
Abstract
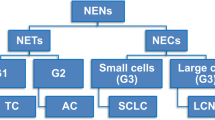
Pulmonary neuroendocrine neoplasms (NENs) are a heterogeneous group of tumors of the neuroendocrine cells, which are diffusely interspersed throughout the respiratory system. Two families of NEN are established according to histological grading: the well-differentiated neuroendocrine tumors (NETs) and the high-grade neuroendocrine carcinomas (NECs). Four types of NEN are recognized, including two NETs represented by typical carcinoid (TC) and atypical carcinoid (AC) and two NECs represented by large cell neuroendocrine carcinoma (LCNEC) and small cell lung carcinoma (SCLC).
These malignancies show different morphological, immunohistochemical, molecular, and imaging features. Such heterogeneity is reflected by extremely different clinical and biological behaviors, ranging from the indolent course of TC to the highly aggressive behavior of SCLC.
This chapter provides a detailed overview on the epidemiologic and histologic features of the pulmonary NENs, including staging, prognostic, and therapeutic aspects. Noteworthy, the chapter will focus on imaging characteristics of NENs, with detailed description of thoracic (local and nodal involvement) and extrathoracic diffusion pathways.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pulmonary neuroendocrine neoplasm
- Carcinoid
- Large cell neuroendocrine carcinoma
- Small cell lung carcinoma
- Chest X-ray
- Chest CT
- Diffusion pathways
5.1 Overview and Classification
Neuroendocrine neoplasms (NEN) are a heterogeneous group of malignancies, with different morphological, immunohistochemical, and molecular characteristics rendered into a noticeably different clinical and biological behavior [1]. The NENs are represented by cells with neuroendocrine phenotype similar to the normal cells of the diffuse neuroendocrine system [2]. Since this system is ubiquitous throughout the body, any organ can be involved by such category of malignancies. The respiratory system is the second most common site of NEN, after the gastrointestinal tract [3]. The global incidence of pulmonary NENs is about 0.2–2/1,000,000/year [4]. Despite rare, NENs showed an average frequency increase of 6% per year in the last 30 years [5, 6], namely, from 0.31 to 1.61/100.000 in the USA from 1973 to 2012 [5]. Particularly, the well-differentiated types of NENs seem to be on the rise, as opposed to the decrease in most aggressive type [6, 7]. This figure is probably due to the improvement in diagnostic tools and diffusion of lung cancer screening programs with computed tomography (CT) [4, 8]. Furthermore, the decrease in smoking rate, extended use of filtered cigarettes, and changes in the pathological diagnostic criteria could have played a role in the relative reduction of poorly differentiated types [9].
Pulmonary NENs arise in the bronchial and bronchiolar epithelium, likely from the pluripotent bronchial epithelial stem cells. They are composed of tumor cells similar in phenotype to the endodermal pulmonary neuroendocrine cells, also known as Kulchitsky cells [10,11,12].
According to the 2015 World Health Organization (WHO) classification, there are four main types of pulmonary NEN: typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC), and small cell lung carcinoma (SCLC) [13]. A correlation between smoking history and the onset of all these neoplasms has been clearly shown, with the exception of TC [1]. The major epidemiological aspects of lung NENs are summarized in ◘ Table 5.1. Pulmonary NENs account for approximately 25% of all invasive primary lung cancers [14], mostly consisting in SCLC (75%), followed by LCNEC (15%), TC (9%), and AT (1%) [4]. This classification was updated in 2015, and each of the aforesaid four histologic variants reflects a unique type of neuroendocrine cell proliferation, with different histological grades. The histological grade of NENs ranges from low-grade (TC), to intermediate-grade (AC) [15], to high-grade and poorly differentiated malignancies (LCNEC and SCLC) [16]. The prognosis and management are widely variable among NENs according to such heterogeneous histological features [13, 17]. Noteworthy, the 2018 consensus proposal of the International Agency for Research on Cancer (IARC) and WHO forwarded a uniform classification framework for NENs from any organ, with the aim of providing consistent taxonomy. Hierarchical classification of NEN is established into two families: neuroendocrine tumor (NET) and neuroendocrine carcinoma (NEC). The term NET is intended to designate a family of well-differentiated neoplasms whose potential for dissemination depends on tumor site, type, and grade. Conversely, NEC is clearly indicative of high-grade malignant histology and biological behavior. In pulmonary NENs, the NET family encompasses the well-differentiated TC and AC. Otherwise, the poorly differentiated and high-grade LCNEC and SCLC are assigned in the NEC family (◘ Table 5.2) [18].
According to the WHO classification [13], the diagnosis of NEN is based on two parameters: the mitotic index per 2 mm2 of viable area and the presence/absence of necrosis (◘ Table 5.3). The highest mitotic index and the most extensive areas of necrosis are typical of the high-grade poorly differentiated carcinomas, such as LCNEC and SCLC [13].
Furthermore, the last WHO classification mentions Ki-67 index for stratification of NENs [13, 19]. Interestingly, a new classification of lung NENs has been published [20], which combines mitosis count (≤2, 3–47, >47), necrosis proportion (absent, <10%, or >10%), and Ki-67 cellular proliferation index (<4%, 4–24%, 25%). Such composite score correlated with prognosis and allowed stratification of AC and LCNEC.
Cytological (cell size and nuclear morphology) and architectural features are additional characteristics which can be particularly useful to distinguish between LCNEC and SCLC [16]. The typical immunohistochemical markers for identifying NENs are chromogranin A, CD56, and synaptophysin. Such immunohistochemical pattern is usually required for diagnosis of lung NEN [7], especially in case of LCNEC that should be distinguished from undifferentiated large cell carcinoma, which is not classified as a NEN [16].
The staging system of NEN was quite heterogeneous in the past, while it is currently deemed within the standard of AJCC/UICC (American Joint Committee on Cancer/Union for International Cancer Control) eighth edition of TNM system for lung cancer [21].
5.2 Carcinoid
5.2.1 Epidemiology and Clinical Presentation
Lung carcinoid shows an annual incidence of 2.3–2.8 cases per million [22]; it accounts for almost 25% of all carcinoids throughout the body [6, 8] and 2.2% of all lung neoplasms [23].
From among all lung carcinoids, 80–90% are classified as TC and the remaining 10–20% as AC [23, 24].
The majority of carcinoids occurs as sporadic disease, while 5–10% of carcinoids are found in association with multiple endocrine neoplasia 1 (MEN 1) [25, 26]. Historically, TC occurs more frequently in women while AC in men. However, it seems that gender distribution is quite dynamic as it was recently observed that TC might be increasing in men [1]. The mean age at diagnosis is 45 years for TC and 55 years for AC (◘ Table 5.1) [1, 27]. Carcinoid represents the most common primary pulmonary neoplasm in children and young adults [28]. Carcinoid was reported to occur more frequently in African-American subjects [5].
At the time of diagnosis, lung carcinoid might be asymptomatic or result in nonspecific respiratory symptoms; usually the clinical pattern follows the tumor location [15]. Central carcinoid presents with cough, hemoptysis, dyspnea, wheezing, recurrent infections, or obstructive pneumonia. Conversely, peripheral carcinoid usually appears as asymptomatic incidental imaging finding [29]. Most carcinoids are located centrally, in the main (10%) or lobar bronchi (75%). A centrally located tumor is more frequently a TC than an AC; therefore, TC tends to manifest about 10 years earlier because of signs and symptoms due to central airway obstruction.
Lung carcinoids are mostly nonfunctional (90%) [29]; hence, paraneoplastic syndrome is seldom reported [30].
TC has better prognosis than AC both because of their earlier symptoms and more indolent behavior. Indeed, both nodal involvement and distant metastatic spread are substantially lower in TC than AC at the time of diagnosis [31]. The 5-year survival of patients with TC ranges from 87% to 100%, while AT yields worse outcome ranging from 44% to 88% [28]. AC also shows a higher recurrence rate when compared to TC (33% vs. 3.6%) [32]. The major differences in the prognosis of carcinoids are reported in ◘ Table 5.4.
In a cohort of patients with metastatic disease (both TC and AC), the median survival is about 24 months, with a 5-year overall survival rate of 32% [33]. Negative prognostic factors for carcinoid are high grading and TNM stage, high tumor burden, peripheral location, higher age at diagnosis, poor performance status, and absence of uptake on somatostatin receptor scintigraphy [34, 35].
Surgical resection is the optimal treatment for localized lung carcinoid [28].
In the suspect of AC, the tumor resection should be completed with parenchymal resection associated with extended lymph node dissection. Furthermore, adjuvant chemotherapy should be considered. Although carcinoid tumors generally show indolent behavior, they could recur. Relapses in patients with TC and AC usually occur within 10 and 5 years, respectively. Therefore, surveillance is recommended for a minimum of 20 years [36]. Metastatic and unresectable tumors may be treated with chemotherapy, immunotherapy (particularly with everolimus), or radiolabeled agents such as somatostatin analogs [28, 34].
5.2.2 Pathology
Carcinoids are usually rounded tumors, with an average size of 2–3 cm. The definition of carcinoid relies on size: a carcinoid is intended to be ≥5 mm in diameter [13], whereas any proliferation of neuroendocrine cells <5 mm is called tumorlet. The tumorlet pattern is typically an incidental finding in lung specimens showing various inflammatory or fibrotic conditions; nevertheless, it can also be found in the rare condition known as diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) [37]. Tumorlets show metastatic potential despite their limited size, especially when multiple in number and fused in larger size lesions in the context of DIPNECH [38, 39].
The nomenclature of TC and AC inherently reflects the grade, G1 and G2, respectively. Otherwise, the cytoarchitectural features are quite overlapping between TC and AC. Notably, they both display a moderate amount of eosinophilic cytoplasm and nuclei with finely granular chromatin, which may also display a coarse pattern in a minority of AC. Furthermore, inconspicuous nucleoli are seen in most TC, while they happen to be also more prominent in AC [37]. As opposed to TC (◘ Fig. 5.1), AC displays possible focal or spotty (i.e., limited in size) necrosis and/or a mitotic index of 2–10 mitoses per 2 mm2 (◘ Table 5.3). In rare cases, AC morphology might be coupled with more than 10 mitoses per 2 mm2; this scenario falls under the definition of LCNEC [13].

Typical carcinoid (TC). Image H&E×20 (hematoxylin and eosin) shows a trabecular/solid islet structure, void of necrosis and with very limited cell pleomorphism, abundant cytoplasm, and monomorphic nuclei; note the absence of mitosis. Overall, such features identify a well-differentiated neuroendocrine neoplasm
TC and AC are widely and strongly positive for the neuroendocrine markers chromogranin A, synaptophysin, and CD56 N-CAM (neural cell adhesion molecule) and usually for somatostatin receptors subtypes 2 and 5 (SSR2 and SSR5). Conversely, the expression of TTF1 (thyroid transcription factor 1) is limited to peripheral lesions only. The proliferation rate by Ki-67 staining is lower in TC (less than 5%) than in AC (usually 5–20%) [37].
5.2.3 Imaging
Typical and atypical lung carcinoids are indistinguishable from an imaging standpoint.
Chest X-ray is usually nonspecific, as it may show an isolated well-defined hilar or perihilar nodule/mass associated with parenchymal abnormalities, such as atelectasis or obstructive pneumonitis. Isolated signs of endobronchial mucoid impaction might also be depicted into the “finger-in-glove” pattern [1].
CT is the main imaging technique for the detection, localization, and staging of these neoplasms. Size and location of the lesion might somehow suggest the differential between TC and AC, with the latter being relatively larger and more peripheral [28]. Three possible patterns are recognized for carcinoids: hilar or perihilar nodules/masses, endobronchial nodules, and peripheral nodules [40].
In case of hilar or perihilar nodules/masses ranging 2–5 cm in size, the tumor is usually a well-defined round or ovoid lesion, and it can appear with lobulated margins. Such lesions are typically located near the bifurcation of central bronchi [40].
Endobronchial lesion is often seen in central carcinoid. They frequently grow as primary endobronchial lesions with a subsequent extension to the adjacent lung parenchyma. Nonetheless, the endobronchial finding of carcinoid can also happen with opposite dissemination pathway, namely, a prominent extraluminal component can grow on the external aspect of the bronchus and subsequently trespass the bronchial wall into the bronchial lumen. Such scenario is known as “iceberg lesion” (◘ Fig. 5.2) referring to the morphological parallelism between the tip of the iceberg (bronchoscopic vision) and its massive drowning portion (extrabronchial growth). Moreover, some small carcinoids can be entirely located within the bronchial lumen.

Axial and coronal chest CT images at lung window setting a, b show an endoluminal lesion in the right lower lobe bronchus (black arrow) extending into the lung parenchyma. At mediastinal window setting c, d, the solid lesion shows marked contrast enhancement and inner calcifications (white arrow), with distal endobronchial mucoid impaction (arrowhead). Histopathology demonstrated a typical carcinoid (TC)
Carcinoid could also manifest as a solitary pulmonary nodule (SPN) in the lung periphery and may not show any bronchial relationship, usually measuring less than 3 cm in size [41].
Calcifications are seen in about 30% of carcinoids, notably with punctate or diffuse pattern [1, 40].
Both TC and AC show uniform intense contrast enhancement, commonly above 30 Hounsfield Unit (HU) (◘ Fig. 5.2) [1]. However, some carcinoid may lack contrast enhancement and may have irregular contours, the majority of them being AC. Rarely, carcinoids can show cavitation and areas of central macroscopic necrosis; this pattern is expected for larger lesions [1].
Other imaging techniques can be useful in the diagnosis and staging of carcinoid. Scintigraphy with octreotide is helpful in the localization of occult tumors, for staging purpose and estimation of somatostatin receptor density, which is relevant information for treatment selection [15]. On positron emission tomography (PET) with Fluorine18-fluorodeoxyglucose (18F-FDG), most carcinoid tumors do not show increased activity, with the exception of ACs, particularly in case of higher proliferation rate [42].
Recent analysis showed that 68-gallium-radiolabeled PET (68Ga-DOTA-PET) displayed higher uptake in TC rather than in AC. Such pattern of uptake is attributed to the slow metabolism and the increased somatostatin receptor expression of TC. On the other hand, 68Ga-DOTA uptake is decreased in AC due to its more aggressive behavior (◘ Fig. 5.3) [43].

Axial chest CT image in the lung window setting a of a solid nodule with lobulated margins in the anterior basal segment of the right lower lobe, showing both endobronchial (arrow) and extrabronchial intraparenchymal growth. Mediastinal window setting b demonstrates homogeneous contrast enhancement. 18F-FDG-PET/CT c does not show increased activity due to the low proliferation index. 68-Ga-DOTA-PET/CT d exhibits a high uptake due to major somatostatin receptor expression. (Courtesy of Prof. V. Rufini). The nodule was confirmed to be a typical carcinoid (TC) at histopathology
5.2.4 Pathway of Diffusion: Local Spread
Central carcinoids can have an endoluminal component with a focal or broad attachment to the bronchial wall. They usually do not infiltrate the bronchial mucosa, which may be substantially preserved above the lesion [30].
Although some carcinoids grow exclusively within the bronchial lumen, the invasion through the bronchial wall is common and can associate with infiltration of peribronchial soft tissue, cartilaginous rings, and adjacent lung parenchyma [30, 44]. This growth pattern gives a “dumbbell” appearance to the tumor, with variable proportion between the endobronchial and the extrabronchial component (◘ Fig. 5.2) [45, 46].
Peripheral carcinoids usually grow without signs of infiltration despite being nonencapsulated tumors [44]. Infiltrating margins with extension into the adjacent lung parenchyma is reported with low frequency (22.2% of cases according to Ha and coworkers) [44].
AC is typically more aggressive than TC, and it is more likely to invade vascular or lymphatic structures (26.1% and 13%, respectively) [44], followed by increased likelihood of local recurrence and poor prognosis [9, 42, 44].
Direct pleural involvement is uncommon for lung carcinoids, as opposed to the frequent involvement by thymic carcinoid [47]; thus, pleural effusion is quite rare, and it is more likely associated with post-obstructive pneumonia [30].
The most aggressive lung carcinoids may show infiltration of the mediastinal fat, with eventually direct extension to the mediastinal structures (superior vena cava, pericardium, etc.) [40].
5.2.5 Pathway of Diffusion: Lymph Node Involvement
Carcinoids may exhibit lymphatic invasion [44]; they may be associated with hilar or mediastinal lymph node metastatic involvement [48]. Regional lymph node metastases can be found in 5–15% of patients with TC, whereas in AC they can be found in almost 50% of cases [49]. Dissemination to hilar and mediastinal stations is usually depicted by enlarged lymph nodes at imaging.
However, enlargement of thoracic lymph nodes in carcinoids may also represent hyperplasia from recurrent pneumonia or chronic peripheral infection [30]. This setting is more common in case of centrally located carcinoid determining bronchial obstruction.
According to Chong et al. [50], a metastatic involvement of N1 stations was found in 5% and 14% of TCs and ACs, respectively. Further dissemination to N2 stations was observed in 6% of TCs and 10% of ACs. Metastatic spread to distant intrathoracic lymph nodes is also possible, although it is rarely observed, namely about 7% of metastatic lung carcinoids [34]. The most frequent involvement of extrathoracic stations is found in the upper abdomen. According to the TNM staging system for lung cancer [21], these metastatic nodes have to be considered as distant metastasis because their prognostic yield overlaps metastatic disease [34].
5.2.6 Pathway of Diffusion: Distant Metastatic Spread
Carcinoids can evolve with distant metastases; notably, they are found in 25% of patients with AC, compared to 3–5% of TC [49].
The most frequent sites of metastasis from lung carcinoid are the liver, bone, central nervous system, and adrenal glands [24, 34]. More than 50% of patients with metastases from lung carcinoids show multiple sites of metastasis, with the liver affected in most cases [51]. It should be underscored that liver involvement appears to be more common in metastatic lung carcinoids than in non-small-cell lung cancer (NSCLC) [51]. Metastatic disease from ACs is noted primarily in the bones, brain, and liver. Conversely, the rare metastases from TC preferably happen in the liver and bones [50]. For carcinoid tumors, a direct correlation between the size of the primary lesion and the probability of metastasis has been reported for hematogenous dissemination to the liver [52].
Among carcinoid tumors from any organ, lung carcinoids show a higher propensity to metastasize to the bone and to central nervous system. Bone metastases from lung carcinoid display solitary or multiple sclerotic bone lesions. Noteworthy, the lone detection of bone metastases from carcinoid tumors is a predictive factor for primary lung carcinoids, with a frequency of about 34% [52]. Likewise, primary lung carcinoids account for more than 50% of central nervous system metastases by all carcinoids, even in this case being the most common primary origin of metastatic carcinoid. Of note, the propensity of lung carcinoid to such metastatic targets might be also explained by anatomical reasoning: the departure of neoplastic cells from the lung can directly harbor to both bone and brain via the systemic circulation, whereas carcinoids from other regions (e.g., the midgut) are obligated to first pass through the liver and lungs.
Multiple synchronous bilateral lesions are reported, especially for TC. In these cases, it is unclear whether this represents true metastasis, enlarging tumorlets, or a tendency to form multiple primary pulmonary carcinoid tumors. Patients with multiple bilateral pulmonary TCs have been reported to have an excellent prognosis [50].
Unusual sites of metastatic spread can be occasionally seen toward pancreas, soft tissues, breasts, heart, skull, testis, and orbits [47].
The risk of recurrence is higher for resected ATs compared to TCs. Indeed, a smaller number of patients with TC (3.6%) than that with AC (33.3%) experience recurrences, with a median time to recurrence of 4 years, significantly longer than that for AC [32].
Most recurrences of carcinoid tumors of the lung are distant metastases, with local recurrences affecting only 2% of TC and 7% of AC [53, 54]. Metastatic lung carcinoid shows metachronous metastases in up to 50%, mostly in AC [34].
5.3 Large Cell Neuroendocrine Carcinoma
5.3.1 Epidemiology and Clinical Presentation
Pulmonary LCNECs are rare neoplasms representing about 2.1–3.5% of all lung tumors and about 15% of lung NENs. However, this frequency is potentially underestimated because of limitations in diagnosing LCNEC on cytological specimens [55, 56].
This neoplasm involves more frequently men than women (2.5:1), with a heavy smoking habit.
The average age at diagnosis is about 70 years [1, 55] (◘ Table 5.1). This neoplasm shows peripheral location in 66–100% of cases. Centrally located LCNEC is associated with symptoms from bronchial obstruction and compression of vascular structures [57].
Slight predilection for upper lobe location has been reported, in about 63% of LCNEC [58].
LCNEC is rarely associated with paraneoplastic syndromes [29].
Surgical resection is recommended for all nonmetastatic stages, with subsequent adjuvant chemotherapy. Unfortunately, the majority of pulmonary LCNEC are not eligible for radical surgical treatment due to local or systemic spread [59]. The optimal therapeutic approach for systemic treatment is not established for LCNEC, yet [59].
It is increasingly understood that the pattern of response to chemotherapy is quite overlapping between LCNEC and SCLC, although with less chemo-responsivity, so they are frequently treated with the same chemotherapeutic regimens used for SCLC [55].
The prognosis of LCNEC is quite poor, with median overall survival of stages I–II, III, and IV about 32.4, 12.6, and 4.0 months, respectively. These figures are lower than survival rates in NSCLC [60]. Negative prognostic factors are advanced tumor stage, tumor size (greater than 3 cm), and male gender [61].
5.3.2 Pathology
LCNEC usually appears as a circumscribed mass of 3–4 cm, with central foci of hemorrhage and necrosis. LCNEC is by definition a poorly differentiated NEN, with high grading (G3). Diagnostic criteria for LCNEC are neuroendocrine morphology (organoid nesting, palisading, or rosette-like structures), high mitotic rate (greater than 10 mitoses per 2 mm2), and non-small-cell cytologic features (including large cell size and prominent nucleoli) (◘ Fig. 5.4).

Large cell neuroendocrine carcinoma (LCNEC). Image H&E×20b (hematoxylin and eosin) depicts a solid structure with evident necrosis (left upper corner) and is composed by large in size, severely atypical cells with abundant cytoplasm, often prominent nucleoli, and evident mitotic activity. Overall such features identify a poorly differentiated neuroendocrine neoplasm
Diagnostic confirmation of neuroendocrine differentiation is required using immunohistochemical markers such as chromogranin A, synaptophysin, and CD56 or electron microscopy [13, 37]. In addition, TTF1 is usually strongly expressed, while SSR2 and SSR5 may also be present. Ki-67 usually shows a very high proliferation index (50–100% of tumor cells) [37].
LCNEC histology is not easily demonstrated on small biopsies or cytology; in most cases, the diagnosis of LCNEC requires a surgical lung biopsy [37]. Staining for Ki-67 may help in identifying the highly proliferative status of tumor cells.
5.3.3 Imaging
LCNECs share similar imaging characteristics with NSCLC; hence, there is no advantage in guessing the differential by imaging [29].
LCNEC typically manifests as a large peripheral pulmonary mass, with well-defined lobular margins, even though spiculated borders may be observed. Calcifications are uncommon, occurring in less than 9% of cases [28], and air bronchogram is extremely rare [58]. The tumor shows soft tissue density substantially similar to chest wall muscles. The contrast enhancement is quite variable: heterogeneous or peripheral, due to central necrosis in case of large lesions [58]. Extensive necrosis is also possible, but cavitation is rarely seen. Hilar and mediastinal nodal enlargement is frequent [28].
According to one series, pleural effusion has been described in almost a quarter of cases [62].
In case of centrally located lesion, post-obstructive signs can be observed such as mucus plugging, atelectasis, or pneumonia.
The comprehensive imaging of LCNEC includes 18F-FDG-PET/CT, which is helpful for staging purpose. Furthermore, PET-CT yields prognostic information: SUV >13.7 has been related to worse prognosis [63].
Although LCNEC contains somatostatin receptors and can be octreoscan/68Ga-DOTA-PET positive, this imaging modality is not suggested for the evaluation of this malignancy.
5.3.4 Pathway of Diffusion
LCNEC is a high-grade malignancy, with a high propensity to local and distant spread.
Tumor cells usually entirely fill pulmonary alveoli, or they can show a compressive growth pattern. This type of local growth into the lung parenchyma gives the tumor the appearance of a large lesion with well-defined margin. Still, lobulated margins observed on imaging may reflect the interruption of tumor growth by anatomic boundaries [58].
In centrally located lesions, tumors are usually adherent to the bronchial wall, with an associated endobronchial polypoid component that invades the surrounding lung parenchyma. In these cases, an endobronchial growth pattern can be seen, usually associated to invasion of lung parenchyma.
Tumor emboli in the adjacent lung parenchyma can be observed [64].
Vascular and lymphatic invasion are possible, and they can correspond to the radiological evidence of spiculations. Where observed, lymphatic permeation and vascular invasion were significantly associated with poorer outcome [64]. Other causes of spiculations are irregularly protuberant tumor nests or a diffuse desmoplastic response to tumor growth [58].
Lesions show multiple punctate necrosis. In large tumors, necrotic foci may become larger and prone to be confluent in extensive areas of necrosis, reflecting the CT appearance of inhomogeneous enhancement with peripheral rim within the lesion.
Direct pleural tumor seeding of LCNEC cells is uncommon [64]. This involvement may explain at least some of the pleural effusions not associated with pneumonia.
At diagnosis, LCNEC presents high rate of lymph node metastasis (60–80%) [55]. Metastatic involvement of lymph node is usually limited to hilar and mediastinal nodes. Metastatic lymph nodes have been associated with a poor outcome [65].
At diagnosis, distant metastasis can be found in about 40% of patients affected by LCNEC [55].
Most common metastatic sites are liver (47%), bone (32%), and brain (23%), similar to that seen in SCLC [60]. Another possible site of metastatic spread is adrenal gland. Moreover, multiple case reports have also described metastasis in atypical locations, including skin, scalp, and breast, substantially similar to other lung NENs.
Some patients may have oligometastatic disease (defined as ≤3 metastatic lesions) at the time of diagnosis. Despite radical treatment, recurrence is quite common; it is reported in almost 40% of patients, with over 90% of cases occurring within 2 years [61].
Local or regional recurrence has been reported in 27–35% of cases. Local recurrence usually affects bronchial stump in patients undergoing surgery, while lymph node recurrence mostly involves mediastinal and/or supraclavicular lymph nodes.
Distant recurrence is observed more frequently than local recurrence. This happens in about 56–60% of cases, with most cases involving brain and liver, followed by bone and lung [66, 67].
5.4 Small Cell Lung Carcinoma
5.4.1 Epidemiology and Clinical Presentation
Small cell lung carcinoma (SCLC) is the most common lung NEN; it represents 15% of any lung cancer [68]. It is strongly associated with cigarette smoking, with 95% of all patients having a history of heavy tobacco exposure. Other risk factors include exposures to agent including radon, halogenated ethers, arsenic, and asbestos.
SCLC typically affects patients between the ages of 60 and 70 years [69]. It has been more frequently observed in men than in women (ratio 2.6:1) (◘ Table 5.1), even if women smokers are more likely to develop SCLC [70].
At the time of diagnosis, early-stage SCLC is very rare, representing only 5% of all cases [71]. The low incidence of early-stage SCLC is mainly attributable to its highly aggressive biological behavior, rapid tumor growth, and absence of early symptoms [71].
Patients typically present with a short duration of rapidly progressive symptoms.
Most patients present with advanced disease, and only 5% of cases of SCLC can display as a solitary lung nodule, usually incidentally discovered [37] or during CT lung cancer screening programs. The aggressive behavior of SCLC hampers early diagnosis and survival advantage of SCLC in lung cancer screening programs by low-dose CT [72].
Patients of any disease stage may present with a variety of paraneoplastic syndromes, such as endocrine ones: the syndrome of inappropriate antidiuretic hormone (SIADH) and Cushing’s syndrome due to ectopic adrenocorticotroph hormone (ACTH) production. Furthermore, neurologic syndromes, as Lambert-Eaton syndrome, limbic encephalitis, and encephalomyelitis, can precede cancer diagnosis [9, 68].
5.4.2 Staging, Treatment, and Prognosis
According to the Veterans Administration Lung Study Group (VALSG) system, SCLC patients are classified as having limited-stage (LS) or extensive-stage (ES) disease [73]. The definition of LS disease includes patients with disease limited to one hemithorax, with hilar and mediastinal nodes that can be encompassed within one tolerable radiotherapy portal, and possibly with ipsilateral supraclavicular lymph nodes. LS-SCLC is deemed a potentially curable disease. Conversely, ES disease is characterized by neoplastic invasion beyond the aforementioned boundaries.
The eighth TNM staging system replaces the VALSG [21]. According to TNM, stages I–III correspond to LS-SCLC, while stage IV corresponds to ES-SCLC.
Surgical resection with mediastinal lymph node dissection, followed by adjuvant chemotherapy, is indicated only for true stage I patients, which unfortunately are very rare (<5%). Most patients with LS receive a combination of cisplatin/carboplatin and etoposide plus thoracic radiation therapy (RT) [74].
Prophylactic cranial irradiation (PCI) is recommended for patients with good performance status who have responded to chemotherapy [75].
ES-SCLC is approached by chemotherapy with palliative purpose. Patients with a good response to therapy should be considered for PCI and/or thoracic RT [74].
SCLC is typically very responsive to chemotherapy; therefore, chemotherapy can prolong survival [76]. Unfortunately, relapse with chemoresistant disease is inevitable, with fewer than 10% of patients with ES alive after 2 years [74]. The overall survival rate is only about 5%: patients with ES-SCLC have a mean survival of about 8–10 months and a 2-year survival rate of 10%, against the 15–20 months in the LS disease, with 20–25% of long-term survival [77]. The 5-year survival rates of SCLC and LCNEC are reported in ◘ Table 5.5.
Negative prognostic factors include extensive disease, poor general conditions, weight loss, and markers such as lactic dehydrogenase. Conversely, positive prognostic factors include younger age, good general conditions, and single metastatic site in patients with extensive disease [78].
5.4.3 Pathology
SCLCs are by definition poorly differentiated NENs. Tumor cells are round to fusiform, with scant cytoplasm, and measure less than the diameter of three resting lymphocytes, with finely granular nuclear chromatin. Nucleoli are inconspicuous or absent. Tumor cells grow in sheets and nests with frequent necrosis, often extensive. Lesions exhibit a high mitotic rate (60–80 per 2 mm2) (◘ Fig. 5.5). Unlike LCNEC, SCLC can readily be diagnosed in small specimens such as bronchoscopic biopsies, fine-needle aspirates, or core biopsies [37].

Small cell lung carcinoma (SCLC). Image H&E×20 shows a solid structure with evident necrosis (left and center of the micrograph) and severely atypical cells with large nuclei and a thin rim of cytoplasm, overall resulting in small size; mitoses are evident often with atypical mitotic figures. As seen in LCNEC, such features identify a poorly differentiated neuroendocrine neoplasm
Up to 90% of SCLCs are positive for at least one neuroendocrine marker such as CD56 N-CAM, chromogranin A, synaptophysin, and TTF1 [79]. SSR2 and SSR5 are usually negative or only focally positive in about 20% of investigated cases. Ki-67 is very helpful in distinguishing SCLC from carcinoids, because it shows a very high proliferation rate, of about 80–100% [37]. The main anatomic and histological features of SCLC with respect to those of other lung NENs are summarized in ◘ Table 5.6.
5.4.4 Imaging
About 90–95% of SCLC is centrally located, arising in lobar bronchi or main bronchus [80]. The most common appearance of SCLC is a large mass with central location, bulky hilar, and mediastinal adenopathies invariably associated. Due to the close relationship with the tracheobronchial tree, SCLC can determine distal lung parenchyma atelectasis.
Chest X-ray usually shows an ill-defined hilar mass, with possible associated enlargement of the mediastinum and pulmonary volume loss due to obstruction and pleural effusion. Often the primary tumor is overshadowed by the mediastinal component, appearing as a poor-defined mediastinal mass involving at least one hilum [9, 29].
Among imaging techniques, CT allows an accurate assessment of intrathoracic involvement. Chest CT scan with contrast medium injection often shows the involvement of the major pulmonary vessels and mediastinal structure (including the superior vena cava), with common evidence of encasement of these structures by the tumor, as well as pleural effusion [9].
Rare manifestations include airspace opacities and consolidation. Thickening of the interlobular septa is reported as a sign of lymphangitic carcinomatosis [81]. Calcifications can be seen in up to 23% of the cases [1].
A peripheral primary tumor is rare. In approximately 5% of cases, SCLC may manifest as an solitary pulmonary nodule (SPN) [37], with well-defined lobulated margins or spiculated appearance. There are no imaging features that help to distinguish between SCLC and NSCLC when presenting as an SPN [1, 82].
The comprehensive clinical staging includes 18F-FDG-PET/CT. Otherwise, MRI is rarely used in the evaluation of SCLC, except when there are contraindications for the administration of intravenous iodine contrast material. To date, MRI of the brain for staging of SCLC shows better sensitivity than CT, particularly in neurologically asymptomatic patients [83]. MRI might be also useful for detecting and characterizing bone or liver metastases.
18F-FDG-PET/CT is extremely helpful in staging and restaging SCLC (with the exception of brain metastases). It turned out to be more sensitive and specific than other imaging techniques in the detection of metastatic disease, in prognosis evaluation – high standardized uptake seems to be significantly associated with poor survival – and, particularly, in the assessment of treatment response, due to the high metabolic activity degree of SCLC [29].
5.4.5 Pathway of Diffusion: Local Spread and Mediastinal Involvement
SCLC malignant cells exhibit heightened invasive properties and increased motility, resulting in rapid tumor growth, high local invasiveness, and early metastatic spread to numerous organs.
The primary site is typically a submucosal endobronchial lesion of the proximal airways such as the lobar or main bronchi. The tumor itself is highly cellular and determines a limited fibrotic or inflammatory response. Consequently, it can rapidly spread through lymphatic and blood vessels even at an early stage of the disease, resulting in early nodal and distant metastatic deposits [84].
Kazawa et al. [81] have described eight different patterns of SCLC local diffusion according to the extension and spreading pattern recognized on CT scan. The most frequent patterns were the central perihilar type, when a central perihilar mass confined within the ipsilateral mediastinal tissue was seen; the central plus mediastinal extension type, with the mass extending to contralateral mediastinum (the most common pattern among their series, occurring in about 30% of cases); the peripheral type, in case of cancer located only in the peripheral lung parenchyma; and peripheral plus mediastinal extension type, in case of a peripheral tumor associated with a hilar/ipsilateral mediastinal lymph node metastasis (◘ Fig. 5.6).

Axial chest CT images at lung a and mediastinal b window settings depict a small (15 mm) nodule, with spiculated margins and necrotic appearance, in the posterior segment of the right upper lobe. Axial and coronal images at mediastinal window setting c–f show confluent and infiltrative adenopathies in the right hilum, in the right paratracheal and subcarinal zones, and in the superior mediastinum, with encasement/infiltration of the vascular structures and the right bronchial tree. Sagittal reconstruction with bone window setting g shows multiple bone metastases of the spine. Histopathology revealed a small cell lung carcinoma
Peripheral SCLC typically appears on CT scan as a spiculated nodule, representing the vascular, lymphatic, or intra-alveolar invasion by nest-forming tumor cells, with surrounding ground-glass opacity determined by edema, parenchymal hemorrhage, and intra-alveolar spread [85].
The centrilobular nodular or branching abnormalities – which usually represent bronchogenic diffusion of inflammation and disease – are usually not observed in SCLC.
Another possible identified pattern was the lymphangitic-spread type, with the presence of lymphangitic carcinomatosis, represented by thickening of the bronchovascular bundles and/or nodular or smooth thickening of the interlobular septa, usually associated with pleural effusion.
Other uncommon manifestations of SCLC local spread include the lobar replacement type, when tumor appears as a huge mass-like lesion that replaces an entire lobe, and the airspace consolidation type, in case of an ill-defined consolidation with cystic or tubular air bronchogram within the lesion [81].
Due to the easy spread via the lymphatic system, SCLC can also disseminate into the pleural space, appearing as multiple subpleural nodules or, less commonly, masses associated with pleural thickening and malignant pleural effusion (pleural dissemination type) [9, 81].
SCLC diffusion often causes main airway stenosis, due to peribronchial extension of the disease, particularly in the submucosal tissue, with almost complete preservation of bronchial inner wall. Stenosis typically involves trachea and main bronchi (in about 50% of cases) and less likely lobar/segmental bronchi or subsegmental branches [81].
Mediastinum is by far the most common site of detected disease (up to 92% of cases), both for the presence of the primary malignancy and associated lymphadenopathies. Indeed, mediastinal and hilar lymph node metastases are detected in 92% and 84% of cases, respectively [1]. Mediastinal masses determine displacement, narrowing, and invasion of mediastinal structures, particularly great vessels, in about 68% of cases [9]. The superior vena cava, the pulmonary arteries, and the main pulmonary trunk are more frequently involved, followed by pulmonary veins and thoracic aorta, ranging from mild stenosis with wall irregularity to severe stenosis, luminal invasion with tumor abutting into vessels, and complete encasement (◘ Fig. 5.7) [81].

Axial CT image at lung window setting a shows a mass in the anterior segment of the right upper lobe. Contrast-enhanced axial image b displays the extensive ipsilateral mediastinal involvement with encasement and infiltration of the right upper lobe artery and superior vena cava. Coronal images c, d better depict the longitudinal extension of the occlusion of superior vena cava, the encasement of the right main bronchus, the distal right main pulmonary artery and the interlobar artery, which look patent, and the occlusion of the right superior pulmonary vein (arrow) close to the left atrium. Note the elevation of the right hemidiaphragm as a sign of the right phrenic nerve infiltration by the mass. Transbronchial biopsy through the anterior segmental bronchus of the right upper lobe revealed a small cell lung carcinoma
Also the azygos vein, the esophagus, the phrenic nerves, and the recurrent laryngeal nerve can be involved. Pericardial spread is relatively common, manifesting as pericardial effusion and/or irregular thickening of the pericardium. Even compression or direct extension to the heart may be seen, particularly to the left atrium [9, 81].
5.4.6 Pathway of Diffusion: Distant Metastatic Spread
Once the tumor reaches access to the mediastinum, there are pathways of spread downward into the abdomen via the aortic and esophageal hiatus, both through lymphatic vessels and direct diffusion. This can result in intra-abdominal lymphadenopathies primarily along the celiac vessels resulting in celiac, periportal, peripancreatic, and para-aortic lymph node metastases. Disease may also reach the gastrohepatic ligament either by spread along vascular planes or through the esophageal hiatus.
SCLC can also directly spread upward via the cervicothoracic hiatus, resulting in supraclavicular and, less commonly, lower neck adenopathies and possible involvement of the lower neck structure (cervical esophagus, hypopharynx, thyroid, etc.) [86].
Metastatic spread to distant organs is also frequent in SCLC. The most common metastatic sites at diagnosis are bone (19–38%), liver (17–34%), adrenal glands (10–17%), and brain (up to 14%, whose 10–15% in neurologically asymptomatic patients) [9]. Particularly, SCLC has been associated with an increased metastatic potential to liver and brain with respect to NSCLC [87].
Nodules or masses in the contralateral lung can be also found as well as other uncommon metastatic sites, such as pancreas, prostate, spinal cord, and oropharynx [81]. Lastly, metastatic involvement of bone marrow has been detected in 10–15% of patients. However, less than 5% of these patients have bone marrow as isolate distant metastatic site [88].
5.5 Conclusions
Pulmonary neuroendocrine neoplasms (NENs) are a heterogeneous group of tumors, with different morphological, immunohistochemical, and molecular characteristics and noticeably different clinical and biological behavior, ranging from the indolent course of TC to the highly aggressive behavior of the SCLC. Also imaging characteristics of NENs are quite different among the two well-differentiated TC and AC, currently classified as NETs, and the two poorly differentiated LCNEC and SCLC, classified as NECs, reflecting the different pathways of diffusion of these neoplasms. Knowledge of the more common morphologic characteristics and diffusion pathways of these neoplasms might help in recognizing NENs. CT and PET/CT are the most useful imaging modalities for diagnosing and staging these tumors, while MR has specific indication for brain, bone, and liver involvement, which is typical of the extensive stage of SCLC.
References
Chong S, Lee KS, Chung MJ, Han J, Kwon OJ, Kim TS. Neuroendocrine tumors of the lung: clinical, pathologic, and imaging findings. Radiographics. 2006;26(1):41–57.
Rossi G, Bisagni A, Cavazza G. High-grade neuroendocrine carcinoma. Curr Opin Pulm Med. 2014;20:332–9.
Gustafsson BI, Kidd M, Modlin IM. Neuroendocrine tumors of the diffuse neuroendocrine system. Curr Opin Oncol. 2008;20(1):1–12.
Filosso PL, Falcoz PE, Solidoro P, Pellicano D, Passani S, Guerrera F, et al. The European Society of Thoracic Surgeons (ESTS) lung neuroendocrine tumors (NETs) database. J Thorac Dis. 2018;10(Suppl 29):S3528–32.
Leoncini E, Boffetta P, Shafir M, Aleksovska K, Boccia S, Rindi G. Increased incidence trend of low-grade and high-grade neuroendocrine neoplasms. Endocrine. 2017;58(2):368–79.
Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934–59.
Caplin ME, Baudin E, Ferolla P, Filosso P, Garcia-Yuste M, Lim E, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoid. Ann Oncol. 2015;26(8):1604–20.
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–72.
Carter BW, Glisson BS, Truong MT, Erasmus JJ. Small cell lung carcinoma: staging, imaging, and treatment considerations. Radiographics. 2014;34(6):1707–21.
Paladugu RR, Benfield JR, Pak HY, Ross RK, Teplitz RL. Bronchopulmonary Kulchitzky cell carcinoma: a new classification scheme for typical and atypical carcinoids. Cancer. 1985;55:1303–11.
Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, editors. WHO classification of tumours. Pathology and genetics of tumours of the lung, pleura, thymus and heart. World Health Organization classification tumours. Lyon: IARC Press; 2004.
Ito T, Udaka N, Okudela K, Yazawa T, Kitamura H. Mechanisms of neuroendocrine differentiation in pulmonary neuroendocrine cells and small cell carcinoma. Endocr Pathol. 2003;14(2):133–9.
Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO classification of tumours of the lung, pleura, thymus and heart. World Health Organization classification tumours. 4th ed. Lyon: IARC Press; 2015.
Yesner R. Small cell tumors of the lung. Am J Surg Pathol. 1983;7:775–85.
Hendifar AE, Marchevsky AM, Tuli R. Neuroendocrine tumors of the lung: current challenges and advances in the diagnosis and management of well-differentiated disease. J Thorac Oncol. 2017;12(3):425–36.
Righi L, Gatti G, Volante M, Papotti M. Lung neuroendocrine tumors: pathological characteristics. J Thorac Dis. 2017;9(Suppl 15):S1442–7.
Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–60.
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol. 2018;31(12):1770–86.
Pelosi G, Rindi G, Travis WD, Papotti M. Ki-67 antigen in lung neuroendocrine tumors: unravelling a role in clinical practice. J Thorac Oncol. 2014;9(3):273–84.
Rindi G, Klersy C, Inzani F, Fellegara G, Ampollini L, Ardizzoni A, et al. Grading the neuroendocrine tumors of the lung: an evidence-based proposal. Endocr Relat Cancer. 2014;21(1):1–16.
Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The eighth edition lung cancer stage classification. Chest. 2017;151(1):193–203.
Morandi U, Casali C, Rossi G. Bronchial typical carcinoid tumors. Semin Thorac Cardiovasc Surg. 2006;18(3):191–8.
Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134(11):1628–38.
Dincer HE, Podgaetz E, Andrade RS. Pulmonary neuroendocrine tumors. Part I. Spectrum and characteristics of tumors. J Bronchology Interv Pulmonol. 2015;22(3):267–73.
Wolin EM. Challenges in the diagnosis and management of well-differentiated neuroendocrine tumors of the lung (typical and atypical carcinoid): current status and future considerations. Oncologist. 2015;20(10):1123–31.
Phan AT, Oberg K, Choi J, Harrison LH Jr, Hassan MM, Strosberg JR, et al. NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the thorax (includes lung and thymus). Pancreas. 2010;39(6):784–98.
Gosain R, Mukherjee S, Yendamuri SS, Iyer R. Management of typical and atypical pulmonary carcinoids based on different established guidelines. Cancers. 2018;10(12):pii:E510.
Benson RE, Rosado-de-Christenson ML, Martínez-Jiménez S, Kunin JR, Pettavel PP. Spectrum of pulmonary neuroendocrine proliferations and neoplasms. Radiographics. 2013;33(6):1631–49.
Detterbeck FC. Clinical presentation and evaluation of neuroendocrine tumors of the lung. Thorac Surg Clin. 2014;24(3):267–76.
Rosado de Christenson ML, Abbott GF, Kirejczyk WM, Galvin JR, Travis WD. Thoracic carcinoids: radiologic-pathologic correlation. Radiographics. 1999;19(3):707–36.
Zagurovskaya M, Tran-Harding K, Gibbs R. Primary lung carcinoid metastatic to the breast. Radiol Case Rep. 2017;12(2):223–8.
Asamura H, Kameya T, Matsuno Y, Noguchi M, Tada H, Ishikawa Y, et al. Neuroendocrine neoplasms of the lung: a prognostic spectrum. J Clin Oncol. 2006;24(1):70–6.
Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017;3(10):1335–42.
Robelin P, Hadoux J, Forestier J, Planchard D, Hervieu V, Berdelou A, et al. Characterization, prognosis, and treatment of patients with metastatic lung carcinoid tumors. J Thorac Oncol. 2019;13:S1556-0864(19)30103-0.
Huang Y, Yang X, Lu T, Li M, Zhao M, Yang X, et al. Assessment of the prognostic factors in patients with pulmonary carcinoid tumor: a population-based study. Cancer Med. 2018;7(6):2434–41.
Detterbeck FC. Management of carcinoid tumors. Ann Thorac Surg. 2010;89(3):998–1005.
Travis WD. Pathology and diagnosis of neuroendocrine tumors: lung neuroendocrine. Thorac Surg Clin. 2014;24:257–66.
Rossi G, Cavazza A, Spagnolo P, Sverzellati N, Longo L, Jukna A, et al. Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia syndrome. Eur Respir J. 2016;47(6):1829–41.
Gorshtein A, Gross DJ, Barak D, Strenov Y, Refaeli Y, Shimon I, et al. Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia and the associated lung neuroendocrine tumors: clinical experience with a rare entity. Cancer. 2012;118(3):612–9.
Jeung MY, Gasser B, Gangi A, Charneau D, Ducroq X, Kessler R, et al. Bronchial carcinoid tumors of the thorax: spectrum of radiologic findings. Radiographics. 2002;22(2):351–65.
Baxi AJ, Chintapalli K, Katkar A, Restrepo CS, Betancourt SL, Sunnapwar A. Multimodality imaging findings in carcinoid tumors: a head-to-toe spectrum. Radiographics. 2017;37(2):516–36.
Erasmus JJ, McAdams HP, Patz EF Jr, Coleman RE, Ahuja V, Goodman PC. Evaluation of primary pulmonary carcinoid tumors using FDG PET. AJR Am J Roentgenol. 1998;170(5):1369–73.
Jiang Y, Hou G, Cheng W. The utility of 18F-FDG and 68Ga-DOTA-Peptide PET/CT in the evaluation of primary pulmonary carcinoid: a systematic review and meta-analysis. Medicine (Baltimore). 2019;98(10):e14769.
Ha SY, Lee JJ, Cho J, Hyeon J, Han J, Kim HK. Lung parenchymal invasion in pulmonary carcinoid tumor: an important histologic feature suggesting the diagnosis of atypical carcinoid and poor prognosis. Lung Cancer. 2013;80(2):146–52.
Colby TV, Koss MN, Travis WD. Carcinoid and other neuroendocrine tumors. In: Colby TV, Koss MN, Travis WD, editors. Atlas of tumor pathology: tumors of the lower respiratory tract, fasc. 13, ser. 3. Washington, DC: Armed Forces Institute of Pathology; 1995. p. 287–317.
Salyer DC, Salyer WR, Eggleston JC. Bronchial carcinoid tumors. Cancer. 1975;36:1522–37.
Scarsbrook AF, Ganeshan A, Statham J, Thakker RV, Weaver A, Talbot D, et al. Anatomic and functional imaging of metastatic carcinoid tumors. Radiographics. 2007;27(2):455–77.
Naidich DP. CT/MR correlation in the evaluation of tracheobronchial neoplasia. Radiol Clin N Am. 1990;28:555–71.
Travis WD. Advances in neuroendocrine lung tumors. Ann Oncol. 2010;21(suppl 7):vii65–71.
Chong CR, Wirth LJ, Nishino M, Chen AB, Sholl LM, Kulke MH, et al. Chemotherapy for locally advanced and metastatic pulmonary carcinoid tumors. Lung Cancer. 2014;86(2):241–6.
Riihimäki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. The epidemiology of metastases in neuroendocrine tumors. Int J Cancer. 2016;139(12):2679–86.
Bhosale P, Shah A, Wei W, Varadhachary G, Johnson V, Shah V, et al. Carcinoid tumours: predicting the location of the primary neoplasm based on the sites of metastases. Eur Radiol. 2013;23(2):400–7.
Wolin EM. Advances in the diagnosis and management of well-differentiated and intermediate-differentiated neuroendocrine tumors of the lung. Chest. 2017;151(5):1141–6.
Lou F, Sarkaria I, Pietanza C, Travis W, Roh MS, Sica G, et al. Recurrence of pulmonary carcinoid tumors after resection: implications for postoperative surveillance. Ann Thorac Surg. 2013;96(4):1156–62.
Fasano M, Della Corte CM, Papaccio F, Ciardiello F, Morgillo F. Pulmonary large-cell neuroendocrine carcinoma: from epidemiology to therapy. J Thorac Oncol. 2015;10(8):1133–41.
Fernandez FG, Battafarano RJ. Large-cell neuroendocrine carcinoma of the lung: an aggressive neuroendocrine lung cancer. Semin Thorac Cardiovasc Surg. 2006;18(3):206–10.
Sulai NH, Kim ES, Ananthanarayanan V. Large cell carcinoma of the lung. In: Raghavan D, Ahluwalia MS, Blanke CD, Brown J, Kim ES, Reaman GH, Sekeres MA, editors. Textbook of uncommon cancer. Hoboken: Wiley; 2017. p. 293–304.
Oshiro Y, Kusumoto M, Matsuno Y, Asamura H, Tsuchiya R, Terasaki H, et al. CT findings of surgically resected large cell neuroendocrine carcinoma of the lung in 38 patients. AJR Am J Roentgenol. 2004;182(1):87–91.
Welter S, Aigner C, Roesel C. The role of surgery in high grade neuroendocrine tumours of the lung. J Thorac Dis. 2017;9(Suppl 15):1474–83.
Derks JL, Hendriks LE, Buikhuisen WA, Groen HJ, Thunnissen E, van Suylen RJ, et al. Clinical features of large cell neuroendocrine carcinoma: a population-based overview. Eur Respir J. 2016;47(2):615–24.
Naidoo J, Santos-Zabala ML, Iyriboz T, Woo KM, Sima CS, Fiore JJ, et al. Large cell neuroendocrine carcinoma of the lung: clinico-pathologic features, treatment, and outcomes. Clin Lung Cancer. 2016;17(5):121–9.
Shin AR, Shin BK, Choi JA, Oh YW, Kim HK, Kang EY. Large cell neuroendocrine carcinoma of the lung: radiologic and pathologic findings. J Comput Assist Tomogr. 2000;24(4):567–73.
Chong S, Lee KS, Kim BT, Choi JY, Yi CA, Chung MJ, et al. Integrated PET/CT of pulmonary neuroendocrine tumors: diagnostic and prognostic implications. AJR Am J Roentgenol. 2007;188(5):1223–31.
Jung KJ, Lee KS, Han J, Kwon OJ, Kim J, Shim YM, et al. Large cell neuroendocrine carcinoma of the lung: clinical, CT, and pathologic findings in 11patients. J Thorac Imaging. 2001;16(3):156–62.
Kinoshita T, Yoshida J, Ishii G, Aokage K, Hishida T, Nagai K. The differences of biological behavior based on the clinicopathological data between resectable large-cell neuroendocrine carcinoma and small-cell lung carcinoma. Clin Lung Cancer. 2013;14(5):535–40.
Takei H, Asamura H, Maeshima A, Suzuki K, Kondo H, Niki T, et al. Large cell neuroendocrine carcinoma of the lung: a clinicopathologic study of eighty-seven cases. J Thorac Cardiovasc Surg. 2002;124(2):285–92.
Sarkaria IS, Iyoda A, Roh MS, Sica G, Kuk D, Sima CS, et al. Neoadjuvant and adjuvant chemotherapy in resected pulmonary large cell neuroendocrine carcinomas: a single institution experience. Ann Thorac Surg. 2011;92(4):1180–6.
Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017;17(12):725–37.
Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113(1):5–21.
Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24(28):4539–44.
Käsmann L, Bolm L, Janssen S, Rades D. Prognostic factors and treatment of early-stage small-cell lung cancer. Anticancer Res. 2017;37(3):1535–7.
Silva M, Galeone C, Sverzellati N, Marchianò A, Calareso G, Sestini S, et al. Screening with low-dose computed tomography does not improve survival of small cell lung cancer. J Thorac Oncol. 2016;11(2):187–93.
Zelen M. Keynote address on biostatistics and data retrieval. Cancer Chemother Rep 3. 1973;4(2):31–42.
Kalemkerian GP, Schneider BJ. Advances in small cell lung cancer. Hematol Oncol Clin North Am. 2017;31(1):143–56.
Früh M, De Ruysscher D, Popat S, Crinò L, Peters S, Felip E, ESMO Guidelines Working Group. Small-cell lung cancer (SCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):99–105.
Onn A, Vaporciyan A, Chang J, Komaki R, Roth J, Herbst R. Cancer of the lung. In: Kufe DW, Bast Jr RC, Hait WN, et al., editors. Cancer medicine. Hamilton: American Association for Cancer Research; 2006. p. 1179–24.
Lu HY, Wang XJ, Mao WM. Targeted therapies in small cell lung cancer. Oncol Lett. 2013;5(1):3–11.
Foster NR, Mandrekar SJ, Schild SE, Nelson GD, Rowland KM Jr, Deming RL, et al. Prognostic factors differ by tumor stage for small cell lung cancer: a pooled analysis of North Central Cancer Treatment Group trials. Cancer. 2009;115(12):2721–31.
Swarts DR, Ramaekers F, Speel E-JM. Molecular and cellular biology of neuroendocrine lung tumors: evidence for separate biological entities. Biochim Biophys Acta (BBA) Rev Cancer. 2012;1826(2):255–71.
Rosado-de-Christenson ML, Templeton PA, Moran CA. Bronchogenic carcinoma: radiologic-pathologic correlation. Radiographics. 1994;14(2):429–46.
Kazawa N, Kitaichi M, Hiraoka M, Togashi K, Mio N, Mishima M, et al. Small cell lung carcinoma: eight types of extension and spread on computed tomography. J Comput Assist Tomogr. 2006;30(4):653–61.
Kreisman H, Wolkove N, Quoix E. Small cell lung cancer presenting as a solitary pulmonary nodule. Chest. 1992;101(1):225–31.
Seute T, Leffers P, ten Velde GP, Twijnstra A. Detection of brain metastases from small cell lung cancer: consequences of changing imaging techniques (CT versus MRI). Cancer. 2008;112(8):1827–34.
Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366(9494):1385–96.
Yabuuchi H, Murayama S, Sakai S, Hashiguchi N, Murakami J, Muranaka T, et al. Resected peripheral small cell carcinoma of the lung: computed tomographic-histologic correlation. J Thorac Imaging. 1999;14(2):105–8.
Ravenel JG. Small cell carcinoma. In: Wheless L, James Brashears J, editors. Lung cancer imaging. New York: Humana Press; 2013. p. 79–88.
Riihimäki M, Hemminki A, Fallah M, Thomsen H, Sundquist K, Sundquist J, et al. Metastatic sites and survival in lung cancer. Lung Cancer. 2014;86(1):78–84.
Levitan N, Byrne RE, Bromer RH, Faling LJ, Caslowitz P, Pattern DH, et al. The value of the bone scan and bone marrow biopsy staging small cell lung cancer. Cancer. 1985;56(3):652–4.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Larici, A.R. et al. (2020). Pulmonary Neuroendocrine Neoplasms. In: Sverzellati, N., Silva, M. (eds) The Thorax. Cancer Dissemination Pathways. Springer, Cham. https://doi.org/10.1007/978-3-030-27233-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-27233-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27232-6
Online ISBN: 978-3-030-27233-3
eBook Packages: MedicineMedicine (R0)