
Abstract
The heat shock protein 90 (Hsp90) is a ubiquitous molecular chaperone that is abundantly expressed in cancer cells and plays a pivotal role in correct folding and functions of a variety of oncogenic clients. Hsp90 is up-regulated in response to cellular stresses that cancer cells encounter, such as heat, hypoxia and nutrient deprivation, conditions commonly associated with the tumor microenvironment. P53 is the tumor suppressor gene that is mutated in nearly 50% of all human cancers. When mutated p53 not only lose its tumor suppressive function but also gain novel oncogenic activities via gain-of-function mechanisms leading to increased genomic instability, chemoresistance, and metastasis, which promote tumor progression. In contrast to wild-type p53, mutant p53 is protected from degradation via interaction with Hsp90 leading to marked stabilization of mutant p53 protein in cancer cells. Recent in vivo studies unequivocally have proven that the stabilization of mutant p53 is crucial pre-requisite for its oncogenic functions. The pharmacological targeting the pathways involved in the stabilization of mutant p53, in particular, the Hsp90 chaperone complex, recently attracted a lot of attention as a promising therapeutic approach to treat mutant p53 harboring cancers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The tumor suppressor protein, p53, was initially discovered in the late 1970s, where it was wrongfully identified as the middle antigen of SV40 virus (Kress et al. 1979; Lane and Crawford 1979; Linzer and Levine 1979). Later, p53 was found to be expressed in the cells in response to viral infections and that it accumulates in the nucleus of tumor cells (Kress et al. 1979). Initially, p53 was characterized as an oncoprotein (Eliyahu et al. 1984; Jenkins et al. 1984; Parada et al. 1984). Over the years, starting in the mid-1980s, TP53 was firmly established as tumor suppressor gene, cell cycle regulator, and cell sensor in response multiple stresses that the cell may encounter (Gatz and Wiesmuller 2006; Menon and Povirk 2014; Soussi 2010).
The tumor suppressive functions of p53 were shown to be inhibited by various mechanisms, such as epigenetic modulations, miRNA, or by a dysregulated CCCTC-binding factor (Saldana-Meyer and Recillas-Targa 2011). However, mutations in TP53 gene are the most frequent inactivating genetic events in most types of cancer ((http://p53.iarc.fr/TP53SomaticMutations.aspx) (Petitjean et al. 2007; Vogelstein et al. 2000)). Unlike other tumor suppressors that are usually inactivated by deletion, 75% of all p53 genetic alterations in TP53 gene are missense mutations within the DNA-binding domain, predominantly located in six most frequent ‘hotspots’ (http://p53.iarc.fr/TP53SomaticMutations.aspx) (Baugh et al. 2018; Bouaoun et al. 2016). TP53 missense mutations hinder p53 binding to DNA response elements precluding its tumor suppressive transcription program. Clinical data suggest that p53 behaves as a classic “two-hit” tumor suppressor so that a point mutation in one allele of p53 at early stages is followed by loss of heterozygosity (LOH) in the second allele later on during tumor progression (Levine et al. 1991).
Mutant p53 exerts its oncogenic activities either by dominant negative (DN) or by gain-of-function (GOF) mechanisms (Petitjean et al. 2007; Schulz-Heddergott and Moll 2018). In early stages of cancers that are heterozygous for p53, mutant p53 may suppress the expression and/or function of wild-type p53 through DN mechanism (Milner et al. 1991; Willis et al. 2004). Mutant p53 was shown to form heterodimers with wild-type p53 that can cause conformational changes to wild-type p53 or may directly inhibit the DNA-binding activity of wild-type p53 on target genes. Similarly, mutant p53 can interact with other p53 family members- TAp63 and TAp73, to inhibit their tumor suppressive functions (Lang et al. 2004; Muller et al. 2009). Although, in vitro studies greatly support DN effect of mutant p53 (Milner et al. 1991; Willis et al. 2004), in mouse models it was shown to be highly tissue-specific and often is manifested only upon stabilization of mutant p53 after genotoxic stress, e.g., epithelium-specific heterozygous expression of mutant p53 (R270H) exerted DN effect on tumor latency, multiplicity, and progression only after UV exposure but not spontaneous tumors (Wijnhoven et al. 2007). Study on R246S mutant p53 knock-in mouse model has shown that the DN effect on transactivation is detectable after acute p53 activation only (Lee et al. 2012). Also, DNA damage increased the DN activity of mutant p53 in various tissues of heterozygous mutant p53 mice (Lang et al. 2004; Olive et al. 2004).
On the other hand, GOF is commonly defined as “any activity of mutant p53 exerted in the absence of co-expressed wild-type p53” (Oren and Rotter 2010). In accord with this notion, we recently demonstrated that p53 LOH is a necessary prerequisite for mutant p53 GOF in vivo (Alexandrova et al. 2017a). The GOF concept was first introduced by Dittmer et al., where they showed the ability of mutant p53, but not wild-type p53, to transform p53-null cells in vitro and in vivo (Dittmer et al. 1993). Since then, numerous publications have confirmed that missense mutations in the TP53 gene not only obliterate wild-type p53 tumor suppressive function, but mutant p53 protein also gains novel oncogenic activities. By inducing multiple oncogenic pathways (Adorno et al. 2009; Li et al. 2014; Muller et al. 2009; Yallowitz et al. 2015), microRNA and altering cell metabolism, transcriptional regulation, chromatin structure (Blandino and Di Agostino 2018; Oren and Rotter 2010), mutant p53 promotes oncogenic reprogramming and cancer progression.
1.1 Stability of Mutant p53 Protein in Cancer Cells
The stability of a wild-type p53 protein is tightly regulated through degradation by E3 ligase mouse double-minute 2 (MDM2) (Haupt et al. 1997; Lukashchuk and Vousden 2007). As wild-type p53 target gene, MDM2 itself is regulated through a negative feedback loop maintaining a very low and steady level of both proteins under normal condition (Wu et al. 1993). In normal and unstressed cancer cells wild-type p53 protein level is very low due to a rapid turnover by MDM2. This tight control is lost in tumors carrying mutant p53. While initially the stability of mutant p53 was thought to be due to the loss of p53-mediated transactivation of MDM2, it was later shown that mutant p53 is inherently unstable in normal cells, and only tumors but not normal tissues display constitutive stabilization of mutant p53 (Lang et al. 2004; Olive et al. 2004; Terzian et al. 2008). Mutant p53 knock-in models demonstrated that MDM2 is still expressed in a p53-independent mechanism in normal tissues and additional alteration(s) occur upon malignant conversion that stabilizes mutant p53 (Lang et al. 2004; Olive et al. 2004).
Similar to wild-type p53, mutant p53 protein fully preserves its ability to interact with MDM2, and MDM2 protein levels can be easily detectable in mutant p53 expressing cancer cells (Li et al. 2011b). Nevertheless, the profound lack of ubiquitination of mutant p53 indicates a severe degradation defect, due to inactivation of MDM2 enzymatic activity (Li et al. 2011b). Mechanistically, it has been shown that mutant p53 stimulates the formation of triple MDM2-p53-Hsp90 complexes, whereby blocking ubiquitin-protein isopeptide ligase activity of MDM2 and resulting in the stabilization of both mutant p53 and MDM2 (Peng et al. 2001). This evidence indicates that Hsp90 is essential for the stabilization of mutant p53 in cancer cells.
Strikingly, it appears that cancer cells become addicted to high levels of mutant p53 protein, as an acute depletion of mutant p53 markedly reduces cells growth in vitro and in xenografts (Li et al. 2011b), suppresses invasion (Adorno et al. 2009; Li et al. 2011b; Muller et al. 2009; Yan et al. 2008), restores normal mammary architecture in 3D culture in breast cancer cell lines (Freed-Pastor et al. 2012), inhibits metastases in vivo (Morton et al. 2010; Weissmueller et al. 2014), and suppresses mammary stem cells (Yallowitz et al. 2015). Mutant p53 depletion in allotransplanted and autochthonous mouse T/B-lymphoma model curbs tumor growth and extends the survival of mice (Alexandrova et al. 2015). Similarly, the genetic ablation of R248Q mutant p53 inhibits tumor growth and invasion in AOM/DSS-Induced colorectal cancer mouse models. Mechanistically, Schulz-Heddergott et al. demonstrated that mutant p53 protein binds and enhances activating phosphorylation of Stat3, while conditional depletion of mutant p53 from established tumors suppresses Jak2/Stat3 signaling, tumor growth, and invasiveness (Schulz-Heddergott et al. 2018). As other evidence supporting the importance of highly stabilized mutant p53 for the manifestation of GOF activity, we found that in heterozygosity wild-type p53 allele suppresses mutant p53 stabilization in cancer cells in vivo, whereas p53 LOH is associated with stabilization of mutant p53 in cancer cells and, most importantly, with mutant p53 GOF features (Alexandrova et al. 2017a).
Collectively, this proof-of-principal from in vivo studies demonstrate that high levels of mutant p53 protein is essential for the manifestation of mutant p53 GOF and underscore the translational significance of highly stabilized mutant p53 as a potentially attractive therapeutic target. However, targeting mutant p53 is a very challenging task, since mutant p53 is neither a typical enzyme nor localized to the cell surface. On the other hand, the addiction of cancer cells to highly stabilized mutant p53 can be exploited by therapeutic approaches aiming to eliminate mutant p53 from cancer cells. Thus, selectively targeting the mechanisms underlying cancer specific stabilization of mutant p53 protein is the feasible strategy to combat mutant p53 harboring cancers.
1.2 Hsp90 in Cancer
A role for Hsp90 in cancer was first implicated in breast tumors where an association of Hsp90 and steroid receptors was observed (Pratt 1987; Shyamala et al. 1989), and in ovarian cancer where increased expression of Hsp90 mRNA was detected (Mileo et al. 1990). To date, it is well known that Hsp90 is ubiquitously expressed in almost all cancer types. The high-level of Hsp90 was shown to be associated with more aggressive phenotypes and poor prognosis in breast cancer patients. Also, the high levels Hsp90 were shown to correlate with more advanced stages of melanomas, leukemia (Chatterjee and Burns 2017) and colon cancer (Milicevic et al. 2008). There are two major isoforms of Hsp90: constitutive Hsp90α and inducible Hsp90β, which together comprise 1–3% of total cellular protein. Although Hsp90 is also expressed in normal tissues, it is activated in cancer cells by various mechanisms: modulation of activity by translational modifications (acetylation, phosphorylation, ubiquitination, S-nitrosylation); activation by interaction with clients; activation of transcription; and subcellular localization (cytoplasm vs. cell surface) (Barrott and Haystead 2013).
As a molecular chaperone , Hsp90 plays an essential role in maintaining proteome homeostasis in cancer cells by facilitating folding and intracellular trafficking of its numerous oncogenic clients. Although Hsp90 is very abundant protein, it gets further upregulated by different stresses experienced by tumor cells such as acidic pH, shortage of nutrients and fluctuating oxygen levels, which commonly occur in cancer (Gabai et al. 1995). It appears that Hsp90 is essential to cancer cells survival by stabilizing and preventing aggregation of many mutated aberrantly folded oncogenic proteins, which drive tumor progression. There are over 400 Hsp90 client proteins (for updated client protein list www.picard.ch/downloads/Hsp90interactors.pdf), with a large number of them playing important roles in different aspects of tumor development such as tumor growth, apoptotic evasion, differentiation and metastasis (Neckers and Workman 2012). The pharmacological inhibition of Hsp90 leads to rapid inhibition of clients activity, with their subsequent degradation leading to depletion of multiple oncoproteins and down-regulation of oncogenic signaling pathways, and ultimately resulting in the modulation of the malignant phenotype (Jackson 2013). The reliance of cancer cells on oncogenic pathways and proteins that are stabilized by Hsp90 is the reason for the sensitivity of these cells to Hsp90 inhibition (Neckers and Workman 2012; Whitesell and Lindquist 2005; Workman et al. 2007). Also, Hsp90 has been found to be secreted by cancer cells, and the role of the secreted Hsp90 has been garnering some attention for its role in cancer cell invasion and metastasis (Eustace and Jay 2004; Eustace et al. 2004; Nolan et al. 2015). Overall, given the clinical importance of numerous oncogenic Hsp90 clients in different types of cancer, targeting Hsp90 and its respective co-chaperones has huge potential for utilization in cancer therapy.
1.3 Mutant p53-Hsp90 Axis
Commonly, mutations in TP53 cause an alteration of mutant p53 protein conformation (e.g., residues R249, G245, R282, R175), lower melting temperature, and lead to aberrant protein folding (Schulz-Heddergott and Moll 2018). To prevent protein aggregation and cell death from proteotoxic stress, mutant p53 proteins interact with a number of molecular chaperones, heat shock proteins (HSP) that are essential for stabilization aggregation-prone proteins. Mutant p53 was shown to interact with several HSP: Hsp90 (Blagosklonny et al. 1996), Hsp70 (Sturzbecher et al. 1988), HSC70 (Hinds et al. 1987) and HSP40/DNAJ complex (Parrales et al. 2016). Recently, mutant p53- Hsp90 interaction was extensively studied, as it was shown that pharmacological Hsp90 inhibition destabilized mutant p53, but not wild-type p53 in cancer cells in vitro (Blagosklonny et al. 1996; Li et al. 2011b; Peng et al. 2001) and in several mouse models (Alexandrova and Marchenko 2015; Schulz-Heddergott et al. 2018). The long-term Hsp90 inhibition by specific inhibitor ganetespib extended the survival of mutant p53 R248Q/− and R172H/R172H knock-in mice by 59% and 48%, respectively, but not p53−/− littermates. Importantly, mutant p53-dependent survival benefits correlated with mutant p53 degradation, apoptosis and prevention of T-cell lymphomagenesis in vivo (Alexandrova et al. 2015). Treating mice with an Hsp90 inhibitor 17AAG suppressed mutant p53 levels and colorectal tumor growth in p53 R248Q/−mice treated with AOM/DSS compared with vehicle controls. Although 17AAG showed some tumor inhibitory effect in p53−/− control mice, it was much less pronounced than in mutant p53 mice (Schulz-Heddergott et al. 2018). Also, ganetespib synergized with a sub-effective dose of the DNA-alkylating cytotoxic agent cyclophosphamide in mutant p53 R248Q lymphoma mouse model by suppressing tumor growth and extending survival. Importantly, ganetespib/cyclophosphamide combinatorial treatment was more beneficial for mutant p53 than p53−/− mice (Alexandrova et al. 2017b). Together, these rigorous in vivo and in vitro studies implicate the potential clinical utility of targeting mutant p53-Hsp90 axis and the mechanisms regulating this interaction to degrade mutant p53 GOF in cancer.
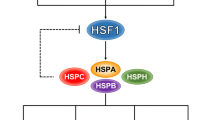
The transcription of inducible HSP is regulated by the master transcription regulator of heat shock response- heat shock factor 1 (HSF1) (Gomez-Pastor et al. 2018). HSF1 induces the transcription of Hsp90α, Hsp70, members of the HSP40/DNAJ family, and numerous co-chaperones (Gomez-Pastor et al. 2018). In normal cells, HSF1 is transiently active in conditions of proteotoxic stress, while tumor cells that under permanent proteotoxic stress due to adverse tumor environment require permanent activation of HSF1. Importantly, activation of HSF1 also appears to be essential for malignant transformation, as shown in several mouse cancer models (Dai et al. 2012; Dai et al. 2007). In addition to regulation of heat shock response, HSF1 drives broad cancer-specific transcriptional programs required for numerous oncogenic processes, such as aberrant cell cycle regulation, signal transduction, translation, metabolism, and invasion (Dai et al. 2007; Mendillo et al. 2012; Min et al. 2007; Zhao et al. 2011). Importantly, recent studies on HSF1 knockout mouse model provide compelling genetic evidence for the existence of crucial cooperation between mutant p53 and HSF1 in malignancy (Dai et al. 2007; Min et al. 2007). Given the importance of both HSF1 and mutant p53 in cancer, we identified the mechanism of interaction between HSF1 and mutant p53 in cancer cells; a novel gain-of-function of mutant p53 as a promoter of HSF1 activity (Li et al. 2014). We found that mutant p53 via stimulation of HSF1 transcriptional activity induces a feed-forward loop to Hsp90, which further stabilizes mutant p53 protein. Mechanistically, mutant p53, through enhanced recycling and/or stability of ErbB2, augments MAPK and PI3K signaling, leading to phospho-activation of HSF1 at Ser326. Furthermore, mutant p53 via direct interaction with phospho-activated HSF1 facilitates the binding of HSF1 to its DNA-binding sites, thereby stimulating the transcription of HSP. In turn, heat shock proteins more potently stabilize their clients ErbB2, EGFR, mutant p53, and HSF1 (and possibly other oncogenes), thus reinforcing tumor progression (Li et al. 2014). This feed-forward loop renders cancer cells resistant to proteotoxic stress, providing a distinct survival advantage. This feed-forward axis was also confirmed in vivo, where we defined its physiological consequences in ErbB2-driven breast cancer mouse model (Yallowitz et al. 2015).
1.4 Translational Significance of Mutant p53–Hsp90 Axis in Cancer
Targeting the pathways involved in the stabilization of mutant p53, in particular, the Hsp90 family, has attracted a lot of attention over many years. Hsp90 has proven to be a promising target for cancer treatment and is actively pursued by drug discovery companies (Neckers and Workman 2012; Travers et al. 2012). As discussed above, Hsp90 inhibition was shown to be more efficacious in mutant p53 harboring cells in vitro (Li et al. 2011b) and in vivo (Alexandrova et al. 2015, 2017b; Schulz-Heddergott and Moll 2018) compared to p53null and wild-type p53 counterparts. The first Hsp90 inhibitor identified is benzo-quinone ansamycin geldanamycin (Whitesell et al. 1994). The majority of currently available Hsp90 inhibitors, and all that have been clinically assessed (reviewed in (Butler et al. 2015)), have a common mechanism of action: bind to the nucleotide binding pocket of the N-terminal domain and block the processing of client proteins by preventing ATP binding and hydrolysis (Prodromou et al. 1997; Roe et al. 1999), thus preventing the completion of the Hsp90 chaperone cycle, and clients are subsequently targeted for proteasomal degradation by E3 ubiquitin ligases (Connell et al. 2001; Ehrlich et al. 2009; Xu et al. 2002). As promising as these inhibitors, however variable limitations to their use have risen due to several factors such as insufficient drug dose, the frequency of administration, variable pharmacokinetics, suboptimal formulation and dose-limiting toxicities, including hepatotoxicity (Butler et al. 2015). Additionally, another problem that frequently arises in using N-terminal domain inhibitors is the development of drug resistance, since blocking N-terminal domain of Hsp90 often leads to the activation of HSF1 which in turn induces more Hsp90 and other HSP (Sauvage et al. 2017). However, Hsp90 N-terminal domain inhibitors demonstrated encouraging results in clinical trials in HER-positive breast cancer and non-small cell lung carcinoma, which have high prevalence of p53 mutations- 72% in HER2 positive breast cancer, 81% in squamous cell carcinomas, and 68% in large cell carcinomas (Neckers and Workman 2012; Shepherd et al. 2017).
Further investigating the mechanisms protecting mutant p53 from degradation in cancer cells, we demonstrated that similar to Hsp90 inhibitors, histone deacetylase (HDAC) inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA) destabilizes mutant p53 at the level of protein degradation. We found that SAHA via inhibition of HDAC6, an essential positive regulator of Hsp90, releases mutant p53 from Hsp90 inhibitory complex and enables its MDM2- and CHIP-mediated degradation. As a result of mutant p53 degradation, SAHA exhibits preferential cytotoxicity for mutant p53 compared to wild-type and null p53 human cancer cells and strongly chemosensitizes mutant p53 cancer cells for chemotherapy (Li et al. 2011a). As a follow-up of our study, Ingallina et al., demonstrated that HDAC6/Hsp90-dependent mutant p53 accumulation is sustained by RhoA geranylgeranylation downstream of the mevalonate pathway, as well as by RhoA- and actin-dependent transduction providing mechanistic cues to control mutant p53 levels via RhoA/actin cytoskeleton. This study identified statins as a mutant p53 destabilizing compounds that disrupt the SREBP-mevalonate/RhoA signaling pathway, which controls Hsp90-mediated mutant p53 stabilization (Ingallina et al. 2018). As SAHA (vorinostat) and statins are FDA-approved drugs, these mechanistic studies can be instantly translated to the clinic. Indeed, following these publications, a Phase I clinical trial was initiated, which confirmed that simultaneous use of vorinostat (a mutant p53 destabilizing drug) and Pazopanib (an anti-angiogenic VEGFR inhibitor) in metastatic sarcoma and colorectal cancer has preferential response in mutant p53 (45%) vs. wild-type p53 patients (16%) (Fu et al. 2015). With regard to statins, a recent study by Turrell et al. demonstrated that statins sensitivity is dependent on the type of p53 mutation. They identified a p53R270H-specific sensitivity to simvastatin, but not in p53R172H, in murine KrasG12D lung cancer model. Importantly, the “statin-sensitivity” transcriptional signature is also present in human lung tumors suggesting the clinical relevance of this data (Turrell et al. 2017).
As discussed above, we demonstrated that ErbB2 signaling via downstream transduction cascade controls HSF1 activity, HSP expression and as a result, mutant p53 stability (Li et al. 2014; Yallowitz et al. 2015). Following this study, we found that pharmacological interception of this circuit by the dual ErbB2/EGFR kinase inhibitor (lapatinib) downregulates mutant p53 in vitro and in vivo. Inhibition of ErbB2 by lapatinib inhibits transcription factor HSF1, and its target Hsp90, followed by mutant p53 degradation in MDM2 dependent manner. We speculate that mutant p53 sensitizes cancer cells to lapatinib via two complementary mechanisms: mutant p53 mediated amplification of ErbB2 signaling, and simultaneous annihilation of both potent oncogenic drivers, ErbB2 and mutant p53. This conclusion is strongly supported by a meta-analysis of the COSMIC drug sensitivity database of 226 human cancer cell lines (representing breast, as well as other cancers) (http://www.cancerrxgene.org/translation/Drug/119). Specifically, we found that mutant p53 human cell lines were more sensitive to lapatinib than wild-type p53 cells (p = 0.0408). Consistently, we found higher sensitivity to lapatinib in R172H/R172H;ErbB2 mammary epithelial cells than their p53null;ErbB2 counterparts (Li and Marchenko 2017). Figure 7.1 is an illustration summarizing the pathways involved in augmenting the mutant p53-Hsp90 axis in cancer and the different druggable targets that have been identified within this pathway.

Targeting mutant p53–Hsp90 axis in cancer. Blue line: cell membrane. Above the line represents extracellular material, and below the line represents cell cytoplasm. “Y”: different receptor tyrosine kinases. Blocked red lines: drug inhibition. Black arrows: activation. Black broken arrows: compensatory activation pathways. Blocked black line: protein-protein inhibition
Adding to the complexity of modulating Hsp90 and its related pathways in treating cancer, a recent follow-up study by our lab (Yallowitz et al. 2018) attempted to identify a mechanism for lapatinib resistance (D’Amato et al. 2015). Several mechanisms of lapatinib resistance have been described in the literature, and they primarily involve compensatory activation of several receptor tyrosine kinases (RTKs) (Stuhlmiller et al. 2015). This and the substantial heterogeneity among adaptive RTKs that exists in different cell lines in response to lapatinib represent a major hurdle for the development of successful combinatorial strategies to reverse and/or prevent lapatinib resistance (Stuhlmiller et al. 2015). In our study, we found that lapatinib-resistant cells show chronically activated HSF1 and its transcriptional targets, heat shock proteins, and consequently, higher tolerance to proteotoxic stress. Importantly, lapatinib-resistant tumors and cells retained sensitivity to Hsp90 and HSF1 inhibitors, both in vitro and in vivo. Indeed, HSF1 inhibition simultaneously downregulated ErbB2, adaptive RTKs, and mutant p53, and notably, its combination with lapatinib prevented the development of lapatinib resistance in vitro (Yallowitz et al. 2018). Recently, a novel drug curaxin CBL0137, which has been shown to suppress HSF1 (Neznanov et al. 2011), entered a clinical trial (phase 1) in patients with metastatic or unresectable advanced solid cancers (Incuron) (Burkhart et al. 2014). Hence, pharmacological inhibition of HSF1 opens up a new therapeutic possibility for the clinical application of HSF1 inhibitors to prevent and/or delay the onset of lapatinib resistance in 72% ErbB2 positive breast cancer patients carrying mutant p53.
2 Conclusions
Although Hsp90 inhibitors have recently shown some clinical success, it becomes increasingly evident that patient selection and predictive biomarkers seem to be the main hurdle to the successful clinical utilization of Hsp90 inhibitors. Despite nearly 400 proteins interacting with Hsp90 have been identified, the clinical value of these interactions remains unclear. As mutant p53 ablation in cancer cells shows a significant survival benefit in multiple in vivo models, here we discussed the therapeutic potential of targeting mutant p53-Hsp90/HDAC6 axis to achieve mutant p53 depletion in cancer cells. Future retrospective and prospective clinical studies are needed to confirm these preclinical studies and establish mutant p53-Hsp90/HDAC6 axis as an exploitable target.
Abbreviations
- DN:
-
dominant negative
- GOF:
-
gain-of-function
- HDAC:
-
histone deacetylase
- HSF1:
-
heat shock factor 1
- HSP:
-
heat shock protein
- LOH:
-
loss of heterozygosity
- MDM2:
-
mouse double-minute 2
- RTK:
-
receptor tyrosine kinases
- SAHA:
-
suberoylanilide hydroxamic acid
References
Adorno M, Cordenonsi M, Montagner M et al (2009) A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137:87–98
Alexandrova EM, Marchenko ND (2015) Mutant p53 – heat shock response oncogenic cooperation: a new mechanism of cancer cell survival. Front Endocrinol (Lausanne) 6:53
Alexandrova EM, Yallowitz AR, Li D et al (2015) Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523:352–356
Alexandrova EM, Mirza SA, Xu S, Schulz-Heddergott R, Marchenko ND, Moll UM (2017a) p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis 8:e2661
Alexandrova EM, Xu S, Moll UM (2017b) Ganetespib synergizes with cyclophosphamide to improve survival of mice with autochthonous tumors in a mutant p53-dependent manner. Cell Death Dis 8:e2683
Barrott JJ, Haystead TA (2013) Hsp90, an unlikely ally in the war on cancer. FEBS J 280:1381–1396
Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS (2018) Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 25:154–160
Blagosklonny MV, Toretsky J, Bohen S, Neckers L (1996) Mutant conformation of p53 translated in vitro or in vivo requires functional Hsp90. Proc Natl Acad Sci USA 93:8379–8383
Blandino G, Di Agostino S (2018) New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cancer Res 37:30
Bouaoun L, Sonkin D, Ardin M et al (2016) TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 37:865–876
Burkhart C, Fleyshman D, Kohrn R et al (2014) Curaxin CBL0137 eradicates drug resistant cancer stem cells and potentiates efficacy of gemcitabine in preclinical models of pancreatic cancer. Oncotarget 5:11038–11053
Butler LM, Ferraldeschi R, Armstrong HK, Centenera MM, Workman P (2015) Maximizing the therapeutic potential of Hsp90 inhibitors. Mol Cancer Res 13:1445–1451
Chatterjee S, Burns TF (2017) Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci 18:1978
Connell P, Ballinger CA, Jiang J et al (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 3:93–96
D’Amato V, Raimondo L, Formisano L et al (2015) Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev 41:877–883
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018
Dai C, Santagata S, Tang Z et al (2012) Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 122:3742–3754
Dittmer D, Pati S, Zambetti G et al (1993) Gain of function mutations in p53. Nat Genet 4:42–46
Ehrlich ES, Wang T, Luo K et al (2009) Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci USA 106:20330–20335
Eliyahu D, Raz A, Gruss P, Givol D, Oren M (1984) Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 312:646–649
Eustace BK, Jay DG (2004) Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 3:1098–1100
Eustace BK, Sakurai T, Stewart JK et al (2004) Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol 6:507–514
Freed-Pastor WA, Mizuno H, Zhao X et al (2012) Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148:244–258
Fu S, Hou MM, Naing A et al (2015) Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Ann Oncol 26:1012–1018
Gabai VL, Mosina VA, Budagova KR, Kabakov AE (1995) Spontaneous overexpression of heat-shock proteins in Ehrlich ascites carcinoma cells during in vivo growth. Biochem Mol Biol Int 35:95–102
Gatz SA, Wiesmuller L (2006) p53 in recombination and repair. Cell Death Differ 13:1003–1016
Gomez-Pastor R, Burchfiel ET, Thiele DJ (2018) Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol 19:4–19
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387:296–299
Hinds PW, Finlay CA, Frey AB, Levine AJ (1987) Immunological evidence for the association of p53 with a heat shock protein, hsc70, in p53-plus-ras-transformed cell lines. Mol Cell Biol 7:2863–2869
Ingallina E, Sorrentino G, Bertolio R et al (2018) Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat Cell Biol 20:28–35
Jackson SE (2013) Hsp90: structure and function. Top Curr Chem 328:155–240
Jenkins JR, Rudge K, Currie GA (1984) Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 312:651–654
Kress M, May E, Cassingena R, May P (1979) Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 31:472–483
Lane DP, Crawford LV (1979) T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263
Lang GA, Iwakuma T, Suh YA et al (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119:861–872
Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, Sabapathy K (2012) Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 22:751–764
Levine AJ, Momand J, Finlay CA (1991) The p53 tumour suppressor gene. Nature 351:453–456
Li D, Marchenko ND (2017) ErbB2 inhibition by lapatinib promotes degradation of mutant p53 protein in cancer cells. Oncotarget 8:5823–5833
Li D, Marchenko ND, Moll UM (2011a) SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ 18:1904–1913
Li D, Marchenko ND, Schulz R et al (2011b) Functional inactivation of endogenous MDM2 and CHIP by Hsp90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res 9:577–588
Li D, Yallowitz A, Ozog L, Marchenko N (2014) A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis 5:e1194
Linzer DI, Levine AJ (1979) Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43–52
Lukashchuk N, Vousden KH (2007) Ubiquitination and degradation of mutant p53. Mol Cell Biol 27:8284–8295
Mendillo ML, Santagata S, Koeva M et al (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562
Menon V, Povirk L (2014) Involvement of p53 in the repair of DNA double strand breaks: multifaceted roles of p53 in homologous recombination repair (HRR) and non-homologous end joining (NHEJ). Subcell Biochem 85:321–336
Mileo AM, Fanuele M, Battaglia F et al (1990) Selective over-expression of mRNA coding for 90 KDa stress-protein in human ovarian cancer. Anticancer Res 10:903–906
Milicevic Z, Bogojevic D, Mihailovic M, Petrovic M, Krivokapic Z (2008) Molecular characterization of hsp90 isoforms in colorectal cancer cells and its association with tumour progression. Int J Oncol 32:1169–1178
Milner J, Medcalf EA, Cook AC (1991) Tumor suppressor p53: analysis of wild-type and mutant p53 complexes. Mol Cell Biol 11:12–19
Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF (2007) Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 26:5086–5097
Morton JP, Timpson P, Karim SA et al (2010) Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA 107:246–251
Muller PA, Caswell PT, Doyle B et al (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139:1327–1341
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18:64–76
Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV (2011) Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget 2:209–221
Nolan KD, Franco OE, Hance MW, Hayward SW, Isaacs JS (2015) Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J Biol Chem 290:8271–8282
Olive KP, Tuveson DA, Ruhe ZC et al (2004) Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119:847–860
Oren M, Rotter V (2010) Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2:a001107
Parada LF, Land H, Weinberg RA, Wolf D, Rotter V (1984) Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312:649–651
Parrales A, Ranjan A, Iyer SV et al (2016) DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol 18:1233–1243
Peng Y, Chen L, Li C, Lu W, Chen J (2001) Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 276:40583–40590
Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M (2007) TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 26:2157–2165
Pratt WB (1987) Transformation of glucocorticoid and progesterone receptors to the DNA-binding state. J Cell Biochem 35:51–68
Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1997) Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90:65–75
Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42:260–266
Saldana-Meyer R, Recillas-Targa F (2011) Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 6:1068–1077
Sauvage F, Messaoudi S, Fattal E, Barratt G, Vergnaud-Gauduchon J (2017) Heat shock proteins and cancer: how can nanomedicine be harnessed? J Control Release 248:133–143
Schulz-Heddergott R, Moll UM (2018) Gain-of-Function (GOF) mutant p53 as actionable therapeutic target. Cancers (Basel) 10:188
Schulz-Heddergott R, Stark N, Edmunds SJ et al (2018) Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits Stat3-mediated tumor growth and invasion. Cancer Cell 34:298–314 e7
Shepherd FA, Lacas B, Le Teuff G et al (2017) Pooled analysis of the prognostic and predictive effects of TP53 comutation status combined with KRAS or EGFR mutation in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol 35:2018–2027
Shyamala G, Gauthier Y, Moore SK, Catelli MG, Ullrich SJ (1989) Estrogenic regulation of murine uterine 90-kilodalton heat shock protein gene expression. Mol Cell Biol 9:3567–3570
Soussi T (2010) The history of p53. A perfect example of the drawbacks of scientific paradigms. EMBO Rep 11:822–826
Stuhlmiller TJ, Miller SM, Zawistowski JS et al (2015) Inhibition of Lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep 11:390–404
Sturzbecher HW, Addison C, Jenkins JR (1988) Characterization of mutant p53-hsp72/73 protein-protein complexes by transient expression in monkey COS cells. Mol Cell Biol 8:3740–3747
Terzian T, Suh YA, Iwakuma T et al (2008) The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 22:1337–1344
Travers J, Sharp S, Workman P (2012) Hsp90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today 17:242–252
Turrell FK, Kerr EM, Gao M et al (2017) Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes Dev 31:1339–1353
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408:307–310
Weissmueller S, Manchado E, Saborowski M et al (2014) Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 157:382–394
Whitesell L, Lindquist SL (2005) Hsp90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772
Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM (1994) Inhibition of heat shock protein Hsp90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA 91:8324–8328
Wijnhoven SW, Speksnijder EN, Liu X et al (2007) Dominant-negative but not gain-of-function effects of a p53.R270H mutation in mouse epithelium tissue after DNA damage. Cancer Res 67:4648–4656
Willis A, Jung EJ, Wakefield T, Chen X (2004) Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23:2330–2338
Workman P, Burrows F, Neckers L, Rosen N (2007) Drugging the cancer chaperone Hsp90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci 1113:202–216
Wu X, Bayle JH, Olson D, Levine AJ (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev 7:1126–1132
Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L (2002) Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA 99:12847–12852
Yallowitz AR, Li D, Lobko A, Mott D, Nemajerova A, Marchenko N (2015) Mutant p53 amplifies epidermal growth factor receptor family signaling to promote mammary tumorigenesis. Mol Cancer Res 13:743–754
Yallowitz A, Ghaleb A, Garcia L, Alexandrova EM, Marchenko N (2018) Heat shock factor 1 confers resistance to lapatinib in ERBB2-positive breast cancer cells. Cell Death Dis 9:621
Yan W, Liu G, Scoumanne A, Chen X (2008) Suppression of inhibitor of differentiation 2, a target of mutant p53, is required for gain-of-function mutations. Cancer Res 68:6789–6796
Zhao Y, Liu H, Liu Z et al (2011) Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res 71:4585–4597
Acknowledgements
This work was supported by the Department of Defense grant W81XWH-16-1-0448 (BC151569) and the Carol Baldwin Breast Cancer Research Fund to N. Marchenko.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ghaleb, A., Marchenko, N. (2019). p53-Hsp90 Axis in Human Cancer. In: Asea, A., Kaur, P. (eds) Heat Shock Protein 90 in Human Diseases and Disorders. Heat Shock Proteins, vol 19. Springer, Cham. https://doi.org/10.1007/978-3-030-23158-3_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-23158-3_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-23157-6
Online ISBN: 978-3-030-23158-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)