
Abstract
Patients of all ages are diagnosed with mastocytosis, and in children, the manifestations primarily involve the skin. Although children are mainly diagnosed with cutaneous mastocytosis, they can experience generalized symptoms involving other organ systems such as the GI tract, neurologic, and musculoskeletal systems. Most patients with systemic disease have a somatically acquired activating mutation in the KIT oncogene. Pediatric-onset disease differs from adult-onset disease by variant, disease manifestations, therapy, and prognosis to include resolution. This chapter encapsulates the disease spectrum of mastocytosis in children, with an overview of the clinical features and the approach to diagnosis, evaluation, management, and prognosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mastocytosis in the skin (MIS)
- Systemic mastocytosis
- Allele-specific qPCR
- Tryptase
- Organomegaly
- Anaphylaxis
- Vaccines
- KIT D816V
Spectrum of Disease in Pediatrics
All categories of mastocytosis share clinical features caused by the overproduction of mast cell (MC)-dependent mediators. The skin is often the first visible sign of the disease, but the gastrointestinal tract, lymph nodes, liver, spleen, BM, and skeletal system all express manifestations of MC burden. In children, the skin is the most common organ involved and may be the only manifestation of the disease. Interestingly, patients with mastocytosis do not suffer from recurrent bacterial, fungal, or viral infections, even though MCs release mediators such as histamine that inhibit immune responses in vitro. In 2007, a proposed addition to the nomenclature was introduced to clarify the pre-diagnostic state before a more definite diagnosis is made prior to a BM biopsy known as mastocytosis of the skin (MIS) [1]. The typical exanthema is considered the major criterion, and one of the two minor criteria based on abnormal MCs in clusters (>15) or >20 cells scattered per high power field (HPF) and/or detection of a KIT mutation at codon 816 is needed for the diagnosis. Thus, the term “cutaneous mastocytosis” (CM) is reserved for cutaneous disease only and subdivided into maculopapular CM (MPCM) or urticaria pigmentosa (UP), diffuse cutaneous mastocytosis (DCM) , and mastocytoma.
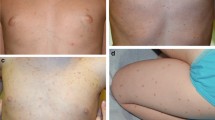
The most common skin manifestation in children (Fig. 6.1 and 6.2) is MPCM or UP, but the size and number are more variable in children with CM and more uniform in adults [2]. UP lesions are seen in almost all children with indolent systemic mastocytosis (ISM). A few studies have documented a regression of the skin lesions with a decrease in serum tryptase and severity of the disease in adults [2, 3]. The typical appearance of UP are yellow-tan to reddish-brown macules or slightly raised papules scattered mainly on the trunk and legs with generally less involvement of the sun-exposed areas. The palms, soles, face, and scalp are generally spared, especially in adults. Dermatologic symptoms include pruritus, flushing, and blistering, with the latter symptom almost uniquely seen in children. Darier’s sign is the local whealing of a lesion induced by friction and, when present, can be diagnostic but may not be consistently elicited.

Maculopapular cutaneous mastocytosis (MPCM), monomorphic-characteristic small red-brown, mainly uniform-sized lesions

Maculopapular cutaneous mastocytosis (MPCM), polymorphic-larger, varied-sized lesions that are asymmetric with hyperpigmentation
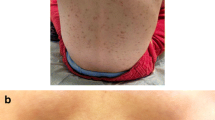
Diffuse cutaneous mastocytosis and mastocytoma have an onset almost exclusively in childhood. Although DCM may persist into adulthood, mastocytomas usually regress spontaneously. DCM is characterized by thickened skin and may exhibit a peau d’orange appearance with a reddish-brown discoloration without characteristic lesions (Fig. 6.3) but may also have scattered nodules similar in appearance to mastocytomas. The skin may be dermatographic, and the formation of hemorrhagic blisters is common. Solitary mastocytomas are red-brown or yellow-orange nodules, which, when traumatized, may cause systemic symptoms such as flushing and hypotension (Fig. 6.4). The onset is generally before the age of 6 months, and it is unusual to develop subsequent skin lesions more than 2 months after the presentation of the initial lesion [4]. UP and DCM are associated with pruritus of varied intensity, which may be exacerbated by changes in climatic temperature; skin friction; and ingestion of hot beverages, spicy foods, alcohol, or certain drugs. Bullous formation is a characteristic limited to pediatric-onset cutaneous disease and usually is associated with lesional skin. Bullae may erupt spontaneously or in association with infection and immunization. This feature is mostly limited to the first few years of life and may need to be distinguished from other bullous diseases of childhood.

Diffuse cutaneous mastocytosis (DCM) – typical skin manifestations with erythematous thickened skin and “peau d’orange” texture. Dermographism is characteristic

Mastocytoma – the lesion usually presents as a reddish brown or dark pink nodule and is typically seen as a single lesion. The consensus group for mastocytosis notes that a maximum of three lesions can be present with this diagnosis [20]
Systemic mastocytosis (SM) can occur in both adults and children and is diagnosed on the basis of BM histopathology outlined by the WHO consensus panel [5]. ISM, the most common variant of systemic disease, is diagnosed when criteria for mastocytosis are met, and there is no evidence of an associated clonal hematologic disorder or severe liver disease, hypersplenism, or significant lymphadenopathy. Isolated BM mastocytosis is a sub-variant of ISM with a low BM burden of MCs, a lower tryptase value, and the absence of skin lesions. The skin lesions in systemic disease may present with the typical monomorphic pattern seen in adults is also seen in pediatrics (Fig. 6.5) or the polymorphic pattern seen in children with cutaneous disease (Fig. 6.6). Several clinical conditions should heighten suspicion of this variant such as idiopathic anaphylaxis, venom anaphylaxis, unexplained osteoporosis, or chronic diarrhea [1001,1002,1003,9]. This variant of systemic disease has not been reported in children. There are case reports in the literature of children with mast cell leukemia [10, 11] and other associated hematologic diseases [8,9,10,15]; however, these are rare associations. The majority of children with ISM have a good prognosis as shown in a study of long-term follow-up of children with mastocytosis [3, 11, 16].

MPCM , monomorphic pattern in a child with indolent systemic mastocytosis. Skin lesions are red-brown in color with mainly small uniform-sized lesions

MPCM , polymorphic pattern in a child with indolent systemic mastocytosis. Skin lesions vary in size with a red-brown vascular appearance
Disease Onset
Mastocytosis in children usually presents as a cutaneous rash together with variable type and degree of symptoms secondary to the effect of a wide variety of proinflammatory mediators released from activated MCs [17, 18]; more rarely, anaphylaxis in the absence of skin lesions may be the clinical presentation of the disease in a small subset of pediatric patients. The vast majority of children who show skin lesions of mastocytosis are assumed to have CM [1] even though BM studies to rule out SM are not routinely performed at the pediatric age. In children with signs or symptoms suggesting systemic involvement such as megalies or cytopenias and in those who have persistently increased serum tryptase levels >20 μg/L, SM should be suspected [1, 3].
Although pediatric mastocytosis can arise at any age, the onset usually occurs during the first 6 months of life, and in some patients, the disease is already present at birth [16, 15,16,21]. Importantly, the age of disease onset has been suggested to show prognostic impact in terms of persistence of the disease into adulthood; thus, children who have a late onset of the disease appear to have lower probability of spontaneous remission of mastocytosis than those with onset at early ages [18,19,24].
Classically, the WHO has recognized three main subtypes of CM (or MIS): (1) maculo-papular cutaneous mastocytosis (MPCM) , formerly known as urticaria pigmentosa (UP), (2) diffuse cutaneous mastocytosis (DCM) , and (3) mastocytoma of the skin [5, 25]. Despite the unquestionable value of the WHO classification of CM as a tool for the distinction among the most prevalent clinical forms of cutaneous involvement by mastocytosis, this classification shows several pitfalls that mainly include terminological and conceptual issues. Some of these limitations have already been addressed by an international task force involving experts from the European Union (EU) and the United States (USA) [20], while others remain to be solved.
The most recent version of the WHO classification of mastocytosis, which was updated in 2016 [5], still accepts the term “urticaria pigmentosa” as a synonym of MPCM. The classical term “urticaria pigmentosa” was coined in 1878 by the English dermatologist Alfred Sangster and described as “an anomalous mottled rash accompanied by pruritus, factitious urticaria, and pigmentation” [26]; 9 years later, the German dermatologist Paul Gerson Unna documented for the first time the presence of MCs in skin biopsies of UP lesions [27]. Given the fact that skin lesions of mastocytosis fail to show the typical transient course of urticaria and, in many cases, mastocytosis skin lesions do not contain the melanic pigment that would define the term “pigmentosa” of UP, the international task force of experts have recently suggested to use the more descriptive term “maculopapular cutaneous mastocytosis” to refer to this subtype of CM [20].
MPCM is characterized by brownish to reddish oval or round macules and papules with variable sizes, distribution, and density, although, in some children, plaques and/or nodules can also be observed or even be the predominant skin lesions; in fact, an early proposal of classification of CM in 2002 already included nodular and plaque-type presentations of CM as independent entities separated from MPCM [28]. It should be noted that the macroscopic appearance of skin lesions can vary during the course of the disease in children, usually from nodules or plaques at disease onset to macules and papules after several years [20]. The heterogeneity of MPCM has also been emphasized in the recent classification proposed by the EU/US consensus group, where MPCM is divided into two categories: (1) monomorphic MPCM, characterized by the presence of skin lesions that show a similar shape and size, usually round and small, which, although is the clinical presentation typically associated with adult-onset mastocytosis (with cutaneous involvement), can be also found in a subset of pediatric mastocytosis; and (2) polymorphic MPCM, which is almost exclusively seen in children and consists of the coexistence of skin lesions displaying different sizes and shapes where large, nodule, or plaque-mimicking lesions frequently predominate [20].
DCM is a rare subtype of CM mostly seen in newborns and infants, defined by a generalized erythema and thickening of almost the entire skin without identifiable individual skin lesions, which shows a typical appearance of orange’s peel (“peau d’orange” in French) or elephant skin (“pachyderma”) [20, 25]. As per definition, DCM shows a very extensive cutaneous involvement by mastocytosis (typically >90% of the whole-body surface area). According to the etymology of the term “diffuse” (i.e., spread over a wide area), patients with extensive MPCM can be misdiagnosed as DCM; hence, the 2015 US/European consensus classification of CM tackles this issue accurately and highlights that the lack of hyperpigmented individualized lesions is a condition sine qua non for the diagnosis of DCM [20]. Moreover, some authors prefer the term “erythrodermic mastocytosis” over DCM to prevent misinterpretations of the adjective diffuse of DCM [29, 30].
Mastocytoma usually presents as a brownish to yellowish, large and solitary nodular-like skin lesion; in other patients, mastocytoma lesions are smaller and less elevated, resembling a solitary form of MPCM. Moreover, both the US/European task force as well as the 2016 WHO classification of mastocytosis still recognize under the denomination of mastocytoma the presence of up to three skin lesions, provided that they show the nodular appearance typically associated with mastocytoma [20]; accordingly, it has been also recommended to change the classical term “solitary mastocytoma” to “cutaneous mastocytoma” [20].
Clinical Symptoms
Clinical manifestations of pediatric mastocytosis mostly include a wide variety of symptoms secondary to the effect of different proinflammatory mediators released from MCs upon their activation; exceptionally, signs and symptoms of end-organ damage due to tissue infiltration by MCs can also occur in cases with advanced mastocytosis, although these are rarely seen in children.
Regarding MC mediator release symptoms, itching, redness, and swelling of lesional skin are common features of CM in children. Urtication together with an erythematous halo can be reproduced by firmly rubbing the skin lesions in what is known as the Darier’s sign, which is considered as pathognomonic of mastocytosis [31]. Frequently, local MC activation of lesional skin results in the development of blisters, particularly during the first months following the onset of the disease. Severe and extensive spontaneous blistering is associated with extensive cutaneous involvement including MPCM cases with >90% of body surface area affected and patients with DCM, where the disease commonly presents itself with generalized blistering [2, 16, 19, 32]; this translates into the need for making a differential diagnosis in these latter cases with other bullous skin diseases of infancy such as bullous pemphigoid, epidermolysis bullosa, or staphylococcal scalded skin syndrome, among other entities. In a few cases of DCM, the content of blisters becomes hemorrhagic, which can be accompanied by some degree of anemization [20]. Of note, extensive blistering has been regarded by some authors as a predictor of massive MC activation and severe complications in pediatric mastocytosis [2, 32]. In fact, markedly increased levels of both total and mature serum tryptase as well as high tryptase levels in blister fluid have been documented in children with extensive blistering in association with other MC-activation symptoms who required hospitalization and emergency therapy [19]; moreover, the rare fatal outcomes of pediatric mastocytosis reported in the literature are practically restricted to children with DCM who developed severe systemic MC activation symptoms preceded by extensive blistering [11, 33]. Other cutaneous manifestations of pediatric CM are dermographism, urticarial rash, and exaggerated local reactions to insect sting/bite.
Flushing is also a relatively common finding in pediatric mastocytosis, which consists of a sudden warmth and reddening of the face and the upper chest caused by increased cutaneous blood flow as a result of the vasodilatory effect of certain MC mediators (e.g., histamine) on the thin and superficial dermal capillaries of these areas of the skin; thus, despite flushing is still largely considered as a cutaneous manifestation of mastocytosis, it should be actually recognized as an early primary vascular event which might precede the development of more severe symptoms including hypotensive collapse in some cases [32].
Other MC mediator release symptoms that can be observed in children with mastocytosis include gastrointestinal complaints (i.e., abdominal cramping, nausea/vomiting, and diarrhea) and, less frequently, dyspnea, fatigue, headache, or neuropsychiatric symptoms such as irritability and attention deficit/hyperactivity-like syndromes.
Overall, anaphylaxis or anaphylactoid reactions are rarely seen in pediatric mastocytosis. In a study by Brockow et al., 4 out of 46 children with mastocytosis (9%) had suffered from anaphylaxis [34]; similarly, the Spanish Network on Mastocytosis (REMA) has reported an incidence of severe MC mediator release symptoms requiring emergency therapy and hospitalization among children with mastocytosis of 11% (12/111 patients) [19]. Of note, in both studies, the severity of MC mediator-related symptoms was closely related to the extent of cutaneous involvement and also with the levels of serum tryptase.
Common elicitors of symptoms in pediatric mastocytosis include the friction of lesional skin, heat and hot water, fever, irritability, and teething [19]. Vaccines are also a relevant trigger for MC activation in children with mastocytosis, particularly among those with DCM [19, 35, 36]; for this reason, an appropriate premedication of vaccines has been recommended by some authors in such cases. Similarly, although the risk of MC mediator release symptoms during anesthesia is relatively low in children with mastocytosis (i.e., 4% in a series reported by the Spanish group including 42 patients undergoing different anesthetic procedures), such risk is clearly increased compared to that found in the general population; thus, it seems also reasonable to adopt preventative measures in this setting, as well as in other procedures associated with increased risk for MC activation such as the administration of iodinated contrast media [37]. Regarding anaphylaxis, idiopathic cause constitutes the most frequent trigger in pediatric mastocytosis; in turn, in contrast to adult-onset mastocytosis, insect-induced anaphylaxis appears to be exceptional in children [19, 34, 38].
Other Associated Diseases (Allergy)
Although it might be hypothesized that mastocytosis could confer an increased risk for other MC-mediated diseases and conditions, different studies have shown controversial results. Despite an early study by Caplan in 1963 suggesting that the prevalence of atopy could be doubled among patients with mastocytosis as compared to the general population (44% vs. 20%) [4], further studies have failed to demonstrate an association between mastocytosis and atopy. In 1990, a Swiss study showed no significant differences in the overall prevalence of atopic diseases (i.e., allergic rhinitis, bronchial asthma, and atopic dermatitis) between a series of 33 patients with mastocytosis and a control group of 52 blood donors (21% vs. 16%) [39]. More recently, a prospective analysis of 67 patients diagnosed with mastocytosis at the National Institutes of Health (NIH) showed a history of atopic diseases including atopic dermatitis, allergic rhinoconjunctivitis, allergic asthma, and food allergy in 31% of the cases [2]. Moreover, in this study, the density (but not the extent) of skin lesions seemed to inversely correlate with the coexistence of atopy in adults (but not in children) [2]. A further study by Brockow et al. in 74 adults and 46 children with mastocytosis revealed the presence of atopic eczema, allergic rhinoconjunctivitis, or allergic asthma in 28% and 11%, respectively [34]. Similarly, a study carried out by the REMA in 210 patients with mastocytosis including 163 adults and 47 children revealed that the prevalence of allergy as defined by clinical symptoms in association with specific IgE was 23.9% and 17%, respectively [38]. Altogether, these observations support that the prevalence of atopy or allergy in patients with mastocytosis does not significantly differ from that found in the general population and that the rate of allergen sensitization might be lower in children than in adults; nevertheless, there are no prospective studies so far that compare the prevalence of allergic diseases in children with mastocytosis versus non-mastocytosis individuals.
Overview of the Differences from Adult Disease
Mastocytosis constitutes a paradigmatic example of a complex and heterogeneous disease, which shows highly variable presentations in terms of age at onset, clinical manifestations, biological features, and outcomes.
Despite the fact that the vast majority of children with mastocytosis are not routinely studied in depth, principally as regard to BM examination, clear differences between pediatric-onset mastocytosis and cases arising in the adulthood have been largely established. First, the clinical spectrum of cutaneous involvement in children is broader than that in adults; thus, although MPCM is the most common subtype of cutaneous disease at any age, mastocytoma of the skin and, less frequently, DCM can be also found in children, whereas these latter clinical forms are rarely seen in adult patients [20]. In addition, MPCM appears to be more heterogeneous among children, in whom both the monomorphic and polymorphic variants of MPCM can occur; by contrast, adult-onset mastocytosis is typically characterized by skin lesions, consistent with the monomorphic variant of MPCM [20]. On the other hand, mastocytosis without skin involvement is relatively frequent among adults, particularly in patients suffering from anaphylaxis with a cardiovascular profile (e.g., hypotension, dizziness, loss of consciousness) in the absence of cutaneo-mucosal symptoms [40], but it is extremely uncommon in children.
Second, it is widely accepted that mastocytosis is a clonal systemic disease in nature when it arises in the adulthood but mostly restricted to the skin at the pediatric age. However, the assumption that children with serum tryptase levels below 20 μg/L are more likely to have CM [1] seems arbitrary and is not based on prospective studies. Taking into account the limitations mentioned above about the lack of complete BM studies in most children, the concept of “pure” CM in children is, at least, questionable, provided that dermal MCs derive from a precursor cell originated in the BM [41]. In the largest cohort of children with mastocytosis who underwent a BM study published so far, more than one-third of patients (19 out of 53) were shown to have SM [3]; however, it should be noted that these 53 children had been selected from a total of 105 patients for the BM analysis on the basis of severe MC mediator release symptoms and/or organomegalies. Interestingly, this study revealed that the presence of organomegalies was the most robust predictor of systemic involvement in children with mastocytosis, as all the patients with organomegalies who were studied but none of those without them had systemic disease [3]. Another fact to consider regarding systemic involvement in mastocytosis is a potentially higher prevalence of the so-called “well-differentiated SM” (WDSM) in children than in adults. This biologically unique variant of SM is characterized by a clonal expansion of MCs in the BM that typically displays an apparently normal morphology together with the lack of CD25 (and CD2) immunophenotypic expression in the absence of the D816V KIT mutation [42, 43], which means that three out of the four minor diagnostic criteria for SM according to the WHO are actually missing in a substantial subset of patients with WDSM; furthermore, these characteristics make the diagnosis of WDSM particularly challenging, since, frequently, only those patients with a significant BM MC infiltration would meet criteria for SM according to the WHO, whereas most of the remaining cases would be misdiagnosed as CM. The skin lesions in WDSM have been noted to present with two patterns. One pattern is more typical of DCM with diffusely thickened skin (Figs. 6.7 and 6.8) and the other with a pattern similar to that of MCPM with distinct red-brown macules and papules (Fig. 6.9). To overcome the diagnostic limitations, specific minor criteria for the diagnosis of WDSM have been recently proposed, which include several biological features together with the onset of mastocytosis skin lesions during childhood [43] (Table 6.1). Because this latter clinical finding accounts for more than 90% of adult patients with WDSM [43], it could be hypothesized at least almost an equal prevalence of such SM variant in children and adults. Moreover, considering the fact that WDSM is typically associated with subtypes of CM rarely seen in adults, such as DCM or polymorphic MPCM [20], it would be expected even a higher frequency of WDSM cases in children vs. adults; in any case, future investigations are warranted in order to establish the true prevalence of WDSM among the pediatric population.

DCM variant of WDSM: The skin is thickened with exaggerated skin folds particularly in the axilla

DCM variant of WDSM: The skin is thickened with exaggerated skin folds and a lack of hyperpigmented skin lesions

The MPCM variant of WDSM-skin lesions is usually small (<0.5 cm) reddish-brown with both macules and papules. The trunk and neck are the main areas of involvement with relative sparing of the extremities
Other differential feature of pediatric versus adult-onset mastocytosis is the higher frequency of mutations involving exons other than 17 of the KIT gene in children. In a study by the French group published in 2010, where the entire KIT sequence was analyzed in skin biopsies from 50 children with mastocytosis, a mutation of codon 816 (exon 17) was found in 42% of cases, whereas mutations outside exon 17 were detected in 44% [44]; these findings contrast with an overall frequency of detection of the D816V KIT mutation in >90% of adult patients, provided that highly sensitive molecular assays are applied [45, 46]. Moreover, the results in terms of frequency of mutations involving the extracellular membrane and transmembrane domains (exons 8–10) of KIT reported in this study together with the well-known close association of this type of KIT mutations with WDSM [43, 47] also support the fact that pediatric WDSM is probably underestimated.
Regarding MC mediator release symptoms, there are clinical manifestations typically seen in children that never occur in adults such as blistering and others that are much more common in adults than in children such as anaphylaxis. Also, the different triggers that can potentially activate MCs appear to play different roles depending on the age of the patients. Thus, whereas insect sting/bite is by far the most common elicitor of anaphylaxis in adult patients with SM [40], such trigger has virtually no clinical relevance in pediatric mastocytosis. By contrast, triggering factors such as fever, teething, and vaccines seem to be almost exclusively restricted to children with mastocytosis.
In terms of outcome, adult-onset mastocytosis is widely considered a chronic and incurable disease. Although the life expectancy of the vast majority of adults with indolent SM (ISM) does not differ from that of the general population [48], a small fraction of patients may evolve with time to advanced forms of the disease, including aggressive SM (ASM), SM with an associated hematological neoplasm (SM-AHN), or MC leukemia (MCL). The most important prognostic factors associated with an increased risk of disease progression in adult patients diagnosed with ISM are the presence of high levels of β2-microglobulin at diagnosis and the demonstration of multilineal involvement of hematopoiesis by the D816V KIT mutation [49]. It has been recently suggested that the crucial event that finally determines progression to advanced mastocytosis is the acquisition of genetic mutations in genes commonly involved in the pathogenesis of other hematological neoplasms, such as SRSF2, ASXL1, or RUNX1 [50, 51]. By contrast, the natural history of pediatric mastocytosis is toward spontaneous regression in most cases, generally before puberty, with only a minority of children remaining with persistent disease in the adulthood, either as conventional SM or as WDSM. Although no definitive factors predicting the persistence of pediatric-onset mastocytosis in adult age have been identified so far, it has been suggested that the aberrant expression of CD25 on cutaneous MCs could be highly predictive of SM in adults [52]. Thus, if such hypothesis was extrapolated to children, then it might be speculated that the expression of this immunophenotypic marker in skin biopsies from children with mastocytosis could be of relevance in the persistence of the disease into the adulthood as a classical CD25+ SM. Regarding WDSM, the only factor that has been suggested to play a role in the persistence of mastocytosis after puberty is the gender, since more than 85% of patients diagnosed with WDSM at adult age who had a previous history of pediatric mastocytosis reported in the literature are women [42, 43, 49,50,55].
Diagnostic Evaluation
Mastocytosis is diagnosed on the basis of history, clinical manifestations, histopathology, and laboratory evaluation and classified based on the WHO criteria for mastocytosis [5]. A history of daily and episodic symptoms should be obtained along with possible triggers, alleviating and exacerbating factors. A thorough physical exam should include details of the skin lesions, lymph node examination, and careful abdominal exam to assess possible organomegaly.
Maculopapular cutaneous mastocytosis (MPCM) is usually seen in two patterns, namely polymorphic and monomorphic [20], and has been shown to be associated with prognosis. Patients with a polymorphic pattern tend to have an earlier onset of disease and resolution of skin manifestations over time, whereas the monomorphic pattern, similar to adult presentation, is associated with a more prolonged course and systemic disease [3]. MPCM lesions have MCs in increased numbers in the dermal papillae beneath macules and papules, particularly near blood vessels in the upper dermis [56]. A band-like infiltrate of MCs is distributed in the papillary dermis or appears as nodular infiltrates from the papillary dermis to subcutaneous tissues. Typically, there is a 15- to 20-fold increase in MCs beneath those lesions, but in some patients, only a twofold to fourfold increase in MCs is found (Fig. 6.10). Thus, it is important to correlate the gross skin examination with skin MC numbers and to avoid the diagnosis of UP exclusively on the basis of small increases in dermal MCs. MCs may also be found in increased numbers in the normal-appearing skin between lesions of UP [56]. The differences in the histologic pattern in cutaneous disease are generally based on the density of the MC infiltrate. In patients with DCM, the skin is typically thickened and described as “peau d’ orange” and diffuse red-brown color. These lesions are more prone to blistering with hemorrhagic crusts with minor friction. There have been two subtypes described, one with plaque-like lesions interspersed with normal-appearing skin and a diffuse thickening or pachydermic pattern [57]. The former has a better prognosis for complete resolution. MCs are observed around blood vessels and throughout the dermis. These band-like infiltrates may be indistinguishable from some lesions of MPCM or from biopsies obtained from mastocytomas. Mastocytomas typically present as a single lesion that is raised with a reddish-brown color, flesh colored, or yellowish color. It is quite sensitive to friction and can be associated with total body flushing. Darier’s sign has been used as a diagnostic expression of CM. A wheal and flare response to rubbing or scratching the lesions with a blunt object is characteristic of MC infiltration. Since mastocytomas have an abundance of MCs and can cause a significant release of mediators, this diagnostic test should be avoided in these patients.

Histopathology of DCM (40×), shown here stained with tryptase antibody demonstrating band-like infiltrates of MCs that extend into the papillary dermis
Laboratory assays that are helpful in the management are complete blood count and differential, liver function tests, vitamin D level, baseline serum tryptase, and IgE if there is a suspected allergic disease. Most laboratory tests are in the normal range with the exception of eosinophilia, lymphocytosis, and thrombocytosis that have not been shown to be of clinical significance and resolved without intervention. If there is a strong suspicion for systemic disease with organomegaly, elevated serum tryptase (>20 ng/ml), and/or severe mast cell mediator symptoms, a peripheral blood allele-specific assay for the KIT D816V mutation is helpful to guide decision for a BM study. The assay is specific for the D816V and may be negative in patients with other mutations in KIT or those patients with a low allelic burden [58]. An abdominal ultrasound is helpful when organomegaly is suspected.
CM is confirmed by a lesional skin biopsy demonstrating characteristic skin histopathology. Blind skin biopsies are not recommended, since other skin conditions including eczema and recurrent episodes of anaphylaxis may be associated with a twofold to fourfold increase in dermal mast cells [56, 59]. In addition, MCs may also be increased at skin sites involved in scleroderma [60], chronic urticaria [61], and prolonged antigenic contact [62]. CM must also be distinguished from other diseases with similar cutaneous characteristics as those of mastocytosis and are included in Table 6.2.
Currently, the standard for the diagnosis of SM is by performing a BM biopsy and aspirating with demonstration of the major criterion, consisting of multifocal dense MC aggregates, and one minor criterion or if three minor criteria are present (Table 6.1). The most useful stain for MCs is tryptase using a monoclonal antibody. In trephine core BM biopsies, decalcification interferes with subsequent attempts to visualize MC granules with metachromatic stains, thereby making tryptase the stain of choice. In addition, immunohistochemistry and/or flow cytometry to identify CD25+ MCs is useful since this parameter has been shown to correlate with the presence of activating mutations in KIT [63].
Other tissue specimens such as lymph nodes, spleen, liver, and gastrointestinal mucosa delineate the extent of MC involvement but are not typically sampled. Gastrointestinal biopsies are obtained only if a gastrointestinal workup is indicated, and lymph nodes are biopsied only if lymphoma is considered. When biopsies have been obtained of involved tissue such as the GI tract, the histopathologic pattern of MC aggregates or sheets is similar to that seen in the BM and is often CD25 positive [60,61,66].
In patients suspected of having mastocytosis, the diagnosis of a carcinoid tumor or pheochromocytoma should be ruled out. Importantly, patients with mastocytosis do not excrete increased amounts of 5-HIAA. Patients with carcinoid tumor or pheochromocytoma do not have histologic evidence of significant MC proliferation and should have normal serum tryptase levels [67].
Follow-Up and Management
Patients can typically be followed by their primary care providers if the above assays are obtained on an annual basis or in association with an acute illness or severe mediator symptoms. Patients should receive routine vaccines since the incidence of unexpected adverse reactions is low [68]. Children with mastocytosis, unlike adults, have not been reported to have an increased risk of anaphylaxis in association with venomous insect stings. Annual serum tryptase values along with peripheral blood allele-specific qPCR in children with suspected or confirmed systemic disease is helpful to determine if a more aggressive workup is needed. Patients with organomegaly should have a repeat ultrasound every 1–2 years until resolution or with acute enlargement.
A referral to a specialist, allergist/immunologist or hematologist, familiar with MC diseases is warranted for the following:
-
The diagnosis is questionable and needs tissue confirmation.
-
Symptoms are not sufficiently controlled with an anti-mediator therapy.
-
There is a suspicion for systemic disease.
-
A persistently elevated or increasing baseline serum tryptase.
-
A peripheral blood assay that is positive for the KIT D816V mutation.
-
Other signs and symptoms of a myeloproliferative disease.
Therapy for mastocytosis is based on the amelioration of symptoms and applies to patients with CM and SM. Cytoreductive therapy for MCs is typically reserved for more aggressive variants such as smoldering SM (SSM) or aggressive SM (ASM). These variants are rarely seen in the pediatric population. Therefore, cytoreductive agents are discussed in detail in other chapters. The most prominent complaints are cutaneous and gastrointestinal problems. Cutaneous symptoms such as flushing, blistering, and pruritus are proportional to the skin MC burden, with more frequent and severe symptoms in patients with DCM and mastocytoma than in patients with MPCM. In a previous study, it was noted that these cutaneous symptoms could be present through the adolescent period, although less severe [3]. Gastrointestinal symptoms were also distinguished by variants noting that patients with ISM had more problems such as diarrhea and reflux, but patients in all variants complained of abdominal cramping. Of note, during an acute flushing and/or blistering event, many patients complained of associated abdominal cramping and diarrhea. Headaches were due to non-mastocytosis-related complaints such as migraines and possibly mastocytosis-related events such as vasodilatation. The therapy is based on the underlying etiology. Musculoskeletal complaints were mainly unrelated to the diagnosis of mastocytosis or unknown etiology, and thus, the approach was again based on the etiology. Therapeutic options are summarized in Table 6.3.
Prognosis and Disease Resolution
Children generally have a favorable prognosis and a measurable resolution, especially when the onset of disease is prior to the age of 2 years. Patients with the onset of disease after the age of 5 years tend to have a more persistent pattern, but there is not much information in the literature regarding the progression to a more severe variant. Resolution tends to occur in late adolescence [1, 3, 16] without reoccurrence of disease. Patients diagnosed with cutaneous disease in adolescence are more likely to have SM; however, children can be diagnosed with systemic disease as early as infancy. These patients mainly expressed the KIT D816V mutation, and the prognosis is dependent on the degree of involvement, but patients are reported to have a normal life expectancy. Further, in a long-term follow-up study of children with systemic disease, the overall clinical outcome reflected an improvement of cutaneous manifestations, organomegaly, and serum tryptase values [3].
In summary, children with CM and SM have a good prognosis. Patients with cutaneous disease only can have complete resolution of the disease, with minimum or no symptoms in most patients . Those with systemic disease will have a chronic pattern with varying degrees of resolution based on the initial presentation. This allows for a conservative approach to management without a need for cytoreductive therapy.
Abbreviations
- BM:
-
Bone marrow
- CM:
-
Cutaneous mastocytosis
- DCM:
-
Diffuse cutaneous mastocytosis
- ISM:
-
Indolent systemic mastocytosis
- MC:
-
Mast cell
- MIS:
-
Mastocytosis in the skin
- MPCM:
-
Maculopapular cutaneous mastocytosis
- NIH:
-
National Institutes of Health
- PB:
-
Peripheral blood
- qPCR:
-
quantitative polymerase chain reaction
- REMA:
-
Spanish Network on Mastocytosis
- SM:
-
Systemic mastocytosis
- UP:
-
Urticaria pigmentosa
- WHO:
-
World Health Organization
References
Valent P, Akin C, Escribano L, Fodinger M, Hartmann K, Brockow K, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Investig. 2007;37:435–53.
Brockow K, Akin C, Huber M, Metcalfe DD. Assessment of the extent of cutaneous involvement in children and adults with mastocytosis: relationship to symptomatology, tryptase levels, and bone marrow pathology. J Am Acad Dermatol. 2003;48:508–16.
Carter MC, Clayton ST, Komarow HD, Brittain EH, Scott LM, Cantave D, et al. Assessment of clinical findings, tryptase levels, and bone marrow histopathology in the management of pediatric mastocytosis. J Allergy Clin Immunol. 2015;136:1673–9 e3.
Caplan R. The nautral course of urticaria pigmentosa. Arch Dermatol. 1963;87:146–57.
Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129:1420–7.
Lidor C, Frisch B, Gazit D, Gepstein R, Hallel T, Mekori YA. Osteoporosis as the sole presentation of bone marrow mastocytosis. J Bone Miner Res. 1990;5(8):871–6.
Floman Y, Amir G. Systemic mastocytosis presenting with severe spinal osteopenia and multiple compression fractures. J Spinal Disord. 1991;4(3):369–73.
Bonadonna P, Perbellini O, Passalacqua G, Caruso B, Colarossi S, Dal Fior D, et al. Clonal mast cell disorders in patients with systemic reactions to Hymenoptera stings and increased serum tryptase levels. J Allergy Clin Immunol. 2009;123(3):680–6.
Rueff F, Przybilla B, Bilo MB, Muller U, Scheipl F, Aberer W, et al. Predictors of severe systemic anaphylactic reactions in patients with Hymenoptera venom allergy: importance of baseline serum tryptase-a study of the European Academy of Allergology and Clinical Immunology Interest Group on Insect Venom Hypersensitivity. J Allergy Clin Immunol. 2009;124(5):1047–54.
Chantorn R, Shwayder T. Death from mast cell leukemia: a young patient with longstanding cutaneous mastocytosis evolving into fatal mast cell leukemia. Pediatr Dermatol. 2012;29:605–9.
Meni C, Bruneau J, Georgin-Lavialle S, Le Sache de Peufeilhoux L, Damaj G, Hadj-Rabia S, et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol. 2015;172:642–51.
Gadage VS, Kadam Amare PS, Galani KS, Mittal N. Systemic mastocytosis with associated acute myeloid leukemia with t (8; 21) (q22; q22). Indian J Pathol Microbiol. 2012;55:409–12.
Intzes S, Wiersma S, Meyerson HJ. Myelomastocytic leukemia with t(8;21) in a 3-year-old child. J Pediatr Hematol Oncol. 2011;33:e372–5.
Mahadeo KM, Wolgast L, McMahon C, Cole PD. Systemic mastocytosis in a child with t(8;21) acute myeloid leukemia. Pediatr Blood Cancer. 2011;57:684–7.
Tzankov A, Sotlar K, Muhlematter D, Theocharides A, Went P, Jotterand M, et al. Systemic mastocytosis with associated myeloproliferative disease and precursor B lymphoblastic leukaemia with t(13;13)(q12;q22) involving FLT3. J Clin Pathol. 2008;61:958–61.
Ben-Amitai D, Metzker A, Cohen HA. Pediatric cutaneous mastocytosis: a review of 180 patients. Isr Med Assoc J. 2005;7:320–2.
Carter MC, Metcalfe DD. Paediatric mastocytosis. Arch Dis Child. 2002;86:315–9.
Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol. 2011;12:259–70.
Alvarez-Twose I, Vano-Galvan S, Sanchez-Munoz L, Morgado JM, Matito A, Torrelo A, et al. Increased serum baseline tryptase levels and extensive skin involvement are predictors for the severity of mast cell activation episodes in children with mastocytosis. Allergy. 2012;67:813–21.
Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Alvarez-Twose I, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35–45.
Lange M, Niedoszytko M, Renke J, Glen J, Nedoszytko B. Clinical aspects of paediatric mastocytosis: a review of 101 cases. J Eur Acad Dermatol Venereol. 2013;27:97–102.
Heinze A, Kuemmet TJ, Chiu YE, Galbraith SS. Longitudinal study of pediatric urticaria pigmentosa. Pediatr Dermatol. 2017;34:144–9.
Meni C, Georgin-Lavialle S, Le Sache de Peufeilhoux L, Jais JP, Hadj Rabia S, Bruneau J, et al. Pediatric mastocytosis: long term follow up of 53 patients with whole sequencing of KIT. A prospective study. Br J Dermatol. 2018;179:925.
Uzzaman A, Maric I, Noel P, Kettelhut BV, Metcalfe DD, Carter MC. Pediatric-onset mastocytosis: a long term clinical follow-up and correlation with bone marrow histopathology. Pediatr Blood Cancer. 2009;53:629–34.
Horny HPAC, Metcalfe DD, Escribano L, Bennett JM, Valent P, Bain BJ. In: Swerdlow S, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. World Health Organization (WHO) Classification of Tumours. Pathology & genetics: tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2008. p. 54–63.
A S. An anomalous mottled rash accompanied by pruritus, factitious urticaria and pigmentation, “urticaria pigmentosa”? Trans Med Soc Lond 1878:161–3.
PG U. Beitrage zur anatomic und pathogenese der urticaria simplex und pigmentosa. Mscch Prakt Dermatol Suppl Dermatol Stud. 1887:3–9.
Hartmann K, Henz BM. Classification of cutaneous mastocytosis: a modified consensus proposal. Leuk Res. 2002;26:483–4.. author reply 5-6
Olgun N, Oren H, Oren B, Irken G, Polat M, Cevik N. Diffuse erythrodermic cutaneous mastocytosis with bone marrow infiltration. Dermatology. 1993;187:127–9.
Torrelo A, Alvarez-Twose I, Escribano L. Childhood mastocytosis. Curr Opin Pediatr. 2012;24:480–6.
Skrabs CC. Darier sign: a historical note. Arch Dermatol. 2002;138:1253–4.
Brockow K, Ring J, Alvarez-Twose I, Orfao A, Escribano L. Extensive blistering is a predictor for severe complications in children with mastocytosis. Allergy. 2012;67:1323–4.
Koga H, Kokubo T, Akaishi M, Iida K, Korematsu S. Neonatal onset diffuse cutaneous mastocytosis: a case report and review of the literature. Pediatr Dermatol. 2011;28:542–6.
Brockow K, Jofer C, Behrendt H, Ring J. Anaphylaxis in patients with mastocytosis: a study on history, clinical features and risk factors in 120 patients. Allergy. 2008;63:226–32.
Bankova LG, Walter JE, Iyengar SR, Lorenzo ME, Hornick JL, Castells MC. Generalized bullous eruption after routine vaccination in a child with diffuse cutaneous mastocytosis. J Allergy Clin Immunol Pract. 2013;1:94–6.
Hudson A, Finlayson L. Diffuse cutaneous bullous mastocytosis and disseminated intravascular coagulation postvaccination: a case report. J Cutan Med Surg. 2016;20:596–9.
Matito A, Morgado JM, Sanchez-Lopez P, Alvarez-Twose I, Sanchez-Munoz L, Orfao A, et al. Management of anesthesia in adult and pediatric mastocytosis: a study of the Spanish Network on Mastocytosis (REMA) based on 726 anesthetic procedures. Int Arch Allergy Immunol. 2015;167:47–56.
Gonzalez de Olano D, de la Hoz Caballer B, Nunez Lopez R, Sanchez Munoz L, Cuevas Agustin M, Dieguez MC, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy. 2007;37:1547–55.
Muller U, Helbling A, Hunziker T, Wuthrich B, Pecoud A, Gilardi S, et al. Mastocytosis and atopy: a study of 33 patients with urticaria pigmentosa. Allergy. 1990;45:597–603.
Alvarez-Twose I, Gonzalez de Olano D, Sanchez-Munoz L, Matito A, Esteban-Lopez MI, Vega A, et al. Clinical, biological, and molecular characteristics of clonal mast cell disorders presenting with systemic mast cell activation symptoms. J Allergy Clin Immunol. 2010;125:1269–78 e2.
Rodewald HR, Dessing M, Dvorak AM, Galli SJ. Identification of a committed precursor for the mast cell lineage. Science. 1996;271:818–22.
Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103:3222–5.
Alvarez-Twose I, Jara-Acevedo M, Morgado JM, Garcia-Montero A, Sanchez-Munoz L, Teodosio C, et al. Clinical, immunophenotypic, and molecular characteristics of well-differentiated systemic mastocytosis. J Allergy Clin Immunol. 2016;137:168–78 e1.
Bodemer C, Hermine O, Palmerini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804–15.
Jara-Acevedo M, Teodosio C, Sanchez-Munoz L, Alvarez-Twose I, Mayado A, Caldas C, et al. Detection of the KIT D816V mutation in peripheral blood of systemic mastocytosis: diagnostic implications. Mod Pathol. 2015;28:1138–49.
Kristensen T, Vestergaard H, Bindslev-Jensen C, Moller MB, Broesby-Olsen S, Mastocytosis Centre OUH. Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89:493–8.
Alvarez-Twose I, Matito A, Morgado JM, Sanchez-Munoz L, Jara-Acevedo M, Garcia-Montero A, et al. Imatinib in systemic mastocytosis: a phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget. 2017;8:68950–63.
Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90:250–62.
Escribano L, Alvarez-Twose I, Sanchez-Munoz L, Garcia-Montero A, Nunez R, Almeida J, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol. 2009;124:514–21.
Jawhar M, Schwaab J, Schnittger S, Meggendorfer M, Pfirrmann M, Sotlar K, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016;30:136–43.
Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122:2460–6.
Hollmann TJ, Brenn T, Hornick JL. CD25 expression on cutaneous mast cells from adult patients presenting with urticaria pigmentosa is predictive of systemic mastocytosis. Am J Surg Pathol. 2008;32:139–45.
Chan EC, Bai Y, Kirshenbaum AS, Fischer ER, Simakova O, Bandara G, et al. Mastocytosis associated with a rare germline KIT K509I mutation displays a well-differentiated mast cell phenotype. J Allergy Clin Immunol. 2014;134:178–87.
de Melo Campos P, Machado-Neto JA, Scopim-Ribeiro R, Visconte V, Tabarroki A, Duarte AS, et al. Familial systemic mastocytosis with germline KIT K509I mutation is sensitive to treatment with imatinib, dasatinib and PKC412. Leuk Res. 2014;38:1245–51.
Huang L, Wang SA, Konoplev S, Bueso-Ramos CE, Thakral B, Miranda RN, et al. Well-differentiated systemic mastocytosis showed excellent clinical response to imatinib in the absence of known molecular genetic abnormalities: a case report. Medicine (Baltimore). 2016;95:e4934.
Garriga MM, Friedman MM, Metcalfe DD. A survey of the number and distribution of mast cells in the skin of patients with mast cell disorders. J Allergy Clin Immunol. 1988;82:425–32.
Heide R, Zuidema E, Beishuizen A, Den Hollander JC, Van Gysel D, Seyger MM, et al. Clinical aspects of diffuse cutaneous mastocytosis in children: two variants. Dermatology. 2009;219:309–15.
Carter MC, Desai A, Komarow HD, Bai Y, Clayton ST, Clark AS, et al. A distinct biomolecular profile identifies monoclonal mast cell disorders in patients with idiopathic anaphylaxis. J Allergy Clin Immunol. 2018;141:180–8 e3.
Brockow K, Akin C, Huber M, Scott LM, Schwartz LB, Metcalfe DD. Levels of mast-cell growth factors in plasma and in suction skin blister fluid in adults with mastocytosis: correlation with dermal mast-cell numbers and mast-cell tryptase. J Allergy Clin Immunol. 2002;109:82–8.
Nishioka K, Kobayashi Y, Katayama I, Takijiri C. Mast cell numbers in diffuse scleroderma. Arch Dermatol. 1987;123:205–8.
Elias J, Boss E, Kaplan AP. Studies of the cellular infiltrate of chronic idiopathic urticaria: prominence of T-lymphocytes, monocytes, and mast cells. J Allergy Clin Immunol. 1986;78:914–8.
Mitchell EB, Crow J, Williams G, Platts-Mills TA. Increase in skin mast cells following chronic house dust mite exposure. Br J Dermatol. 1986;114:65–73.
Teodosio C, Garcia-Montero AC, Jara-Acevedo M, Sanchez-Munoz L, Alvarez-Twose I, Nunez R, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol. 2010;125:719–26, 26 e1–26 e4
Cherner JA, Jensen RT, Dubois A, O’Dorisio TM, Gardner JD, Metcalfe DD. Gastrointestinal dysfunction in systemic mastocytosis. A prospective study. Gastroenterology. 1988;95:657–67.
Hahn HP, Hornick JL. Immunoreactivity for CD25 in gastrointestinal mucosal mast cells is specific for systemic mastocytosis. Am J Surg Pathol. 2007;31:1669–76.
Sokol H, Georgin-Lavialle S, Canioni D, Barete S, Damaj G, Soucie E, et al. Gastrointestinal manifestations in mastocytosis: a study of 83 patients. J Allergy Clin Immunol. 2013;132:866–73.e1–3.
Schwartz LB, Sakai K, Bradford TR, Ren S, Zweiman B, Worobec AS, et al. The alpha form of human tryptase is the predominant type present in blood at baseline in normal subjects and is elevated in those with systemic mastocytosis. J Clin Invest. 1995;96:2702–10.
Parente R, Pucino V, Magliacane D, Petraroli A, Loffredo S, Marone G, et al. Evaluation of vaccination safety in children with mastocytosis. Pediatr Allergy Immunol. 2017;28:93–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Alvarez-Twose, I., Carter, M.C. (2020). Pediatric Mastocytosis. In: Akin, C. (eds) Mastocytosis. Springer, Cham. https://doi.org/10.1007/978-3-030-27820-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-27820-5_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27822-9
Online ISBN: 978-3-030-27820-5
eBook Packages: MedicineMedicine (R0)