
Abstract
Silica aerogels are the most commercially relevant aerogel materials. By volume, supercritically dried blanket composites are still leading global sales by a substantial amount; however, particle-based aerogels such as granulate and powder are cost-competitive alternatives. This chapter summarizes the last three decades of research and industrial process development in the field of “low-cost,” sodium silicate-based aerogel preparation by means of ambient pressure drying, illustrating key developments and milestones in both academic research and process engineering fields. Key process steps such as gelation, aging, hydrophobization, and APD drying are analyzed in detail in the context of feasibility, simplicity, product quality, and scalability. The chapter finishes with a brief discussion of key process parameters and their effect on the physical properties of the obtained aerogel materials as well as a current snapshot of the most promising applications for particle-based aerogels.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Sodium silicate
- Waterglass
- Ambient pressure drying
- Hydrophobization
- Parameter optimizations
- Commercialization
1 Introduction
Commercially available aerogel materials today are primarily of the silica variety. The importance of this particular chemistry rests on two main pillars, namely the availability of inexpensive silica precursors and compatible silane hydrophobes as well as their early scientific discovery and academic relevance. Silica aerogels were first prepared by S. S. Kistler at the College of the Pacific, in 1931, using sodium silicate as a precursor and supercritical drying [1]. However, the more elaborate and time-consuming supercritical drying procedure, coupled with the discovery of simpler preparation routes for porous silicates, caused people to lose interest in aerogels for roughly three decades. In 1968, a team of researchers led by Prof. S. J. Teichner at the University of Lyon, France, pioneered the alkoxide-based chemistry for silica aerogels using tetraethoxysilane (TEOS) and tetramethoxysilane (TMOS) to prepare sols and gels. Aerogels could now be prepared within by high temperature supercritical drying from their respective alcohol pore fluids in only 1 day which was significantly faster than Kistler’s original method [2]. The major simplification was the omission of an additional solvent exchange step when using alcohol as a main solvent. In 1995, Prakash et al. reported the first preparation of ambient pressure-dried sodium silicate-based films by surface chemical modification of wet silica films prior to drying [3, 4]. Soon thereafter, Deshpande [5] and Schwertfeger [6, 7] were able to produce aerogels from a waterglass precursor through Na+ ion exchange followed by surface chemical modification and an ambient pressure drying (APD) protocol. This exciting period marked the birth of the two archetypal silica aerogel preparative techniques, namely alcohol-based alkoxide and aqueous sodium silicate chemistries. These are the technological foundation of today’s two major aerogel production companies Aspen Aerogels and Cabot.
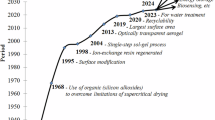
Both systems have their advantages: the alkoxide precursor route may appear more costly when comparing only raw materials cost, but processing is simplified because the pore fluid is already an organic solvent, which in theory allows a solvent exchange-free process. Though sodium silicate is the cheaper precursor, the use of an aqueous gel medium requires more cumbersome solvent exchange and drying process technology. Following the cornerstone works by Schwertfeger and Desphande, the early 2000s sparked a strong academic interest in ambient pressure dried sodium silicate aerogels [6, 8]. A large number of early studies described the effect of specific synthetic parameters on the final material properties while simultaneously targeting process simplifications as shown in Fig. 16.1. After 2012 and with the publishing of the first edition of the Aerogel handbook, researchers began to expand their studies into three main areas, namely i) more efficient “one-pot” synthesis routes geared towards process improvement and commercialization [9,10,11,12], ii) the development of aerogel composites with properties tailored towards specific applications [13,14,15,16,17,18,19,20,21,22], and iii) efforts to source the raw materials for aerogel preparation from silica-rich bio- or industrial wastes through suitable recycling protocols [23, 24]. The commercial significance of silica aerogels derived from low-cost sodium silicate precursors was recognized early on by Kistler, yet even now – a decade and a half into the second attempt to aerogel commercialization – there is still room for improvement and the need to develop a faster and cheaper production method is bigger than ever. In this chapter we are describing the chemistry and preparation of water-soluble silicate-based aerogel materials, their technical relevance as well as their applications in industry.

Histogram of the published history of the ambient pressure dried (APD) waterglass-based silica aerogels
1.1 Sodium Silicate Chemistry and Technical Relevance
Sodium silicate is the common name of hydrated alkali silicate compounds with the formula of Na2O ∙ n SiO2; it is well known as waterglass or liquid glass. Traditionally, sodium silicate is used in the area of adhesives, passive fire protection, anticorrosion paints, ceramics technology, detergent and binder auxiliaries for textile and lumber processing and of course also as a precursor for the preparation of silica gels. The applications of sodium silicates rely significantly on its composition, which follows the SiO2-Na2O-H2O ternary phase diagram, shown in Fig. 16.3 [25]. A well-known member is sodium meta-silicate, Na2SiO3, which is an anhydrate with 50% SiO2 and Na2O. As far as aerogel synthesis is concerned, high sodium ion contents are undesirable in terms of final materials properties because sodium ions are undesirable guest species in the final aerogel. Hence a solution with a low molar ratio (e.g., SiO2:Na2O = 1:1), consisting primarily of monomers (SiO44−) and dimers (e.g., Si2O52−), is less suitable for aerogel preparation. Sodium silicate solutions with a molar ratio above 3:1 (Fig. 16.2), and thus a higher proportion of polysilicate species, are more commonly used for aerogel synthesis [26]. Commercially available sodium silicate solution typically have SiO2: Na2O molar ratio is in the range of 1.5:1–3.5:1(region A in Fig. 16.3). Chemically speaking, these silicate hydrates are between true solutions (region C, such as Na2SiO3), and colloidal suspensions (region B), which consist of a mixture of polymeric and oligomeric silicates (depending on the molar ratio of SiO2:Na2O) and will be largely protonated owing to the weakness of silicic acid [25]. The solution pH also depends on SiO2/Na2O molar ratio. A saturated waterglass solution with relatively high SiO2/Na2O ratio is a viscous liquid with a density around 1.4 g cm−3 and a pH value around 12.5.

Histogram of SiO2:Na2O molar ratio used in the literature for silica aerogel synthesis

Three-component phase diagram for the system SiO2–Na2O–H2O showing the range of compounds, anhydrates, and solutions stable at room temperature, together with commercial products [25]. (Modified from Copyright 2002, Elsevier B.V)
Commercially, waterglass is synthesized by the solid-state reaction of quartz sand with sodium hydroxide, sodium carbonate, and/or sodium sulfate at elevated temperature, which leads to the solid form of sodium silicate or cullet. In a second step, cullet is dissolved into aqueous sodium hydroxide, the stoichiometry between cullet and base defining the SiO2:Na2O molar ratio of the waterglass solution. Given the wide abundance and inexpensive nature of the reactants, waterglass is among the least expensive commercial, soluble sources for silica. Besides, it is easy to handle and does not have the flammability hazards of their metal organic silicon alkoxide counterparts such as TEOS or TMOS. It is also chemically long-term stable under standard conditions of use and storage. Consequently, waterglass is an ideal precursor for silica gel and aerogel manufacture at industrial scale.
In order to make aerogels more competitive with other conventional insulation materials, cost reduction is a major challenge for industrial manufacturers and possible newcomers to this market. There are two main cost drivers for aerogel materials, namely i) raw materials cost and ii) the process cost which is basically the sum of discounted capital investments into the manufacturing line (CAPEX) and the cost of operation (OPEX). Process cost depends largely on the different process steps, where a large part of industrial R&D is focusing. For silica aerogels, raw materials typically account for roughly half the total manufacturing cost. The lion’s share goes to the silica gel precursor whereas hydrophobization agents typically come second. Note that for an industrial process to be viable, all washing and exchange solvents must be completely reused and that losses of solvent through volatile organic carbon (VOC) emissions also need to be addressed. It is only through completely closed process design that cost-effective manufacturing of aerogels is even theoretically conceivable.
Currently, the Cabot Nanogel product and JIOS powder products (Chap. 64) are manufactured from waterglass precursors in a process RIa/HAe and IRe/HAb process according to the definition defined in Table 16.1 and the graphical representation shown below in Fig. 16.4 [9].

Schematic showing the generally accepted pathways to APD silica aerogel preparation from sodium silicate through gelation to aging, solvent exchange, and drying
2 Comparison of Commonly Used Synthetic Methods
2.1 Evolution of Different Sodium Silicate-Based APD Aerogels
At this point, it seems worth mentioning that the chemistry of silicates in aqueous solution is rather complex. In some published work, the alkoxysilane sol–gel chemistry language had been used to describe the formation of hydrogels from waterglass or ion exchanged waterglass (silicic acid oligomers). In waterglass chemistry for example there is no hydrolysis step and hence the use of this term is technically incorrect. Despite the obvious similarities between waterglass and alkoxide-based silica gel systems, the limitation of such analogies and the respective mechanistic differences made must always be kept in mind. The main challenges in the synthesis of aerogels from sodium silicate are linked to the removal of sodium ions on the one hand and the introduction of the hydrophobe on the other hand. Both actions can be done at various stages of the synthesis process, triggered correspondingly at the sol, hydrogel, aged hydrogel, or organogel state. Common strategies for sodium ion removal (IR) and hydrophobe addition (HA), respectively, are IRa, IRc, IRd, IRe and HAb, HAc, HAd, and HAe, respectively, which will be elucidated in more detail in the following discussion.
As previously mentioned, sodium silicate is an ideally suited and commonly used precursor for sol–gel materials. In order to better understand its chemistry, let us discuss the different process steps for sodium silicate-based APD aerogels. Figure 16.4 shows the process steps i) through v) from sodium silicate to the final aerogel material, which are:
-
(i)
Preparation of a silicate sol by dilution of a sodium silicate solution and optional ion exchange
-
(ii)
Gelation of the gel, typically initiated by neutralization (or base addition in the case of an ion exchanged sol)
-
(iii)
Aging of the gel to improve mechanical strength needed for APD
-
(iv)
Solvent exchange into an organic solvent system suitable for APD
-
(v)
Ambient pressure drying of the gel by evaporation of the pore liquid
Let us begin with the discussion of the original method described by S. S. Kistler in his pioneering work in 1931 [27]. His early works describe gel formation (step ii) from sodium silicate (specific gravity 1.15) adjusted to equivalent SiO2 concentration between 8 and 24 weight percent (step i). Following aging (step iii), the aqueous pore fluid was exchanged to ethanol or methanol (step iv) and the gel was dried by high temperature supercritical drying directly from ethanol (step v). The process simplification which allows for effective APD was developed by Desphande and Schwertfeger and consists primarily in combining the solvent exchange together with a surface modification treatment to render the gel surfaces hydrophobic (HAe). HAe is probably the most common hydrophobe addition mode in modern waterglass-APD aerogel synthesis today. Yet, from the time of their discovery and first patents, substantial developments have been made to further simplify and improve process efficiency, which eventually helped enable the recent industrialization of aerogels. Of particular relevance for scalable processes is the reduction of the cycle time. Hence, the combination of aging (including optional ion removal by washing), solvent exchange, and surface modification, and drying – steps iii) through v) – are where development efforts are focusing and there is most room for improvements. Yet, also the specific conditions used to initiate the sol–gel transition have a strong influence on the final materials. In the following, we shall compare the most commonly used synthetic methods step by step and elaborate on their differences, with a view on key developments toward reducing process time and improving efficiency.
2.1.1 Neutralization and Sodium Ion Removal
In alkoxysilane chemistry (Chap. 13), the gelation process is initiated by the hydrolysis of alkoxy groups Si-OR, which leads to the formation of condensable silanol groups. By definition, hydrolysis is a chemical reaction in which chemical bonds in a molecule are broken and a water molecule enters to become a part of the final product, typically resulting in the formation of -OH functional groups. In the waterglass case, no actual hydrolysis takes place, but the gelation of basic sodium silicate is triggered by simple acid base chemistry, that is, through partial neutralization of Si-O− Na+ ion pairs according to

where HX is a Bronsted acid and X− its corresponding base. Typical acid sources used to initiate gelation by neutralization are mineral acids or low-molecular organic carboxylic acids such as acetic, oxalic, or citric acid. Alternatively, sodium ions can be removed by ion exchange at the sol-stage (IRa) that is before initiating the gelation. Ion exchange of Na+ by H+ converts the sodium silicate into a silicic acid solution and causes the pH of the sol to drop from around 12.5 to values between 1 and 2.5. Such a sodium-free silicic acid sol are then gelled by neutralization with small amounts of base (typically ammonia or sodium hydroxide). Whether one starts from sodium silicate sols or from ion exchanged silicic acid sols, the partial neutralization initiates condensation reactions, whose gelation kinetics are fastest at intermediate pH. Mechanistically, both the formation of free Si–OH covalent bonds and the overcoming of electrostatic repulsion interactions between the oligomeric silica species are prerequisites for aggregation of colloidal silicate particles and gel formation.
In this context, let us focus on the different sodium ion removal process options: When it comes to the final aerogel properties, the covalent Si-O-Si bonding character of the silicate network is quite essential for the final material properties. It is found that even relatively low residual Na+ levels cause large macropore fractions and the resulting aerogels turn white and structurally unstable. In other words, a suitable sodium ion removal strategy is the foundation of any sodium silicate-based aerogel synthesis route. Sodium-free gels are always obtained when using an ion exchanged silicic acid as a precursor (IRa, Fig. 16.4). This is the most straightforward synthetic method used for the preparation of monoliths and high quality “academic” samples [6, 28, 29]. However, the ion exchange process adds significant process complexity and production cost and is a bottleneck for the large-scale commercial production of silica aerogels. Alternatively gels can be prepared from sodium silicate and the sodium ions removed later on by washing with water or steam [30,31,32,33] after gelation (IRc) or aging (IRd) of the hydrogels. In yet another variation, the so-called single-step solvent exchange surface modification or combined solvent exchange and hydrophobization (CSH) method IRe, sodium ions are extracted together with the aqueous phase during solvent exchange and hydrophobization [9,10,11, 34].
But why is sodium ion removal so crucial for obtaining high-quality materials? Aside from experimental observations that residual Na+ negatively affect pore size distributions and mechanical strength, there is only little published work on this topic. Yet, from a conceptual standpoint there are at least three factors that affect the mesostructure of a silica gel network:
-
First, at the molecular level, visualized in Fig. 16.5a, the presence of sodium ions reduces the fraction of covalent bonds formed and consequently also the overall framework connectivity. Pauly et al. reported [43] that the covalent, “non-ionic” assembly pathways typically lead to more stable silica gel mesostructures in comparison to electrostatically assembled analogues. For a SiO2 to Na2O ratio 3:1, at least one oxygen in each silicate tetrahedron is negatively charged and the average connectivity between Q2 and Q3. This is considerably lower than for a sodium-free network where practically observed average connectivities are between Q3 and Q4. As it is known, for example in organosilane-derived aerogels (Chap. 15), a lower connectivity (e.g., T2/T3 in the case of methyltriethoxysilane (MTES) or methyltrimethoxysilane (MTMS) derived aerogels) generally leads to reduced mechanical strength and higher macroporosity.
-
Secondly, at the primary particle level (Fig. 16.5a, b), the nucleation and growth of small colloids is affected by both the reduced Si-O-Si bonding order and the ionic interactions of Na+ cations [44], which can influence structural features and mesoporosity of the gels.
-
Thirdly, interactions of colloids are also relevant at secondary particle level and again affected by surface charge and size of the primary particles [45], as well as the ionic strength and nature of the solvents. Although the presence of sodium ions will undoubtedly alter the gel structure at molecular, primary, and secondary particle levels, the dynamic nature of silicate bonded networks seems to be rather forgiving. The fact that virtually identical aerogels are obtained from ion-exchanged sols and non-ion exchanged ones with subsequent Na+ removal suggests that the gel structure for the two systems seems rather comparable in the end. This means that the structure forming effects of Na+ are either rather insignificant or completely reversible, that is, that removal of Na+ ions after gelation makes the gels converge toward the same particle network structure with primarily covalent bonding character.

Schematic showing (a) the general silica formation from sodium silicate through oligomers to primary particles, (b) TEM image showing the primary particle, and (c) SEM image showing the secondary particle network
2.1.2 Condensation (Gelation)
The chemistry of aqueous silicates and silicic acid systems is quite complex: repulsion and ion pairing are known to govern intermolecular interactions in alkali silicate solutions [46]. More than 20 different oligomer species of waterglass containing 2–8 silicon atoms have been identified by means of 29Si nuclear magnetic resonance (NMR) spectroscopy [47]. Alkaline silicates tend to form ring and cage-like structures. The condensation kinetics depend strongly on the pH value since they are governed by electrostatic interactions of charged molecular species and clusters: At pH >10, that is, where sodium silicate is the prevalent species, condensation reactions are slow, because of negative charge repulsion, lack of Si-OH species for condensation and increased competition with the backward reaction (dissolution). In addition, the condensation of sodium silicate species with themselves or a partially neutralized silicic acid/silicate molecule is disfavored, instead of this, the dimerization of two molecules of ortho-silicic acid has substantially smaller kinetic barrier, because H4SiO4 is electroneutral (the isoelectric point of bulk silica ranges from 2 to 4) compared to silicate, there is significantly less electrostatic repulsion, and the reaction is also favored from an entropy point of view (formation of H2O as a reaction product). This is why a waterglass solution is rather stable with long shelf life and does not gel unless partially neutralized. This is certainly a simplified view and the complete picture is far more complex, but the example of the dimer formation should serve an illustrative purpose and help the general discussion.
At pH <4, waterglass exists mainly in its protonated form that is as silicic acid oligomers. Even at low pH, ortho-silicic acid tends to dimerize, as shown in Fig. 16.6. Trimers such as H8Si3O10 are formed by addition of another molecule of silicic acid [49]. The stepwise condensation of small ortho-silicic acid oligomers is believed to produce mostly ring-like structures with three to six silicon atoms and a largest possible number of Si–O–Si linkages. Numerical calculations [50] predict a constant increase in the enthalpy of formation of about 17 kJ/mol for each additional molecule of H4SiO4 added. As a result, the nucleation of colloidal silica particles is believed to be occurring around a critical cluster size containing three to six silicon atoms. Now let us take a look at what happens at more neutral pH values and why condensation of smaller colloids happens primarily in that range. So far, one has established that silicon oxides exist in the form of small molecular clusters at high and low pH. In alkaline solution, electrostatic repulsion and the lack of free hydroxyl leaving groups prevent the formation of larger polycondensates and network structures. In the case of silicic acids, polycondensation in solution does also occur, but there is significant competition in the chemical equilibrium reaction which makes the overall reaction slow: the formation (forward reaction) is outweighed by the dissolution of larger silicate species because of tremendous competition of free protons for oxide groups (Si-OH and Si-O-Si). A second factor that helps explain slower polycondensation rates altogether is the lack of anionic Si–O− groups at low pH, which are far better nucleophiles than free silanol groups. Hence it follows that condensation reactions and thus gelation of silica gels are fastest in the pH range from 5 to 9. Note that gelation of silicic acid oligomers can also be induced and/or influenced by the addition of other ions, particularly earth alkali ions as used in cementitious systems. However for the preparation of aerogel materials, the use of any type of ionic guest species is highly undesirable – hence the lengthy discussion about sodium ion removal.

Mechanism of condensation of silicic acid species to generate linear and cyclic silica structures [48]. (Modified from Copyright 2012, NIH Belton)
2.1.3 Aging
Freshly prepared gels are usually very weak but also have the tendency to get progressively stronger with time when kept in their mother liquor. But what exactly is happening at the colloidal level to explain these changes? The aging process generally strengthens interparticle necks in colloidal particle type gels, which at the time of gelation are mere point contacts. Depending on the exact conditions, aging can be accompanied by a coarsening or microscopic phase separation of the gel phase. Yet, when carried out ideally, the aging process does not strongly influence a gels mesopore structure and surface area, but simply boosts its strength and final stiffness. In the case of a sodium silicate-based gel, aging is the result of deposition of residual soluble silicates in the mother liquor immediately after gelation. Yet strengthening of the gel also is observed much later on, so at later times, soluble silicates no longer come from the pore liquid but are dissolved from the gel structure. Long-term aging is hence best described by Ostwald ripening, where soluble matter is transported between different parts of a solid phase in solution, following a continuous dissolution-precipitation loop as symbolized in Fig. 16.7. The driving force for the material transport is the difference in solubility: while small particles within the gel structure or colloids have a large surface free energy [29, 51] favoring dissolution, interparticle necks have large negative curvatures and lower surface free energy hence favoring reprecipitation. In common practice, waterglass gels are aged for several hours, typically at elevated temperatures between room temperature and 70 °C leading to a significant strengthening of the gel network. A certain degree of aging is necessary for the gels to build up sufficient strength to resist capillary stresses experienced during later APD. Both temperature and pH (e.g., addition of acid) are the two factors known to have the strongest influence on the aging treatment of waterglass-based hydrogels [52, 53].

Pictorial illustration of Ostwald ripening [48]. (Modified from Copyright 2012, NIH Belton)
2.1.4 Solvent Exchange and Surface Modification (Hydrophobization)
Up until now, ambient pressure-dried silica aerogels must always be dried from organic solvents. Hydrocarbons typically serve the double purpose of drying and exchange solvents and simultaneous delivery host for hydrophobization agents to reach the gels inner surfaces. Drying from alcohol and alcohol/hydrophobe mixtures has been successfully demonstrated in the case of alkoxide-based silica aerogels. However, the most important prerequisite for APD is hydrophobization or surface modification – those terms can be used synonymously. The treatment is essential to minimize condensation of the gels during drying and to prevent structural collapse (Chap. 3).
Commonly used washing and drying procedures for waterglass are quite similar to silicon alkoxide-derived aerogels, starting from virtually identical silica hydrogels or alcogels, respectively. The main difference is in the pore fluid. Waterglass gels are typically synthesized in purely aqueous systems and alkoxysilane gels are synthesized in alcohol media, where the ethanol content is typically >95%. The choice of pore liquid affects not only the aging process (higher water content impacts kinetics of dissolution and reprecipitation of silica from and onto the gel network) but also the surface chemistry and solvent exchange step itself. As described earlier, solvent exchange, surface modification, and optional Na+ ion removal (Fig. 16.4 iv, HAe, IRe) can be done as three different treatments separately, much in the same way as ion exchange can be done during sol preparation (HAb). In general, a process with separate steps is more time-consuming and less desirable, at least if the same performance can be achieved. This is also where researchers are being creative and are finding new ways to combine and simplify process steps, eventually without taking a noticeable reduction in performance. Some of the more recent studies [37,38,39,40, 54, 55] combine solvent exchange and surface modification into a single process step, typically by a treatment with a solution containing an alcohol, a saturated linear hydrocarbon, and a hydrophobization agent, for example, IPA/n-hexane/TMCS [38]. For the preparation of aerogel powders from sodium silicate, solvent exchange and surface modification are even directly combined with gelation and aging steps, that is, n-hexane/HNO3/HMDZ [9, 56, 57].
The hydrophobization of silica gel surfaces is well documented and understood and has been described already earlier [58]. A schematic of the surface modification of silica with trimethylsilyl (TMS) groups is shown in Fig. 16.8b: Silanol groups on the gel surface serve as active sites for covalent grafting (endcapping) with TMS groups through reaction with suitable silane coupling agents (TMCS, HMDSO, or HMDZ).

(a) Histograms of agents used for silica aerogel modification, (b) hydrophobization of a surface silanol group in a silica gel with trimethylsilane (TMS) [154]. (Modified from Copyright 2018, Elsevier B.V)
However, if gels are modified with a large excess of hydrophobe, the resulting gels and aerogels should display rather similar chemistries despite their different precursors (waterglass versus alkoxides), hydrophobization agents (HMDZ, TMCS, HMDSO), as well as gelation and APD drying solvents (water, ethanol/heptane). Solid-state MAS NMR spectroscopy has proven a valuable tool to quantify the hydrophobe content of the gel for different treatments, TMS contents are typically in the range of 22–27 wt.% [59]. Another surprising fact is that the exchange kinetics of hydroxyl bearing species (water / alcohol) seems rather fast under the typical conditions used for solvent exchange (room temperature – 70 °C, atmospheric pressure). Consequently, for pure hydrogels exchanged with alcohol-containing solvent mixtures over longer periods of time, replacement of silanol by alkoxy groups was observed (Fig. 16.9a), and identified/quantified by means of 1H−29Si heteronuclear solid-state NMR (Fig. 16.9b, c) [59].


(a) Schematic illustration of a silica nanoparticle: the calculations assume that the surface layer consists of Q3 (with either hydroxyl or ethoxy groups) and Q4 attached to TMS groups; the inside of the particle consists exclusively of Q4 with four siloxane neighbors, (b) 1H−29Si heteronuclear solid-state NMR spectra of sodium silicate-based silica aerogels together with their 1H and 29Si projections and quantitative 1H and 29Si spectra, the aerogels prepared from TMCS/n-heptane modification without or with ethanol solvent [59]. (Copyright 2015, American Chemical Society)
2.1.5 Drying
The APD drying mechanism of silica aerogels in general is rather well understood. In a first step, pore fluid is evaporating from the macroscopic gel surfaces. This leads to a shrinkage of the gel and building up of internal strain – similar to charging a spring – up to the point where it can no longer contract further. At this point, further evaporation of pore fluid causes a gradual emptying of the mesopores. It is during this stage where gas, liquid, and solid phase coexist within the gel and stresses on the gel are maximal. As the pores are nearing the point of complete emptying, the internal strain competes against capillary forces, eventually allowing the gel to retrieve most of its original pore volume and dimensions. This reversible shrinkage is also called spring-back effect [4]. The exact extent of the maximum deformation state of the surface-modified gel structure depends on a number of factors, namely the solvent type, vapor pressure, surface tension, etc. Together with the drying rate and this will influence the structure and final form (monolith, granule, powder) of the products. Note that high surface coverage of the inner gel surface with hydrophobic groups to suppress condensation reactions in the shrunk state is an essential prerequisite for APD.
The reversible shrinkage mechanism that we discussed above apply to both alkoxide-based and sodium silicate-based SiO2 aerogels. In the cases of the inorganic aerogels, for example, zeolite [60, 61] or alumina [62], if their inner surfaces are modified, they can also be dried from ambient pressure atmosphere. Hence, in the next section, the uniqueness of drying routes for sodium silicate aerogel shall be discussed in more details.
In the literature, four forms of APD waterglass aerogel materials have been reported, that is, monolithic, granulate, bead/sphere, and powder. Monolithic samples are normally dried slowly and in two stages: first, the majority of the pore fluid is evaporated at close to ambient temperatures (25–75 °C, Fig. 16.10a) or in a nearly closed chamber with small pinholes [41], the step is typically lasting over 10 h (Fig. 16.10b). For granulate and powder materials, the first predrying step is typically done over 60 °C or completely omitted. The second step aims to cross the springback regime as quickly as possible and is done at higher temperatures between 150 °C and 200 °C. These higher drying temperatures increase the rate of removal of solvents by improved heat and mass transfer (convection), which generally helps maximize the spring-back by preventing an unnecessarily long remaining of the gel in the shrunk state where condensation reactions are competing against spring-back. Considering much faster heat and mass transfer in a small size sample, the overall drying time of powder and granulate is much shorter than monolithic samples, for example, 30–60 min drying reported by Bhagat [31, 37] and Joung [9]. Overall, regarding to the drying efficiency of the sodium silicate aerogels, powder and granulate are much higher as compared to monolithic samples.

Histograms of (a) initial drying temperature, and (b) total drying time for the preparation of four different forms of aerogel materials, powder: tens to several hundred μm; sphere/bead: several hundred μm to 1–2 mm; granulate: several mm; monolith: over several cm
More recently, Shiller et al. have reported an alternative APD mechanism for sodium silicate-based gels by means of a pore gas generated through a chemical generation which is then used to displace the pore fluid [63].
2.2 Summary of Commonly Used Synthetic Methods
2.2.1 Multistep Solvent Exchange and Hydrophobization
One of the classical waterglass-based routes goes through a multistep solvent exchange: In this approach, the pore fluid is first exchanged from water to alcohol or acetone, and then, in a second step into an organic hydrocarbon mixture, also containing a reactive hydrophobe. Typical final solvent and hydrophobe systems are linear hydrocarbons or isomer mixtures thereof such as heptane of hexane and trimethylchlorosilane (TMCS), respectively. As mentioned previously, the overall procedure is time consuming, yet the prior removal of water helps to accelerate the hydrophobization reaction. With most of the pore water and alcohol removed from the system, the hydrophobization agent TMCS [54] reacts only with surface silanol groups and is not consumed by side reactions (16.1) and (16.2).



2.2.2 Combined Solvent Exchange and Hydrophobization (CSH)
The more industrially relevant CSH method combines solvent exchange and hydrophobization into a single step. To do so, the hydrogel is immersed into a mixture of an organic solvent/hydrophobe mixture, typically (hexane or heptane)/TMCS [38,39,40]. To facilitate the transfer from the organic into the aqueous phase, amphiphilic molecules such as isopropanol (IPA), ethanol, or methanol are typically added to serve as phase transfer catalysts. For system with very fast sol–gel and exchange kinetics, primarily aerogel powder slurries, alkaline hydrophobes (hexamethyldisilazane, HMDZ) can even be added together with a hydrocarbon solvent together with the sol and the two phase mixture gelled by neutralization [37]. In such a minimal process all necessary process steps gelation/aging/solvent exchange/surface modification/sodium ion removal (steps c, d, e, i, and j) can be combined into a single one, in which case the overall sol-to hydrophobic gel process time can be significantly reduced to just a few hours [9,10,11,12, 56, 57].
Note that in the case of CSH routes, the equilibration of pore liquids inside a gel is controlled by diffusion of solvent and/or reagent like in any solvent exchange. The treatment time necessary to ensure a complete exchange and chemical reaction increases with the square of the smallest dimension of a given gel body. For this reason, it is generally far more economical to produce smaller particles or granular samples rather than larger monoliths. Certainly, the largest process time savings can be achieved already when replacing three consecutive exchanges with a single step (CSH) process. This is the reason why industrial CSH is the only viable option for the preparation of sodium silicate-based aerogels. On the other hand, there is the question of the higher hydrophobe consumption in CSH. At 4–8 US$/kg, hydrophobization agents such as HMDZ or TMCS are the most expensive components in the aerogel synthesis and losses are to be avoided at all cost. Nevertheless if used in aqueous or mixed aqueous/alcohol media, they are known to hydrolyze rather quickly to form different hydrolysates (TMS-OH, TMS-OR) as well as the silylether TMS-dimer hexamethyldisiloxane (HMDSO) as shown in (16.1) and (16.2). In the case of TMCS, during the exchange of the pore water, the water and alcohol solvent both consume TMCS. If large amounts of pore water are present, the formation of silanols is the preferred reaction [(16.1), R = H]. The hydrolysates TMS-OH and TMS-OR and the alcohol are amphiphilic molecules which promote the phase transfer and hence promote solvent exchange from the aqueous to organic medium while actively hydrophobizing the inner gel surfaces. Depending on the partition function in the organic and aqueous phases, respectively, a significant fraction of the hydrophobe typically ends up in the aqueous mixed phase. Since a large excess (>10× based on the total number of the gel’s accessible silanol groups) of hydrophobe must be used to guarantee sufficient hydrophobization to allow for APD, hydrolysis and silylether formation must be well understood and the side reaction products recirculated at industrial scale. Note that the silylether HMDSO can also be hydrolyzed under acidic conditions, thus allowing for continuous regeneration of reactive trimethylsilanol species in the reaction mixture (reaction 16.3), which can then go on to react with free silanol groups on the gel surface at the hydrocarbon/water solvent interface.
Despite the more challenging handling of hydrophobe streams in both aqueous and organic phases, CSH is always the method of choice in an industrial process because of the much higher space yield for a given size installation.
2.3 Waterglass-Based Aerogel Composites
As with many other sol–gel systems, sodium silicate gels and aerogels can be readily combined with selected inorganic or organic materials to produce aerogel composites. This is a research topic of great popularity, as it holds the promise to overcome the mechanical limitations (friability, poor tensile strength, dustiness) of pure silica aerogels. For sodium silicate-based gels, formulations are generally limited to water-soluble guest compounds as shown in Table 16.2. Aside from mechanical reinforcement, the addition of new specific chemical function is also desirable, often allowing for synergistic enhancement of some of silica aerogels outstanding thermal properties or introduction of further functional properties for use in sensing, detection, catalysis, and sorption applications.
3 Materials Properties
In the following, we are going to discuss selected materials properties of APD sodium silicate-based aerogels as published in the literature. To do so we have analyzed the key works (91 publications) starting from the introduction of APD method by Schwertfeger in 1998 until today.
Today, the APD process yields aerogels that offer properties virtually identical to supercritically dried ones. This is well reflected in the reported density, surface area, and thermal conductivity histograms as shown in Fig. 16.11: published densities range from 0.05 to 0.25 g cm−3 and surface areas from 300 to 800 m2 g−1. Again, these values compare rather well with alkoxide-based APD aerogels but also with supercritically dried waterglass and alkoxide-based silica aerogels. The corresponding histograms of APD waterglass aerogels for density and surface area are given in Fig. 16.11a, b, respectively. Surprisingly, the thermal conductivity data (Fig. 16.11c) from these collected studies is rather confusing, yet even misleading. First of all, the spread of published values is rather large but more strikingly, the published numbers are systematically much higher than one would expect. This can be attributed to three main reasons – listed here in order of relevance as perceived by the authors – namely i) poor accuracy of thermal conductivity measurement values and use of unsuitable measurement methods for thermal characterization of aerogel specimens, particularly in the form of complex shapes (granules, powder) obtained by the APD process, ii) general lack of thermal characterization equipment resulting in a comparably low fraction of specimens being characterized in terms of thermal conductivity compared to density and nitrogen sorption which also may lead to a statistical misrepresentation of high density/high thermal conductivity samples (which are easier to characterize), and iii) a large spread of materials prepared, some of the published samples exhibiting higher density and thus also higher thermal conductivity values. Although the different experimental parameters tested during process and method development activities clearly lead to dissimilar materials properties and performance, the fact that a majority of specimens are in the density range between 0.075 and 0.175 and published surface area values are also in the right range, one would indeed expect a majority population also to have thermal conductivities in the 20 mW/(m k) range, as there is a direct correlation between density/mesoporosity and thermal conductivity of silica aerogel materials. Hence the generally much too high reported thermal conductivity values presented in the histogram in Fig. 16.11 are likely to originate from difficulties and lack of experience related to the thermal characterization.

Histograms of (a) envelope density, (b) specific surface area, and (c) thermal conductivity for sodium silica aerogels [1, 6, 11,12,13,14,15,16,17,18,19,20,21,22,23, 28, 30,31,32, 34, 36,37,38,39, 45, 54, 65, 66, 68,69,70,71,72, 75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158]
In the following let us further analyze the influence of different synthetic parameters on the final materials properties.
3.1 Effect of the Silica Concentration in the Sol
In the literature, we find different definitions for the silica precursor content used in silica gel preparations, for example, the sodium silicate concentration [87], the SiO2 concentration [6], the sodium silicate to H2O molar ratio [159], or the specific density or specific gravity of sodium silicate solution used [28]. For a direct comparison with alkoxide-based aerogel works, the SiO2 concentration is probably the most relevant definition for total silicate loading in the sol. To facilitate the following discussion, silica contents from different studies have been converted to SiO2 concentration, with a histogram of typical sol concentrations given in Fig. 16.12. Similar to the alkoxide-based aerogel [160], the silica concentration in the sol is the most central parameter for the synthesis of silica aerogels. It determines the amount of silica per added sol volume and hence also the “packing density” of silica units in the gel network. So a variation of the precursor concentration under optimized processing conditions will lead to a range of materials with controllable density. Note that the processing conditions themselves can also change the properties and be valid only for a certain range of gel densities and at lower gel density values cause the collapse of the gel structure. As mentioned, key process parameters are, for example, gelation pH, aging procedure [161], the final surface coverage of the gel surface with TMS groups after the hydrophobization treatment [162] as well as residual sodium ions present in the silicate network structure [30]. Despite the dependence of the final materials properties on the entire preparation history, the following discussion is a simplified one in terms of the single variable “SiO2 concentration in the starting sol.”

Histogram of the SiO2 concentration used in the literature for sodium silicate-based aerogel synthesis
An early study by Shewale et al. [159] varied the silica mass concentration from 1 to 4 wt.% (H2O/Na2SiO3 molar ratio from 83.3 to 333.3). Acetic acid was used for neutralization, and the amount was proportionally adjusted to the silicate concentration. Gels were then aged for 2 h at 50 °C in water, followed by an ion removal step by water vapor extraction. Gels were then solvent exchanged into MeOH, and hydrophobized using a MeOH:TMCS:n-hexane modification solution (volume ratio 1:1:1) for 24 h at 50 °C. Hydrophobized gels were then dried under ambient pressure for (24 h, 1 h, and 1 h at room temperature, 50 °C, and 200 °C, respectively). It is observed that with decreasing silica concentration in the sol, the gelation time tg increases from 1 min to 6 h. This is due to the fact that with increasing dilution (H2O/SiO2 molar ratio), the colloidal particle collision frequency decreases (the mean free path of colloids increases) and thus the rate of formation of the three-dimensional “aggregated particle” network – the gel. Correspondingly, the bulk density of the final aerogel generally increases with the waterglass sol concentration. At very low silica concentrations, however, this trend is broken because in these highly dilute systems, the gel is so fragile that it cannot withstand capillary pressure and collapses during ambient pressure drying. Both the dependence of the final APD aerogel density on the silica concentration in the sol and the need for a minimal density and strength of the gel needed to withstand APD stresses are also conceptually valid for other gel or aerogel systems.
3.2 Effect of Sol pH
As explained earlier, gelation in waterglass-based gel systems is driven by condensation reactions and hence gelation kinetics depend on the pH of the hydrosol. In silicate systems, condensation rates are known to be fastest at moderate pH values. The addition of an acid to a sodium silicate or a base to a silicic acid solution leads to neutralization which then explains the acceleration in condensation and network formation. Rao et al. [90] studied the influence of the pH of the silica hydrosol on gelation time, porosity density, and optical transparency of the final product as shown in Table 16.3. The synthesis protocol started from a sodium silicate solution which was diluted to 1 wt.% SiO2 (H2O/SiO2 molar ratio 370 [90]) and ion exchanged to form a silicic acid solution with a pH value around 2. The pH of the sol was then adjusted to values from 3 to 8 by addition of 1 M NaOH solution. Wet gels were aged and processed by a consecutive washing, exchange (methanol), and hydrophobization (TMCS/n-hexane) process and dried at ambient pressure.
The gelation time tg decreased with increasing pH within the studied range. This is consistent with the highest condensation kinetics in silica hydrosols typically occurring at a pH value around 9. At a pH of 4 or lower, the colloidal silica building blocks still carry significant positive surface charges [163, 164], resulting in columbic interparticle repulsion. Two other factors that further hinder gelation at low pH are i) the strong competing effect of H+ as a catalyst, which shifts the equilibrium from the condensation reaction toward the side of hydrolysis of siloxane bonds and ii) the neutralization of deprotonated silanols (Si–O−), which is a much better nucleophile than free silanols. We also note that at low pH, a change in pH therefore has the largest effect on the gelation time. Under identical processing (solvent exchange, hydrophobization, APD) conditions, it was also found that density decreases and porosity increases with increasing pH. Best performance of the aerogels was found at pH values from 5 to 8, with porosities above 95% and envelope densities of 0.1 g/cm3 or lower. The transparency follows the density trend and can be readily explained by a pore size argument (higher density materials feature smaller mean pore sizes which also means that the fraction of scattering centers with feature sizes on the order of half the wavelength of visible light decreases substantially).
These findings are consistent with the classic formation model for silica gel originally formulated by Scherer and Brinker [165]: the gelation of acidic silica hydrosols is dominated by hydrolysis (the rate of hydrolysis kH is much larger than the rate of condensation kC) and the gelation mechanism is understood to proceed through aggregation of charged colloids (Cluster-Cluster model). Corresponding gels are weak and show a more porous primary particle structure with very high -OH functional group coverage (Fig. 16.13b). When the gelation occurs in basic media, the condensation rate kC is much higher than the hydrolysis rate and dominates the process. In this case, gelation is believed to proceed through the condensation of small molecular oligomers onto existing clusters – forming some sort of a glue, which binds cluster together. This mechanism (Monomer-Cluster model) leads to a much coarser, more aggregated primary particle structure. Such gels form more quickly and they are much stronger than acid-derived analogues, but feature also lower porosity as well as a lower -OH surface coverage.

Structural model of silica-based aerogels synthesized with different catalytic conditions [165]. (Copyright 1990, Elsevier B.V)
Most importantly, the microstructural gelation model helps to explain the results summarized in Table 16.3: Low pH hydrogels formed through the Cluster-Cluster aggregation method are initially more porous but also much weaker mechanically. Under identical aging conditions, the low-pH gels are therefore significantly weaker before being solvent exchanged and hydrophobized. It is primarily the difference in gel mechanics that explains why the low pH aerogels end up with high density and lower porosity, as they cannot withstand the stresses and collapse significantly during APD.
To reach the general conclusion that low-pH gels cannot yield satisfactory APD aerogels with low densities would be incorrect. However, for low-pH hydrogels to be converted into good quality aerogels, the aging and hydrophobization treatments must be addressed separately.
3.3 Effect of Aging (ta) Period
Gel aging is of particular relevance for waterglass APD materials as discussed above. Most typical aging processes involve keeping the gel in its mother liquor for some period of time, preferably at intermediate temperatures in the 50 °C to 70 °C range. Alternative approaches involve aging in dilute silane monomer solutions (TEOS/TMOS) [161]; however, the substantial amount of newly added silicate leads to a significant increase in the gel and aerogel (typically >0.2 g/cm3) density. The main function of the aging treatment is to strengthen interparticle necks and thereby reduces the risk of structural collapse during the subsequent processing steps, particularly APD [81, 166].
Sarawade et al. [81] studied the aging influence on the silica hydrogels synthesized from an 8%w sodium silicate solution which was ion exchanged and then gelled by addition of 1 M NH4OH. Fresh gels were aged for various periods of time from 6 to 24 h in 6 h intervals at four different temperatures (25 °C, 40 °C, 60 °C, 80 °C). It is observed that with an increase in the aging period, the density of aerogel decreases first up to 16 h aging and then increases again toward 24 h. This trend is more pronounced at higher temperature which may suggest that evaporation of water may have led to a partial drying and densification (particularly 80 °C aged samples). After combined solvent exchange and hydrophobization CSH with MeOH/n-hexane/TMCS, the gels were predried at room temperature and then dried for 2 h and 1 h, respectively, at 80 °C and 200 °C, respectively. The densities of all aerogel samples made in this way were in the range of 0.1 to 0.07 g/cm3.
Mechanistically, polycondensation of silica species still continues after the gelation with the speciation population being dominated by Q2 at the gelation point, Qn denoting the individual siloxane bonding order of any given silicon atom with n bridging Si–O–Si bonds. 29Si NMR is a well-established tool to study gel aging and can be used to follow the increase in average bonding order, commensurate with an increase of Q3 and Q4 speciation with aging time. In analogy to crosslinking reactions in polymer chemistry, an increase in Qn speciation increases both strength and stiffness of the gel network. Aging under pH neutral to slightly basic conditions also causes a coarsening of the gel network leading to the growth of thicker inter-particle necks due to an Ostwald ripening effect.
The observed APD densities are the result of an interplay between i) strengthening of the gel network which prevents structural collapse during APD – and thus maintains the low densities defined by the silica concentration in the hydrosol – and ii) a partial contraction (shrinkage) during the drying itself, due to increasing connectivity at the molecular level (average Qn value) [167]. The latter then explains the higher shrinkage and hence higher aerogel density, which are accompanied by longer aging [30, 159]. Notably, the exact aging conditions must be optimized for each particular system and also depend strongly on the sol/aging liquid pH, solvent system, etc.
3.4 Effect of Solvent Exchange
As hydrophobization agents (TMCS, HMDSO, HMDZ) are insoluble in water the pore fluid of a hydrogel needs to be exchanged into an organic solvent system before APD as discussed and illustrated in Sect. 16.2. When switching from a protic to an aprotic (hydrocarbon) medium, alcohols are ideal intermediary exchange solvents, as their bifunctional nature (polar hydroxyl group/nonpolar hydrocarbon chain) promotes miscibility of water and organic phase. What seems at first surprising is the fact that the choice of the alcohol exchange solvent in a two-step, non-CSH solvent exchange has a tremendous effect on the pore structure and also on the final materials properties: Rao et al. [168] investigated the influence of different low-molecular alcohols during solvent exchange on the silicic acid (ion exchanged WG) derived aerogels. Aged hydrogels were first exchanged with water, then with a given intermediary alcohol solvent, and finally modified with a mixture of alcohol, TMCS, and hexane. The results are summarized in Table 16.4.
The data suggests that the density and volumetric shrinkage of APD aerogels are influenced by the types of alcohols as solvent or cosolvent used in a first solvent exchange step. Note that in this study, one is looking at the combined effect of the alcohol as both first exchange solvent and APD drying cosolvent. Isopropanol and methanol seem to produce the best aerogels overall based on density. The chain length of the hydrocarbon end influences both vapor pressure and the surface tension and wetting behavior of the solvent/gel interface which is particularly relevant during APD. Generally, low vapor pressure and low surface tension solvents decrease the surface tension in the gel and produce less shrinkage. With increasing chain length of the alcohol, the hydrophobic part becomes dominant over the hydrophilic one, which reduces the exchange efficiency with water.
In conclusion, lower vapor pressure solvents and compact hydrocarbon groups (-CH3, -CH(CH3)2) are better suited for APD drying as they allow for a more efficient removal of the pore water from the hydrogel. Long chain alcohols are non-ideal drying cosolvents because of their poor performance in extracting pore water coupled with significantly higher boiling point. In CSH, methanol, ethanol, and isopropanol are the most commonly used cosolvent systems which are in full agreement with these findings.
3.5 Silylating Agents and the Hydrophobization Treatment Duration
Regardless of their sol precursor, the preparation history or the type of hydrophobe used, a successful surface modification with an excess of hydrophobization agent, and optimized synthesis process will always display remarkably similar physical properties and near-identical TMS contents in the 22–27 wt.% range [59]. From an industrial manufacturing standpoint, it is essential to know the minimum amount of silylating agent needed per equivalent of SiO2 for each preparation method.
To study the effect of the concentration and exposure time of the silylating agent on the physical properties of aerogels, an early model study employing the HMDZ/n-hexane with a consecutive solvent exchange was reported by Shewale et al. [169]: sodium silicate hydrogels were obtained by neutralization with acetic acid and Na+ ions removed by water washing. Aged gels were first exchanged into methanol, then into n-hexane, and finally into a hydrophobization mixture consisting of HMDZ/n-hexane. During the initial solvent exchange in the water/methanol solvent system, the gels sank to the bottom of the beaker (Fig. 16.14, left) [85]. Yet, after being left immersed in the hydrophobization mixture, the modified gel began to float atop the organic phase (Fig. 16.14, middle). Note that during CSH in a two-phase aqueous/organic solvent mixture, a transfer of the hydrophobized from the bottom aqueous to the top organic phase is always observed.

Image of hydrogels during the initial methanol exchange (a) and after a longer immersion period in the heptane/HMDZ hydrophobization mixture (b). A contact angle above 140° indicates a hydrophobic surface of the APD aerogel (c) [85]. (Modified from Copyright 2008, IOPScience)
In the case of the multistep solvent exchange example [29, 35, 170], a variation of the HMDZ/SiO2 molar ratio from 1.8 to 6.9 shows a noticeable effect in the APD aerogel properties: from 1.8 to 3.5, the final aerogel densities are in the desired range and show a systematic decrease from 0.117 down to 0.095 g/cm3 with increasing molar ratio and the optical transparency from 70% to 60% as shown in Fig. 16.15. The systematic decrease observed in this range is attributed to a more complete hydrophobization at higher HMDZ/SiO2 molar ratios, resulting in higher TMS surface coverage and thus better protection of the silica surfaces from direct silanol contacting which minimizes volume shrinkage during drying. For HMDZ contents above five equivalents, the aerogel properties rapidly deteriorate, yielding a density >0.2 g/cm3 and transparency below 20% for a molar ratio of 7. This trend can be explained by the solvent systems phase diagram which suggests phase separation into micelle-like HMDZ-rich subphases. This effectively lowers the surface chemical compatibility of the hydrophobe phase with the native silica surface and decreases in the surface modification efficiency.

Effect of (a) HMDZ/SiO2 molar ratio and (b) silylation period on bulk density and optical transmission of aerogels [85]. (Copyright 2008, IOPScience)
Aside from the hydrophobe amount, the duration of the silylation treatment also plays a significant role in the surface modification treatment. In the same study, the HMDZ/SiO2 molar ratio was kept fixed at 3.6, and the hydrophobization time varied from 6 to 36 h in 6 h intervals while keeping all other synthesis parameters the same. From 6 to 30 h, the bulk density of the resulting APD aerogels declined monotonically from 0.12 g/cm3 to a minimum value of about 0.085 g/cm3 and then increased again further toward 36 h. In other words, the product of the diffusion of the hydrophobe mixture into the most inner pores of the gel body combined and the kinetics of the surface modification reaction is quite slow. Other silylation agents such as TMCS are more reactive and are expected to react more quickly [171]. Furthermore, other parameters are known to accelerate the hydrophobization reaction speed, namely a reduction in gel body size (which addresses the diffusion component), an increase in temperature (which addresses both diffusion and chemical reaction kinetics) as well as the choice of the hydrophobe system itself (which primarily influences the chemical reaction kinetics). In the literature, HMDZ and TMCS are the most commonly used hydrophobes used for the preparation of silica aerogels as shown in Fig. 16.8a. They react reasonably fast with surface silanol groups and can be easily handled at laboratory scale to yield good performance materials. When targeting economically scalable process technologies geared toward industrialization [56, 63, 172], alternative silane and hydrophobization chemistries are growing fields of development efforts worldwide.
3.6 General Comments About Parameter Optimizations
So far, we have analyzed a number of parameters of the whole fabrication process of aerogels from silicates without going into too much detail. The preparation of sodium silicate aerogels by APD contains a total of 2–9 main steps (depending on whether or how ion removed, solvent exchanged, and hydrophobization or CSH are used), each of which comprises at least one main process variable. Hence, it is extremely challenging to optimize all steps involved in the preparation, or in other words, the devil is in the details. Another fact often neglected is that the ideal parameter combination for one particular step, such as for example the hydrophobization, may not be generally valid and depends on the previous history, that is, on the choice of preparation steps, methods, and the set of parameters used before. Just like in human psychology, everything is connected to everything else. Furthermore, industrial scale-up of potential attractive routes cannot always be viewed as a simple linear combination of its individual steps. Particularly the mass balance and the enrichment of different components in the various process liquid streams is to be well understood when attempting the design of feasible production schemes. Yet, the rewards can be promising: when done properly, scale-up studies coupled with systematic, fundamental research efforts have the potential to coin next generation aerogels with low thermal conductivity, high optical transparency, improved mechanical properties, and reduced dustiness accessible to the masses. This is what we are all waiting for, so please innovate!
4 Applications and Commercialization
When it comes to technical applications of aerogel materials, silica has long been the material of choice. Monsanto set up the first commercial production of Kistler’s original waterglass-based aerogels in the form of granulate and powders under the trade name Santocel in Massachusetts during WWII. This first generation of products quickly found its way into the classical insulation application, primarily for cryogenic transport and storage vessels. Other applications used aerogel powder as an additive and rheology modifier in coatings, medical, and defense applications. Santocel was also used in silicone rubber formulations and in neat form as a “physically acting” insecticide – being a strong desiccant, it essentially disrupted the water balance in insects upon contact. But what happened after Santocel was discontinued in the late 1960s? Clearly, around that time interest in aerogels faded due to lacking competitiveness as alternative insulation materials and micro/mesoporous pyrogenic and precipitated silicas became commercially available at much lower cost.
Though tremendous academic research efforts and discoveries were made in silica aerogels during the 1980s and 1990s, it took until the early 2000s for Cabot and Aspen Aerogels to decide to attempt commercialization of silica aerogel-based materials and products for the second time. Cabot is the only large-scale manufacturer of aerogel granulates that is using a sodium silicate-based APD process. The technology was acquired from the Hoechst team in the late 1990s with its pioneering works largely led by Schwertfeger. Cabot’s product is mostly larger granulate with smaller particle fractions (powders) available as a second tier product during the production. Newcomer companies such as Svenska and Jios aerogel have been focusing on smaller particles and powders exclusively. All three companies are using non-ion exchanged waterglass chemistry but a different hydrophobization and drying protocols. For a more detailed discussion on commercial manufacturers and products, the reader is referred to the industrial applications chapters (Chap. 64).
Despite the inherent advantage of sodium silicate-based aerogels in granulate and powder forms linked to the small particle size and associated rapid exchange and process times, these materials are to be thought of as semifinished products or even raw materials and require substantial product development and system integration efforts to become usable for end customers. Over the past decade a number of product developments have been made in collaboration with abovementioned aerogel manufacturers and system integrators. A few selected examples of such products include:
-
Daylighting applications in both window and GFRP (glass fiber reinforced polymer) structural cavities filled with aerogel granulate
-
Interior insulation/finishing ETICS system “StoTherm In Aevero” developed, produced, and marketed by Sto AG
-
Various aerogel particle-based blanket products (e.g., Cabot “Aeroclad,” Jios/Armacell “Armagel”) with application focusing on high-temperature insulation
-
Aerogel-based insulating plasters and other paste-like minerally bonded wet applied mortar products such as the “Fixit 222” insulation render developed by Fixit AG and Empa (Swiss Federal Laboratories for Material Science and Technology)
Given the general potential of sodium silicate aerogels as a low-cost raw materials source, the development and commercialization of additional products in different markets is essential to the survival of this fascinating class of materials. Today’s optimized manufacturing technology could allow a significant reduction in price levels from roughly 2000–2500 $/m3 (at a target density of 100 kg/m3) to less than half through improvements and economy of scale, however the main factor holding back commercial development is the lack of good products and the still prohibitively high cost for market adoption in the building and construction sector. In 2018, particle-based sodium silicate powder and granulate markets account for a total annual market volume between 25 M and 30 M US$. A recent market study predicts a rapid drop of aerogel powder and granulate prices beyond the year 2021 to less than half of today’s levels [173]. If this prediction ends up coming true, this would open doors for many new applications of silica aerogel materials, components and systems in an extended range of industries.
5 Summary
Sodium silicate aerogels are approaching their first century anniversary and are now finally quite well established, both academically and industrially. This is due to a large extent to the technical revival in the 1990s which largely coined modern production processes. From an academic perspective, the different process steps for making sodium silicate aerogels in all its colorful variations are better understood than any other aerogel synthesis procedure. Furthermore, they offer a tremendous market potential as a result of their straightforward and reliable ambient pressure drying protocols. In terms of academic progress, the room for improvement and innovation is fairly limited which is why many researchers are now moving toward hybrid gel systems (e.g., with biopolymers, see also Chap. 25).
Today’s commercial products have now been on the market for over 10 years and successfully applied to various different use cases. In the coming years, granulate and powder aerogels will still remain an important product morphologies, given the cheap and abundant raw material, fast and continuous production options, and resulting low production cost.
However, SiO2 aerogel granulates and powders are still semifinished products. In that sense, large-scale implementation primarily requires more product development efforts. Since the first APD aerogel developed by Deshpande and Schwertfeger in the mid-1990s, there were no real breakthroughs in the field anymore. Although the current adaptation of CSH-APD processes is faster and less costly, it is offset however by a partial loss of thermal and optical transparency performance. Furthermore, new competition is arising from recent progress in the fields of polymethylsiloxane [172] (see also Chap. 15) and polymer [174, 175] aerogels. In conclusion, the proliferation of sodium silicate-based aerogel materials and technologies in real-life applications will depend on the development of derived products as well as competing technologies.
References
Kistler, S.S.: Coherent expanded-aerogels. J. Phys. Chem. 36(1), 52–64 (1932)
Nicolaon, G., Teichner, S.: On a new process of preparation of silica xerogels and aerogels and their textural properties. Bull. Soc. Chim. Fr. 5, 1900 (1968)
Smith, D.M., Deshpande, R., Brinke, C.J.: Preparation of low-density aerogels at ambient pressure. MRS Online Proc. Libr. Arch. 271, 567–572 (1992)
Prakash, S.S., Brinker, C.J., Hurd, A.J., Rao, S.M.: Silica aerogel films prepared at ambient pressure by using surface derivatization to induce reversible drying shrinkage. Nature. 374(6521), 439–443 (1995)
Deshpande, R., Smith, D.M., Brinker, C.J.: Preparation of high porosity xerogels by chemical surface modification. US Patent 5,565,142, 1996
Schwertfeger, F., Frank, D., Schmidt, M.: Hydrophobic waterglass based aerogels without solvent exchange or supercritical drying. J. Non-Cryst. Solids. 225, 24–29 (1998)
Schwertfeger, F.: Process for producing organically modified aerogel. Patent WO1998005591 A1, 1998, Patents
Deshpande, R., Smith, D.M., Brinker, C.J.: Preparation of high porosity xerogels by chemical surface modification. Google Patents, 1996
Joung, Y.C., Roe, M.J., Yoo, Y.J., Park, J.C., Choi, H.J., Kim, M.W.: Method of preparing silica aerogel powder. US Patent 8,961,919, 2015
Lee, K.J., Kim, Y.H., Lee, J.K., Hwang, H.J.: Fast synthesis of spherical silica aerogel powders by emulsion polymerization from water glass. ChemistrySelect. 3(4), 1257–1261 (2018)
Júlio, M.D.F., Ilharco, L.M.: Superhydrophobic hybrid aerogel powders from waterglass with distinctive applications. Microporous Mesoporous Mater. 199, 29–39 (2014)
Rida, M.A., Harb, F.: Synthesis and characterization of amorphous silica nanoparticles from aqueous silicates using cationic surfactants. J. Metals Mater. Miner. 24(1), 37–42 (2014)
Sung Lee, J., Hong, S.K., Hur, N.J., Seo, W.-S., Hwang, H.J.: Fabrication of spherical silica aerogel/magnetite nanocomposite particles. Mater. Lett. 112, 153–157 (2013)
Lee, J.S., Lee, E.J., Hwang, H.J.: Synthesis of Fe3O4-coated silica aerogel nanocomposites. Trans. Nonferrous Metals Soc. China. 22, s702–s706 (2012)
Wang, B., Song, K., Han, Y., Zhang, T.: Synthesis and characterization of multi-walled carbon nanotube doped silica aerogels. J. Wuhan Univ. Technol. Mater. Sci. Ed. 27(3), 512–515 (2012)
Kim, Y.N., Shao, G.N., Jeon, S.J., Imran, S.M., Sarawade, P.B., Kim, H.T.: Sol–gel synthesis of sodium silicate and titanium oxychloride based TiO2–SiO2 aerogels and their photocatalytic property under UV irradiation. Chem. Eng. J. 231, 502–511 (2013)
Schäfer, H., Milow, B., Ratke, L.: Synthesis of inorganic aerogels via rapid gelation using chloride precursors. RSC Adv. 3(35), 15263 (2013)
Amirkhani, L., Moghaddas, J., Jafarizadeh, H.: Effect of surface modification agent and calcination process on the preparation of the hydrophobic magnetic silica aerogels by ambient pressure drying method. Bulg. Chem. Commun. 47, 82–88 (2015)
Amirkhani, L., Moghaddas, J., Jafarizadeh-Malmiri, H.: Candida rugosa lipase immobilization on magnetic silica aerogel nanodispersion. RSC Adv. 6(15), 12676–12687 (2016)
Yousefi Amiri, T., Moghaddas, J.: Cogeled copper–silica aerogel as a catalyst in hydrogen production from methanol steam reforming. Int. J. Hydrog. Energy. 40(3), 1472–1480 (2015)
Motahari, S., Abolghasemi, A.: Silica aerogel–glass fiber composites as fire shield for steel frame structures. J. Mater. Civ. Eng. 27(10), 04015008 (2015)
Huang, Y., Gong, L., Pan, Y., Li, C., Zhou, T., Cheng, X.: Facile construction of the aerogel/geopolymer composite with ultra-low thermal conductivity and high mechanical performance. RSC Adv. 8(5), 2350–2356 (2018)
Kow, K.-W., Yusoff, R., Aziz, A.R.A., Abdullah, E.C.: From bamboo leaf to aerogel: preparation of water glass as a precursor. J. Non-Cryst. Solids. 386, 76–84 (2014)
Shi, F., Liu, J.-X., Song, K., Wang, Z.-Y.: Cost-effective synthesis of silica aerogels from fly ash via ambient pressure drying. J. Non-Cryst. Solids. 356(43), 2241–2246
Garcı́a, N.J., Ingram, M.D., Bazán, J.C.: Ion transport in hydrated sodium silicates (water glasses) of varying water content. Solid State Ionics. 146(1–2), 113–122 (2002)
Garcia-Lodeiro, I., Palomo, A., Fernández-Jiménez, A.: Crucial insights on the mix design of alkali-activated cement-based binders. In: Handbook of Alkali-Activated Cements, Mortars and Concretes, pp. 49–73. Elsevier (2015)
Kistler, S.S.: Coherent expanded aerogels and jellies. Nature. 127, 741–741 (1931)
Parvathy Rao, A., Rao, A.V., Pajonk, G.M., Shewale, P.M.: Effect of solvent exchanging process on the preparation of the hydrophobic silica aerogels by ambient pressure drying method using sodium silicate precursor. J. Mater. Sci. 42(20), 8418–8425 (2007)
Einarsrud, M.-A., Nilsen, E.: Strengthening of water glass and colloidal sol based silica gels by aging in TEOS. J. Non-Cryst. Solids. 226(1–2), 122–128 (1998)
Gurav, J.L., Rao, A.V., Rao, A.P., Nadargi, D.Y., Bhagat, S.D.: Physical properties of sodium silicate based silica aerogels prepared by single step sol–gel process dried at ambient pressure. J. Alloys Compd. 476(1–2), 397–402 (2009)
Bhagat, S.D., Park, K.-T., Kim, Y.-H., Kim, J.-S., Han, J.-H.: A continuous production process for silica aerogel powders based on sodium silicate by fluidized bed drying of wet-gel slurry. Solid State Sci. 10(9), 1113–1116 (2008)
Li, T., Wang, T.: Preparation of silica aerogel from rice hull ash by drying at atmospheric pressure. Mater. Chem. Phys. 112(2), 398–401 (2008)
Shewale, P.M., Rao, A.V., Rao, A.P.: Effect of different trimethyl silylating agents on the hydrophobic and physical properties of silica aerogels. Appl. Surf. Sci. 254(21), 6902–6907 (2008)
Rajanna, S.K., Kumar, D., Vinjamur, M., Mukhopadhyay, M.: Silica aerogel microparticles from rice husk ash for drug delivery. Ind. Eng. Chem. Res. 54(3), 949–956 (2015)
Rao, A.P., Rao, A.V., Pajonk, G.M.: Hydrophobic and physical properties of the ambient pressure dried silica aerogels with sodium silicate precursor using various surface modification agents. Appl. Surf. Sci. 253(14), 6032–6040 (2007)
Sarawade, P.B., Quang, D.V., Hilonga, A., Jeon, S.J., Kim, H.T.: Synthesis and characterization of micrometer-sized silica aerogel nanoporous beads. Mater. Lett. 81, 37–40 (2012)
Bhagat, S.D., Kim, Y.-H., Suh, K.-H., Ahn, Y.-S., Yeo, J.-G., Han, J.-H.: Superhydrophobic silica aerogel powders with simultaneous surface modification, solvent exchange and sodium ion removal from hydrogels. Microporous Mesoporous Mater. 112(1–3), 504–509 (2008)
Lee, C., Kim, G., Hyun, S.: Synthesis of silica aerogels from waterglass via new modified ambient drying. J. Mater. Sci. 37(11), 2237–2241 (2002)
Hwang, S.W., Kim, T.Y., Hyun, S.H.: Optimization of instantaneous solvent exchange/surface modification process for ambient synthesis of monolithic silica aerogels. J. Colloid Interface Sci. 322(1), 224–230 (2008)
Hwang, S.-W., Jung, H.-H., Hyun, S.-H., Ahn, Y.-S.: Effective preparation of crack-free silica aerogels via ambient drying. J. Sol-Gel Sci. Technol. 41(2), 139–146 (2007)
Venkateswara Rao, A., Nilsen, E., Einarsrud, M.A.: Effect of precursors, methylation agents and solvents on the physicochemical properties of silica aerogels prepared by atmospheric pressure drying method. J. Non-Cryst. Solids. 296(3), 165–171 (2001)
Michael, G., Kasack, V., Nowak, R.: Hydrophobic silica. US Patent 7,282,236, 2007
Pauly, T.R., Petkov, V., Liu, Y., Billinge, S.J., Pinnavaia, T.J.: Role of framework sodium versus local framework structure in determining the hydrothermal stability of MCM-41 mesostructures. J. Am. Chem. Soc. 124(1), 97–103 (2002)
Yang, L., Wang, Y., Luo, G., Dai, Y.: A new ‘pH-induced rapid colloid aggregation’ method to prepare micrometer-sized spheres of mesostructured silica in water-in-oil emulsion. Microporous Mesoporous Mater. 94(1–3), 269–276 (2006)
Kobayashi, M., Juillerat, F., Galletto, P., Bowen, P., Borkovec, M.: Aggregation and charging of colloidal silica particles: effect of particle size. Langmuir. 21(13), 5761–5769 (2005)
Provis, J.L., Duxson, P., Lukey, G.C., Separovic, F., Kriven, W.M., Van Deventer, J.S.: Modeling speciation in highly concentrated alkaline silicate solutions. Ind. Eng. Chem. Res. 44(23), 8899–8908 (2005)
Swaddle, T.W., Salerno, J., Tregloan, P.A.: Aqueous aluminates, silicates, and aluminosilicates. Chem. Soc. Rev. 23(5), 319–325 (1994)
Belton, D.J., Deschaume, O., Perry, C.C.: An overview of the fundamentals of the chemistry of silica with relevance to biosilicification and technological advances. FEBS J. 279(10), 1710–1720 (2012)
Icopini, G.A., Brantley, S.L., Heaney, P.J.: Kinetics of silica oligomerization and nanocolloid formation as a function of pH and ionic strength at 25 C. Geochim. Cosmochim. Acta. 69(2), 293–303 (2005)
West, J.K., Hench, L.L.: Molecular orbital models of silica rings and their vibrational spectra. J. Am. Ceram. Soc. 78(4), 1093–1096 (1995)
Einarsrud, M.-A., Britt Kirkedelen, M., Nilsen, E., Mortensen, K., Samseth, J.: Structural development of silica gels aged in TEOS. J. Non-Cryst. Solids. 231(1–2), 10–16 (1998)
Hæreid, S., Anderson, J., Einarsrud, M., Hua, D., Smith, D.: Thermal and temporal aging of TMOS-based aerogel precursors in water. J. Non-Cryst. Solids. 185(3), 221–226 (1995)
Smitha, S., Shajesh, P., Aravind, P.R., Kumar, S.R., Pillai, P.K., Warrier, K.G.K.: Effect of aging time and concentration of aging solution on the porosity characteristics of subcritically dried silica aerogels. Microporous Mesoporous Mater. 91(1–3), 286–292 (2006)
Hwang, S.-W., Kim, T.-Y., Hyun, S.-H.: Effect of surface modification conditions on the synthesis of mesoporous crack-free silica aerogel monoliths from waterglass via ambient-drying. Microporous Mesoporous Mater. 130(1–3), 295–302 (2010)
Shi, F., Wang, L., Liu, J.: Synthesis and characterization of silica aerogels by a novel fast ambient pressure drying process. Mater. Lett. 60(29–30), 3718–3722 (2006)
Jantke, D., Hindelang, K., Weidner, R.: Economic process for the manufacture of organically modified aerogels, or lyophilizates. Patent DE201510211812, 2016
Hindelang, K., Gottschalk-Gaudig, T., Jantke, D., Weidner, R.: A process for producing organically modified aerogels. Patent DE201510207939, 2016
Prakash, S.S., Brinker, J.C., Hurd, A.J., Rao, S.M.: Silica aerogel films prepared at ambient pressure by using surface derivatization to induce reversible drying shrinkage. Nature. 375, 431–431 (1995)
Malfait, W.J., Zhao, S., Verel, R., Iswar, S., Rentsch, D., Fener, R., Zhang, Y., Milow, B., Koebel, M.M.: Surface chemistry of hydrophobic silica aerogels. Chem. Mater. 27(19), 6737–6745 (2015)
Adebajo, M.O., Frost, R.L., Kloprogge, J.T., Carmody, O., Kokot, S.: Porous materials for oil spill cleanup: a review of synthesis and absorbing properties. J. Porous. Mater. 10(3), 159–170 (2003)
Sakthivel, T., Reid, D.L., Goldstein, I., Hench, L., Seal, S.: Hydrophobic high surface area zeolites derived from fly ash for oil spill remediation. Environ. Sci. Technol. 47(11), 5843–5850 (2013)
Wu, L., Huang, Y., Wang, Z., Liu, L., Xu, H.: Fabrication of hydrophobic alumina aerogel monoliths by surface modification and ambient pressure drying. Appl. Surf. Sci. 256(20), 5973–5977 (2010)
Han, X., Hassan, K.T., Harvey, A., Kulijer, D., Oila, A., Hunt, M.R., Šiller, L.: Bioinspired synthesis of monolithic and layered aerogels. Adv. Mater. 30, 1706294 (2018)
Kim, G., Hyun, S.: Effect of mixing on thermal and mechanical properties of aerogel-PVB composites. J. Mater. Sci. 38(9), 1961–1966 (2003)
Shinozaki, N., Takahashi, R., Sato, S., Sodesawa, T.: Strength and elasticity of bimodal porous silica prepared from water glass. J. Sol-Gel Sci. Technol. 43(3), 275–282 (2007)
Setyawan, H., Balgis, R.: Mesoporous silicas prepared from sodium silicate using gelatin templating. Asia Pac. J. Chem. Eng. 7(3), 448–454 (2012)
Amiri, T.Y., Moghaddas, J., Khajeh, S.R.: Silica aerogel-supported copper catalyst prepared via ambient pressure drying process. J. Sol-Gel Sci. Technol. 77(3), 627–635 (2016)
Li, Y., Cheng, X., Cao, W., Gong, L., Zhang, R., Zhang, H.: Fabrication of adiabatic foam at low temperature with sodium silicate as raw material. Mater. Des. 88, 1008–1014 (2015)
Mazrouei-Sebdani, Z., Khoddami, A., Hadadzadeh, H., Zarrebini, M.: Synthesis and performance evaluation of the aerogel-filled PET nanofiber assemblies prepared by electro-spinning. RSC Adv. 5(17), 12830–12842 (2015)
Shao, Z., He, X., Niu, Z., Huang, T., Cheng, X., Zhang, Y.: Ambient pressure dried shape-controllable sodium silicate based composite silica aerogel monoliths. Mater. Chem. Phys. 162, 346–353 (2015)
Liu, G., Liu, Y.: Rapid synthesis of super insulation silica aerogel composites strengthened with mullite fibers. In: International Conference on Civil, Transportation and Environment (ICCTE 2016), pp. 1300–1304. Atlantis Press (2016)
Dervin, S., Lang, Y., Perova, T., Hinder, S.H., Pillai, S.C.: Graphene oxide reinforced high surface area silica aerogels. J. Non-Cryst. Solids. 465, 31–38 (2017)
Nazeran, N., Moghaddas, J.: Synthesis and characterization of silica aerogel reinforced rigid polyurethane foam for thermal insulation application. J. Non-Cryst. Solids. 461, 1–11 (2017)
Vedenin, A., Vityaz, P., Ivanova, I., Mazalov, Y.A., Pustovgar, A., Sudnik, L.: Experimental investigation of thermal insulating aerogel composites of hydrothermal reactor for biomass-to-hydrogen conversion. Int. J. Hydrog. Energy. 43(14), 6899–6903 (2018)
Wang, L., Li, Z., Jing, Q., Liu, P.: Synthesis of composite insulation materials – expanded perlite filled with silica aerogel. J. Porous. Mater. 25(2), 373–382 (2018)
Heinrich, T., Klett, U., Fricke, J.: Aerogels – nanoporous materials part I: sol-gel process and drying of gels. J. Porous. Mater. 1(1), 7–17 (1995)
Yoldas, B., Annen, M., Bostaph, J.: Chemical engineering of aerogel morphology formed under nonsupercritical conditions for thermal insulation. Chem. Mater. 12(8), 2475–2484 (2000)
Knoblich, B., Gerber, T.: The arrangement of fractal clusters dependent on the pH value in silica gels from sodium silicate solutions. J. Non-Cryst. Solids. 296(1–2), 81–87 (2001)
Marzouk, S., Rachdi, F., Fourati, M., Bouaziz, J.: Synthesis and grafting of silica aerogels. Colloids Surf. A Physicochem. Eng. Asp. 234(1–3), 109–116 (2004)
Venkateswara Rao, A., Parvathy Rao, A., Kulkarni, M.M.: Influence of gel aging and Na2SiO3/H2O molar ratio on monolithicity and physical properties of water–glass-based aerogels dried at atmospheric pressure. J. Non-Cryst. Solids. 350, 224–229 (2004)
Sarawade, P.B., Kim, J.-K., Park, J.-K., Kim, H.-K.: Influence of solvent exchange on the physical properties of sodium silicate based aerogel prepared at ambient pressure. Aerosol Air Qual. Res. 6(1), 93–105 (2006)
Bhagat, S.D., Kim, Y.-H., Moon, M.-J., Ahn, Y.-S., Yeo, J.-G.: A cost-effective and fast synthesis of nanoporous SiO2 aerogel powders using water-glass via ambient pressure drying route. Solid State Sci. 9(7), 628–635 (2007)
Chun-Xi, Z.: Recent advances in waterglass sand technologies. China Foundry. 4, 13–17 (2007)
Shewale, P.M., Rao, A.V., Gurav, J.L., Rao, A.P.: Synthesis and characterization of low density and hydrophobic silica aerogels dried at ambient pressure using sodium silicate precursor. J. Porous. Mater. 16(1), 101–108 (2007)
Bangi, U.K., Venkateswara Rao, A., Parvathy Rao, A.: A new route for preparation of sodium-silicate-based hydrophobic silica aerogels via ambient-pressure drying. Sci. Technol. Adv. Mater. 9(3), 035006 (2008)
Chandradass, J., Kang, S., Bae, D.-s.: Synthesis of silica aerogel blanket by ambient drying method using water glass based precursor and glass wool modified by alumina sol. J. Non-Cryst. Solids. 354(34), 4115–4119 (2008)
Kim, C.E., Yoon, J.S., Hwang, H.J.: Synthesis of nanoporous silica aerogel by ambient pressure drying. J. Sol-Gel Sci. Technol. 49(1), 47–52 (2008)
Kim, T., Hwang, S., Hyun, S.: Development of a continuous manufacturing process for silica sols via the ion-exchange of a waterglass. Ind. Eng. Chem. Res. 47(18), 6941–6948 (2008)
Parvathy Rao, A., Rao, A.V.: Microstructural and physical properties of the ambient pressure dried hydrophobic silica aerogels with various solvent mixtures. J. Non-Cryst. Solids. 354(1), 10–18 (2008)
Rao, A.P., Rao, A.V., Bangi, U.K.H.: Low thermalconductive, transparent and hydrophobic ambient pressure dried silica aerogels with various preparation conditions using sodium silicate solutions. J. Sol-Gel Sci. Technol. 47(1), 85–94 (2008)
Aravind, P.R., Shajesh, P., Soraru, G.D., Warrier, K.G.K.: Ambient pressure drying: a successful approach for the preparation of silica and silica based mixed oxide aerogels. J. Sol-Gel Sci. Technol. 54(1), 105–117 (2010)
Gao, G.-M., Liu, D.-R., Zou, H.-F., Zou, L.-C., Gan, S.-C.: Preparation of silica aerogel from oil shale ash by fluidized bed drying. Powder Technol. 197(3), 283–287 (2010)
Rani, T.S., Subha, M.C.S., Venkata Reddy, G., Kim, Y.-H., Ahn, Y.-S.: Synthesis of water-glass-based silica aerogel powder via with and without squeezing of hydrogels. J. Appl. Polym. Sci. 115(3), 1675–1679 (2010)
Rao, A.V., Bangi, U.K.H., Kavale, M.S., Imai, H., Hirashima, H.: Reduction in the processing time of doped sodium silicate based ambient pressure dried aerogels using shaker. Microporous Mesoporous Mater. 134(1–3), 93–99 (2010)
Sarawade, P.B., Kim, J.-K., Hilonga, A., Kim, H.T.: Influence of aging conditions on textural properties of water-glass-based silica aerogels prepared at ambient pressure. Korean J. Chem. Eng. 27(4), 1301–1309 (2010)
Sarawade, P.B., Kim, J.-K., Hilonga, A., Kim, H.T.: Production of low-density sodium silicate-based hydrophobic silica aerogel beads by a novel fast gelation process and ambient pressure drying process. Solid State Sci. 12(5), 911–918 (2010)
Sinkó, K.: Influence of chemical conditions on the nanoporous structure of silicate aerogels. Materials. 3(1), 704–740 (2010)
Venkateswara Rao, A., Ganbavle, V.V., Bangi, U.K.H., Dhere, S.L.: Influence of preparation conditions on nanoporous structure and optical transmission of sodium silicate based ambient pressure dried aerogels employing shaking. J. Porous. Mater. 18(6), 751–759 (2010)
Yuan, M.-R., Lu, J.-T., Kong, G.: Effect of SiO2:Na2O molar ratio of sodium silicate on the corrosion resistance of silicate conversion coatings. Surf. Coat. Technol. 204(8), 1229–1235 (2010)
Rao, A.V., Pajonk, G.M., Bangi, U.K.H., Rao, A.P., Koebel, M.M.: Sodium silicate based aerogels via ambient pressure drying. In: Aerogels Handbook, pp. 103–124. Springer (2011)
Sarawade, P.B., Kim, J.-K., Hilonga, A., Quang, D.V., Jeon, S.J., Kim, H.T.: Synthesis of sodium silicate-based hydrophilic silica aerogel beads with superior properties: effect of heat-treatment. J. Non-Cryst. Solids. 357(10), 2156–2162 (2011)
Sarawade, P.B., Kim, J.-K., Hilonga, A., Quang, D.V., Kim, H.T.: Synthesis of hydrophilic and hydrophobic xerogels with superior properties using sodium silicate. Microporous Mesoporous Mater. 139(1–3), 138–147 (2011)
Ding, Y., Yin, G., Liao, X., Huang, Z., Chen, X., Yao, Y.: Key role of sodium silicate modulus in synthesis of mesoporous silica SBA-15 rods with controllable lengths and diameters. Mater. Lett. 75, 45–47 (2012)
Essien, E.R., Olaniyi, O.A., Adams, L.A., Shaibu, R.O.: Sol-gel-derived porous silica: economic synthesis and characterization. J. Miner. Mater. Charact. Eng. 11(10), 976 (2012)
Hong, S.K., Yoon, M.Y., Hwang, H.J.: Synthesis of spherical silica aerogel powder by emulsion polymerization technique. J. Ceram. Process. Res. 13, s145–s148 (2012)
Jung, I.-K., Gurav, J.L., Ha, T.-J., Choi, S.G., Baek, S., Park, H.-H.: The properties of silica aerogels hybridized with SiO2 nanoparticles by ambient pressure drying. Ceram. Int. 38, S105–S108 (2012)
Liu, G., Zhou, B., Ni, X., Shen, J., Wu, G., Du, A., Zu, G.: Influence of thermal process on microstructural and physical properties of ambient pressure dried hydrophobic silica aerogel monoliths. J. Sol-Gel Sci. Technol. 62(2), 126–133 (2012)
Pouretedal, H., Kazemi, M.: Characterization of modified silica aerogel using sodium silicate precursor and its application as adsorbent of Cu 2+, Cd 2+, and Pb 2+ ions. Int. J. Ind. Chem. 3(1), 20 (2012)
Tadjarodi, A., Haghverdi, M., Mohammadi, V.: Preparation and characterization of nano-porous silica aerogel from rice husk ash by drying at atmospheric pressure. Mater. Res. Bull. 47(9), 2584–2589 (2012)
Yamagata, C., Elias, D.R., Paiva, M.R.S., Misso, A.M., Mello, S.R.H.: Influence of the precursor concentration on the characteristics of silica powder obtained from Na2SiO3 by a facile low temperature synthesis process. J. Mater. Sci. Eng. A. 2(8A), 429 (2012)
Dourbash, A., Motahari, S., Omranpour, H.: Effect of water content on properties of one-step catalyzed silica aerogels via ambient pressure drying. J. Non-Cryst. Solids. 405, 135–140 (2014)
Gao, T., Sandberg, L.I.C., Jelle, B.P.: Nano insulation materials: synthesis and life cycle assessment. Procedia CIRP. 15, 490–495 (2014)
He, P., Gao, X.-d., Li, X.-m., Jiang, Z.-w., Yang, Z.-h., Wang, C.-l., Gu, Z.-y.: Highly transparent silica aerogel thick films with hierarchical porosity from water glass via ambient pressure drying. Mater. Chem. Phys. 147(1–2), 65–74 (2014)
Nazriati, N., Setyawan, H., Affandi, S., Yuwana, M., Winardi, S.: Using bagasse ash as a silica source when preparing silica aerogels via ambient pressure drying. J. Non-Cryst. Solids. 400, 6–11 (2014)
Rizkiyanto, D., Putri, W.E., Setyawan, H., Affandi, S.: Preparation of thermal insulator of silica from waterglass using electrophorectic deposition. AIP Conf. Proc. 1586, 101–103 (2014)
Shi, H.-X., Cui, J.-T., Shen, H.-M., Wu, H.-K.: Preparation of silica aerogel and its adsorption performance to organic molecule. Adv. Mater. Sci. Eng. 2014, 1–8 (2014)
Ui, S.-W., Choi, I.-S., Choi, S.-C.: Synthesis of high surface area mesoporous silica powder using anionic surfactant. ISRN Mater. Sci. 2014, 1–6 (2014)
Yun, S., Luo, H., Gao, Y.: Superhydrophobic silica aerogel microspheres from methyltrimethoxysilane: rapid synthesis via ambient pressure drying and excellent absorption properties. RSC Adv. 4(9), 4535–4542 (2014)
Cui, S., Yu, S.-w., Lin, B.-l., Shen, X.-d., Gu, D.: Preparation of SiO2 aerogel from rice husk ash. RSC Adv. 5(81), 65818–65826 (2015)
He, S., Li, Z., Shi, X., Yang, H., Gong, L., Cheng, X.: Rapid synthesis of sodium silicate based hydrophobic silica aerogel granules with large surface area. Adv. Powder Technol. 26(2), 537–541 (2015)
Laskowski, J., Milow, B., Ratke, L.: The effect of embedding highly insulating granular aerogel in cellulosic aerogel. J. Supercrit. Fluids. 106, 93–99 (2015)
Liu, S.-W., Wei, Q., Cui, S.-P., Nie, Z.-R., Du, M.-H., Li, Q.-Y.: Hydrophobic silica aerogel derived from wheat husk ash by ambient pressure drying. J. Sol-Gel Sci. Technol. 78(1), 60–67 (2015)
Mahesh, S., Joshi, S.C.: Thermal conductivity variations with composition of gelatin-silica aerogel-sodium dodecyl sulfate with functionalized multi-walled carbon nanotube doping in their composites. Int. J. Heat Mass Transf. 87, 606–615 (2015)
Minju, N., Abhilash, P., Nair, B.N., Mohamed, A.P., Ananthakumar, S.: Amine impregnated porous silica gel sorbents synthesized from water–glass precursors for CO2 capturing. Chem. Eng. J. 269, 335–342 (2015)
Mohammadi, A., Moghaddas, J.: Synthesis, adsorption and regeneration of nanoporous silica aerogel and silica aerogel-activated carbon composites. Chem. Eng. Res. Des. 94, 475–484 (2015)
Wa, L., Fengyun, L., Fanlu, Z., Mengjing, C., Qiang, C., Jue, H., Weijun, Z., Mingwei, M.: Preparation of silica aerogels using CTAB/SDS as template and their efficient adsorption. Appl. Surf. Sci. 353, 1031–1036 (2015)
Yu, H., Liang, X., Wang, J., Wang, M., Yang, S.: Preparation and characterization of hydrophobic silica aerogel sphere products by co-precursor method. Solid State Sci. 48, 155–162 (2015)
Zong, S., Wei, W., Jiang, Z., Yan, Z., Zhu, J., Xie, J.: Characterization and comparison of uniform hydrophilic/hydrophobic transparent silica aerogel beads: skeleton strength and surface modification. RSC Adv. 5(68), 55579–55587 (2015)
Cheng, Y., Xia, M., Luo, F., Li, N., Guo, C., Wei, C.: Effect of surface modification on physical properties of silica aerogels derived from fly ash acid sludge. Colloids Surf. A Physicochem. Eng. Asp. 490, 200–206 (2016)
Cui, S., Yu, S., Lin, B., Shen, X., Zhang, X., Gu, D.: Preparation of amine-modified SiO2 aerogel from rice husk ash for CO2 adsorption. J. Porous. Mater. 24(2), 455–461 (2016)
Du, M.-H., Wei, Q., Nie, Z.-R., Cui, S.-P., Liu, S.-W., Li, Q.-Y.: A rapid and low solvent/silylation agent-consumed synthesis, pore structure and property of silica aerogels from dislodged sludge. J. Sol-Gel Sci. Technol. 81(2), 427–435 (2016)
Firoozmandan, M., Moghaddas, J., Yasrebi, N.: Performance of water glass-based silica aerogel for adsorption of phenol from aqueous solution. J. Sol-Gel Sci. Technol. 79(1), 67–75 (2016)
García-Torres, B.A., Aguilar-Elguezabal, A., Román-Aguirre, M., Álvarez-Contreras, L.: Synthesis of silica aerogels microspheres prepared by ink jet printing and dried at ambient pressure without surface hydrophobization. Mater. Chem. Phys. 172, 32–38 (2016)
Maleki, H., Duraes, L., Garcia-Gonzalez, C.A., Del Gaudio, P., Portugal, A., Mahmoudi, M.: Synthesis and biomedical applications of aerogels: possibilities and challenges. Adv. Colloid Interf. Sci. 236, 1–27 (2016)
Mazrouei-Sebdani, Z., Khoddami, A., Hadadzadeh, H., Zarrebini, M., Karimi, A., Shams-Ghahfarokhi, F.: The effect of the nano-structured aerogel powder on the structural parameters, water repellency, and water vapor/air permeability of a fibrous polyester material. Mater. Chem. Phys. 177, 99–111 (2016)
Morsi, R.M.M., Basha, M.A.F., Morsi, M.M.: Synthesis and physical characterization of amorphous silicates in the system SiO2-Na2O– RO (R=Zn, Pb or Cd). J. Non-Cryst. Solids. 439, 57–66 (2016)
Pisal, A.A., Rao, A.V.: Comparative studies on the physical properties of TEOS, TMOS and Na2SiO3 based silica aerogels by ambient pressure drying method. J. Porous. Mater. 23(6), 1547–1556 (2016)
Sorour, M.H., Hani, H.A., Al-Bazedi, G.A., El-Rafei, A.M.: Hydrophobic silica aerogels for oil spills clean-up, synthesis, characterization and preliminary performance evaluation. J. Porous. Mater. 23(5), 1401–1409 (2016)
Yang, H., Xu, H., Frank, J.K., Xu, G., Huan, W., Ni, C., Yang, Y.: Influences of SiO2/Na2O molar ratio on aging and chemical modification of water glass. Open J. Inorg. Chem. 06(02), 125–134 (2016)
Yücel, S., Karakuzu, B., Terzioğlu, P., Temel, T.M.: Influence of gel aging time on the properties of various silica aerogels synthesized from water glass. Mater. Sci. Forum. 866, 176–180 (2016)
Abdul Halim, Z.A., Yajid, M.A.M., Idris, M.H., Hamdan, H.: Effects of silica aerogel particle sizes on the thermal–mechanical properties of silica aerogel – unsaturated polyester composites. Plast. Rubber Compos. 46(4), 184–192 (2017)
Gu, B., Ko, D., Ahn, S., Choon Hyun, D., Lee, H.-K., Kim, J.: Synthesis of high purity silica from low cost water glass via sol–gel process and soxhlet extraction. J. Sol-Gel Sci. Technol. 82(3), 675–681 (2017)
Karamahmut Mermer, N., Yilmaz, M.S., Ozdemir, O.D., Piskin, M.B.: The synthesis of silica-based aerogel from gold mine waste for thermal insulation. J. Therm. Anal. Calorim. 129(3), 1807–1812 (2017)
Li, L., Wang, Z., Huang, J., Ji, S., Mei, Y., Wang, Y., Fang, Y.: Comparison of silica leaching behaviors from the acid-leached residue of catalytic gasification and combustion. Energy Fuel. 31(10), 10745–10751 (2017)
Mazrouei-Sebdani, Z., Javazmi, L., Khoddami, A., Shams-Ghahfarokhi, F., Low, T.: Fabrication of a silica aerogel and examination of its hydrophobic properties via contact angle and 3M water repellency tests. IOP Conf. Ser.: Mater. Sci. Eng. 204, 012014 (2017)
Nah, H.-Y., Parale, V.G., Jung, H.-N.-R., Lee, K.-Y., Lim, C.-H., Ku, Y.S., Park, H.-H.: Role of oxalic acid in structural formation of sodium silicate-based silica aerogel by ambient pressure drying. J. Sol-Gel Sci. Technol. 85(2), 302–310 (2017)
Pan, Y., He, S., Cheng, X., Li, Z., Li, C., Huang, Y., Gong, L.: A fast synthesis of silica aerogel powders-based on water glass via ambient drying. J. Sol-Gel Sci. Technol. 82(2), 594–601 (2017)
Pan, Y., He, S., Gong, L., Cheng, X., Li, C., Li, Z., Liu, Z., Zhang, H.: Low thermal-conductivity and high thermal stable silica aerogel based on MTMS/water-glass co-precursor prepared by freeze drying. Mater. Des. 113, 246–253 (2017)
Temel, T.M., İkizler, B.K., Terzioğlu, P., Yücel, S., Elalmış, Y.B.: The effect of process variables on the properties of nanoporous silica aerogels: an approach to prepare silica aerogels from biosilica. J. Sol-Gel Sci. Technol. 84(1), 51–59 (2017)
Yun, S., Guo, T., Zhang, J., He, L., Li, Y., Li, H., Zhu, X., Gao, Y.: Facile synthesis of large-sized monolithic methyltrimethoxysilane-based silica aerogel via ambient pressure drying. J. Sol-Gel Sci. Technol. 83(1), 53–63 (2017)
Feng, Q., Chen, K., Ma, D., Lin, H., Liu, Z., Qin, S., Luo, Y.: Synthesis of high specific surface area silica aerogel from rice husk ash via ambient pressure drying. Colloids Surf. A Physicochem. Eng. Asp. 539, 399–406 (2018)
Gao, H., Zhang, Z., Shi, Z., Zhang, J., Zhi, M., Hong, Z.: Synthesis of high-temperature resistant monolithic zirconia-based aerogel via facile water glass assisted sol–gel method. J. Sol-Gel Sci. Technol. 85(3), 567–573 (2018)
Jia, G., Li, Z., Liu, P., Jing, Q.: Preparation and characterization of aerogel/expanded perlite composite as building thermal insulation material. J. Non-Cryst. Solids. 482, 192–202 (2018)
Li, Z., Cheng, X., Gong, L., Liu, Q., Li, S.: Enhanced flame retardancy of hydrophobic silica aerogels by using sodium silicate as precursor and phosphoric acid as catalyst. J. Non-Cryst. Solids. 481, 267–275 (2018)
Wu, X., Fan, M., Shen, X., Cui, S., Tan, G.: Silica aerogels formed from soluble silicates and methyl trimethoxysilane (MTMS) using CO 2 gas as a gelation agent. Ceram. Int. 44(1), 821–829 (2018)
Zhou, T., Cheng, X., Pan, Y., Li, C., Gong, L., Zhang, H.: Mechanical performance and thermal stability of glass fiber reinforced silica aerogel composites based on co-precursor method by freeze drying. Appl. Surf. Sci. 437, 321–328 (2018)
Kistler, S.S., Caldwell, A.G.: Thermal conductivity of silica aerogel. Ind. Eng. Chem. 26(6), 658–662 (1934)
Fricke, J.: Aerogels – highly tenuous solids with fascinating properties. J. Non-Cryst. Solids. 100(1–3), 169–173 (1988)
Shewale, P.M., Rao, A.V., Gurav, J.L., Rao, A.P.: Synthesis and characterization of low density and hydrophobic silica aerogels dried at ambient pressure using sodium silicate precursor. J. Porous. Mater. 16(1), 101–108 (2009)
Wong, J.C.H., Kaymak, H., Brunner, S., Koebel, M.M.: Mechanical properties of monolithic silica aerogels made from polyethoxydisiloxanes. Microporous Mesoporous Mater. 183, 23–29 (2014)
Scherer, G.W., Hæeid, S., Nilsen, E., Einarsrud, M.-A.: Shrinkage of silica gels aged in TEOS. J. Non-Cryst. Solids. 202(1–2), 42–52 (1996)
Malfait, W.J., Verel, R., Koebel, M.M.: Hydrophobization of silica aerogels: insights from quantitative solid-state NMR spectroscopy. J. Phys. Chem. C. 118(44), 25545–25554 (2014)
Gerber, T., Himmel, B., Hübert, C.: WAXS and SAXS investigation of structure formation of gels from sodium water glass. J. Non-Cryst. Solids. 175(2–3), 160–168 (1994)
Knoblich, B., Gerber, T.: Aggregation in SiO2 sols from sodium silicate solutions. J. Non-Cryst. Solids. 283(1–3), 109–113 (2001)
Brinker, C.J., Scherer, G.W.: Sol-Gel Science. The Physics and Chemistry of Sol-Gel Processing. Elseiver (1990)
Iswar, S., Malfait, W.J., Balog, S., Winnefeld, F., Lattuada, M., Koebel, M.M.: Effect of aging on silica aerogel properties. Microporous Mesoporous Mater. 241, 293–302 (2017)
Brinter, C., Scherer, G.: The physics and chemistry of sol-gel processing. In: Sol-Gel Science, p. 373. Academic, New York (1990)
Rao, A.P., Rao, A.V., Gurav, J.: Effect of protic solvents on the physical properties of the ambient pressure dried hydrophobic silica aerogels using sodium silicate precursor. J. Porous. Mater. 15(5), 507–512 (2008)
Shewale, P.M., Rao, A.V., Rao, A.P., Bhagat, S.: Synthesis of transparent silica aerogels with low density and better hydrophobicity by controlled sol–gel route and subsequent atmospheric pressure drying. J. Sol-Gel Sci. Technol. 49(3), 285–292 (2009)
Lee, S., Cha, Y.C., Hwang, H.J., Moon, J.-W., Han, I.S.: The effect of pH on the physicochemical properties of silica aerogels prepared by an ambient pressure drying method. Mater. Lett. 61(14–15), 3130–3133 (2007)
Li-Jiu, W., Shan-Yu, Z., Mei, Y.: Structural characteristics and thermal conductivity of ambient pressure dried silica aerogels with one-step solvent exchange/surface modification. Mater. Chem. Phys. 113(1), 485–490 (2009)
Zu, G., Shimizu, T., Kanamori, K., Zhu, Y., Maeno, A., Kaji, H., Shen, J., Nakanishi, K.: Transparent, superflexible doubly cross-linked polyvinylpolymethylsiloxane aerogel superinsulators via ambient pressure drying. ACS Nano. 12(1), 521–532 (2018)
Collins, R.: Aerogels 2017–2027: Technologies, Markets and Players. 2017. https://www.idtechex.com/research/reports/aerogels-2017-2027-technologies-markets-and-players-000507.asp
Takeshita, S., Yoda, S.: Translucent, hydrophobic, and mechanically tough aerogels constructed from trimethylsilylated chitosan nanofibers. Nanoscale. 9(34), 12311–12315 (2017)
Nguyen, B.N., Meador, M.A.B., Scheiman, D., McCorkle, L.: Polyimide aerogels using triisocyanate as cross-linker. ACS Appl. Mater. Interfaces. 9(32), 27313–27321 (2017)
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rao, A.V., Zhao, S., Pajonk, G.M., Bangi, U.K.H., Rao, A.P., Koebel, M.M. (2023). Sodium Silicate-Based Aerogels by Ambient Pressure Drying. In: Aegerter, M.A., Leventis, N., Koebel, M., Steiner III, S.A. (eds) Springer Handbook of Aerogels. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-030-27322-4_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-27322-4_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27321-7
Online ISBN: 978-3-030-27322-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)