
Abstract
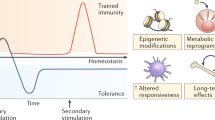
The long-standing dogma that immunological memory is the exclusive prerogative of the adaptive immune system has been challenged by emerging evidence that innate immunity can also maintain memory of past events. Such immunological imprinting takes two forms, trained innate immunity and tolerance. Trained immunity involves metabolic and epigenetic adaptations in innate immune cells and their progenitors in the bone marrow upon exposure to certain microbial and/or inflammatory stimuli so that the “trained” cells would be poised to respond much faster and stronger to a subsequent challenge (e.g., a new infection that is not necessarily the same as the earlier one). Conversely, tolerance leads to attenuated immune responses to secondary stimuli. This review focuses on trained immunity and discusses evidence for its existence from lower organisms to humans, its mechanistic underpinnings, and its translational ramifications. Although trained immunity can be considered as an evolutionarily conserved beneficial response against reinfections, in the setting of modern societies with high prevalence of chronic mucosal and systemic inflammatory diseases, trained immunity could also promote maladaptive immune responses that aggravate pathology. Thus, depending on context, innate immune memory could be therapeutically manipulated using defined agonists to either promote innate immune responses (particularly useful for the treatment of infections or chemotherapy-induced myelosuppression) or suppress excessive inflammation in inflammatory and autoimmune diseases.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others
Introduction
Historically, the adaptive arm of immunity (etymologically derived from immunis; Latin for “exempt”) has been described in contrasting terms with regard to innate immunity. Features, such as receptor repertoire diversity, specificity, and memory, which underlie the concept of vaccination, were considered to be the exclusive prerogative of adaptive immunity (Roitt et al. 1989). For instance, whereas innate immune receptors are encoded in the germline resulting in limited diversity, adaptive immune receptors are generated randomly by gene recombination and somatic hypermutation, thereby leading to an unlimited receptor repertoire. Adaptive immunity is exquisitely specific being able to discriminate even between individual microbes of the same species (strain-dependent antigenic variability). Moreover, immunological memory, the ability of the adaptive immune system to “remember” past encounters with pathogens, enables the immune system to swiftly and specifically recognize a subsequent challenge with the same pathogen and initiate a faster and stronger immune response that is often protective for the host. Lack of immunological memory in innate immunity meant that the innate immune response is invariable and thus not enhanced upon repeated encounters with a pathogen; the perceived significance of the innate response lied in its ability to act fast and “buy” time until the adaptive immunity becomes activated.
However, the defining differences between the adaptive and innate forms of immunity are not as unambiguously defined as previously thought. For instance, innate immunity is endowed with adequate specificity to distinguish between different classes of microorganisms as well as to discriminate between healthy and damaged/stressed host cells (Miyake 2007; Fearon 1997; Medzhitov and Janeway 2000). Moreover, recent advances show that, despite lacking the fine antigen specificity, clonality, and longevity of adaptive lymphocytes, innate immune cells can retain “memory” of earlier challenges (e.g., infection or vaccination) and thereby display increased responsiveness upon rechallenge with the same or even unrelated pathogen (Netea et al. 2016; Goodridge et al. 2016).
This enhanced state of immune activation that is based on innate immune memory is known as “trained innate immunity” or more briefly “trained immunity” (Netea and van der Meer 2017). As implied above, innate immune memory lacks specificity to the initial infection (or other inflammatory stimuli); indeed, exposure to a certain pathogen can confer enhanced protection against a future challenge with a different pathogen even from a different kingdom (e.g., the original challenge could be a fungal pathogen and the secondary one could be a bacterial or viral pathogen). Innate immune memory is not necessarily expressed as trained immunity but also as “innate immune tolerance.” In this regard, prior exposure of myeloid cells such as monocytes/macrophages to relatively high doses of bacterial lipopolysaccharide (LPS) induces a state of reduced ability to elicit proinflammatory cytokines to subsequent restimulation with LPS (homologous tolerance) or other proinflammatory stimuli (heterologous tolerance) (Jacinto et al. 2002). In this review, we will focus on trained immunity and will discuss its evolutionary origins, its functional consequences at mucosal barrier sites, as well as the mechanisms that underlie the induction of a trained immune phenotype and how this can be exploited therapeutically.
Trained Immunity, an Evolutionary Conserved Function for Broad-Based Protection Against Infections
Trained innate immunity is seemingly an evolutionarily conserved way of remembering past challenges as it is not present only in vertebrates but also in non-vertebrate animals and in plants (Penkov et al. 2019). Although plants lack adaptive immunity, they use sophisticated mechanisms to anticipate and effectively fight infections. Systemic acquired resistance (SAR) of plants is a mechanism of inducible defense that provides long-lasting protection (weeks to months) against a broad range of microbial pathogens (Durrant and Dong 2004). SAR spreads from the site of the infection to the entire tissues of the plant through production of mobile signals, accumulation of salicylic acid (which, in plants, functions as a defense factor), increased expression of pattern-recognition receptors, and secretion of antimicrobial proteins, thereby establishing protection against secondary infections (Fu and Dong 2013; Reimer-Michalski and Conrath 2016). Thus, SAR endows the plant with memory to the initial infection by priming even remote tissues for enhanced defense to subsequent infections. In a manner analogous to immunization in vertebrates, plants inoculated with attenuated microorganisms acquire long-term protection against subsequent infections but in a relatively nonspecific manner (cross-protection against bacterial, fungal, and viral pathogens) (Durrant and Dong 2004; Fu and Dong 2013). Importantly, SAR involves adaptations in the chromatin state and thus can be transmitted to progeny through epigenetic mechanisms (Fu and Dong 2013; Reimer-Michalski and Conrath 2016), suggesting that the enhanced innate resistance of plants to reinfection involves memory mechanisms similar to those of trained immunity in jawed vertebrates (Netea and van der Meer 2017).
Adaptive immunity is thought to have arisen in the first jawed vertebrates (gnathostomes; Greek for jawed mouth), i.e., the placoderm fish (Flajnik and Kasahara 2010). Therefore, the investigation of the presence of innate immune memory can be better studied in invertebrates, such as insect models, that lack adaptive immune memory. A number of studies have demonstrated that the immune system of insects can be primed for long-lasting immune responses that can often afford protection against future infections. A few examples will be given here. Priming of Drosophila melanogaster with a sublethal dose of Streptococcus pneumoniae protected the insect for life against an otherwise-lethal reinfection with the same pathogen (Pham et al. 2007). This protective effect required the Toll pathway and phagocytes (Pham et al. 2007). When initially challenged with LPS, larvae of the mealworm beetle, Tenebrio molitor, elicited a long-lasting antimicrobial response that protected the larvae against a subsequent exposure to an entomopathogenic fungus (Moret and Siva-Jothy 2003). Anopheles gambiae mosquitoes infected with Plasmodium falciparum were shown to be primed for a long-lasting enhanced antibacterial response that could efficiently reduce Plasmodium survival upon reinfection (Rodrigues et al. 2010). The primed state was induced when plasmodium ookinetes breached the gut barrier and bacteria gained access to injured epithelial cells, leading to induction of quantitative changes and qualitative differentiation of hemocytes (insect equivalent of leukocytes) that lasted for the lifespan of the Anopheles mosquito (Rodrigues et al. 2010).
Besides infection itself, other cues that may confer protection against secondary microbial challenge is apoptotic corpse engulfment. Indeed, a recent study has shown that innate immune memory can be induced in Drosophila embryonic macrophages (hemocytes) following phagocytosis of apoptotic cells (Weavers et al. 2016). This priming is triggered by calcium-induced JNK signaling, which in turn up-regulates the damage receptor Draper and the cell is thus poised to respond more rapidly to subsequent infection or injury. Unlike primed or “trained” hemocytes, naive hemocytes that have not engulfed apoptotic cells are not efficient in detecting sterile tissue damage in vivo and migrating accordingly. Moreover, untrained hemocytes are also inefficient to recognize infection with Escherichia coli and respond by phagocytosing the pathogen (Weavers et al. 2016).
Emerging evidence suggests that the free-living nematode (roundworm) Caenorhabditis elegans also can build long-lasting and even transgenerational innate immune memory (Penkov et al. 2019). This memory allows the worm to effectively avoid or resist secondary pathogen encounters. For instance, brief exposure to virulent or avirulent strains of enteropathogenic E. coli (EPEC) primes C. elegans to survive an otherwise-lethal re-encounter with EPEC (Anyanful et al. 2009). C. elegans worms exposed to Pseudomonas aeruginosa were “trained” to avoid this pathogenic bacterium; however, in the absence of relevant training, the worms failed to develop aversive learning behavior against pathogenic P. aeruginosa (Lee and Mylonakis 2017). Intriguingly, moreover, second-generation C. elegans feeding on pathogenic bacteria can avoid bacterial infection by entering diapause, a hibernation-like state that arrests development (Palominos et al. 2017).
These findings have called for a reevaluation of the earlier assumption that invertebrate innate immunity lacks memory qualities and the current burden of proof is consistent with the presence of innate immune memory in lower organisms. Together, the studies discussed in this section indicate that innate immune memory is a conserved function that has persisted in evolution.
Trained Immunity in Mammals: Protection Against Mucosal and Systemic Infection
Candida albicans is a commensal fungus at mucosal surfaces of healthy individuals but behaves as an opportunistic pathogen in immunocompromised patients, where it frequently causes superficial infections in the oral cavity and the vagina (Williams et al. 2013). In immunocompromised individuals, moreover, C. albicans (and other Candida species) can also cause systemic infections, which constitute the fourth leading cause of nosocomial bloodstream infection in modern hospitals (50,000 new cases of systemic candidiasis per year in the U.S.) with high morbidity and mortality (Lionakis 2014). C. albicans is also frequently detected in high numbers in the biofilm that develops on the tooth surfaces (“dental plaque”) of toddlers with early childhood caries, a severe form of tooth decay where this fungus contributes to its pathogenesis (Hajishengallis et al. 2017).
C. albicans is a prototype organism used to consolidate the concept of trained immunity. In this regard, T- and B-cell–deficient mice that were previously “trained” with low-dose C. albicans or purified β-glucan (major fungal cell wall constituent) displayed increased protection, as compared to untrained controls, against reinfection with C. albicans through enhanced production of cytokines and phagocytic killing (Quintin et al. 2012). Thus, trained innate immune cells, such as monocytes/macrophages, acquire enhanced responsiveness after secondary stimulation with the fungus, which can thereby be eliminated more readily by innate immune means, irrespective of adaptive immunity. As alluded to above, innate immune memory is not specific, as adaptive immune memory is, since exposure to one pathogen can afford enhanced protection against a second, unrelated pathogen. Indeed, systemic infection with attenuated C. albicans protected mice not only against secondary challenge with virulent C. albicans but also against Staphylococcus aureus in a macrophage-dependent manner (Bistoni et al. 1986). Innate immune memory is not an exclusive feature of the monocytic lineage but also resides within other innate immune cell lineages, such as natural killer cells, which rapidly degranulate and produce cytokines upon reactivation with cytomegalovirus (Sun et al. 2009).
Epidemiological studies on the effectiveness of vaccines showed that specific immunizations had off-target beneficial effects, i.e., were associated with protection against diseases that were not intended to fight (Netea and van der Meer 2017; Goodridge et al. 2016). For instance, a randomized trial of bacillus Calmette–Guérin (BCG) vaccination suggested that BCG has nonspecific protective affects against neonatal sepsis and infections of the respiratory mucosa (Aaby et al. 2011). Moreover, the off-target effects of BCG include protection against malignant tumors (Falk et al. 1973; Martinez-Pineiro and Muntanola 1977; Simmons and Rios 1971). This “bonus” protection can be explained by vaccine-induced trained immunity. In this regard, besides protein immunogens, vaccines contain adjuvants either intrinsically (whole microbial cells contain inherent adjuvant substances) or extrinsically, such as alum, the most commonly used adjuvant in vaccine formulations. Adjuvants can directly or indirectly (through the release of danger-associated molecular pattern-containing molecules) activate pattern-recognition receptors on innate immune cells (Sanders and Feavers 2011; Mizel and Bates 2010; Marrack et al. 2009). Thus, the off-target effects of vaccines may be mediated, at least in part, by adjuvant-dependent induction of trained innate immunity (Arts et al. 2018; Kleinnijenhuis et al. 2012). Indeed, BCG vaccination of healthy adults resulted in elevated ex vivo induction of monocyte-derived cytokines (TNF and IL-1β) in response to stimulation with unrelated bacterial or fungal pathogens (Kleinnijenhuis et al. 2012). This enhanced responsiveness of monocytes was dependent on the NOD2 receptor and persisted for at least 3 months after vaccination (Kleinnijenhuis et al. 2012). Moreover, a randomized placebo-controlled human challenge study showed that BCG vaccination confers protection against experimental infection with an attenuated vaccine strain of yellow fever virus, as evidenced by reduced viremia that correlated with elevated IL-1β (Arts et al. 2018).
The findings discussed in the previous section that apoptotic cell phagocytosis by hemocytes in Drosophila primes these cells for enhanced responses to subsequent infection or injury (Weavers et al. 2016) seems to deviate from the functional consequences of apoptotic cell phagocytosis (efferocytosis) in mammalian macrophages. In this regard, efferocytosis activates liver X receptor (LXR) signaling and reprograms the macrophage toward a pro-resolving phenotype (Kourtzelis et al. 2017, 2019; Ravichandran and Lorenz 2007). However, the effects of LXR on inflammation in mammals appear to be context dependent. For instance, LXR signaling inhibits LPS-induced TLR4 activation in macrophages when LPS and LXR agonists are added together (simulating conditions when efferocytosis occurs at the declining stage of infection and initiation of resolution). However, LXR signaling enhances LPS-induced TLR4 activation if the macrophages are pretreated with LXR agonist 48 h prior to LPS challenge (Fontaine et al. 2007; Rigamonti et al. 2008), a priming scenario analogous to the experimental design in the Drosophila study (Weavers et al. 2016). In other words, LXR signaling primes or trains the macrophage for heightened antimicrobial responses upon subsequent encounters with pathogens; however, if the infection has already occurred, LXR signaling acts to mitigate associated inflammation and promote transition to resolution.
Mechanisms of Trained Immunity
Recent studies have shown that the acquisition of a trained immunity state by innate immune cells involves metabolic, epigenetic, and transcriptional reprogramming, which is sustained for months (Bekkering et al. 2018; Arts et al. 2016a; Saeed et al. 2014; Cheng et al. 2014; Norata et al. 2015; Penkov et al. 2019). Before discussing this evidence in greater detail, it would be instructive to briefly discuss the importance of epigenetic rewiring in the context of trained immunity.
Many loci encoding inflammatory genes are in a repressed state in myeloid cells. Upon activation with a proinflammatory stimulus, important changes occur in the genomic elements (enhancers and promoters) that regulate gene expression. Specifically, there is increased histone acetylation and chromatin opening and enhanced recruitment of certain transcription factors (e.g., NF-κB, AP-1, and STAT proteins) and RNA polymerase II (Glass and Natoli 2016; Saccani et al. 2001). In other words, chromatin becomes more accessible to the transcriptional machinery. Importantly, such enhanced accessibility persists over time as the acquisition of specific chromatin marks (histone acetylation or methylation) is well maintained (only partially lost after stimulus elimination). For instance, a latent enhancer, which may be unmarked epigenetically in unstimulated cells, may acquire histone modifications typical of active enhancers (e.g., H3K4me1; monomethylation of histone H3 at K4) (Ostuni et al. 2013). This epigenetic adaptation underlies trained immunity as it could be maintained well after the removal of the inductive stimulus and promotes more efficient induction of genes and protection in response to future challenges (Netea et al. 2016). In this regard, BCG vaccination-induced training that protects against experimental infection with attenuated yellow fever virus was correlated with BCG-induced genome-wide epigenetic adaptations in monocytes and increased production of IL-1β (Arts et al. 2018).
Similar to trained innate immunity, innate immune tolerance, the other side of the coin in terms of innate immune memory, is also regulated by chromatin modifications, which, however, silence genes encoding proinflammatory mediators (Foster et al. 2007). Epigenetic modifications also underlie the fact that myeloid cells, such as macrophages, exhibit tissue-specific functions that are largely instructed by micro-environmental stimuli (Stout et al. 2009; Matzinger 2007). Indeed, the local micro-environment shapes the enhancer landscape of macrophages beyond what could be attributed to developmental origin, thus contributing to their plasticity in a tissue-specific context (Lavin et al. 2014).
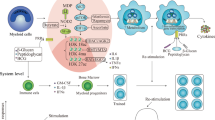
Training induced by C. albicans or β-glucan in monocytes/macrophages is mediated through binding to the C-type lectin receptor dectin-1 that activates a noncanonical Raf-1–dependent pathway, which in turn leads to genome-wide alterations in epigenetic marks, such as H3K4me1, H3K4me3, and H3K27Ac (Netea et al. 2016). The mechanisms by which epigenetic changes are induced and how these in turn regulate trained immunity involve complex cross-talk interactions between metabolism, the epigenome, and the immune response (Fig. 1). Resting monocytes and macrophages primarily use oxidative phosphorylation (OXPHOS). However, during β-glucan–induced trained immunity, macrophages undergo a rapid shift toward aerobic glycolysis (also known as the Warburg effect), a process that can swiftly accommodate the increased metabolic needs associated with cell activation and expansion, specifically in terms of energy and building blocks required for cell membranes (cholesterol and phospholipids) and DNA (purines) (Cheng et al. 2014; Norata et al. 2015). This metabolic switch from OXPHOS to aerobic glycolysis during β-glucan–induced training involves activation of the Akt–mTOR–HIF1α signaling pathway (Cheng et al. 2014).


Immuno-epigeno-metabolic cross talk in trained innate immunity. Outline of major mechanisms by which innate immune signaling-triggered metabolic pathways induce epigenetic changes and how these prime for heightened secondary immune responses or trained immunity, which also cross talks with metabolism. See text for details
The β-glucan-induced metabolic alterations are functionally connected to the epigenetic rewiring. In this regard, Krebs cycle metabolites have the ability to regulate the function of key epigenetic enzymes. Whereas citrate is exported to the cytoplasm for synthesis of cell membrane building blocks (cholesterol and phospholipids), fumarate accumulates in response to β-glucan stimulation of macrophages (owing to glutamine replenishment of the Krebs cycle and glutaminolysis) (Arts et al. 2016b). Fumarate in turn inhibits KDM5 histone (H3K4) demethylases, thus bringing about epigenetic reprogramming by increasing histone methylation (Arts et al. 2016b). Another example of a link between trained immune signaling, cellular metabolism, and epigenetic reprogramming involves the acetyl coenzyme A (acetyl-CoA), which serves as an acetyl donor to the Krebs cycle for oxidation and energy production. Acetyl-CoA also provides the acetyl group for histone acetylation, a histone mark that promotes gene transcription (Gut and Verdin 2013). Moreover, lactate, the end product of anaerobic glycolysis, can inhibit the activity of histone deacetylases (HDAC) and, hence, promote gene accessibility and transcription (Latham et al. 2012). The cholesterol synthesis pathway also contributes to trained immunity as the latter can be inhibited in myeloid cells by statins. However, the essential effector of the trained phenotype is not cholesterol itself but the intermediate metabolite mevalonate, which via the insulin-like growth factor-1 receptor and downstream mTOR signaling leads to histone modifications in inflammatory pathways (Bekkering et al. 2018). In summary, metabolites exert a significant impact on the epigenetic landscape by serving as signaling molecules or as substrates and/or cofactors for chromatin-modifying enzymes. The reverse may also be true since the modified chromatin structure in turn can regulate the expression of metabolic genes (Hino et al. 2013). Overall, innate immune cells exposed to appropriate stimuli modify their metabolism and remodel their chromatin in a manner that can elicit an “educated” (much faster and stronger) response to subsequent encounter with the same or different stimuli. However, this concept also generated a paradox.
The long-term effects of trained immunity on circulating monocytes have been puzzling, as these cells have a relatively short lifespan (Yona et al. 2013) in the circulation. This paradox was resolved by two recent reports that appeared in the same issue of Cell (Kaufmann et al. 2018; Mitroulis et al. 2018). These studies have shown for the first time that the processes induced by trained immunity involve long-term adaptations (metabolic, epigenetic, and transcriptional) in hematopoietic progenitor cells that give rise to lines of differentiated innate immune cells in the bone marrow (BM) (Kaufmann et al. 2018; Mitroulis et al. 2018). For instance, β-glucan reprograms several pathways associated with cell proliferation, glycolysis, and cholesterol biosynthesis and induces the expansion of hematopoietic stem and progenitor cells (HSPCs) in the BM (Mitroulis et al. 2018) (Fig. 2). Appropriately enough, β-glucan increases the frequencies of myeloid-biased CD41+ long-term hematopoietic stem cells (LT-HSCs) and of multipotent progenitors (MPP) that are biased toward myelopoiesis, namely the MPP3 subset. These effects do not involve direct β-glucan contact with hematopoietic progenitors but are instead mediated by β-glucan–induced innate immune mediators in the BM microenvironment, namely IL-1β and granulocyte-macrophage colony-stimulating factor (GM-CSF). This β-glucan–induced trained phenotype persists for at least several weeks, can be adoptively transferred to untrained recipient mice, and protects against (a) DNA damage in LT-HSCs induced by a secondary challenge with LPS and (b) chemotherapy-induced myelosuppression (Mitroulis et al. 2018) (Fig. 2). Similarly, BCG can also induce expansion of HSCs and promote myelopoiesis leading to the generation of trained monocytes/macrophages that protect against Mycobacterium tuberculosis infection (Kaufmann et al. 2018). This BCG-induced training is dependent on IFN-γ signaling. Thus, trained innate immunity is initiated by modulation of the progenitors of myeloid cells in the bone marrow in a manner that enhances the replenishment of innate immune cell populations upon stress associated with infectious, inflammatory, or chemotherapeutic challenges and leads to protective immunity.


Trained innate immunity acts at the level of myeloid progenitors. (a) Trained immunity as induced by β-glucan mediates a myelopoiesis bias in the bone marrow. CMP common myeloid progenitor, GMP granulocyte-monocyte progenitor, HSC hematopoietic stem cell, MPP multipotent progenitor. (b) The β-glucan–dependent myelopoiesis bias is mediated by an early activated IL-1β pathway associated with enhanced glycolysis and lipid metabolism changes in hematopoietic stem and progenitor cells (HSPC) resulting in enhanced cholesterol-rich membrane microdomains and thereby enhanced GM-CSF/CD131 signaling, which drives myelopoiesis. (c) Induction of trained immunity in HSPCs protects them from LPS-induced DNA damage and enables beneficial responses of the bone marrow to a secondary LPS challenge as well as to chemotherapy-induced myelosuppression
Maladaptive Trained Immunity
Although trained innate immunity can protect against subsequent systemic or mucosal infections or chemotherapy-induced myeloablation (Mitroulis et al. 2018; Kaufmann et al. 2018; Quintin et al. 2012; Arts et al. 2018; Kleinnijenhuis et al. 2012), there may be settings in which trained immunity can do more harm than good. As reasoned above, trained immunity may have evolved as a form of innate immune memory to provide broad cross-protection against reinfections. However, in modern societies, trained immunity may be a potential contributory factor to age-related chronic inflammatory diseases. Such maladaptive trained immunity could be inappropriately induced by microbial or endogenous stimuli (damage-associated molecular patterns; DAMPs) and lead to exaggerated immune responses that drive or exacerbate inflammatory or autoimmune diseases. In other words, if the immune system is epigenetically trained (due to earlier infection, vaccination, or injury) to elicit a heightened immune response, this enhanced responsiveness may aggravate existing inflammatory/autoimmune diseases or prove deleterious in hosts that are genetically susceptible to inflammatory/autoimmune diseases. These notions are consistent with clinical observations or experimental studies in animal models.
As mentioned above, mevalonate has been implicated as an inducer of the trained immunity phenotype. Intriguingly, monocytes of patients with hyper immunoglobulin D syndrome (HIDS), who accumulate mevalonate due to mevalonate kinase deficiency, constitutively exhibit a trained phenotype both epigenetically and immunologically (Bekkering et al. 2018). This finding may explain, at least in part, the susceptibility of these patients to autoinflammatory disorders. Indeed, HIDS patients suffer from recurrent febrile episodes often associated with lymphadenopathy, abdominal and joint pain, hepatomegaly, splenomegaly, and skin rash among other inflammatory symptoms (van der Meer et al. 1984).
Low concentrations of oxidized low-density lipoprotein (oxLDL) were shown to induce a long-lasting proinflammatory phenotype in monocytes associated with increased histone methylation (H3K4me3) in the promoter regions of several pro-atherogenic genes; the proinflammatory IL6, MCP1, IL8, TNF, MMP2, and MMP9; and the scavenger receptors CD36 and scavenger receptor-A (Bekkering et al. 2014). Upon rechallenge with TLR agonists, the oxLDL-trained monocytes showed increased production of pro-atherogenic cytokines, whereas upon rechallenge with oxLDL, the trained monocytes express higher levels of scavenger receptors and show increased foam cell formation (Bekkering et al. 2014). This study suggests that the proinflammatory nature of high-fat western diets (WD) could induce maladaptive trained immunity. Using LDL receptor (Ldlr)-deficient mice on a WD as a model of metabolic syndrome-induced autoinflammation, a recent study showed that WD induces long-lasting inflammasome-mediated trained immunity in myeloid cells (Christ et al. 2018). Mice that were doubly deficient in both Ldlr and the Nlrp3 inflammasome did not exhibit a trained phenotype and developed significantly reduced systemic inflammation and atherosclerotic plaque size in response to WD feeding (Christ et al. 2018).
In a possibly analogous manner involving nonimmune cells and high glucose as an endogenous proinflammatory stimulus with potential for epigenetic alterations (Yun et al. 2011), transient hyperglycemia was associated with persistent up-regulation of NFκB-p65 gene expression in human vascular endothelial cells attributed to chromatin modifications (enhanced H3K4 and reduced H3K9 methylation) (Brasacchio et al. 2009; El-Osta et al. 2008). This so-called “hyperglycemic memory” (Brasacchio et al. 2009) may represent a form of maladaptive trained immunity that could contribute to the pathogenesis of diabetes. This study also suggests that the trained immunity phenotype may not be restricted to professional immune cells and additional examples will be given in later sections.
“Microglial priming” in neurodegenerative disorders refers to the property of microglia (the resident macrophages of the central nervous system) to elicit an exaggerated inflammatory response to a secondary inflammatory stimulus, even if that would be a normal subthreshold challenge (Norden et al. 2015; Perry, Holmes 2014). Interestingly, the initial priming stimulus and the secondary challenge may be separated temporally. For instance, an inflammatory challenge in utero may affect microglial reactivity later in life (Knuesel et al. 2014). The microglia can develop a primed state with aging or after stress, traumatic brain injury, or neurodegenerative disease, and the functional consequences of subsequent exaggerated inflammatory responses include development of cognitive dysfunction, compromised synaptic plasticity, and accelerated neurodegeneration (Norden et al. 2015; Perry and Holmes 2014).
A recent review of relevant literature suggests that the priming of microglia, which are unusually long-lived tissue-resident cells (Reu et al. 2017), is mediated by epigenetic mechanisms similar to those of trained immunity involving other types of myeloid cells (histone modifications such as H3K4me1 deposition at latent enhancer sites) (Haley et al. 2017). In other words, exposure to inflammatory stimuli might lead to accumulation of epigenetic alterations in the microglia that confer a trained/hyperactive phenotype that could contribute to destructive immune responses associated with neurodegenerative conditions. As alluded to above, aging is a risk factor contributing to microglia priming. Indeed, activation of the peripheral innate immune system by intraperitoneal injection of LPS leads to increased microglia-dependent neuroinflammation in old as compared with young adult mice (Godbout et al. 2005). This form of maladaptive trained immunity might contribute to the behavioral deficits that frequently follow systemic infections in the elderly.
In a recent study, peripherally administered inflammatory stimuli induced either immune training or tolerance in the brain depending on the frequency of stimulus application. Specifically, a single intraperitoneal injection of LPS-induced tolerance, whereas four consecutive daily intraperitoneal injections of LPS-induced training; these outcomes were associated with differential epigenetic reprogramming of the microglia (Wendeln et al. 2018). This imprinted memory persisted for at least 6 months and, in the form of immune training, exacerbated cerebral inflammation and β-amyloidosis in a mouse model of Alzheimer’s pathology. In contrast, pathology in the same model was alleviated when tolerance was induced (Wendeln et al. 2018). Thus immune memory in the brain, induced by peripheral stimuli, represents an important modifier of neuroinflammatory pathology. Although it has long been known that the frequency or dosing of LPS may differentially induce priming or tolerance, this cannot entirely explain the manner by which LPS regulates host immune responses to secondary challenges. Indeed, it was earlier established that tolerogenic LPS-induced chromatin modifications silence genes encoding proinflammatory mediators but not genes encoding antimicrobial effectors, which are instead primed by the same LPS stimulus (Foster and Medzhitov 2009).
Trained Immunity, β-glucan, and Mucosal Dysbiotic Diseases
As mentioned above, C. albicans (and other Candida species) are commensal yeasts in mucosal surfaces of healthy individuals but may cause systemic infections in immunocompromised patients (Lionakis 2014). At least in principle, at mucosal sites colonized by C. albicans or other fungal species, locally produced β-glucans could induce regional trained immunity in mature myeloid cells and the continued presence of β-glucans could ensure a sustained trained innate immune status. Periodontitis and colitis are mucosal inflammatory diseases with a dysbiotic component (Saleh and Trinchieri 2011; Stecher et al. 2013; Lamont and Hajishengallis 2015; Hajishengallis 2015). Thus although tissue damage is primarily inflicted by an exaggerated or dysregulated host immune response, suboptimal host immunity is also an unfavorable situation as it may facilitate transition from a symbiotic to a dysbiotic, hence, disease-provoking microbiota. It is therefore not easy to predict the effects of trained immunity, e.g., as induced by β-glucan, on mucosal dysbiosis-driven inflammatory disease.
β-Glucans produced by fungi at mucosal sites, such as the lung, can enter the circulation where they can persist and reach remote sites, such as the joints where β-glucans were shown to activate synovial cells (Obayashi et al. 1995; Yasuoka et al. 1996; Yoshitomi et al. 2005). Moreover, β-glucan from C. albicans could substitute for Freund’s complete adjuvant to cause collagen-induced arthritis in DBA/1 mice (Hida et al. 2005). Of course, rheumatoid arthritis primarily represents “sterile” inflammation (Chen and Nunez 2010) and if disseminated β-glucans indeed induce trained immunity at remote sites, the trained phenotype would likely exacerbate existing rheumatoid arthritis (or perhaps trigger arthritis in susceptible individuals). However, the above-discussed findings (Obayashi et al. 1995; Yasuoka et al. 1996; Yoshitomi et al. 2005) suggest a hypothetical, if not plausible, scenario by which β-glucans released locally at mucosal barrier sites may reach the bone marrow at concentrations sufficient enough to train myeloid progenitors. Alternatively, C. albicans-derived β-glucan may act on local macrophages and induce IL-1β (and/or other cytokines), which in turn may reach the bone marrow to mediate trained immunity.
Although fungal pathogenesis has primarily been associated with immunocompromised states, even in systemically healthy individuals, C. albicans appears to contribute to the pathogenesis of periodontal disease. In this regard, although the main reservoir of C. albicans is the buccal mucosa, tongue, and palate, these fungal organisms can co-aggregate with bacteria in subgingival biofilms and also adhere to and infect gingival epithelial cells (Sardi et al. 2010; Sztukowska et al. 2018; Dongari-Bagtzoglou et al. 2005). C. albicans was shown to colonize the periodontal pockets of approximately 15–20% chronic periodontitis patients, and, in fact, hyphae were found within the underlying gingival connective tissue (Reynaud et al. 2001; Urzua et al. 2008; Jarvensivu et al. 2004). Importantly, the presence of C. albicans in the periodontal pockets was associated with the severity of chronic periodontitis (Canabarro et al. 2013). However, the underlying mechanisms for this association, whether causal or not, are uncertain. Equally uncertain is whether the presence of C. albicans in periodontal pockets is linked to induction of trained innate immunity and how this affects periodontal disease.
Gut colonization by C. albicans was shown to increase the incidence of allergic diarrhea in sensitized BALB/c mice, promote limb joint inflammation in collagen-induced arthritis in DBA/1J mice, and exacerbate contact hypersensitivity in NC/Nga mice (Sonoyama et al. 2011). This model involves persistent C. albicans gut colonization by a single intragastric inoculation in immunocompetent adult mice in the absence of antibiotics or immunosuppressants, and thus preventing systemic disseminated infections by C. albicans; this model therefore mimics immunocompetent humans with chronic, latent intestinal colonization by C. albicans (Yamaguchi et al. 2005; Sonoyama et al. 2011). The aggravation of allergic diarrhea by C. albicans gut colonization was attributed to increased infiltration of the colon with eosinophils and mast cells (Sonoyama et al. 2011). Moreover, the intestinal C. albicans-associated aggravation of hapten-induced contact hypersensitivity and collagen-induced arthritis mice was associated with increased myeloperoxidase activity, a marker of neutrophil recruitment in the inflamed tissues. Whether these enhanced host immune responses derive, at least in part, through C. albicans-induced trained immunity is not known but is a plausible possibility. Even more challenging is to explain the mechanisms connecting C. albicans gut colonization with exacerbation of inflammatory pathology in tissues distant from the gut.
In models of colitis, β-glucan was shown to have variable effects. Mice orally pretreated with β-glucans for 14 days before initiation of dextran sulphate sodium (DSS)-induced colitis exhibited worsened colitis with increased colonic levels of inflammatory cytokines and chemokines, compared to vehicle-treated mouse controls (Heinsbroek et al. 2015). The authors speculated that this might be due to training of the immune system by β-glucan treatment (Heinsbroek et al. 2015). In contrast, another study found that β-glucan given to C57BL/6J mice as part of their diet for 26 days attenuated DSS-induced colitis, which was initiated 5 days before the end of the experimental period (Zhou et al. 2014). In another investigation, oral administration of β-glucan also mitigated DSS-induced colitis associated with improved structural integrity of tight junctions and intestinal permeability and with decreased levels of myeloperoxidase, eosinophil peroxidase, and N-acetyl-β-d-glucosaminidase (Han et al. 2017). Interestingly, group 2 innate lymphoid cells (ILC2s) were shown to protect against DSS-induced colitis through production of amphiregulin, a molecule that promotes restoration of tissue integrity after acute or chronic inflammation-induced damage (Monticelli et al. 2015; Zaiss et al. 2015). Whether β-glucan can also prime the immune system for enhanced homeostatic/tissue repair responses in the gut is not known. However, at least in some settings, training has been shown to promote tissue repair (below).
Trained Tissue Repair
Tissue repair during inflammation resolution is a complex process that depends on the regenerative capacity of the tissue and the participation at the site of injury of inflammatory cells, e.g., macrophages, which contribute to wound debridement and produce growth factors, chemokines, and other metabolites that stimulate fibroblasts and other cells involved in wound healing (White and Mantovani 2013; Eming et al. 2017). Epithelial cells are not only involved in repairing the epithelial barrier but also contribute to the general tissue repair process by regulating the proliferation of fibroblasts and their production of collagen required for wound healing (Zhang et al. 2017).
A recent study has shown that the healing capacity of skin epithelial cells is enhanced by previous exposure to inflammatory stimuli (chemical [imiquimod], mechanical, or microbial [C. albicans]) (Naik et al. 2017). These findings strongly indicate that pre-conditioning due to previous inflammatory events is not an exclusive property of professional immune cells. Specifically, after acute inflammation, epithelial stem cells can accelerate barrier restoration following future tissue damage. Importantly, resident skin macrophages and T cells are not required for the enhanced wound repair following secondary inflammation in this model. This epithelial cell “memory” was associated with alterations at the chromatin level as determined by Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq). Following the initial inflammatory stimulus, the trained epithelial stem cells maintained chromatin accessibility at key stress response genes, which were thus transcribed faster upon a secondary challenge (Naik et al. 2017). To identify downstream effectors of the epithelial stem cell memory that confer tissue repair advantage, the authors performed pathway analysis of the rapid-response transcripts and identified “inflammasome signaling” among the top featured terms. Gain- and loss-of-function experiments with AIM2, CASP1, IL1β, and IL1R1 confirmed the involvement of the inflammasome pathway in mediating enhanced wound repair to inflammation-trained epithelial stem cells (Naik et al. 2017). In this context, gut epithelial cells can also detect tissue damage by expressing AIM2 and activating the inflammasome (Hu et al. 2016). Therefore, it would be interesting to know if epithelial stem cells in tissues other than the skin share a similar AIM2 inflammasome-associated memory of earlier inflammatory assaults. It should be noted, however, that such memory may not always be beneficial as, at least in principle, it could amplify tissue damage upon a secondary assault (e.g., in the setting of inflammatory or autoimmune disorders) rather than promoting tissue repair.
Summary and Perspective
Trained immunity has recently emerged as a new concept that endows innate immunity with immunological memory of past inflammatory events so that the host is poised to respond rapidly and robustly to subsequent challenges. As innate immune memory is not as specific as adaptive immune memory, it can provide cross-protection against different infections. However, by the same mechanism, maladaptive trained immunity can prime the host to become more susceptible to a range of distinct inflammatory or autoimmune disorders. Cellular metabolic pathways and chromatin modifications involved in the induction of trained immunity could be therapeutically modulated to better treat genetic or acquired immunodeficiencies (through restoration or enhancement of immune function), counteract the adverse effects of chemotherapy-induced myelosuppression, or to alleviate autoimmune and inflammatory disorders. Among the primary targets of therapeutic trained immunity are likely infants and the elderly, who are particularly vulnerable to infectious diseases although the elderly are also susceptible to inflammatory and degenerative conditions. Here, it would be important to understand which chronic inflammatory disorders are strongly linked to a trained immune phenotype and how this could be therapeutically manipulated to better treat the disease.
Although the demonstration that trained innate immunity can act at the level of the myeloid progenitor cells has resolved the paradox of long-lasting innate immune memory despite the short life of mature myeloid cells in circulation, there are still many unanswered questions. For instance, although epigenetic adaptations are strongly correlated with trained immunity, formal and specific cause-and-effect connection is currently lacking. Moreover, we are far from an in-depth understanding of the molecular mechanisms of trained immunity and we do not understand the precise mechanisms that allow trained cells to maintain their chromatin in an open state at select loci for a long time (e.g., months). During DNA replication and cell division, chromatin is disassembled and reassembled, which begs the question as to how chromatin landmarks associated with trained immunity are retained or even transmitted in the context of transgenerational innate immune memory. Of course, a low rate of cell division would facilitate maintenance of chromatin memory as compared to high-rate cell division. Another great challenge ahead is to develop ways for targeted pharmacologic manipulation of the metabolic pathways or epigenetic landscapes involved in regulation of trained immunity in order to promote optimal gene expression patterns.
References
Aaby, P., Roth, A., Ravn, H., Napirna, B. M., Rodrigues, A., Lisse, I. M., et al. (2011). Randomized trial of BCG vaccination at birth to low-birth-weight children: Beneficial nonspecific effects in the neonatal period? The Journal of Infectious Diseases, 204(2), 245–252. https://doi.org/10.1093/infdis/jir240.
Anyanful, A., Easley, K. A., Benian, G. M., & Kalman, D. (2009). Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host & Microbe, 5(5), 450–462. https://doi.org/10.1016/j.chom.2009.04.012.
Arts, R. J., Joosten, L. A., & Netea, M. G. (2016a). Immunometabolic circuits in trained immunity. Seminars in Immunology, 28(5), 425–430. https://doi.org/10.1016/j.smim.2016.09.002.
Arts, R. J., Novakovic, B., Ter Horst, R., Carvalho, A., Bekkering, S., Lachmandas, E., et al. (2016b). Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metabolism, 24(6), 807–819. https://doi.org/10.1016/j.cmet.2016.10.008.
Arts, R. J. W., Moorlag, S., Novakovic, B., Li, Y., Wang, S. Y., Oosting, M., et al. (2018). BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe, 23(1), 89–100 e5. https://doi.org/10.1016/j.chom.2017.12.010.
Bekkering, S., Quintin, J., Joosten, L. A., van der Meer, J. W., Netea, M. G., & Riksen, N. P. (2014). Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arteriosclerosis, Thrombosis, and Vascular Biology, 34(8), 1731–1738. https://doi.org/10.1161/ATVBAHA.114.303887.
Bekkering, S., Arts, R. J. W., Novakovic, B., Kourtzelis, I., van der Heijden, C., Li, Y., et al. (2018). Metabolic induction of trained immunity through the mevalonate pathway. Cell, 172(1–2), 135–46 e9. https://doi.org/10.1016/j.cell.2017.11.025.
Bistoni, F., Vecchiarelli, A., Cenci, E., Puccetti, P., Marconi, P., & Cassone, A. (1986). Evidence for macrophage-mediated protection against lethal Candida albicans infection. Infection and Immunity, 51(2), 668–674.
Brasacchio, D., Okabe, J., Tikellis, C., Balcerczyk, A., George, P., Baker, E. K., et al. (2009). Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes, 58(5), 1229–1236. https://doi.org/10.2337/db08-1666.
Canabarro, A., Valle, C., Farias, M. R., Santos, F. B., Lazera, M., & Wanke, B. (2013). Association of subgingival colonization of Candida albicans and other yeasts with severity of chronic periodontitis. Journal of Periodontal Research, 48(4), 428–432. https://doi.org/10.1111/jre.12022.
Chen, G. Y., & Nunez, G. (2010). Sterile inflammation: Sensing and reacting to damage. Nature Reviews. Immunology, 10(12), 826–837. https://doi.org/10.1038/nri2873.
Cheng, S. C., Quintin, J., Cramer, R. A., Shepardson, K. M., Saeed, S., Kumar, V., et al. (2014). mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science, 345(6204), 1250684. https://doi.org/10.1126/science.1250684.
Christ, A., Gunther, P., Lauterbach, M. A. R., Duewell, P., Biswas, D., Pelka, K., et al. (2018). Western diet triggers NLRP3-dependent innate immune reprogramming. Cell, 172(1–2), 162–75 e14. https://doi.org/10.1016/j.cell.2017.12.013.
Dongari-Bagtzoglou, A., Villar, C. C., & Kashleva, H. (2005). Candida albicans-infected oral epithelial cells augment the anti-fungal activity of human neutrophils in vitro. Medical Mycology, 43(6), 545–549.
Durrant, W. E., & Dong, X. (2004). Systemic acquired resistance. Annual Review of Phytopathology, 42, 185–209. https://doi.org/10.1146/annurev.phyto.42.040803.140421.
El-Osta, A., Brasacchio, D., Yao, D., Pocai, A., Jones, P. L., Roeder, R. G., et al. (2008). Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. The Journal of Experimental Medicine, 205(10), 2409–2417. https://doi.org/10.1084/jem.20081188.
Eming, S. A., Wynn, T. A., & Martin, P. (2017). Inflammation and metabolism in tissue repair and regeneration. Science, 356(6342), 1026–1030. https://doi.org/10.1126/science.aam7928.
Falk, R. E., Mann, P., & Langer, B. (1973). Cell-mediated immunity to human tumors. Abrogation by serum factors and nonspecific effects of oral BCG therapy. Archives of Surgery, 107(2), 261–265.
Fearon, D. T. (1997). Seeking wisdom in innate immunity. Nature, 388(6640), 323–324.
Flajnik, M. F., & Kasahara, M. (2010). Origin and evolution of the adaptive immune system: Genetic events and selective pressures. Nature Reviews. Genetics, 11(1), 47–59. https://doi.org/10.1038/nrg2703.
Fontaine, C., Rigamonti, E., Nohara, A., Gervois, P., Teissier, E., Fruchart, J.-C., et al. (2007). Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circulation Research, 101(1), 40–49. https://doi.org/10.1161/circresaha.106.135814.
Foster, S. L., & Medzhitov, R. (2009). Gene-specific control of the TLR-induced inflammatory response. Clinical Immunology, 130(1), 7–15.
Foster, S. L., Hargreaves, D. C., & Medzhitov, R. (2007). Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature, 447(7147), 972–978. https://doi.org/10.1038/nature05836.
Fu, Z. Q., & Dong, X. (2013). Systemic acquired resistance: Turning local infection into global defense. Annual Review of Plant Biology, 64, 839–863. https://doi.org/10.1146/annurev-arplant-042811-105606.
Glass, C. K., & Natoli, G. (2016). Molecular control of activation and priming in macrophages. Nature Immunology, 17(1), 26–33. https://doi.org/10.1038/ni.3306.
Godbout, J. P., Chen, J., Abraham, J., Richwine, A. F., Berg, B. M., Kelley, K. W., et al. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. The FASEB Journal, 19(10), 1329–1331. https://doi.org/10.1096/fj.05-3776fje.
Goodridge, H. S., Ahmed, S. S., Curtis, N., Kollmann, T. R., Levy, O., Netea, M. G., et al. (2016). Harnessing the beneficial heterologous effects of vaccination. Nature Reviews. Immunology, 16(6), 392–400. https://doi.org/10.1038/nri.2016.43.
Gut, P., & Verdin, E. (2013). The nexus of chromatin regulation and intermediary metabolism. Nature, 502(7472), 489–498. https://doi.org/10.1038/nature12752.
Hajishengallis, G. (2015). Periodontitis: From microbial immune subversion to systemic inflammation. Nature Reviews. Immunology, 15(1), 30–44. https://doi.org/10.1038/nri3785.
Hajishengallis, E., Parsaei, Y., Klein, M. I., & Koo, H. (2017). Advances in the microbial etiology and pathogenesis of early childhood caries. Molecular Oral Microbiology, 32(1), 24–34. https://doi.org/10.1111/omi.12152.
Haley, M. J., Brough, D., Quintin, J., & Allan, S. M. (2017). Microglial priming as trained immunity in the brain. Neuroscience, 405, 47. https://doi.org/10.1016/j.neuroscience.2017.12.039.
Han, F., Fan, H., Yao, M., Yang, S., & Han, J. (2017). Oral administration of yeast β-glucan ameliorates inflammation and intestinal barrier in dextran sodium sulfate-induced acute colitis. Journal of Functional Foods, 35, 115–126.
Heinsbroek, S. E., Williams, D. L., Welting, O., Meijer, S. L., Gordon, S., & de Jonge, W. J. (2015). Orally delivered beta-glucans aggravate dextran sulfate sodium (DSS)-induced intestinal inflammation. Nutrition Research, 35(12), 1106–1112. https://doi.org/10.1016/j.nutres.2015.09.017.
Hida, S., Miura, N. N., Adachi, Y., & Ohno, N. (2005). Effect of Candida albicans cell wall glucan as adjuvant for induction of autoimmune arthritis in mice. Journal of Autoimmunity, 25(2), 93–101. https://doi.org/10.1016/j.jaut.2005.06.002.
Hino, S., Nagaoka, K., & Nakao, M. (2013). Metabolism-epigenome crosstalk in physiology and diseases. Journal of Human Genetics, 58(7), 410–415. https://doi.org/10.1038/jhg.2013.57.
Hu, B., Jin, C., Li, H. B., Tong, J., Ouyang, X., Cetinbas, N. M., et al. (2016). The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science, 354(6313), 765–768. https://doi.org/10.1126/science.aaf7532.
Jacinto, R., Hartung, T., McCall, C., & Li, L. (2002). Lipopolysaccharide- and lipoteichoic acid-induced tolerance and cross-tolerance: Distinct alterations in IL-1 receptor-associated kinase. Journal of Immunology, 168(12), 6136–6141.
Jarvensivu, A., Hietanen, J., Rautemaa, R., Sorsa, T., & Richardson, M. (2004). Candida yeasts in chronic periodontitis tissues and subgingival microbial biofilms in vivo. Oral Diseases, 10(2), 106–112.
Kaufmann, E., Sanz, J., Dunn, J. L., Khan, N., Mendonca, L. E., Pacis, A., et al. (2018). BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell, 172(1–2), 176–90 e19. https://doi.org/10.1016/j.cell.2017.12.031.
Kleinnijenhuis, J., Quintin, J., Preijers, F., Joosten, L. A., Ifrim, D. C., Saeed, S., et al. (2012). Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proceedings of the National Academy of Sciences of the United States of America, 109(43), 17537–17542. https://doi.org/10.1073/pnas.1202870109.
Knuesel, I., Chicha, L., Britschgi, M., Schobel, S. A., Bodmer, M., Hellings, J. A., et al. (2014). Maternal immune activation and abnormal brain development across CNS disorders. Nature Reviews. Neurology, 10(11), 643–660. https://doi.org/10.1038/nrneurol.2014.187.
Kourtzelis, I., Mitroulis, I., von Renesse, J., Hajishengallis, G., & Chavakis, T. (2017). From leukocyte recruitment to resolution of inflammation: The cardinal role of integrins. Journal of Leukocyte Biology, 102, 677–683. https://doi.org/10.1189/jlb.3MR0117-024R.
Kourtzelis, I., Li, X., Mitroulis, I., Grosser, D., Kajikawa, T., Wang, B., et al. (2019). DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nature Immunology, 20(1), 40–49. https://doi.org/10.1038/s41590-018-0249-1.
Lamont, R. J., & Hajishengallis, G. (2015). Polymicrobial synergy and dysbiosis in inflammatory disease. Trends in Molecular Medicine, 21(3), 172–183. https://doi.org/10.1016/j.molmed.2014.11.004.
Latham, T., Mackay, L., Sproul, D., Karim, M., Culley, J., Harrison, D. J., et al. (2012). Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Research, 40(11), 4794–4803. https://doi.org/10.1093/nar/gks066.
Lavin, Y., Winter, D., Blecher-Gonen, R., David, E., Keren-Shaul, H., Merad, M., et al. (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell, 159(6), 1312–1326. https://doi.org/10.1016/j.cell.2014.11.018.
Lee, K., & Mylonakis, E. (2017). An intestine-derived neuropeptide controls avoidance behavior in Caenorhabditis elegans. Cell Reports, 20(10), 2501–2512. https://doi.org/10.1016/j.celrep.2017.08.053.
Lionakis, M. S. (2014). New insights into innate immune control of systemic candidiasis. Medical Mycology, 52(6), 555–564. https://doi.org/10.1093/mmy/myu029.
Marrack, P., McKee, A. S., & Munks, M. W. (2009). Towards an understanding of the adjuvant action of aluminium. Nature Reviews. Immunology, 9(4), 287–293. https://doi.org/10.1038/nri2510.
Martinez-Pineiro, J. A., & Muntanola, P. (1977). Nonspecific immunotherapy with BCG vaccine in bladder tumors: A preliminary report. European Urology, 3(1), 11–22.
Matzinger, P. (2007). Friendly and dangerous signals: Is the tissue in control? Nature Immunology, 8(1), 11–13.
Medzhitov, R., & Janeway, C., Jr. (2000). Innate immunity. The New England Journal of Medicine, 343(5), 338–344.
Mitroulis, I., Ruppova, K., Wang, B., Chen, L. S., Grzybek, M., Grinenko, T., et al. (2018). Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell, 172(1–2), 147–61 e12. https://doi.org/10.1016/j.cell.2017.11.034.
Miyake, K. (2007). Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Seminars in Immunology, 19(1), 3–10. https://doi.org/10.1016/j.smim.2006.12.002.
Mizel, S. B., & Bates, J. T. (2010). Flagellin as an adjuvant: Cellular mechanisms and potential. Journal of Immunology, 185(10), 5677–5682. https://doi.org/10.4049/jimmunol.1002156.
Monticelli, L. A., Osborne, L. C., Noti, M., Tran, S. V., Zaiss, D. M., & Artis, D. (2015). IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proceedings of the National Academy of Sciences of the United States of America, 112(34), 10762–10767. https://doi.org/10.1073/pnas.1509070112.
Moret, Y., & Siva-Jothy, M. T. (2003). Adaptive innate immunity? Responsive-mode prophylaxis in the mealworm beetle, Tenebrio molitor. Proceedings of the Biological Sciences, 270(1532), 2475–2480. https://doi.org/10.1098/rspb.2003.2511.
Naik, S., Larsen, S. B., Gomez, N. C., Alaverdyan, K., Sendoel, A., Yuan, S., et al. (2017). Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature, 550(7677), 475–480. https://doi.org/10.1038/nature24271.
Netea, M. G., & van der Meer, J. W. (2017). Trained immunity: An ancient way of remembering. Cell Host & Microbe, 21(3), 297–300. https://doi.org/10.1016/j.chom.2017.02.003.
Netea, M. G., Joosten, L. A., Latz, E., Mills, K. H., Natoli, G., Stunnenberg, H. G., et al. (2016). Trained immunity: A program of innate immune memory in health and disease. Science, 352(6284), aaf1098. https://doi.org/10.1126/science.aaf1098.
Norata, G. D., Caligiuri, G., Chavakis, T., Matarese, G., Netea, M. G., Nicoletti, A., et al. (2015). The cellular and molecular basis of translational immunometabolism. Immunity, 43(3), 421–434. https://doi.org/10.1016/j.immuni.2015.08.023.
Norden, D. M., Muccigrosso, M. M., & Godbout, J. P. (2015). Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology, 96(Pt A), 29–41. https://doi.org/10.1016/j.neuropharm.2014.10.028.
Obayashi, T., Yoshida, M., Mori, T., Goto, H., Yasuoka, A., Iwasaki, H., et al. (1995). Plasma (1-->3)-beta-D-glucan measurement in diagnosis of invasive deep mycosis and fungal febrile episodes. Lancet, 345(8941), 17–20.
Ostuni, R., Piccolo, V., Barozzi, I., Polletti, S., Termanini, A., Bonifacio, S., et al. (2013). Latent enhancers activated by stimulation in differentiated cells. Cell, 152(1–2), 157–171. https://doi.org/10.1016/j.cell.2012.12.018.
Palominos, M. F., Verdugo, L., Gabaldon, C., Pollak, B., Ortiz-Severin, J., Varas, M. A., et al. (2017). Transgenerational diapause as an avoidance strategy against bacterial pathogens in Caenorhabditis elegans. MBio, 8(5). https://doi.org/10.1128/mBio.01234-17.
Penkov, S., Mitroulis, I., Hajishengallis, G., & Chavakis, T. (2019). Immunometabolic crosstalk: An ancestral principle of trained immunity? Trends in Immunology, 40(1), 1–11. https://doi.org/10.1016/j.it.2018.11.002.
Perry, V. H., & Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nature Reviews. Neurology, 10(4), 217–224. https://doi.org/10.1038/nrneurol.2014.38.
Pham, L. N., Dionne, M. S., Shirasu-Hiza, M., & Schneider, D. S. (2007). A specific primed immune response in Drosophila is dependent on phagocytes. PLoS Pathogens, 3(3), e26. https://doi.org/10.1371/journal.ppat.0030026.
Quintin, J., Saeed, S., Martens, J. H. A., Giamarellos-Bourboulis, E. J., Ifrim, D. C., Logie, C., et al. (2012). Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host & Microbe, 12(2), 223–232. https://doi.org/10.1016/j.chom.2012.06.006.
Ravichandran, K. S., & Lorenz, U. (2007). Engulfment of apoptotic cells: Signals for a good meal. Nature Reviews. Immunology, 7(12), 964–974. https://doi.org/10.1038/nri2214.
Reimer-Michalski, E. M., & Conrath, U. (2016). Innate immune memory in plants. Seminars in Immunology, 28(4), 319–327. https://doi.org/10.1016/j.smim.2016.05.006.
Reu, P., Khosravi, A., Bernard, S., Mold, J. E., Salehpour, M., Alkass, K., et al. (2017). The lifespan and turnover of microglia in the human brain. Cell Reports, 20(4), 779–784. https://doi.org/10.1016/j.celrep.2017.07.004.
Reynaud, A. H., Nygaard-Ostby, B., Boygard, G. K., Eribe, E. R., Olsen, I., & Gjermo, P. (2001). Yeasts in periodontal pockets. Journal of Clinical Periodontology, 28(9), 860–864.
Rigamonti, E., Chinetti-Gbaguidi, G., & Staels, B. (2008). Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(6), 1050–1059. https://doi.org/10.1161/ATVBAHA.107.158998.
Rodrigues, J., Brayner, F. A., Alves, L. C., Dixit, R., & Barillas-Mury, C. (2010). Hemocyte differentiation mediates innate immune memory in Anopheles gambiae mosquitoes. Science, 329(5997), 1353–1355. https://doi.org/10.1126/science.1190689.
Roitt, I., Brostoff, J., & Male, D. (1989). Immunology (2nd ed.). London: Gower Medical Publishing.
Saccani, S., Pantano, S., & Natoli, G. (2001). Two waves of nuclear factor kappaB recruitment to target promoters. The Journal of Experimental Medicine, 193(12), 1351–1359.
Saeed, S., Quintin, J., Kerstens, H. H., Rao, N. A., Aghajanirefah, A., Matarese, F., et al. (2014). Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science, 345(6204), 1251086. https://doi.org/10.1126/science.1251086.
Saleh, M., & Trinchieri, G. (2011). Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nature Reviews. Immunology, 11(1), 9–20. . nri2891 [pii]. https://doi.org/10.1038/nri2891.
Sanders, H., & Feavers, I. M. (2011). Adjuvant properties of meningococcal outer membrane vesicles and the use of adjuvants in Neisseria meningitidis protein vaccines. Expert Review of Vaccines, 10(3), 323–334. https://doi.org/10.1586/erv.11.10.
Sardi, J. C., Duque, C., Mariano, F. S., Peixoto, I. T., Hofling, J. F., & Goncalves, R. B. (2010). Candida spp. in periodontal disease: A brief review. Journal of Oral Science, 52(2), 177–185.
Simmons, R. L., & Rios, A. (1971). Immunotherapy of cancer: Immunospecific rejection of tumors in recipients of neuraminidase-treated tumor cells plus BCG. Science, 174(4009), 591–593.
Sonoyama, K., Miki, A., Sugita, R., Goto, H., Nakata, M., & Yamaguchi, N. (2011). Gut colonization by Candida albicans aggravates inflammation in the gut and extra-gut tissues in mice. Medical Mycology, 49(3), 237–247. https://doi.org/10.3109/13693786.2010.511284.
Stecher, B., Maier, L., & Hardt, W. D. (2013). ‘Blooming’ in the gut: How dysbiosis might contribute to pathogen evolution. Nature Reviews. Microbiology, 11(4), 277–284. https://doi.org/10.1038/nrmicro2989.
Stout, R. D., Watkins, S. K., & Suttles, J. (2009). Functional plasticity of macrophages: In situ reprogramming of tumor-associated macrophages. Journal of Leukocyte Biology, 86(5), 1105–1109. https://doi.org/10.1189/jlb.0209073.
Sun, J. C., Beilke, J. N., & Lanier, L. L. (2009). Adaptive immune features of natural killer cells. Nature, 457(7229), 557–561. https://doi.org/10.1038/nature07665.
Sztukowska, M. N., Dutton, L. C., Delaney, C., Ramsdale, M., Ramage, G., Jenkinson, H. F., et al. (2018). Community development between Porphyromonas gingivalis and Candida albicans mediated by InlJ and Als3. MBio, 9(2), e00202. https://doi.org/10.1128/mBio.00202-18.
Urzua, B., Hermosilla, G., Gamonal, J., Morales-Bozo, I., Canals, M., Barahona, S., et al. (2008). Yeast diversity in the oral microbiota of subjects with periodontitis: Candida albicans and Candida dubliniensis colonize the periodontal pockets. Medical Mycology, 46(8), 783–793. https://doi.org/10.1080/13693780802060899.
van der Meer, J. W., Vossen, J. M., Radl, J., van Nieuwkoop, J. A., Meyer, C. J., Lobatto, S., et al. (1984). Hyperimmunoglobulinaemia D and periodic fever: A new syndrome. Lancet, 1(8386), 1087–1090.
Weavers, H., Evans, I. R., Martin, P., & Wood, W. (2016). Corpse engulfment generates a molecular memory that primes the macrophage inflammatory response. Cell, 165(7), 1658–1671. https://doi.org/10.1016/j.cell.2016.04.049.
Wendeln, A. C., Degenhardt, K., Kaurani, L., Gertig, M., Ulas, T., Jain, G., et al. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature, 556, 332. https://doi.org/10.1038/s41586-018-0023-4.
White, E. S., & Mantovani, A. R. (2013). Inflammation, wound repair, and fibrosis: Reassessing the spectrum of tissue injury and resolution. The Journal of Pathology, 229(2), 141–144. https://doi.org/10.1002/path.4126.
Williams, D. W., Jordan, R. P., Wei, X. Q., Alves, C. T., Wise, M. P., Wilson, M. J., et al. (2013). Interactions of Candida albicans with host epithelial surfaces. Journal of Oral Microbiology, 5. https://doi.org/10.3402/jom.v5i0.22434.
Yamaguchi, N., Sonoyama, K., Kikuchi, H., Nagura, T., Aritsuka, T., & Kawabata, J. (2005). Gastric colonization of Candida albicans differs in mice fed commercial and purified diets. The Journal of Nutrition, 135(1), 109–115. https://doi.org/10.1093/jn/135.1.109.
Yasuoka, A., Tachikawa, N., Shimada, K., Kimura, S., & Oka, S. (1996). (1-->3) beta-D-glucan as a quantitative serological marker for Pneumocystis carinii pneumonia. Clinical and Diagnostic Laboratory Immunology, 3(2), 197–199.
Yona, S., Kim, K. W., Wolf, Y., Mildner, A., Varol, D., Breker, M., et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity, 38(1), 79–91. https://doi.org/10.1016/j.immuni.2012.12.001.
Yoshitomi, H., Sakaguchi, N., Kobayashi, K., Brown, G. D., Tagami, T., Sakihama, T., et al. (2005). A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. The Journal of Experimental Medicine, 201(6), 949–960. https://doi.org/10.1084/jem.20041758.
Yun, J. M., Jialal, I., & Devaraj, S. (2011). Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. The Journal of Nutritional Biochemistry, 22(5), 450–458. https://doi.org/10.1016/j.jnutbio.2010.03.014.
Zaiss, D. M. W., Gause, W. C., Osborne, L. C., & Artis, D. (2015). Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity, 42(2), 216–226. https://doi.org/10.1016/j.immuni.2015.01.020.
Zhang, C., Lim, J., Liu, J., Ponugoti, B., Alsadun, S., Tian, C., et al. (2017). FOXO1 expression in keratinocytes promotes connective tissue healing. Scientific Reports, 7, 42834. https://doi.org/10.1038/srep42834.
Zhou, M., Wang, Z., Chen, J., Zhan, Y., Wang, T., Xia, L., et al. (2014). Supplementation of the diet with Salecan attenuates the symptoms of colitis induced by dextran sulphate sodium in mice. The British Journal of Nutrition, 111(10), 1822–1829. https://doi.org/10.1017/S000711451300442X.
Acknowledgement
The authors’ research is supported by U.S. Public Health Service grants from the National Institutes of Health (AI068730, DE024153, DE024716, DE015254 to G. H.; DE026152 to G. H. and T. C.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper
Cite this paper
Hajishengallis, G., Li, X., Mitroulis, I., Chavakis, T. (2019). Trained Innate Immunity and Its Implications for Mucosal Immunity and Inflammation. In: Belibasakis, G.N., Hajishengallis, G., Bostanci, N., Curtis, M.A. (eds) Oral Mucosal Immunity and Microbiome. Advances in Experimental Medicine and Biology, vol 1197. Springer, Cham. https://doi.org/10.1007/978-3-030-28524-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-28524-1_2
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28523-4
Online ISBN: 978-3-030-28524-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)