
Abstract
MicroRNAs (miRNAs) are short RNA sequences actively involved in post-transcriptional gene regulation. Such miRNAs have been discovered in most eukaryotic organisms. They also seem to exist in viruses and perhaps in microbial pathogens to target the host. Drosha is the enzyme which first cleaves the pre-miRNA from the nascent pri-miRNA. Previously, we showed that it is possible to distinguish between pre-miRNAs of different species depending on their evolutionary distance using just k-mers.
In this study, we introduce three new sets of features which are extracted from the precursor sequence and summarize the distance between k-mers. These new set of features, named inter k-mer distance, k-mer location distance and k-mer first-last distance, were compared to k-mer and all other published features describing a pre-miRNA. Classification at well above 80% (depending on the evolutionary distance) is possible with a combination of distance and regular k-mer features.
The novel features specifically aid classification at closer evolutionary distances when compared to k-mers only. K-mer and k-mer distance features together lead to accurate classification for larger evolutionary distances such as Homo sapiens versus Brassicaceae (93% ACC). Including the novel distance features further increases the average accuracy since they are more effective for lower evolutionary distances. Secondary structure-based features were not effective in this study. We hope that this will fuel further analysis of miRNA evolution. Additionally, our approach provides another line of evidence when predicting pre-miRNAs and can be used to ensure that miRNAs detected in NGS samples are indeed not contaminations. In the future, we aim for automatic categorization of unknown hairpins into all species/clades available in miRBase.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Background
Dysregulation of gene expression often defines a disease and microRNAs (miRNAs) are post-transcriptional regulators strongly influencing protein abundance. Mature miRNAs (18–24 nt in length; single stranded) are produced from precursor miRNAs (pre-miRNAs) which are excised from nascent RNA (Erson-Bensan 2014). While miRNAs are described for large parts of the phylogenetic tree, the molecular pathway for plants and animals may have evolved independently (Chapman and Carrington 2007). Both, however, share that pri-miRNAs are transcribed, hairpins (pre-miRNAs) are cleaved from them and that the mature miRNA is incorporated into a protein complex which performs the targeting with the mature sequence as a key element. MicroRNAs have been described for a variety of species ranging from viruses (Grey 2015) to plants (Yousef et al. 2016). Due to involved experimental detection mechanisms, there is reliance on computational approaches to detect miRNAs and many approaches have been developed (Yousef et al. 2006; Allmer and Yousef 2012; Saçar Demirci and Allmer 2014). Many such approaches are based in machine learning and these, with few exceptions (Dang et al. 2008; Yousef et al. 2008; Khalifa et al. 2016), perform two class classification. MicroRNAs and microRNA targets are collected in databases like miRTarBase (Hsu et al. 2014), TarBase (Vergoulis et al. 2012), and MirGeneDB (Fromm et al. 2015) which generally depend on miRBase (Kozomara and Griffiths-Jones 2011) which is the main collection of all miRNAs.
Hundreds of features have been proposed (Saçar Demirci and Allmer 2013) for the parameterization of pre-miRNA sequences. Saçar Demiric et al. (2017) and miRNAfe (Yones et al. 2015) implemented almost all of the published features categorized into sequence, structural, thermodynamic, probabilistic based ones or a mixture of these types which can further be normalized by other features like stem length, number of stems, or similar. The tool, izMiR, evaluated the previously published approaches in terms of their selected feature sets (Saçar Demirci et al. 2017).
Short nucleotide sequences (k-mers) have been used early on for the machine learning-based ab initio detection of pre-miRNAs (Lai et al. 2003). Additionally, we have recently conducted studies to answer the question whether the pre-miRNA sequence (ignoring the secondary structure) can be differentiated among species and may, therefore, contain a hidden message that could influence recognition via the protein machinery of the miRNA pathway. We further investigated whether there is a consistent difference among species taking into account their evolutionary relationship.
In order to answer these questions, we established random forest machine learning models using two class classification with the positive class being pre-miRNAs from one species/clade and the negative pre-miRNAs from a different species/clade (Yousef et al. 2017a) and found that distantly related species can be distinguished on this basis. In another recent study (Yousef et al. 2017b), we corroborated on this approach and introduced information-theoretic features but found that k-mers were sufficient for this type of analysis. Here, we have established novel features based on k-mers and compare the performance results with other type of features. The new k-mer distance features perform slightly better (on average ~1%) than k-mer features and is slightly less effective (on average ~0.6%) when compared to selected features from all categories. Combining the k-mer distance with the simple k-mer feature sets does not improve performance. However, the novel features are more successful for closer evolutionary distances. In conclusion, the usage of the novel k-mer distance feature set can be encouraged in future studies aiming to differentiate among species based on their miRNAs. In the future, we aim to further analyze the importance of the location of k-mers within a miRNA and the distance among k-mers in order to find a biological interpretation and we will establish an automated categorization system which will place pre-miRNA candidates into their clade/species of origin.
2 Methods
2.1 Parameterization of Pre-miRNAs
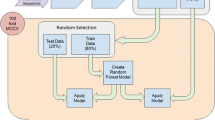
Recently, we have shown that k-mers are sufficient to allow categorization of pre-miRNAs into their species of origin (Yousef et al. 2006). Here we use k-mer features as described in (Yousef et al. 2006). In addition to k-mers we use inter k-mer distance where for each k-mer we find its first occurrence in the sequence and then calculate its distance to each k-mer’s terminal occurrence in the sequence including the subject k-mer. The sum of these distances computes the overall score which is further normalized by the length of the sequence. Another novel set of features is the k-mer first-last distance which is the distance between the first occurrence and last occurrence of a k-mer within the pre-miRNA sequence. The distance is normalized to the length of the pre-miRNA sequence. Finally, we introduce the k-mer location distance which concerns the average of k-mer distances between locations (dl = dl/|loci|). If the k-mer is not found in the sequences the value will be -1 and if it appears only once the value of its feature will be 0. For comparison we also include known secondary structure based features: (1) Number of Base Pairs, Number of Bulges, (2) Number of Loops, (3) Number of bulges with length i (i = 1 to 6), (4) Number of bulges with length greater than 6, (5) Number of loops with length I, i = 1 to 6 (odd number capture asymmetric loops), and (6) Number of loops with length greater than 6. A KNIME workflow (Berthold et al. 2008) was created to extract those features using the secondary structures obtained from the mirBase (Griffiths-Jones 2010).
The data consists of information from 15 clades. The sequences of Homo sapiens were taken out of the data of its clade Hominidae. The process of removing homology sequences (keeping just one representative) consisted of combining all clades and Homo sapiens sequences into one dataset and then applying the USEARCH (Edgar 2010) to clean the data by removing similar sequences. The USEARCH tool clustered the sequences by similarity. From each cluster, one representative was chosen to form a new dataset with non-homologous sequences. The new dataset was then broken into clades without similar sequences between each pair of clades. Cleaning the data ensured that the results were accurate. The following clades and species from miRBase were used: Hominidae, Brassicaceae, Hexapoda, Monocotyledons (Liliopsida), Nematoda, Fabaceae, Pisces (Chondricthyes), Virus, Aves, Laurasiatheria, Rodentia, Homo sapiens, Cercopithecidae, Embryophyta, Malvaceae, Platyhelminthes.
Following the study of (Yousef et al. 2017a), we used the random forest (RF) classifier implemented by the platform KNIME (Berthold et al. 2008). The classifier was trained and tested with a split into 80% training and 20% testing data. Negative and positive examples were forced to equal amounts using stratified sampling while performing a 100-fold Monte Carlo cross-validation (MCCV) (Xu and Liang 2001) for model establishment. For each established model, we calculated a number of statistical measures like the Matthews’s correlation coefficient (Matthews 1975), sensitivity, specificity, and accuracy for evaluation of model performance. All reported performance measures refer to the average of 100-fold MCCVs.
3 Results and Discussion
We have previously shown that k-mers may be sufficient to allow the categorization of miRNAs into species (Yousef et al. 2017b). For this study, we selected pre-miRNAs of a number of species and/or clades to analyze the ability of three new set of features to aid the categorization of pre-miRNAs into their species/clades (see Methods). The selected data represents a range of clades at various evolutionary distances to ensure comprehensive testing. For each pair of species/clades we trained a classifier. We compare k-mer features and all published features with our new feature sets (Table 1).
In summary, secondary structure based features are not as successful when used for categorizing pre-miRNAs into species/clades. Using all published features is as successful as using the best 100 from k-mer and k-mer distance features selected by information gain. K-mer location distance feature is most successful among the k-mer distance features with similar accuracy as using k-mers alone.
4 Conclusions
Here we conducted experiments using a novel transformation of the k-mer features used to parameterize pre-miRNAs for machine learning. Three k-mer distance features, inter k-mer, k-mer first-last, and k-mer location distance were examined and compared to regular k-mer and most published features for pre-miRNA parameterization.
In general, categorization is better for more distant species/clades. The ability of k-mer features to perform accurate categorization at larger evolutionary distances confirms our previous observation (Yousef et al. 2017a, b). K-mer inter and k-mer location distance perform similar to k-mer alone while their combination followed by selection of the best 100 features using information gain leads to a slight increase in average accuracy of 1%. We selected parameters describing the secondary structure of pre-miRNAs in order to understand their contribution for categorization and found that they are on average about 10% less accurate (Table 1). This finding supports the conservation of structure over the conservation of sequence. In conclusion, k-mer and k-mer distance features together lead to accurate categorization for larger evolutionary distances such as Homo sapiens versus Brassicaceae (93% ACC). Including the novel distance features further increases the average accuracy since they are more effective for lower evolutionary distances; while using secondary structure-based features is not as effective. We hope that this will fuel further analysis of miRNA evolution. Additionally, our approach provides another line of evidence when predicting pre-miRNAs and can be used to ensure that miRNAs detected in NGS samples are indeed not contaminations. In the future, we aim for automatic categorization of unknown hairpins into all species/clades available in miRBase.
References
Allmer, J., Yousef, M.: Computational methods for ab initio detection of microRNAs. Front. Genet. 3, 209 (2012). https://doi.org/10.3389/fgene.2012.00209
Berthold, M.R., Cebron, N., Dill, F., et al.: KNIME: the Konstanz information miner. In: Preisach, C., Burkhardt, H., Schmidt-Thime, L., Decker, R. (eds.) Data Analysis, Machine Learning and Applications, pp. 319–326. Springer, Heidelberg (2008). https://doi.org/10.1007/978-3-540-78246-9_38
Chapman, E.J., Carrington, J.C.: Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 8(11), 884–896 (2007). https://doi.org/10.1038/nrg2179
Dang, H.T., Tho, H.P., Satou, K., Tu, B.H.: Prediction of microRNA hairpins using one-class support vector machines. In: 2nd International Conference on Bioinformatics and Biomedical Engineering, iCBBE 2008, pp. 33–36 (2008)
Edgar, R.C.: Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19), 2460–2461 (2010). https://doi.org/10.1093/bioinformatics/btq461
Erson-Bensan, A.E.: Introduction to MicroRNAs in biological systems. In: Yousef, M., Allmer, J. (eds.) miRNomics: MicroRNA Biology and Computational Analysis, 1st edn. Humana Press, New York, pp. 1–14 (2014)
Fromm, B., Billipp, T., Peck, L.E., et al.: A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 49, 213–242 (2015). https://doi.org/10.1146/annurev-genet-120213-092023
Grey, F.: Role of microRNAs in herpesvirus latency and persistence. J. Gen. Virol. 96(4), 739–751 (2015). https://doi.org/10.1099/vir.0.070862-0
Griffiths-Jones, S.: miRBase: microRNA sequences and annotation. Curr. Protoc. Bioinf. 12.9.1–12.9.10 (2010). Chap. 12. Unit. https://doi.org/10.1002/0471250953.bi1209s29
Hsu, S.-D., Tseng, Y.-T., Shrestha, S., et al.: miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 42(Database issue), D78–D85 (2014). https://doi.org/10.1093/nar/gkt1266
Khalifa, W., Yousef, M., Saçar Demirci, M.D., Allmer, J.: The impact of feature selection on one and two-class classification performance for plant microRNAs. PeerJ 4, e2135 (2016). https://doi.org/10.7717/peerj.2135
Kozomara, A., Griffiths-Jones, S.: miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39(Database issue), D152–D157 (2011). https://doi.org/10.1093/nar/gkq1027
Lai, E.C., Tomancak, P., Williams, R.W., Rubin, G.M.: Computational identification of Drosophila microRNA genes. Genome Biol. 4(7), R42 (2003). https://doi.org/10.1186/gb-2003-4-7-r42
Matthews, B.W.: Comparison of the predicted and observed secondary structure of T4 phage lysozyme. BBA - Protein Struct. 405(2), 442–451 (1975). https://doi.org/10.1016/0005-2795(75)90109-9
Saçar Demirci, M.D., Baumbach, J., Allmer, J.: On the performance of pre-microRNA detection algorithms. Nat. Commun. (2017). https://doi.org/10.1038/s41467-017-00403-z
Saçar Demirci, M.D., Allmer, J.: Data mining for microRNA gene prediction: on the impact of class imbalance and feature number for microrna gene prediction. In: 2013 8th International Symposium on Health Informatics and Bioinformatics, pp. 1–6. IEEE (2013)
Saçar Demirci, M.D., Allmer, J.: Machine learning methods for MicroRNA gene prediction. In: Yousef, M., Allmer, J. (eds.) miRNomics: MicroRNA Biology and Computational Analysis SE - 10, 1st edn., pp. 177–187. Humana Press, New York (2014)
Vergoulis, T., Vlachos, I.S., Alexiou, P., et al.: TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 40(Database issue), D222–D229 (2012). https://doi.org/10.1093/nar/gkr1161
Xu, Q.-S., Liang, Y.-Z.: Monte Carlo cross validation. Chemom. Intell. Lab. Syst. 56(1), 1–11 (2001). https://doi.org/10.1016/S0169-7439(00)00122-2
Yones, C.A., Stegmayer, G., Kamenetzky, L., Milone, D.H.: miRNAfe: a comprehensive tool for feature extraction in microRNA prediction. Biosystems 138, 1–5 (2015). https://doi.org/10.1016/j.biosystems.2015.10.003
Yousef, M., Allmer, J., Khalifa, W.: Plant microRNA prediction employing sequence motifs achieves high accuracy (2016)
Yousef, M., Jung, S., Showe, L.C., Showe, M.K.: Learning from positive examples when the negative class is undetermined–microRNA gene identification. Algorithms Mol. Biol. 3, 2 (2008). https://doi.org/10.1186/1748-7188-3-2
Yousef, M., Khalifa, W., Acar, I.E., Allmer, J.: MicroRNA categorization using sequence motifs and k-mers. BMC Bioinf. 18(1), 170 (2017a). https://doi.org/10.1186/s12859-017-1584-1
Yousef, M., Nebozhyn, M., Shatkay, H., et al.: Combining multi-species genomic data for microRNA identification using a Naive Bayes classifier. Bioinformatics 22(11), 1325–1334 (2006). https://doi.org/10.1093/bioinformatics/btl094
Yousef, M., Nigatu, D., Levy, D., et al.: Categorization of species based on their MicroRNAs employing sequence motifs, information-theoretic sequence feature extraction, and k-mers. EURASIP J. Adv. Sig. Process. 2017(70), 1–10 (2017b). https://doi.org/10.1186/s13634-017-0506-8
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Yousef, M., Allmer, J. (2019). Classification of Pre-cursor microRNAs from Different Species Using a New Set of Features. In: Anderst-Kotsis, G., et al. Database and Expert Systems Applications. DEXA 2019. Communications in Computer and Information Science, vol 1062. Springer, Cham. https://doi.org/10.1007/978-3-030-27684-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-27684-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27683-6
Online ISBN: 978-3-030-27684-3
eBook Packages: Computer ScienceComputer Science (R0)