
Abstract
Mastocytosis is defined as a heterogeneous group of disorders characterized by clonal expansion and aberrant activation of mast cells. Most cases are associated with gain-of-function mutations in KIT, the receptor for stem cell factor, the major cytokine involved in mast cell growth and differentiation. While a rare disease, it is increasingly diagnosed due to the refinements in diagnostic criteria and increased physician and patient awareness. Novel therapeutic approaches targeting mast cell survival and activation makes it important to correctly diagnosing the disease. This chapter serves as an introduction on the symptomatology, diagnostic criteria, classification, and treatment of mastocytosis, which are discussed in more detail in other chapters of the book.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Mastocytosis is a disorder characterized by clonal expansion of mast cells. In most cases, it is caused by gain-of-function mutations in the KIT gene, which encodes a critical growth factor receptor involved in mast cell growth, differentiation, and survival [1]. Clonal mast cells can be found in skin, bone marrow, liver, spleen, and gastrointestinal tract. The symptomatology is due to local expansion and accumulation of mast cells, release of vasoactive mediators as well as cytokines from activated mast cells, and, in some patients, presence of an associated hematologic disorder.
Epidemiology
The estimated prevalence is approximately 1 in 20,000. It can be seen in both children and adults [2, 3]. In children, the disease is limited to skin and diagnosed by typical cutaneous lesions usually noted in the first year of life. Ninety percent of childhood-onset disease resolves by adolescence. In contrast, adult-onset mastocytosis involves the bone marrow and is persistent. It is seen in all ethnic populations, although most diagnosed cases in the Western world are Caucasians.
Clinical Presentations
The disease has protean clinical manifestations and may present in one of the clinical scenarios described below:
-
1.
Urticaria pigmentosa (maculopapular cutaneous mastocytosis = MPCM): This is the most common presentation in both children and adults. MPCM consists of hyperpigmented, fixed lesions usually less than 2 cm in diameter, involving trunk and extremities, usually sparing sun-exposed areas in adults (Fig. 2.1a, b). Children generally have lesions of varying sizes (polymorphic MPCM) and may have scalp involvement (Fig. 2.2). Blistering of the lesions may be seen earlier in life, mostly in the first 3 years (Fig. 2.3). The lesions are generally not pruritic at baseline but urticate with friction, temperature changes, fever, emotional stress, and exercise. Darier’s sign, defined by a wheal and flare reaction of the skin lesions upon mechanical rubbing, is a pathognomonic hallmark of cutaneous disease involvement and confirms the diagnosis of mastocytosis in the skin (Fig. 2.4a, b). MPCM is the most common variant. Less common variants include diffuse cutaneous mastocytosis and mastocytomas in children [4] (Fig. 2.4). A skin biopsy confirms the diagnosis.
-
2.
Anaphylaxis and symptoms of mast cell activation: These patients may or may not have skin lesions but come to clinical attention due to recurrent anaphylaxis or other episodic symptoms of mast cell activation including flushing, abdominal cramps, diarrhea, tachycardia, hypotension, and loss of consciousness. Flushing episodes typically last for 15–30 minutes. Acute urticaria and angioedema are uncommon [5, 6].
-
3.
Hematologic disorders: Approximately 10–15% of the patients are diagnosed because of abnormalities in their blood counts, prompting a bone marrow biopsy. These patients show evidence of an associated hematologic disorder, usually myeloproliferative or a myelodysplastic syndrome (MDS), in addition to mastocytosis [7].
-
4.
Bone disease: A small number of patients are initially diagnosed due to bone pain; vertebral compression fractures; and osteopenia, osteoporosis, or rarely sclerotic or even osteolytic lesions shown by imaging studies, prompting a bone biopsy [8].

a, b. Adult-onset maculopapular cutaneous mastocytosis

Typical MPCM skin lesions in pediatric cutaneous mastocytosis

Bullous MPCM in a patient with pediatric cutaneous mastocytosis

Darier’s sign. a. Mastocytoma prior to rubbing. b. Wheal and flare formation after rubbing of the lesion
Diagnosis
Cutaneous disease is diagnosed by observing typical skin lesions, by testing for Darier’s sign, and by a skin biopsy. Systemic disease is diagnosed by a bone marrow biopsy, aspiration, and demonstration of World Health Organization (WHO) diagnostic criteria for systemic mastocytosis (Table 2.1) [9, 10]. These criteria are discussed in more detail in other chapters of this book.
Major Criterion
Multifocal mast cell accumulations of >15 cells per collection. Tryptase, CD117, and CD25 staining of the core biopsy is recommended to evaluate for the presence of the major criterion.
Minor Criteria
-
1.
Aberrant Mast Cell Morphology: Normal mast cells have a round shape and are fully granulated with a central nucleus. Mast cells in mastocytosis are spindle shaped and have cytoplasmic projections and an oval off-center nucleus, which, in advanced disease, may be clefted or multilobated. This abnormal morphology can be demonstrated in bone marrow sections or in aspirate smears. Mast cells in aspirate smears are usually found in or around spicules and are degranulated or hypogranulated [11].
-
2.
CD2 and/or CD25 Expression: Normal mast cells do not express CD2 or CD25. Aberrant CD25 expression can be detectable by immunohistochemistry or flow cytometry. In flow cytometry, detection of mast cells requires acquisition of at least 500,000 or more events and appropriate gating strategies, and they may not be detectable by routine leukemia/lymphoma phenotyping. IHC can be performed in archival paraffin blocks. Serial sections should be evaluated for tryptase and/or CD117-positive mast cells co-expressing CD25. CD2 is also aberrantly expressed, but it may be variably detectable and is especially low or even absent in mast cells in many cases of advanced disease [12].
-
3.
KIT D816V Mutation: KIT encodes for the receptor of stem cell factor, which is the most important growth factor for mast cell growth and development. D816V somatic gain-of-function mutation is found in >90% of adult cases with systemic mastocytosis and approximately 30% of pediatric cutaneous mastocytosis [13]. The most sensitive method to detect this mutation is an allele-specific qPCR in bone marrow aspirate. Peripheral blood may yield wild-type results in patients with low mast cell burden unless very sensitive (not yet widely available) PCR techniques are used [14] and in patients with non-D816V KIT mutations. Therefore, mutation analysis studies should be performed on bone marrow cells when the blood test is negative.
-
4.
Serum Tryptase >20 ng/ml: Tryptase is a relatively specific marker, as this enzyme is primarily synthesized by mast cells. Mature tryptases (mainly beta tryptase) are stored in mast cell granules and are released during mast cell activation [15]. Pro-tryptases (mainly alpha tryptase) are secreted constitutively from mast cells, resulting in a stable baseline serum level that reflects the total body burden of mast cells. Commercially available tryptase assays test for total (pro- and mature tryptases). Normal median serum or plasma baseline tryptase is approximately 5 ng/ml. Values greater than 20 ng/ml are typically found in systemic mastocytosis. Tryptase levels <20 ng/ml can be seen in patients with low mast cell burden, monoclonal mast cell activation syndrome, bone marrow mastocytosis, and cutaneous mastocytosis. An important aspect is that tryptase levels >20 ng/ml can also be seen in conditions other than mastocytosis, including hereditary alpha tryptasemia [16], chronic renal disease [17], and myeloid neoplasms [18]. Therefore, an elevated basal tryptase level is a minor (but not major) criterion of systemic mastocytosis.
Presence of at least the major plus one minor or three minor criteria is required to establish the diagnosis of systemic mastocytosis. In patients with hematologic disease, tryptase criterion is not valid, as it can be elevated due to the hematologic disease itself. Patients presenting mast cell activation symptoms who show CD25 expression in mast cells and/or KIT D816V mutation are termed to have monoclonal mast cell activation syndrome (MCAS) when all MCAS criteria are fulfilled [19,20,21].
Well-Differentiated Systemic Mastocytosis
A histopathologic variant termed “well-differentiated mastocytosis” was described in 2002, and consists of mast cells with a round, fully mature morphology, absence of CD25 expression, and usually lack of KIT mutations. This variant usually satisfies the diagnostic criteria due to the presence of the major criterion, demonstration of clonality by either Kit mutation or HUMARA assay, and elevated tryptase levels [22, 23]. However, the well-differentiated variant of mastocytosis can be detected in all WHO categories of mastocytosis including indolent mastocytosis and mast cell leukemia.
Classification
WHO recognizes seven categories of the disease [9]:
-
1.
Cutaneous Mastocytosis: This category of disease is almost exclusively seen in children and means that the disease is limited to skin [4]. It should be noted that “mastocytosis in skin” is the preferred term for adult patients with skin lesions in whom systemic involvement cannot be ruled out because no bone marrow studies were performed. Cutaneous mastocytosis can present as MPCM, diffuse cutaneous mastocytosis or mastocytoma of the skin (Please see the Chap. 5 by Hartmann et al. in this book for more information).
-
2.
Systemic Mastocytosis: This is the most common category in adults and means that the disease is detectable in an extracutaneous tissue, most often in bone marrow (see above for systemic mastocytosis diagnostic criteria). It has five subcategories.
-
(a)
Indolent Systemic Mastocytosis: This category of disease is characterized by the presence of systemic disease in bone marrow but absence of a hematologic disorder, multiple B findings, any C findings, and less than 20% mast cells in bone marrow aspirate smears. The life expectancy is similar to that in the general population, but mast cell mediator-related symptoms occur frequently. Rate of progression to a more advanced category is low (less than 5%).
-
(b)
Systemic Mastocytosis (SM) with Associated Hematologic Neoplasm (AHN): This category is diagnosed by demonstrating SM criteria as well as another coexisting hematologic disease meeting the WHO criteria. The AHN is often a chronic myeloproliferative disease (MPN-U), MDS, or MDS/MPN (CMML), but occasionally, a lymphoproliferative disease can also be diagnosed. In patients with ISM-AHN, the prognosis depends on the AHN. In advanced SM associated with AHN, the prognosis depends on both the SM and the AHN components of the disease.
-
(c)
Smoldering Systemic Mastocytosis: This category is marked by presence of 2 or 3 so called B-findings indicating large mast cell burden. First B finding is tryptase levels of >200 ng/ml and bone marrow infiltration of >30% mast cells in biopsy sections. As a second B-finding, splenomegaly and/or lymphadenopathy is frequently recorded without liver dysfunction or hypersplenism. Finally, signs of dysplasia or myeloproliferation in non-mast cell lineages may be found without an evidence of an overt hematologic disorder meeting WHO criteria. The smoldering type of SM is considered an intermediate category. Rate of progression to an advanced disease variant may be low but is not precisely known due to the rarity of this category of disease [24].
-
(d)
Aggressive Systemic Mastocytosis: This rare subtype (less than 5% of all cases) presents with C findings, reflecting organ damage (C-findings) resulting from tissue infiltration by immature mast cells. Involved tissues may include bone marrow (cytopenias: absolute neutrophils counts <1000/microliter, hemoglobin <10 g/dl, platelets <100,000/microliter), liver (hepatomegaly , portal hypertension, ascites, elevated liver function tests), spleen (splenomegaly with hypersplenism), bone (lytic lesions >2 cm with pathologic fractures), and gastrointestinal (malabsorption with hypoalbuminemia and weight loss). The C-findings must be due to mast cell infiltration [25]. Sometimes, a bone marrow-related C-finding may be difficult to attribute with certainty to mast cell infiltration in patients with AHN.
-
(e)
Mast Cell Leukemia: This is the rarest category with the poorest prognosis. Bone marrow typically shows diffuse dense infiltration with atypical mast cells. It is characterized by presence of >20% abnormal mast cells in bone marrow aspirate smears [26]. In patients with classical MCL, >10% mast cells are found in peripheral blood smears. If this is not the case (<10% mast cells of all circulating blood leukocytes), the diagnosis is aleukemic MCL.
-
(a)
-
3.
Mast Cell Sarcoma: These are rare isolated solid mast cell tumors consisting of immature mast cells with local invasion. In most patients, mast cell sarcoma progresses to MCL within short time and the prognosis remains poor [27].
Prognosis
Prognosis for cutaneous mastocytosis is excellent. In 90% of the children, skin lesions resolve or improve spontaneously. The remaining 10% is persistent and may later be diagnosed with systemic mastocytosis. SM is suspected in children who keep skin lesions after adolescence, those with persistently elevated tryptase levels >20 ng/ml, hematologic abnormalities, or hepatosplenomegaly. These patients should be evaluated for consideration of a bone marrow biopsy and aspiration [28]. Otherwise, patients with typical childhood-onset mastocytosis do not require bone marrow biopsy. Prognosis for indolent systemic mastocytosis is good, and these patients have a life expectancy comparable to that in the general population [3]. Risk of progression to an advanced variety is rare (<5%). SM-AHN, ASM, and MCL are collectively termed as advanced mastocytosis. Prognosis in SM-AHN is poorer and depends on the AHN. ASM carries a poor prognosis with an estimated 50% survival rate of about 3 years. MCL has the poorest prognosis, with most cases being fatal within a year unless treated with intensive therapy or KIT-targeting drugs [29, 30]. A chronic form of MCL with long-term survival (over 5 years) has been described, which meets histopathologic criteria for MCL but without C-findings [31].
Treatment
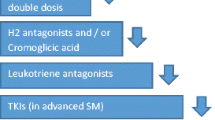
Treatment of mastocytosis is explained in more detail in other chapters of this book. All categories of mastocytosis should be treated for mast cell mediator-related symptoms [32]. H1 antihistamines are used for itching, flushing, and prophylactic treatment of anaphylactic episodes. H2 antihistamines are recommended for those with gastrointestinal symptoms such as abdominal cramping, peptic ulcers, reflux, bloating, and diarrhea. Anti-leukotriene agents may be added in patients with refractory skin or GI symptoms. Oral cromolyn can be used as a mast cell stabilizer especially in patients with gastrointestinal symptoms. Oral steroids may be effective in those with advanced disease with liver involvement or recurrent anaphylaxis. Omalizumab has been reported to be effective in preventing anaphylactic episodes [33, 34]. There is a clearly increased risk for anaphylaxis compared to the healthy background population, and the lifetime risk of anaphylaxis is approximately 40% in adult-onset mastocytosis (<10% in children-onset cases) [35]. Anaphylaxis may be associated with IgE-mediated (hymenoptera stings) sensitization or non-IgE-mediated drug reactions (such as NSAIDs, physical factors including exercise) or idiopathic [5]. Risk factors for anaphylaxis include IgE levels of >15, tryptase <40 ng/ml, absence of skin lesions, and male gender [36]. Self-injectable epinephrine should be prescribed for all patients. All patients with anaphylaxis should be evaluated thoroughly for an underlying, relevant IgE-mediated allergy, and relevant prophylactic measures should be taken (e.g. allergen avoidance or immunotherapy in patients with venom allergy) [37]. In this regard, it is worth noting that in patients with mastocytosis, it is sometimes difficult to document IgE involvement by conventional allergy tests. In patients with severe recurrent anaphylaxis (MCAS) without identifiable cause, treatment with omalizumab may be required to control symptoms. Treatment of advanced disease requires cytoreductive therapies such as IFN-alpha, cladribine [38], or tyrosine kinase inhibitor midostaurin [39]. Newer kinase inhibitors such as avapritinib [40] and a monoclonal antibody targeting Siglec 8 [41], a surface receptor with inhibitory signaling, are under clinical trial at the time of writing this text. Stem cell transplantation may also be considered in select cases [20].
Conclusions
Mastocytosis is a hematopoietic disorder of the mast cell progenitor resulting in pathologic accumulation and activation of mast cells. It has been increasingly recognized and diagnosed owing to increased public awareness, especially of low disease burden states presenting with mast cell activation symptoms or anaphylaxis, and refining of diagnostic criteria including availability of sensitive KIT D816V mutation detection techniques. Emerging therapies include cytoreductive avapritinib targeting KIT D816V mutation as well as those targeting mast cell activation such as omalizumab, midostaurin, or anti-Siglec 8. More research is needed in areas of therapeutics, biomarker discovery, and prognostic markers.
References
Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885–6.
Brockow K. Epidemiology, prognosis, and risk factors in mastocytosis. Immunol Allergy Clin N Am. 2014;34(2):283–95.
Cohen SS, Skovbo S, Vestergaard H, Kristensen T, Møller M, Bindslev-Jensen C, Fryzek JP, Broesby-Olsen S. Epidemiology of systemic mastocytosis in Denmark. Br J Haematol. 2014;166(4):521–8.
Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Alvarez-Twose I, Matito A, Broesby-Olsen S, Siebenhaar F, Lange M, Niedoszytko M, Castells M, Oude Elberink JNG, Bonadonna P, Zanotti R, Hornick JL, Torrelo A, Grabbe J, Rabenhorst A, Nedoszytko B, Butterfield JH, Gotlib J, Reiter A, Radia D, Hermine O, Sotlar K, George TI, Kristensen TK, Kluin-Nelemans HC, Yavuz S, Hägglund H, Sperr WR, Schwartz LB, Triggiani M, Maurer M, Nilsson G, Horny HP, Arock M, Orfao A, Metcalfe DD, Akin C, Valent P. Cutaneous manifestations in patients with mastocytosis: consensus report of the European competence network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European academy of Allergology and clinical immunology. J Allergy Clin Immunol. 2016;137(1):35–45.
Schuch A, Brockow K. Mastocytosis and anaphylaxis. Immunol Allergy Clin N Am. 2017;37(1):153–64.
Matito A, Alvarez-Twose I, Morgado JM, Sánchez-Muñoz L, Orfao A, Escribano L. Anaphylaxis as a clinical manifestation of clonal mast cell disorders. Curr Allergy Asthma Rep. 2014;14(8):450.
Horny HP, Sotlar K, Sperr WR, Valent P. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: a histopathological challenge. J Clin Pathol. 2004;57(6):604–8.
Greene LW, Asadipooya K, Corradi PF, Akin C. Endocrine manifestations of systemic mastocytosis in bone. Rev Endocr Metab Disord. 2016;17(3):419–31.
Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420–7.
Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nuñez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, Parwaresch RM, Austen KF, Lennert K, Metcalfe DD, Vardiman JW, Bennett JM. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25(7):603–25.
Sperr WR, Escribano L, Jordan JH, Schernthaner GH, Kundi M, Horny HP, Valent P. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res. 2001;25(7):529–36.
Teodosio C, Mayado A, Sánchez-Muñoz L, Morgado JM, Jara-Acevedo M, Álvarez-Twose I, García-Montero AC, Matito A, Caldas C, Escribano L, Orfao A. The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J Leukoc Biol. 2015;97(1):49–59.
Bibi S, Langenfeld F, Jeanningros S, Brenet F, Soucie E, Hermine O, Damaj G, Dubreuil P, Arock M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am. 2014;34(2):239–62.
Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S, Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493–8.
Schwartz LB. Diagnostic value of tryptase in anaphylaxis and mastocytosis. Immunol Allergy Clin N Am. 2006;26(3):451–63.
Lyons JJ, Yu X, Hughes JD, Le QT, Jamil A, Bai Y, Ho N, Zhao M, Liu Y, O’Connell MP, Trivedi NN, Nelson C, DiMaggio T, Jones N, Matthews H, Lewis KL, Oler AJ, Carlson RJ, Arkwright PD, Hong C, Agama S, Wilson TM, Tucker S, Zhang Y, McElwee JJ, Pao M, Glover SC, Rothenberg ME, Hohman RJ, Stone KD, Caughey GH, Heller T, Metcalfe DD, Biesecker LG, Schwartz LB, Milner JD. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. 2016;48(12):1564–9.
Sirvent AE, González C, Enríquez R, Fernández J, Millán I, Barber X, Amorós F. Serum tryptase levels and markers of renal dysfunction in a population with chronic kidney disease. J Nephrol. 2010;23(3):282–90.
Sperr WR, El-Samahi A, Kundi M, Girschikofsky M, Winkler S, Lutz D, Endler G, Rumpold H, Agis H, Sillaber C, Jäger U, Valent P. Elevated tryptase levels selectively cluster in myeloid neoplasms: a novel diagnostic approach and screen marker in clinical haematology. Eur J Clin Investig. 2009;39(10):914–23.
Akin C, Scott LM, Kocabas CN, Kushnir-Sukhov N, Brittain E, Noel P, Metcalfe DD. Demonstration of an aberrant mast-cell population with clonal markers in a subset of patients with “idiopathic” anaphylaxis. Blood. 2007;110(7):2331–3.
Ustun C, Reiter A, Scott BL, Nakamura R, Damaj G, Kreil S, Shanley R, Hogan WJ, Perales MA, Shore T, Baurmann H, Stuart R, Gruhn B, Doubek M, Hsu JW, Tholouli E, Gromke T, Godley LA, Pagano L, Gilman A, Wagner EM, Shwayder T, Bornhäuser M, Papadopoulos EB, Böhm A, Vercellotti G, Van Lint MT, Schmid C, Rabitsch W, Pullarkat V, Legrand F, Yakoub-Agha I, Saber W, Barrett J, Hermine O, Hagglund H, Sperr WR, Popat U, Alyea EP, Devine S, Deeg HJ, Weisdorf D, Akin C, Valent P. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol. 2014;32(29):3264–74.
Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, Castells M, Escribano L, Hartmann K, Lieberman P, Nedoszytko B, Orfao A, Schwartz LB, Sotlar K, Sperr WR, Triggiani M, Valenta R, Horny HP, Metcalfe DD. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol. 2012;157(3):215–25.
Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222–5.
Álvarez-Twose I, Jara-Acevedo M, Morgado JM, García-Montero A, Sánchez-Muñoz L, Teodósio C, Matito A, Mayado A, Caldas C, Mollejo M, Orfao A, Escribano L. Clinical, immunophenotypic, and molecular characteristics of well-differentiated systemic mastocytosis. J Allergy Clin Immunol. 2016;137(1):168–78.
Valent P, Akin C, Sperr WR, Horny HP, Metcalfe DD. Smouldering mastocytosis: a novel subtype of systemic mastocytosis with slow progression. Int Arch Allergy Immunol. 2002;127(2):137–9.
Valent P, Akin C, Hartmann K, Nilsson G, Reiter A, Hermine O, Sotlar K, Sperr WR, Escribano L, George TI, Kluin-Nelemans HC, Ustun C, Triggiani M, Brockow K, Gotlib J, Orfao A, Schwartz LB, Broesby-Olsen S, Bindslev-Jensen C, Kovanen PT, Galli SJ, Austen KF, Arber DA, Horny HP, Arock M, Metcalfe DD. Advances in the classification and treatment of Mastocytosis: current status and outlook toward the future. Cancer Res. 2017;77(6):1261–70.
Jawhar M, Schwaab J, Meggendorfer M, Naumann N, Horny HP, Sotlar K, Haferlach T, Schmitt K, Fabarius A, Valent P, Hofmann WK, Cross NCP, Metzgeroth G, Reiter A. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica. 2017;102(6):1035–43.
Ryan RJ, Akin C, Castells M, Wills M, Selig MK, Nielsen GP, Ferry JA, Hornick JL. Mast cell sarcoma: a rare and potentially under-recognized diagnostic entity with specific therapeutic implications. Mod Pathol. 2013;26(4):533–43.
Uzzaman A, Maric I, Noel P, Kettelhut BV, Metcalfe DD, Carter MC. Pediatric-onset mastocytosis: a long term clinical follow-up and correlation with bone marrow histopathology. Pediatr Blood Cancer. 2009;53(4):629–34.
Pardanani A, Lim KH, Lasho TL, Finke CM, McClure RF, Li CY, Tefferi A. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115(1):150–1.
Valent P, Oude Elberink JNG, Gorska A, Lange M, Zanotti R, van Anrooij B, Bonifacio M, Bonadonna P, Gleixner KV, Hadzijusufovic E, Perkins C, Hartmann K, Illerhaus A, Merante S, Elena C, Shoumariyeh K, von Bubnoff N, Parente R, Triggiani M, Schwaab J, Jawhar M, Caroppo F, Fortina AB, Brockow K, Fuchs D, Greul R, Yavuz AS, Doubek M, Mattsson M, Hagglund H, Panse J, Sabato V, Aberer E, Al-Ali HK, Morren MA, Varkonyi J, Zink A, Niedoszytko M, Niederwieser D, Malcovati L, Reiter A, Kennedy V, Gotlib J, Lortholary O, Hermine O, Arock M, Kluin-Nelemans H, Sperr WR, Study Group of the European Competence Network on Mastocytosis (ECNM). The data registry of the European competence network on Mastocytosis (ECNM): set up, projects, and perspectives. J Allergy Clin Immunol Pract. 2019;7(1):81–7.
Valent P, Sotlar K, Sperr WR, Reiter A, Arock M, Horny HP. Chronic mast cell leukemia: a novel leukemia-variant with distinct morphological and clinical features. Leuk Res. 2015;39(1):1–5.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets. 2006;5(1):61–77.
Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GG, Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550–1.
Broesby-Olsen S, Vestergaard H, Mortz CG, Jensen B, Havelund T, Hermann AP, Siebenhaar F, Møller MB, Kristensen TK, Bindslev-Jensen C, Mastocytosis Centre Odense University Hospital (MastOUH). Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: efficacy and safety observations. Allergy. 2018;73(1):230–8.
Broesby-Olsen S, Farkas DK, Vestergaard H, Hermann AP, Møller MB, Mortz CG, Kristensen TK, Bindslev-Jensen C, Sørensen HT, Frederiksen H. Risk of solid cancer, cardiovascular disease, anaphylaxis, osteoporosis and fractures in patients with systemic mastocytosis: a nationwide population-based study. Am J Hematol. 2016;91(11):1069–75.
Gülen T, Ljung C, Nilsson G, Akin C. Risk factor analysis of anaphylactic reactions in patients with systemic Mastocytosis. J Allergy Clin Immunol Pract. 2017;5(5):1248–55.
Niedoszytko M, Bonadonna P, Oude Elberink JN, Golden DB. Epidemiology, diagnosis, and treatment of Hymenoptera venom allergy in mastocytosis patients. Immunol Allergy Clin N Am. 2014;34(2):365–81.
Valent P, Sperr WR, Akin C. How I treat patients with advanced systemic mastocytosis. Blood. 2010;116(26):5812–7.
Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, Awan FT, Hexner E, Mauro MJ, Sternberg DW, Villeneuve M, Huntsman Labed A, Stanek EJ, Hartmann K, Horny HP, Valent P, Reiter A. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530–41.
Evans EK, Gardino AK, Kim JL, Hodous BL, Shutes A, Davis A, Zhu XJ, Schmidt-Kittler O, Wilson D, Wilson K, DiPietro L, Zhang Y, Brooijmans N, LaBranche TP, Wozniak A, Gebreyohannes YK, Schöffski P, Heinrich MC, DeAngelo DJ, Miller S, Wolf B, Kohl N, Guzi T, Lydon N, Boral A, Lengauer C. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9(414):eaao1690.
Kiwamoto T, Kawasaki N, Paulson JC, Bochner BS. Siglec-8 as a drugable target to treat eosinophil and mast cell-associated conditions. Pharmacol Ther. 2012;135(3):327–36.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Akin, C., Broesby-Olsen, S., Valent, P. (2020). Mastocytosis: Overview of Diagnosis and Classification. In: Akin, C. (eds) Mastocytosis. Springer, Cham. https://doi.org/10.1007/978-3-030-27820-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-27820-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27822-9
Online ISBN: 978-3-030-27820-5
eBook Packages: MedicineMedicine (R0)