y., Scott, K. F., Levin, L., Gaitonde, K., Bracken, R. B., et al. (2010). Secretory phospholipase A2-IIa is involved in prostate cancer progression and may potentially serve as a biomarker for prostate cancer. Carcinogenesis, 31(11), 1948–1955.
Dore, E., & Boilard, E. (2019). Roles of secreted phospholipase A2 group IIA in inflammation and host defense. Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids, 1864(6), 789–802.
Duchez, A. C., Boudreau, L. H., Naika, G. S., Bollinger, J., Belleannee, C., Cloutier, N., et al. (2015). Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proceedings of the National Academy of Sciences of the United States of America, 112(27), E3564–E3573.
Dutzan, N., Kajikawa, T., Abusleme, L., Greenwell-Wild, T., Zuazo, C. E., Ikeuchi, T., et al. (2018). A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Science Translational Medicine, 10(463), eaat0797.
Eom, T., Kim, Y. S., Choi, C. H., Sadowsky, M. J., & Unno, T. (2018). Current understanding of microbiota- and dietary-therapies for treating inflammatory bowel disease. Journal of Microbiology, 56(3), 189–198.
Fonteh, A. N., Bass, D. A., Marshall, L. A., Seeds, M., Samet, J. M., & Chilton, F. H. (1994). Evidence that secretory phospholipase A2 plays a role in arachidonic acid release and eicosanoid biosynthesis by mast cells. Journal of Immunology, 152(11), 5438–5446.
Foreman-Wykert, A. K., Weinrauch, Y., Elsbach, P., & Weiss, J. (1999). Cell-wall determinants of the bactericidal action of group IIA phospholipase A2 against Gram-positive bacteria. The Journal of Clinical Investigation, 103(5), 715–721.
Fujita, M., Zhu, K., Fujita, C. K., Zhao, M., Lam, K. S., Kurth, M. J., et al. (2015). Proinflammatory secreted phospholipase A2 type IIA (sPLA-IIA) induces integrin activation through direct binding to a newly identified binding site (site 2) in integrins alphavbeta3, alpha4beta1, and alpha5beta1. The Journal of Biological Chemistry, 290(1), 259–271.
Ganesan, K., Ivanova, T., Wu, Y., Rajasegaran, V., Wu, J., Lee, M. H., et al. (2008). Inhibition of gastric cancer invasion and metastasis by PLA2G2A, a novel beta-catenin/TCF target gene. Cancer Research, 68(11), 4277–4286.
Garcia-Garcia, H. M., & Serruys, P. W. (2009). Phospholipase A2 inhibitors. Current Opinion in Lipidology, 20(4), 327–332.
Ghosh, S. K., Feng, Z., Fujioka, H., Lux, R., McCormick, T. S., & Weinberg, A. (2018). Conceptual perspectives: Bacterial antimicrobial peptide induction as a novel strategy for symbiosis with the human host. Frontiers in Microbiology, 9, 302.
Gibson, F. C., 3rd, Yumoto, H., Takahashi, Y., Chou, H. H., & Genco, C. A. (2006). Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. Journal of Dental Research, 85(2), 106–121.
Granata, F., Petraroli, A., Boilard, E., Bezzine, S., Bollinger, J., Del Vecchio, L., et al. (2005). Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. Journal of Immunology, 174(1), 464–474.
Greer, A., Zenobia, C., & Darveau, R. P. (2013). Defensins and LL-37: A review of function in the gingival epithelium. Periodontology 2000, 63(1), 67–79.
Griveau, A., Wiel, C., Le Calve, B., Ziegler, D. V., Djebali, S., Warnier, M., et al. (2018). Targeting the phospholipase A2 receptor ameliorates premature aging phenotypes. Aging Cell, 17(6), e12835.
Gronroos, J. O., Laine, V. J., & Nevalainen, T. J. (2002). Bactericidal group IIA phospholipase A2 in serum of patients with bacterial infections. The Journal of Infectious Diseases, 185(12), 1767–1772.
Gronroos, J. O., Salonen, J. H., Viander, M., Nevalainen, T. J., & Laine, V. J. (2005). Roles of group IIA phospholipase A2 and complement in killing of bacteria by acute phase serum. Scandinavian Journal of Immunology, 62(4), 413–419.
Hajishengallis, G., Liang, S., Payne, M. A., Hashim, A., Jotwani, R., Eskan, M. A., et al. (2011). Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host & Microbe, 10(5), 497–506.
Hayashi, C., Gudino, C. V., Gibson, F. C., 3rd, & Genco, C. A. (2010). Review: Pathogen-induced inflammation at sites distant from oral infection: Bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Molecular Oral Microbiology, 25(5), 305–316.
Hernandez, M., Fuentes, L., Fernandez Aviles, F. J., Crespo, M. S., & Nieto, M. L. (2002). Secretory phospholipase A(2) elicits proinflammatory changes and upregulates the surface expression of fas ligand in monocytic cells: Potential relevance for atherogenesis. Circulation Research, 90(1), 38–45.
Hosokawa, I., Hosokawa, Y., Komatsuzawa, H., Goncalves, R. B., Karimbux, N., Napimoga, M. H., et al. (2006). Innate immune peptide LL-37 displays distinct expression pattern from beta-defensins in inflamed gingival tissue. Clinical and Experimental Immunology, 146(2), 218–225.
Ibeas, E., Fuentes, L., Martin, R., Hernandez, M., & Nieto, M. L. (2009). Secreted phospholipase A2 type IIA as a mediator connecting innate and adaptive immunity: New role in atherosclerosis. Cardiovascular Research, 81(1), 54–63.
Ilic, D., Bollinger, J. M., Gelb, M., & Mauro, T. M. (2014). sPLA2 and the epidermal barrier. Biochimica et Biophysica Acta, 1841(3), 416–421.
Ishida, H., Shinohara, H., Nagata, T., Nishikawa, S., & Wakano, Y. (1994). Phospholipase A(2) activity in gingival crevicular fluid from patients with periodontal disease: A possible marker of disease activity. Mediators of Inflammation, 3(1), 17–21.
Ishizaki, J., Ohara, O., Nakamura, E., Tamaki, M., Ono, T., Kanda, A., et al. (1989). cDNA cloning and sequence determination of rat membrane-associated phospholipase A2. Biochemical and Biophysical Research Communications, 162(3), 1030–1036.
Jacques, C., Bereziat, G., Humbert, L., Olivier, J. L., Corvol, M. T., Masliah, J., et al. (1997). Posttranscriptional effect of insulin-like growth factor-I on interleukin-1beta-induced type II-secreted phospholipase A2 gene expression in rabbit articular chondrocytes. The Journal of Clinical Investigation, 99(8), 1864–1872.
Jamal, O. S., Conaghan, P. G., Cunningham, A. M., Brooks, P. M., Munro, V. F., & Scott, K. F. (1998). Increased expression of human type IIa secretory phospholipase A2 antigen in arthritic synovium. Annals of the Rheumatic Diseases, 57(9), 550–558.
Jang, S. E., Jeong, J. J., Kim, J. K., Han, M. J., & Kim, D. H. (2018). Simultaneous amelioratation of colitis and liver injury in mice by Bifidobacterium longum LC67 and Lactobacillus plantarum LC27. Scientific Reports, 8(1), 7500.
Jaross, W., Eckey, R., & Menschikowski, M. (2002). Biological effects of secretory phospholipase A(2) group IIA on lipoproteins and in atherogenesis. European Journal of Clinical Investigation, 32(6), 383–393.
Jauregui, C. E., Wang, Q., Wright, C. J., Takeuchi, H., Uriarte, S. M., & Lamont, R. J. (2013). Suppression of T-cell chemokines by Porphyromonas gingivalis. Infection and Immunity, 81(7), 2288–2295.
Jensen, M. D., Sheng, W., Simonyi, A., Johnson, G. S., Sun, A. Y., & Sun, G. Y. (2009). Involvement of oxidative pathways in cytokine-induced secretory phospholipase A2-IIA in astrocytes. Neurochemistry International, 55(6), 362–368.
Ji, S., Kim, Y., Min, B. M., Han, S. H., & Choi, Y. (2007). Innate immune responses of gingival epithelial cells to nonperiodontopathic and periodontopathic bacteria. Journal of Periodontal Research, 42(6), 503–510.
Jiang, J., Neubauer, B. L., Graff, J. R., Chedid, M., Thomas, J. E., Roehm, N. W., et al. (2002). Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. The American Journal of Pathology, 160(2), 667–671.
Kau, A. L., Ahern, P. P., Griffin, N. W., Goodman, A. L., & Gordon, J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature, 474(7351), 327–336.
Kennedy, B. P., Payette, P., Mudgett, J., Vadas, P., Pruzanski, W., Kwan, M., et al. (1995). A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. The Journal of Biological Chemistry, 270(38), 22378–22385.
Khalaf, H., Nakka, S. S., Sanden, C., Svard, A., Hultenby, K., Scherbak, N., et al. (2016). Antibacterial effects of Lactobacillus and bacteriocin PLNC8 alphabeta on the periodontal pathogen Porphyromonas gingivalis. BMC Microbiology, 16(1), 188.
Koduri, R. S., Gronroos, J. O., Laine, V. J., Le Calvez, C., Lambeau, G., Nevalainen, T. J., et al. (2002). Bactericidal properties of human and murine groups I, II, V, X, and XII secreted phospholipases A(2). Journal of Biological Chemistry, 277(8), 5849–5857.
Koll-Klais, P., Mandar, R., Leibur, E., Marcotte, H., Hammarstrom, L., & Mikelsaar, M. (2005). Oral lactobacilli in chronic periodontitis and periodontal health: Species composition and antimicrobial activity. Oral Microbiology and Immunology, 20(6), 354–361.
Kortekangas, P., Aro, H. T., & Nevalainen, T. J. (1994). Group II phospholipase A2 in synovial fluid and serum in acute arthritis. Scandinavian Journal of Rheumatology, 23(2), 68–72.
Kramer, R. M., Hession, C., Johansen, B., Hayes, G., McGray, P., Chow, E. P., et al. (1989). Structure and properties of a human non-pancreatic phospholipase A2. The Journal of Biological Chemistry, 264(10), 5768–5775.
Kugiyama, K., Ota, Y., Takazoe, K., Moriyama, Y., Kawano, H., Miyao, Y., et al. (1999). Circulating levels of secretory type II phospholipase A(2) predict coronary events in patients with coronary artery disease. Circulation, 100(12), 1280–1284.
Kumar, P. S., Griffen, A. L., Moeschberger, M. L., & Leys, E. J. (2005). Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. Journal of Clinical Microbiology, 43(8), 3944–3955.
Kupert, E., Anderson, M., Liu, Y., Succop, P., Levin, L., Wang, J., et al. (2011). Plasma secretory phospholipase A2-IIa as a potential biomarker for lung cancer in patients with solitary pulmonary nodules. BMC Cancer, 11, 513.
Kuwata, H., Sawada, H., Murakami, M., & Kudo, I. (1999). Role of type IIA secretory phospholipase A2 in arachidonic acid metabolism. Advances in Experimental Medicine and Biology, 469, 183–188.
Lambeau, G., & Lazdunski, M. (1999). Receptors for a growing family of secreted phospholipases A2. Trends in Pharmacological Sciences, 20(4), 162–170.
Lamont, R. J., Koo, H., & Hajishengallis, G. (2018). The oral microbiota: Dynamic communities and host interactions. Nature Reviews. Microbiology, 16(12), 745–759.
Lin, M. K., Farewell, V., Vadas, P., Bookman, A. A., Keystone, E. C., & Pruzanski, W. (1996). Secretory phospholipase A2 as an index of disease activity in rheumatoid arthritis. Prospective double blind study of 212 patients. The Journal of Rheumatology, 23(7), 1162–1166.
Madsen, L. M., Inada, M., & Weiss, J. (1996). Determinants of activation by complement of group II phospholipase A2 acting against Escherichia coli. Infection and Immunity, 64(7), 2425–2430.
Maekawa, T., Krauss, J. L., Abe, T., Jotwani, R., Triantafilou, M., Triantafilou, K., et al. (2014). Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host & Microbe, 15(6), 768–778.
Magrioti, V., & Kokotos, G. (2010). Phospholipase A2 inhibitors as potential therapeutic agents for the treatment of inflammatory diseases. Expert Opinion on Therapeutic Patents, 20(1), 1–18.
Magrioti, V., & Kokotos, G. (2013). Phospholipase A2 inhibitors for the treatment of inflammatory diseases: A patent review (2010—present). Expert Opinion on Therapeutic Patents, 23(3), 333–344.
Mallat, Z., Benessiano, J., Simon, T., Ederhy, S., Sebella-Arguelles, C., Cohen, A., et al. (2007). Circulating secretory phospholipase A2 activity and risk of incident coronary events in healthy men and women: The EPIC-Norfolk study. Arteriosclerosis, Thrombosis, and Vascular Biology, 27(5), 1177–1183.
Marshall, J., Krump, E., Lindsay, T., Downey, G., Ford, D. A., Zhu, P., et al. (2000). Involvement of cytosolic phospholipase A2 and secretory phospholipase A2 in arachidonic acid release from human neutrophils. Journal of Immunology, 164(4), 2084–2091.
Maruvada, P., Leone, V., Kaplan, L. M., & Chang, E. B. (2017). The human microbiome and obesity: Moving beyond associations. Cell Host & Microbe, 22(5), 589–599.
Menschikowski, M., Kasper, M., Lattke, P., Schiering, A., Schiefer, S., Stockinger, H., et al. (1995). Secretory group II phospholipase A2 in human atherosclerotic plaques. Atherosclerosis, 118(2), 173–181.
Menschikowski, M., Hagelgans, A., Schuler, U., Froeschke, S., Rosner, A., & Siegert, G. (2013). Plasma levels of phospholipase A2-IIA in patients with different types of malignancies: Prognosis and association with inflammatory and coagulation biomarkers. Pathology Oncology Research, 19(4), 839–846.
Miller, D. P., Wang, Q., Weinberg, A., & Lamont, R. J. (2018). Transcriptome analysis of Porphyromonas gingivalis and Acinetobacter baumannii in polymicrobial communities. Molecular Oral Microbiology, 33(5), 364–377.
Minami, T., Tojo, H., Shinomura, Y., Tarui, S., & Okamoto, M. (1992). Raised serum activity of phospholipase A2 immunochemically related to group II enzyme in inflammatory bowel disease: Its correlation with disease activity of Crohn’s disease and ulcerative colitis. Gut, 33(7), 914–921.
Minami, T., Tojo, H., Shinomura, Y., Matsuzawa, Y., & Okamoto, M. (1994). Increased group II phospholipase A2 in colonic mucosa of patients with Crohn’s disease and ulcerative colitis. Gut, 35(11), 1593–1598.
Minami, T., Shinomura, Y., Miyagawa, J., Tojo, H., Okamoto, M., & Matsuzawa, Y. (1997). Immunohistochemical localization of group II phospholipase A2 in colonic mucosa of patients with inflammatory bowel disease. The American Journal of Gastroenterology, 92(2), 289–292.
Moses, G. S., Jensen, M. D., Lue, L. F., Walker, D. G., Sun, A. Y., Simonyi, A., et al. (2006). Secretory PLA2-IIA: A new inflammatory factor for Alzheimer’s disease. Journal of Neuroinflammation, 3, 28.
Mounier, C., Faili, A., Vargaftig, B. B., Bon, C., & Hatmi, M. (1993). Secretory phospholipase A2 is not required for arachidonic acid liberation during platelet activation. European Journal of Biochemistry, 216(1), 169–175.
Mounier, C. M., Wendum, D., Greenspan, E., Flejou, J. F., Rosenberg, D. W., & Lambeau, G. (2008). Distinct expression pattern of the full set of secreted phospholipases A2 in human colorectal adenocarcinomas: sPLA2-III as a biomarker candidate. British Journal of Cancer, 98(3), 587–595.
Movert, E., Wu, Y., Lambeau, G., Kahn, F., Touqui, L., & Areschoug, T. (2013). Secreted group IIA phospholipase A2 protects humans against the group B streptococcus: Experimental and clinical evidence. The Journal of Infectious Diseases, 208(12), 2025–2035.
Mueller, H. W., Pritzker, C. R., Kubik, A., & Deykin, D. (1993). Characterization of phospholipase A2 secretion from human platelets. Thrombosis Research, 72(6), 519–530.
Murakami, M., Nakatani, Y., & Kudo, I. (1996). Type II secretory phospholipase A2 associated with cell surfaces via C-terminal heparin-binding lysine residues augments stimulus-initiated delayed prostaglandin generation. The Journal of Biological Chemistry, 271(47), 30041–30051.
Murakami, M., Kuwata, H., Amakasu, Y., Shimbara, S., Nakatani, Y., Atsumi, G., et al. (1997a). Prostaglandin E2 amplifies cytosolic phospholipase A2- and cyclooxygenase-2-dependent delayed prostaglandin E2 generation in mouse osteoblastic cells. Enhancement by secretory phospholipase A2. The Journal of Biological Chemistry, 272(32), 19891–19897.
Murakami, M., Nakatani, Y., Atsumi, G., Inoue, K., & Kudo, I. (1997b). Regulatory functions of phospholipase A2. Critical Reviews in Immunology, 17(3–4), 225–283.
Murakami, M., Taketomi, Y., Miki, Y., Sato, H., Hirabayashi, T., & Yamamoto, K. (2011a). Recent progress in phospholipase A(2) research: From cells to animals to humans. Progress in Lipid Research, 50(2), 152–192.
Murakami, M., Taketomi, Y., Sato, H., & Yamamoto, K. (2011b). Secreted phospholipase A2 revisited. Journal of Biochemistry, 150(3), 233–255.
Murakami, M., Taketomi, Y., Miki, Y., Sato, H., Yamamoto, K., & Lambeau, G. (2014). Emerging roles of secreted phospholipase A2 enzymes: The 3rd edition. Biochimie, 107(Pt A), 105–113.
Murakami, M., Nakatani, Y., Atsumi, G. I., Inoue, K., & Kudo, I. (2017). Regulatory functions of phospholipase A2. Critical Reviews in Immunology, 37(2–6), 121–179.
Nevalainen, T. J., & Haapanen, T. J. (1993). Distribution of pancreatic (group I) and synovial-type (group II) phospholipases A2 in human tissues. Inflammation, 17(4), 453–464.
Nevalainen, T. J., Marki, F., Kortesuo, P. T., Grutter, M. G., Di Marco, S., & Schmitz, A. (1993). Synovial type (group II) phospholipase A2 in cartilage. The Journal of Rheumatology, 20(2), 325–330.
Nevalainen, T. J., Aho, H. J., & Peuravuori, H. (1994). Secretion of group 2 phospholipase A2 by lacrimal glands. Investigative Ophthalmology & Visual Science, 35(2), 417–421.
Nevalainen, T. J., Graham, G. G., & Scott, K. F. (2008). Antibacterial actions of secreted phospholipases A2. Review. Biochimica et Biophysica Acta, 1781(1–2), 1–9.
O’Donoghue, M. L., Mallat, Z., Morrow, D. A., Benessiano, J., Sloan, S., Omland, T., et al. (2011). Prognostic utility of secretory phospholipase A(2) in patients with stable coronary artery disease. Clinical Chemistry, 57(9), 1311–1317.
Okita, Y., Shiono, T., Yahagi, A., Hamada, S., Umemura, M., & Matsuzaki, G. (2016). Interleukin-22-induced antimicrobial phospholipase A2 group IIA mediates protective innate immunity of nonhematopoietic cells against Listeria monocytogenes. Infection and Immunity, 84(2), 573–579.
Oliveira, R. R., Fermiano, D., Feres, M., Figueiredo, L. C., Teles, F. R., Soares, G. M., et al. (2016). Levels of candidate periodontal pathogens in subgingival biofilm. Journal of Dental Research, 95(6), 711–718.
Ouellette, A. J. (2011). Paneth cell alpha-defensins in enteric innate immunity. Cellular and Molecular Life Sciences, 68(13), 2215–2229.
Paganelli, F. L., Leavis, H. L., He, S., van Sorge, N. M., Payre, C., Lambeau, G., et al. (2018). Group IIA-secreted phospholipase A2 in human serum kills commensal but not clinical Enterococcus faecium isolates. Infection and Immunity, 86(8), e00180–e00118.
Perez-Chaparro, P. J., Goncalves, C., Figueiredo, L. C., Faveri, M., Lobao, E., Tamashiro, N., et al. (2014). Newly identified pathogens associated with periodontitis: A systematic review. Journal of Dental Research, 93(9), 846–858.
Pernet, E., Guillemot, L., Burgel, P. R., Martin, C., Lambeau, G., Sermet-Gaudelus, I., et al. (2014). Pseudomonas aeruginosa eradicates Staphylococcus aureus by manipulating the host immunity. Nature Communications, 5, 5105.
Pernet, E., Brunet, J., Guillemot, L., Chignard, M., Touqui, L., & Wu, Y. (2015). Staphylococcus aureus adenosine inhibits sPLA2-IIA-mediated host killing in the airways. Journal of Immunology, 194(11), 5312–5319.
Piris-Gimenez, A., Paya, M., Lambeau, G., Chignard, M., Mock, M., Touqui, L., et al. (2005). In vivo protective role of human group IIa phospholipase A2 against experimental anthrax. Journal of Immunology, 175(10), 6786–6791.
Pruzanski, W., Vadas, P., Stefanski, E., & Urowitz, M. B. (1985). Phospholipase A2 activity in sera and synovial fluids in rheumatoid arthritis and osteoarthritis. Its possible role as a proinflammatory enzyme. The Journal of Rheumatology, 12(2), 211–216.
Pruzanski, W., Keystone, E. C., Sternby, B., Bombardier, C., Snow, K. M., & Vadas, P. (1988). Serum phospholipase A2 correlates with disease activity in rheumatoid arthritis. The Journal of Rheumatology, 15(9), 1351–1355.
Pruzanski, W., Bogoch, E., Stefanski, E., Wloch, M., & Vadas, P. (1990). Synthesis and release of phospholipase A2 by unstimulated human articular chondrocytes. The Journal of Rheumatology, 17(10), 1386–1391.
Pruzanski, W., Greenwald, R. A., Street, I. P., Laliberte, F., Stefanski, E., & Vadas, P. (1992). Inhibition of enzymatic activity of phospholipases A2 by minocycline and doxycycline. Biochemical Pharmacology, 44(6), 1165–1170.
Pruzanski, W., Albin-Cook, K., Laxer, R. M., MacMillan, J., Stefanski, E., Vadas, P., et al. (1994). Phospholipase A2 in juvenile rheumatoid arthritis: Correlation to disease type and activity. The Journal of Rheumatology, 21(10), 1951–1954.
Richards, A. M., Abu Kwaik, Y., & Lamont, R. J. (2015). Code blue: Acinetobacter baumannii, a nosocomial pathogen with a role in the oral cavity. Molecular Oral Microbiology, 30(1), 2–15.
Rizzatti, G., Lopetuso, L. R., Gibiino, G., Binda, C., & Gasbarrini, A. (2017). Proteobacteria: A common factor in human diseases. BioMed Research International, 2017, 9351507.
Sanchez-Mejia, R. O., Newman, J. W., Toh, S., Yu, G. Q., Zhou, Y., Halabisky, B., et al. (2008). Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nature Neuroscience, 11(11), 1311–1318.
Schaeffer, E. L., da Silva, E. R., Novaes Bde, A., Skaf, H. D., & Gattaz, W. F. (2010). Differential roles of phospholipases A2 in neuronal death and neurogenesis: Implications for Alzheimer disease. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 34(8), 1381–1389.
Schewe, M., Franken, P. F., Sacchetti, A., Schmitt, M., Joosten, R., Bottcher, R., et al. (2016). Secreted phospholipases A2 are intestinal stem cell niche factors with distinct roles in homeostasis, inflammation, and cancer. Cell Stem Cell, 19(1), 38–51.
Schmitter, T., Fiebich, B. L., Fischer, J. T., Gajfulin, M., Larsson, N., Rose, T., et al. (2018). Ex vivo anti-inflammatory effects of probiotics for periodontal health. Journal of Oral Microbiology, 10(1), 1502027.
Scott, D. L., White, S. P., Browning, J. L., Rosa, J. J., Gelb, M. H., & Sigler, P. B. (1991). Structures of free and inhibited human secretory phospholipase A2 from inflammatory exudate. Science, 254(5034), 1007–1010.
Seilhamer, J. J., Pruzanski, W., Vadas, P., Plant, S., Miller, J. A., Kloss, J., et al. (1989). Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. The Journal of Biological Chemistry, 264(10), 5335–5338.
Shinohara, H., Komatsubara, T., Tojo, H., Okamoto, M., Nishikawa, S., Nagata, T., et al. (1995). Group II phospholipase A(2) in human gingiva with periodontal disease. Mediators of Inflammation, 4(2), 95–97.
Silliman, C. C., Moore, E. E., Zallen, G., Gonzalez, R., Johnson, J. L., Elzi, D. J., et al. (2002). Presence of the M-type sPLA(2) receptor on neutrophils and its role in elastase release and adhesion. American Journal of Physiology. Cell Physiology, 283(4), C1102–C1113.
Sun, G. Y., Xu, J., Jensen, M. D., & Simonyi, A. (2004). Phospholipase A2 in the central nervous system: Implications for neurodegenerative diseases. Journal of Lipid Research, 45(2), 205–213.
Sun, G. Y., Shelat, P. B., Jensen, M. B., He, Y., Sun, A. Y., & Simonyi, A. (2010). Phospholipases A2 and inflammatory responses in the central nervous system. Neuromolecular Medicine, 12(2), 133–148.
Sun, C. Q., Zhong, C. Y., Sun, W. W., Xiao, H., Zhu, P., Lin, Y. Z., et al. (2016). Elevated type II secretory phospholipase A2 increases the risk of early atherosclerosis in patients with newly diagnosed metabolic syndrome. Scientific Reports, 6, 34929.
Takada, Y., & Fujita, M. (2017). Secreted phospholipase A2 type IIA (sPLA2-IIA) activates integrins in an allosteric manner. Advances in Experimental Medicine and Biology, 925, 103–115.
Takeuchi, H., Hirano, T., Whitmore, S. E., Morisaki, I., Amano, A., & Lamont, R. J. (2013). The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-kappaB RelA/p65. PLoS Pathogens, 9(4), e1003326.
Tan, T. L., & Goh, Y. Y. (2017). The role of group IIA secretory phospholipase A2 (sPLA2-IIA) as a biomarker for the diagnosis of sepsis and bacterial infection in adults—A systematic review. PLoS One, 12(7), e0180554.
Teanpaisan, R., Piwat, S., & Dahlen, G. (2011). Inhibitory effect of oral Lactobacillus against oral pathogens. Letters in Applied Microbiology, 53(4), 452–459.
Thomas, G., Bertrand, F., & Saunier, B. (2000). The differential regulation of group II(A) and group V low molecular weight phospholipases A(2) in cultured rat astrocytes. The Journal of Biological Chemistry, 275(15), 10876–10886.
Thwin, M. M., Satyanarayanajois, S. D., Nagarajarao, L. M., Sato, K., Arjunan, P., Ramapatna, S. L., et al. (2007). Novel peptide inhibitors of human secretory phospholipase A2 with antiinflammatory activity: Solution structure and molecular modeling. Journal of Medicinal Chemistry, 50(24), 5938–5950.
Triggiani, M., Granata, F., Oriente, A., Gentile, M., Petraroli, A., Balestrieri, B., et al. (2002). Secretory phospholipases A2 induce cytokine release from blood and synovial fluid monocytes. European Journal of Immunology, 32(1), 67–76.
Triggiani, M., Granata, F., Balestrieri, B., Petraroli, A., Scalia, G., Del Vecchio, L., et al. (2003). Secretory phospholipases A2 activate selective functions in human eosinophils. Journal of Immunology, 170(6), 3279–3288.
Vadas, P., Stefanski, E., & Pruzanski, W. (1985). Characterization of extracellular phospholipase A2 in rheumatoid synovial fluid. Life Sciences, 36(6), 579–587.
Velsko, I. M., Chukkapalli, S. S., Rivera-Kweh, M. F., Zheng, D., Aukhil, I., Lucas, A. R., et al. (2015). Periodontal pathogens invade gingiva and aortic adventitia and elicit inflammasome activation in alphavbeta6 integrin-deficient mice. Infection and Immunity, 83(12), 4582–4593.
Villanueva, E. B., Little, J. P., Lambeau, G., & Klegeris, A. (2012). Secreted phospholipase A(2) group IIA is a neurotoxin released by stimulated human glial cells. Molecular and Cellular Neurosciences, 49(4), 430–438.
Wang, M., Krauss, J. L., Domon, H., Hosur, K. B., Liang, S., Magotti, P., et al. (2010). Microbial hijacking of complement-toll-like receptor crosstalk. Science Signaling, 3(109), ra11.
Wang, X., Huang, C. J., Yu, G. Z., Wang, J. J., Wang, R., Li, Y. M., et al. (2013). Expression of group IIA phospholipase A2 is an independent predictor of favorable outcome for patients with gastric cancer. Human Pathology, 44(10), 2020–2027.
Wang, J., Ji, H., Wang, S., Liu, H., Zhang, W., Zhang, D., et al. (2018). Probiotic Lactobacillus plantarum promotes intestinal barrier function by strengthening the epithelium and modulating gut microbiota. Frontiers in Microbiology, 9, 1953.
Weiss, J. P. (2015). Molecular determinants of bacterial sensitivity and resistance to mammalian Group IIA phospholipase A2. Biochimica et Biophysica Acta, 1848(11 Pt B), 3072–3077.
Wery, J. P., Schevitz, R. W., Clawson, D. K., Bobbitt, J. L., Dow, E. R., Gamboa, G., et al. (1991). Structure of recombinant human rheumatoid arthritic synovial fluid phospholipase A2 at 2.2 A resolution. Nature, 352(6330), 79–82.
Wu, Y., Zhang, Q., Ren, Y., & Ruan, Z. (2017). Effect of probiotic Lactobacillus on lipid profile: A systematic review and meta-analysis of randomized, controlled trials. PLoS One, 12(6), e0178868.
Yagami, T., Ueda, K., Asakura, K., Hata, S., Kuroda, T., Sakaeda, T., et al. (2002). Human group IIA secretory phospholipase A2 induces neuronal cell death via apoptosis. Molecular Pharmacology, 61(1), 114–126.
Yin, X., Heeney, D., Srisengfa, Y., Golomb, B., Griffey, S., & Marco, M. (2018). Bacteriocin biosynthesis contributes to the anti-inflammatory capacities of probiotic Lactobacillus plantarum. Beneficial Microbes, 9(2), 333–344.
Zvaritch, E., Lambeau, G., & Lazdunski, M. (1996). Endocytic properties of the M-type 180-kDa receptor for secretory phospholipases A2. The Journal of Biological Chemistry, 271(1), 250–257.
Acknowledgements
Findings reported in this review were funded by NIH/NIDCR grant DE024804, NIH/NIGMS P20GM103538 and College of Dentistry University of Kentucky. We thank Drs. Yongzheng Wu and Lhousseine Touqi from Institute Pasteur for kindly sharing the hPLA2-IIA-Tg mice as well as Drs. Jorge Frias (U. of Florida) and Sreenatha Kirakodu (U. of Kentucky) for their support in oral microbiome preliminary analysis. We also are grateful to Dr. Brittany Camenisch and Division of Periodontology for supporting the ongoing preliminary clinical study in salivary PLA2-IIA levels.
Conflict of Interest: The authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper
Cite this paper
Gonzalez, O.A., Euzebio-Alves, V., Alimova, Y., Al-Attar, A., Ebersole, J.L. (2019). A Potential Role of Phospholipase 2 Group IIA (PLA2-IIA) in P. gingivalis-Induced Oral Dysbiosis. In: Belibasakis, G.N., Hajishengallis, G., Bostanci, N., Curtis, M.A. (eds) Oral Mucosal Immunity and Microbiome. Advances in Experimental Medicine and Biology, vol 1197. Springer, Cham. https://doi.org/10.1007/978-3-030-28524-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-28524-1_7
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28523-4
Online ISBN: 978-3-030-28524-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)