
Abstract
Over the last two decades, clinicians managing invasive fungal infections in neutropenic cancer patients have encountered many challenges – please see chapter on “Fungal Infections”. Fortunately we have also seen an expansion in our antifungal armamentarium during this time frame as well. This chapter will focus on the antifungal agents which are utilized for the prevention and treatment of invasive fungal infections in the neutropenic cancer patient. Specifically, we will discuss the polyene antifungal amphotericin B, the anti-metabolite flucytosine, select azole antifungals (fluconazole, voriconazole, posaconazole, isavuconazonium sulfate), and the echinocandins (caspofungin, micafungin, anidulafungin).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Polyene Antifungals
- Amphotericin B deoxycholate
- Nystatin
- Flucytosine
- Azole antifungal
- Fluconazole
- Voriconazole
- Posaconazole
- Isavuconazonium sulfate
- Echinocandin Antifungals
- Caspofungin
- Micafungin

Polyene Antifungals
Amphotericin B
Stucture of Amphotericin B [1]

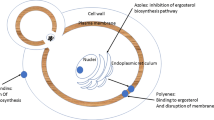
Amphotericin B deoxycholate (Fungizone, AmBD) , also known as “conventional” amphotericin B, is a broad-spectrum, fungicidal polyene antifungal agent which was a life-saving innovative agent when it was FDA approved in 1959 [2]. Amphoterin B binds to ergosterol on the fungal cell membrane then merges into the fungal cell membrane causing a pore which allows for increased passage of fluids into the fungal cell and cell rupture [1,2,3]. Amphotericin B deoxycholate was the primary agent for the treatment of invasive fungal infections for decades but its use was limited by its toxicity which lead to the development of lipid formulations of amphotericin B which have largely replaced the use of AmBD in the neutropenic cancer patient population [2].
Amphotericin B Antifungal Spectrum of Activity
Amphotericin B has broad-spectrum fungicidal activity for a wide variety of invasive fungal infections including but not limited to aspergillosis, candidiasis, fusariosis, mucormycosis, and the endemic mycosis such as blastomycosis, histoplasmosis, coccidiomycosis, paracoccidioidomycois, and sporotrichosis [3, 4]. Amphotericin B resistance is typically associated with a MIC >2 mg/L [4] and has been most commonly reported in Candida lusitanie [4], Aspergillus terreus [5], Scedosporium species [4], and Trichosporin beigelii [6]. More recently, there are increasing reports of amphotericin B resistance among some isolates of Candida auris [7] and Fusarium [8].
Amphotericin B Adverse Reactions
Conventional amphotericin B has been referred to by clinicians and sometimes patients as “Ampho-terrible” due to the frequency of infusion reactions and nephrotoxicity [2].
Amphotericin B associated infusion reactions, sometimes known unofficially as the “Shake and Bake Syndrome,” can cause fevers, chills, and rigors but also nausea and vomiting, headache, arthralgias, myalgias, and ever anaphylactoid reactions resulting in acute dyspnea [2, 3, 9]. Pre-medications such as hydrocortisone (50 mg PO/IV), acetaminophen (650 mg PO), and diphenhydramine (25–50 mg PO/IV) may reduce the incidence and severity of these reactions although quality clinical trial data is lacking [9]. Amphotericin B induced rigors have been treated with opioids such as hydromorphone (0.5 mg IV) or meperidine (25 mg IV), typically allowing for completion of the amphotericin B infusion [2, 9]. One study which randomized patients to receive AmBD over 4 versus 24 h per day demonstrated a more than 50% reduction in the incidence of fever, chills, and rigors with the extended infusion [10].
Amphotericin B induced nephrotoxicity risk increases with cumulative dose and can result in an acute decline in glomerular filtration rate with associated rise in serum creatinine as well as urinary wasting of potassium and magnesium [2, 9]. Saline loading (NS 500 mL IV before and after each dose) may reduce or delay AmBD nephrotoxicity but does not affect electrolyte wasting [9, 11]. Continuous infusion AmBD has also recently been demonstrated to be less nephrotoxic compared to a 4 h daily IV infusion [10].
Amphotericin B induced hypokalemia and hypomagnesemia can be severe and require close laboratory monitoring throughout therapy – in addition to acute replacement with intravenous and/or oral electrolytes, scheduled oral potassium chloride and/or magnesium oxide supplementation is often required [9]. Amiloride 5–10 mg PO BID can also be utilized to reduce AmBD induced potassium wasting but it should be discontinued with or shortly after the completion of amphotericin B therapy to avoid hyperkalemia as the renal tubules recover [12].
Lipid Formulations of Amphotericin B
Three lipid formulations of amphotericin B were developed to reduce toxicity of this agent while preserving its efficacy: Amphotericin B Lipid Complex (Abelcet, ABLC) FDA approved in 1995, Amphotericin B Cholesterol Sulfate or AmB Colloidal Dispersion (Amphotec, ABCD) FDA approved in 1996, and Liposomal Amphotericin B (Ambisome, L-AmB) FDA approved in 1997 [2].
The lipid formulations were primarily designed to reduce nephrotoxicity and, as expected, all 3 agents have been demonstrated to be less nephrotoxic than AmBD [2, 13].. A randomized, double-blind comparative trial in patients with persistent neutropenic fever by Wingard and colleagues compared ABLC to two doses of L-AmB and demonstrated reduced risk for nephrotoxicity in the L-Amb group (14.1% and 14.8% vs. 42.3%, P < 0.01) [14].
Adverse infusion reactions vary by amphotericin B formulation [13]. ABCD has a higher incidence of rate of infusion reactions than AmBD: including chills (53% vs 30%) and fever (27% vs 16%) [15]. Wingard and colleagues demonstrated that L-Amb exhibited a lower rate of chills and rigors than ABLC (18.8% and 23.5% vs 79.5%) on day 1 [14]. While L-Amb appears to have the lowest incidence of infusion-related reactions among the amphotericin B formulations, a minority of patients may experience an atypical hyper-acute and often very dramatic infusion reaction to L-Amb involving one or more of the following adverse effects: “ [1] chest pain, dyspnea, and hypoxia [2]; severe abdomen, flank, or leg pain; and/or [3] flushing and urticaria” [16]. In these rare cases of atypical L-Amb infusion reactions, stop the infusion immediately and consider intravenous diphenhydramine. For future doses, high dose intravenous diphenhydramine (up to 1 mg/kg) has been added to pre-medications prior to subsequent doses [16] or, in our experience, many of these patients can tolerate ABLC with aggressive pre-medication and initially reduced rate of infusion.
Due to their superior safety profile and equivalent efficacy, lipid formulations of amphotericin B are preferred over AmBD [2, 13] in the treatment of invasive fungal infections in neutropenic cancer patients. A direct comparison of continuous infusion AmBD to a lipid formulation could be of value.
Amphotericin B Dosing Recommendations
Amphotericin B Deoxycholate (Fungizone) – use actual body weight for dosing in obese patients [17] | |
Aspergillosis | 1–1.5 mg/kg IV Q24H |
Blastomycosis | 0.7–1 mg/kg IV Q24H, total dose of 2–2.5 g |
Candida esophagitis | 0.3–0.7 mg/kg IV Q24H |
Candidemia or Disseminated Candidasis | 0.7–1 mg/kg IV Q24H |
Coccidioidomycosis | 0.5–0.7 mg/kg IVQ24H, total dose 7–20 mg/kg |
Cryptococcal meningitis | 0.7 mg/kg IV Q24H in combination with flucytosine for a minimum of 2 weeks and CSF is sterile, then fluconazole |
Histoplamsosis | 0.7–1 mg/kg IV Q24H |
Mucormycosis | 1–1.5 mg/kg IV Q24H, total dose of 30–40 mg/kg |
Amphotericin B Lipid Complex (Abelcet) – consider ideal body weight for dosing in obese patients [16] | |
Invasive Fungal Infections | 5 mg/kg IV Q24H |
Liposomal Amphotericin B (Ambisome) – consider ideal body weight for dosing in obese patients [16] | |
Aspergillosis | 3 mg/kg IV Q24H [18] (3 mg/kd/day with same efficacy, less toxicity than 10 mg/kg/day for first 2 weeks of therapy) [18] |
Candidemia | 3–5 mg/kg IV Q24H |
Fusariosis/Mucormycosis | 5 mg/kg IV Q24H, higher doses of 10–15 mg/kg/day have been used in refractory infections |
Persistent neutropenic fever | 3 mg/kg IV Q24H, up to 5 mg/kg IV Q24H |
Antifungal prophylaxis in high risk | 1 mg/kg IV Q24H [19] or 10 mg/kg IV (over 2 h) weekly [20] or |
Neutropenics intolerant to other options | 15 mg/kg IV (over 6 h) every other week [21] |
Antifungal prophylaxis to reduce the risk | 12.5 mg over 30 min via nebulizer |
Of Invasive Pulmonary Aspergillosis | On 2 consecutive days per week [22] |
Nystatin
Structure of nystatin [23]

Nystatin (Mycostatin), like amphotericin B, is a polyene antifungal agent which was discovered in 1950 [24]. The antifungal spectrum of nystatin is comparable, but not identical, to that of amphotericin B [24]. Strains of Candida albicans have been identified that are much more susceptible to nystatin than amphotericin B [24]. Nystatin may also exhibit activity against Candida glabrata, Candida krusei, Geotrichum, and Beauvaria which are amphotericin B resistant [24].
Intravenous nystatin was initially investigated for the treatment of invasive fungal infections but it was never FDA approved due to excessive toxicities including venous sclerosis as well as intolerable infusion related infusions such as severe fever, rigors, and malaise [24]. A liposomal formulation of nystatin, with the goal of reducing toxicity, has been studied in phase 1 and 2 studies primarily [24]. Offner and colleagues studied the use of liposomal nystatin in 26 patients with invasive aspergillosis who either could not tolerate amphotericin B or whose infection was refractory to amphotericin B [25]. In this trial of high risk patients, the overall response rate was 28% with 68% overall mortality and a high incidence (67%) of infusion related reactions which lead to discontinuation in 2 patients [25]. Nephrotoxicity and hypokalemia were manageable in this study [25]. Liposomal nystatin has to date not been submitted to the FDA for review and remains unavailable outside of clinical trials.
Due to lack of significant bioavailability, nystatin can be safely administered swish-and-swallow for the treatment of oral candidiasis or topically for the management of cutaneous and vaginal fungal infections, predominantly for candidiasis [24].
Nystatin dosing [26] | |
Thrush | 500,000–1,000,000 units (5–10 mL) in mouth 35× per day |
Half of suspension in left side of mouth, swish as long as possible before swallowing, then repeat in right side of mouth. | |
Cutaneous candidiasis | Apply cream, ointment, or powder to affected areas BID |
Vaginal candidiasis | 1 vaginal tablet (100,000 units) daily for 2 weeks |
Flucytosine
Structure of flucytosine [27]

Flucytosine (Ancobon, FC) has been available as 250 mg and 500 mg capsules for oral administration since 1968 [28, 29]. Currently FC is the only antimetabolite agent with antifungal activity. Flucytosine inhibits fungal protein synthesis by replacing uracil with 5-flourouracil (5-FU) in fungal RNA as well as inhibiting thymidylate synthetase via 5- fluorodeoxy-uridine monophosphatase which interferes with fungal DNA synthesis [28, 29].
Flucytosine Antifungal Spectrum of Activity
Flucytosine’s activity against yeasts include Candida (except for Candida krusei) and Cryptococcus [29]. Flucytosine is recommended to be used in combination with amphotericin B (either conventional or lipid formulations) for the treatment of Cryptococcal meningitis due to the enhanced rate of CSF clearance as well as improved survival compared to amphotericin B plus fluconazole 800 mg/day [30]. Flucytosine has been used in combination with amphotericin B or fluconazole in patients with refractory invasive Candida infections despite lack of randomized clinical trial data to support this indication [29]. The 2016 Infectious Disease Society of America (IDSA) Candidiasis guidelines note that flucytosine in combination with amphotericin B may be considered for CNS infections, endocarditis, and endopthalmitis [31]. Flucytosine is typically not recommended as monotherapy for invasive yeast infections due to baseline resistance in 7–8% of Candida species and rapid induction of resistance [29]. Flucytosine monotherapy may be considered as an option for the treatment of fluconazole-resistant Candida glabrata urinary tract infections given the high concentrations achieved in the urinary tract with this renally cleared antifungal agent [29, 31].
Flucytosine has in vitro activity against Aspergillus species and had been utilized in the pre-echinocandin era as part of combination therapy for refractory cases of aspergillosis despite lack of data to support this practice [29]. Flucytosine is not, however, mentioned in the 2016 IDSA Aspergillosis guidelines [32].
Flucytosine Adverse Reactions and Financial Toxicity
As may be expected for a pro-drug of the cytotoxic cancer agent 5-fluorouracil, flucytosine’s most clinically significant adverse effect is myelosuppression [29]. Flucytosine drug induced neutropenia and thrombocytopenia more so than anemia is typically observed within the first 2 weeks of therapy in 27% of patients and is usually associated with FC serum concentrations >100 mg/L [29].
Hepatotoxicity may be a significant complication of FC therapy and has been reported to occur in up to 41% of patients [29]. Most commonly this consists of reversible elevated transaminases and alkaline phosphatase [29]. Hyperbilirubinemia is less common than transaminitis and there have been 2 cases of life-threatening liver necrosis attributed to FC [29]. It may be possible to reduce the incidence of FC induced hepatotoxicity by avoiding peak FC concentrations above 100 mg/L [29].
Gastrointestinal side effects such as nausea and diarrhea more so than vomiting and abdominal pain can occur in approximately 6% of patients receiving FC [29].
Unfortunately, the exorbitant cost of flucytosine in the United States is worthy of mention. In 2009, Valeant Pharmaceuticals acutely increased the price of flucytosine to the extent that a 2 week course of FC costs approximately $28,000 for FC alone whereas the cost in the United Kingdom is one-tenth of this price [33]. While this has resulted in enhanced scrutiny of FC utilization in the United States, even at this inflated price the combination of amphotericin B plus flucytosine may be cost-effective for the treatment of Cryptococcal meningitis given superior efficacy compared to amphotericin B plus high dose fluconazole [33].
Flucytosine Dosing [28, 34] & Therapeutic Drug Monitoring [29, 34]
Dose by ideal body weight in obese patients [34] | |
Creatinine Clearance | Dose |
>40 ml/min: | 25 mg/kg PO Q6H |
20–40 ml/min | 25 mg/kg PO Q12H |
10–19 ml/min | 25 mg/kg PO Q24H |
<10 ml | 25 mg/kg PO Q48H |
Hemodialysis | 25–50 mg/kg Q48-72H, dose after HD on HD days |
Goal peak of 30–80 mg/L to be obtained 2 h post dose after 3–5 days of therapy [34]. | |
Alternative recommendation: peak 50–100 mg/L with trough 25–50 mg/L [29]. | |
Azole Antifungals
Commonly utilized systemic azole antifungal agents used in the neutropenic patient population include fluconazole, voriconazole, posaconazole, and isavuconazonium sulfate. Azole antifungals impair the synthesis of ergosterol, a vital component in the fungal cell membrane which is analogous to cholesterol in the mammalian cell membrane [35]. This is accomplished by inhibition of 14α-sterol demethylase which is a cytochrome p450 (CYP450) enzyme – this also explains why CYP450 based drug interactions are so common with these agents [35].
Fluconazole
Structure of Fluconazole [36]

Fluconazole (Diflucan) demonstrates activity against the vast majority of Candida albicans, Candida keyfr, Candida dublinensis, Candida tropicalis, Candida parapsilosis, Candida guillermondii, and Candida lusitaniae [31]. Fluconazole may demonstrate reduced activity against specific strains of Candida glabrata due to their propensity to produce efflux pumps to expel azole antifungals [31]. Depending on the efficacy and density of these efflux pumps, higher doses of fluconazole may be an option [31]. Candida krusei is intrinsically resistant to fluconazole [31].
Fluconazole is highly active against Cryptococcus allowing for its use for maintenance and secondary prophylaxis in the treatment of cryptococcal meningitis [37]. Fluconazole has also been used first-line for mild to moderate pulmonary cryptococcosis and other single sites of infection in the absence of meningitis or cryptococcemia [37].
Fluconazole is generally well tolerated but patients should be monitored for rare cases of hepatotoxicity [38] and QTc prolongation especially in combination with other QTc prolonging drugs including fluoroquinolones [38].
Fluconazole demonstrates excellent bioavailability and therefore the oral route should be utilized unless the patient is unable to tolerate oral medications, such as severe mucositis, or has an active NPO order [31]. Unlike the other azole antifungal agents, fluconazole is renally eliminated which also allows for achievement of urine concentrations 10–20 times the serum concentration [31]. Fluconazole also has the best distribution of the triazoles into the cerebrospinal fluid (CSF) and vitreous fluid with greater than 70% penetration [31].
Fluconazole is considered a mild to moderate inhibitor of CYP450 3A4, 2C9, and 2C19 [39]. Since fluconazole can be administered over a wide range of dosing, please be aware that higher fluconazole doses can result in more clinically significant drug interactions. Examples of agents with potentially clinically significant fluconazole drug interactions include astemizole, certain benzodiazepines (alprazolam, diazepam, midazolam, triazolam), cisapride, clopidogrel, cyclosporine, fentanyl, ifosfamide, lovastatin, oral hypoglycemics, phenytoin, rifabutin, rifampin, simvastatin, sirolimus, tacrolimus, terfenadine, tyrosine kinase inhibitors, and warfarin [40].
Fluconazole dosing – consider adjusted body weight for dosing in obese patients [40] | ||
Candidemia | Loading Dose | 800 mg (12 mg/kg) PO/IV ×1 |
Maintenance Dose | 400 mg (6 mg/kg) PO/IV Q24H | |
Central Nervous System/Infective Endocarditis | 400 (6 mg/kg) to 800 mg (12 mg/kg) PO/IV Q24H | |
Candida glabrata infection | 800 mg (12 mg/kg) PO/IV Q24H | |
(Sensitive dose-dependent, SDD) | ||
Oropharyngeal | 100–200 mg PO/IV Q24H | |
Antifungal prophylaxis | 200–400 mg PO/IV Q24H | |
Renal adjustment | CrCl <50 ml/min | Reduce MD by 50% |
Hemodialysis | 100% of dose after each HD session | |
Voriconazole
Structure of Voriconazole [39]

Voriconazole (Vfend) demonstrates similar activity against yeasts compared to fluconazole except that it maintains activity versus Candida krusei and may be utilized once susceptibility data has been verified as an oral option for the treatment of fluconazole-resistant, voriconazole-sensitive Candida glabrata infections [31]. Voriconazole is FDA approved for the treatment of esophageal candidiasis as well as candidemia and disseminated candidiasis in the skin, abdomen, kidney, bladder wall, and wounds in non-neutropenic patients [31]. Neutropenic patients were excluded from the clinical trial which lead to FDA approval for this indication [41].
Voriconazole has significantly broader antifungal activity than its parent compound fluconazole. Voriconazole is FDA approved for the treatment of invasive aspergillosis and is considered to be the drug of choice for this indication due to lower mortality rates compared to patients randomized to conventional amphotericin B [32]. Voriconazole is also FDA approved for the treatment of invasive fungal infections caused by Scedosporium apiospermum and Fusarium in patients intolerant of, or refractory to other therapy [41]. Aspergillus ustus, which requires amphotericin B therapy, has been reported to cause infections in stem cell transplants receiving voriconazole prophylaxis [42]. In clinical practice, voriconazole is typically considered the drug of choice for Scedosporium apiospermum given superior activity compared to amphotericin products [43]. Voriconazole is often used in combination with other potentially active agents such as lipid formulations of amphotericin B or terbinafine for the treatment of Fusariosis in this high risk patient population [8], at least until antifungal susceptibility results are back. Unfortunately voriconazole resistant Fusarium is being increasingly reported [8].
Voriconazole lacks activity against mucormycosis and must not be used to treat these life-threatening invasive fungal infections [44].
Voriconazole can cause adverse reactions commonly associated with most of the other azole fungals such as hepatotoxicity, rash, and QTc prolongation [41, 45]. But voriconazole exhibits additional side effects that are more unique. Voriconazole can cause visual disturbances such as blurred vision or color perception issues in up to 30% of patients, typically occurring approximately 30 min after a dose and lasting for up to 30 min – “The Rule of 30” [41, 45]. The site of this usually manageable toxicity is the retina and it has been demonstrated via electroretinography to be reversible following discontinuation [41, 45]. Voriconazole prescribing information recommends that patients not drive when their vision is affected or at night [41]. This needs to be distinguished from central nervous system toxicity (hallucinations and encephalopathy) which occurs in 4.3% of patients [41, 45] and can range from vivid dreams to elaborate visual hallucinations which can be upsetting to patients especially if they are not forewarned about this possibility. Risk for encephalopathy and hallucinations are associated with elevated trough concentrations greater than 5.5 mg/L [45, 46]. Voriconazole is unlike the other azole fungals in that it is a photosensitizer which can result in sunburn-like rashes in 2% of patients and with prolonged utilization, it has been reported to increase the risk of cutaneous squamous cell carcinoma or melanoma [45, 47]. Patients taking voriconazole should limit their exposure to sunlight and use UVA and UVB SPF30+ sunscreen and seek medical attention promptly for new skin lesions. Patients receiving prolonged voriconazole therapy may also experience musculoskeletal pain due to fluorosis and periositis [48]. This has not been associated with other triazole antifungals likely because voriconazole contains 3 fluoride ions per molecule compared to 2 in posaconazole, for example [48].
Voriconazole exhibits excellent bioavailability if separated from meals [41] or enteral feedings [49] by at least 1 h. Voriconazole achieves extensive tissue distribution [41] and achieves clinically significant (>50% penetration) CSF and vitreous concentrations [31]. Voriconazole, unlike fluconazole, does not accumulate in the urine and should not be used for Candida urinary tract infections [31].
Voriconazole exhibits substantial variation in hepatic metabolism which inactivates voriconazole primarily via CYP2C19, but also to a lesser extent CYP2C9 and CYP3A4 enzymes [41]. Voriconazole metabolism can demonstrate non-linearity which means that a 50% dose increase can result in a serum concentration increase ranging from 0.4 to 7.7 fold [50].
Genetic polymorphisms in CYP2C19 metabolism appear to represent 30–50% of the inter-patient variability in voriconazole clearance [50]. We are currently evaluating the impact of increasing voriconazole starting dose in rapid CYP2C19 metabolizers and avoiding voriconazole in ultra-rapid CYP2C19 metabolizers [45]. Voriconazole metabolism can be induced by agents such as phenytoin, rifabutin, and rifampin resulting in reduced levels or inhibited by omeprazole resulting in increased levels [41].
Voriconazole is a potent inhibitor of CYP3A4, CYP 2C9, and CYP2C19 which can lead to multiple drug interactions including, but not limited to, the following agents: alprazolam, amlodipine, astemizole, atazanavir, cisapride, clopidogrel, cyclosporine, diazepam, diltiazem, efavirenz, etravirine, felodipine, fentanyl, fosamprenavir, lovastatin, marviroc, midazolam, neviraprine, nifedipine, nisoldipine, omeprazole, oral hypoglycemics, oxycodone, phenytoin, ranolazine, rifabutin,ritonavir, simvastatin, sirolimus, tacrolimus, terfenadine, verapamil, vinblastine, vincristine, and warfarin [45].
Voriconazole dosing – consider adjusted body weight in obese patients [45, 51] | ||
Invasive Fungal Infections | Loading Doses: | 6 mg/kg PO/IV BID ×2 doses |
(Aspergillosis, Candidiasis, Fusariosis, Scediosporosis, etc.) | OR 400 mg PO/IV BID ×2 doses | |
Maintenance Doses: 3–4 mg/kg PO/IV BID | ||
OR 200 mg to 300 mg PO BID | ||
Goal trough of 1–5.5 mg/L or random level of 2–6 mg/L to be obtained after 5–7 days [46, 52]. | ||
Renal dysfunction | Renal insufficiency has no effect on voriconazole elimination [41] | |
Intravenous voriconazole contains a cyclodextrin solubilizing agent which may accumulate in patients with renal insufficiency [41]. As a result, the prescribing information recommends to avoid intravenous voriconazole in patients with creatinine clearance less than 50 ml/min if possible [41]. This is, however, a theoretical concern and no increase in rates of nephrotoxicity, hepatoxicity, or other adverse effects have been observed in the literature to date despite seven separate retrospective studies [53].
Hepatic dysfunction: | Child-Pugh score-based maintenance dose recommendations are found in the voriconazole prescribing information [41]: |
Child Pugh Score A-B: | Reduce voriconazole maintenance dose by 50% |
Child Pugh Score C: | Dose reduction required, but no data to guide by how much. |
Many neutropenic cancer patients experience acute hepatic dysfunction as a result of their underlying malignancy, cytotoxic chemotherapy, or other medications and Child Pugh score has not been validated in this patient population. Therapeutic drug monitoring can be very helpful in this scenario.
Posaconazole
Structure of Posaconazole [54]

Posaconazole (Noxafil) exhibits a spectrum of activity similar to voriconazole with the notable addition of mucromycosis activity [55]. Posaconazole is FDA approved for prevention and treatment of aspergillosis and candidiasis in high risk patients (MDS/AML receiving induction chemotherapy, allogeneic stem cell transplant recipients with graft-versus-host disease requiring high dose corticosteroids) [56]. Posaconazole has been utilized as salvage therapy for invasive fungal infections, especially against aspergillosis and mucormycosis, but the quantity and quality of this data is limited and based on the use of the original and suboptimal oral liquid formulation [57, 58].
One advantage that posaconazole can claim over voriconazole is tolerability. Posaconazole exhibits an adverse reaction profile comparable to fluconazole therefore patients need only be monitored for hepatotoxicity, QTc prolongation, and rash [59]. Intravenous posaconazole can also cause phlebitis with multiple doses via peripheral vein administration, which is why it is not recommended to give more than one dose prior to central line placement [56].
Posaconazole was originally FDA approved in 2006 only as an oral suspension with 200 mg/5 mL concentration [56]. Posaconazole oral suspension needs to be taken with a meal, ideally a fatty meal, or an enteral feeding such as Boost Plus to increase absorption [56]. Posaconazole is also dependent upon gastric pH to achieve adequate absorption which is why it should not be taken concurrently with proton pump inhibitors and why concurrent acidic ginger ale and/or ascorbic acid can increase absorption [56, 60]. This formulation also exhibits saturable absorption which is why the drug was given as 200 mg/5 mL by mouth 3–4 times per day despite a half-life of greater than 24 h [56,57,58,59,60]. The FDA approval of posaconazole delayed release tablets and an intravenous formulation with a cyclodextrin solubilizing agent eliminated the requirements for low gastric pH and concurrent food intake and thereby greatly improved the pharmacokinetic profile of posaconazole [56].
Posconazole has excellent distribution although its CSF penetration appears to be inferior to fluconazole and voriconazole [55, 56]. Posaconazole is primarily metabolized by UDP-glucoronidation and has no major oxidative, CYP450-mediated metabolites [55, 56]. Posaconazole metabolism can be induced by efavirenz, phenytoin, rifabutin, and rifampin resulting in subtherapeutic concentrations [55, 56]. Posaconazole is a strong inhibitor of CYP450 3A4 specifically and unlike fluconazole and voriconazole does not significantly impact on CYP450 2C9 or 2C19 metabolism [4, 5, 55, 56]. This results in nearly as many drug interactions as voriconazole which include but are not limited to the following: alprazolam, amiodarone, amlodipine, astemizole, cisapride, cyclosporine, diazepam, diltiazem, dofetilide, ergot alkaloids, felodipine, irinotecan, lovastatin, midazolam, maraviroc, nifedipine, nisolidipine, oral hypglycemics, ritonavir, sildenafil, simvastatin, sirolimus, tacrolimus, tadalafil, terfenadine, triazolam, vardenafil, verapamil, vinblastine, and vincristine [55, 56, 59].
Posaconazole delayed release 100 mg tablets | |
Take 3 tablets (300 mg) by mouth twice per day on Day 1 | |
Do not omit loading doses, required to rapidly achieve therapeutic concentrations | |
Then take 3 tablets (300 mg) by mouth daily starting on Day 2 | |
Posconazole intravenous: | 300 mg IV Q12H ×2 doses then 300 mg IV Q24H |
To be administered via central line to reduce risk of phlebitis with multiple doses | |
Posaconazole 200 mg/5 mL oral suspension is inferior from a pharmacokinetic standpoint – recommend use of newer formulations if possible. | |
Prophylaxis: | 200 mg/5 mL PO TID with meals or 1 can Boost plus or Ensure plus |
200 mg/5 mL PO QID with meals of 1 can Boost plus or Ensure plus | |
400 mg/10 mL PO BID with meals or 1 can Boost plus or Ensure plus at discharge | |
Consider use of a “posconazole bundle” [60] to maximum absorption: | |
Ascorbic acid 250–500 mg PO with each posaconazole dose in addition to acidic beverage, and heavy snack /meal/nutritional supplement. No concurrent proton pump inhibitors. | |
Renal dysfunction: | No impact on posaconazole clearance [56] |
Due to potential for cyclodextrin accumulation, the prescribing information discourages use of IV posaconazole in patients with CrCl <50 mL/min [56] – note that is a theoretical concern and based on data with IV voriconazole [53], the risk of increased toxicity in patients with renal impairment is probably minor. | |
Hepatic dysfunction : | No dose adjustment is recommended [56] |
Isavuconazonium Sulfate
Structure of isavuconazonium sulfate [61], pro-drug of isavuconazole

Structure of isavuconazole [62], the active antifungal agent

Isavuconazonium sulfate (Cresemba) is a water-soluble pro-drug for isavuconazole (ISV), which exhibits a spectrum of activity very similar to posaconazole [63]. Cresemba was FDA approved for the treatment of invasive aspergillosis and invasive mucormycosis on March 6, 2015 [64]. Phase II clinical trial data is available in the setting of antifungal prophylaxis in neutropenic acute leukemics [65] and treatment of esophageal candidiasis [66].
Cresemba is generally well tolerated and with similar adverse reaction profile to fluconazole or posaconazole [63] and exhibited a slightly lower incidence and severity of ocular toxicity and hepatotoxicity than voriconazole in a randomized trial for aspergillosis [67]. Unlike the other azoles, isavuconazole does not cause QTc prolongation and actually displays a modest QTc shortening effect [63, 64].
Isavuconazonium sulfate is rapidly cleaved to ISV following administration of oral capsules or intravenously, which is why there is no need for a cyclodextrin solubilizing agent [ 63, 64]. Isavuconazonium sulfate may also be prematurely converted to ISV if the product is shaken or sent via a pneumomatic tube system [63, 64]. Cresemba exhibits excellent oral absorption which is not affected by concurrent oral intake or gastric pH [63, 64]. Isavuconazole displays a large volume of distribution, is highly protein bound, and has a prolonged half-life of greater than 100 h [63]. Given the extremely long half-life, the use of the recommended loading doses is critical – without loading, it can take weeks to reach the therapeutic steady state concentration [63]. Isavuconazole should not be used to treat fungal urinary tract infections due to poor urinary penetration [63]. Data is lacking on central nervous penetration – distribution into the brain parenchyma is likely to be superior to penetration into cerebrospinal fluids [68].
Isavuconazole is primarily metabolized by CYP 3A4 [63]. Concurrent strong CYP 3A4 inducers such as rifampin, carbamazepine, St. John’s wort, or barbiturates are contraindicated due to high probability of resulting in subtherapeutic ISV concentrations [63]. Concurrent strong 3A4 inhibitors such as high dose ritonavir (400 mg PO BID) and ketoconazole are not advised due to potential for supratherapeutic ISV concentrations [63].
Isavuconazole is a mild to moderate inhibitor of CYP 3A4 [68] which can be beneficial in patients receiving essential medications that are 3A4 substrates if the only other alternatives would be strong CYP 3A4 inhibitors such as voriconazole and posaconazole. Nonetheless, isavuconazole can result in multiple drug interactions including, but not limited to atorvastatin, buproprion, cyclosporine, digoxin, lopinavir-ritonavir, midazolam, mycophenolate mofetil, sirolimus, and tacrolimus [63].
Isavuconazonium sulfate dosing [ [63]] | |
NOTE: Cresemba (isavuconazonium sulfate) is available in 186 mg capsules or 372 mg vials for IV use. | |
Isavuconazonium sulfate 372 mg = Isavuconazole 200 mg | |
Cresemba 186 mg #2 (372 mg) PO Q8H ×2 days (6 doses) then 186 mg #2 (372 mg) PO Daily | |
Cresemba 372 mg IVPB Q8H ×2 days (6 doses) then 372 mg IVPB Q24H | |
In clinical trials, the maintenance dosing started 12–24 h after the last loading dose | |
Do not omit loading doses, required to achieve therapeutic concentrations in timely manner | |
Swallow capsules whole – do not chew, crush, dissolve, or open the capsules | |
Renal dysfunction: | No dose adjustment |
Hepatic dysfunction : | No dose adjustment required for Child Pugh Class A and B |
No recommendation for Child Pugh Class C | |
Echinocandin Antifungals
Structure of caspofungin acetate [69]

Structure of micafungin [70]

Structure of anidulafungin [71]

There are currently three FDA approved echinocandin antifungal agents: Caspofungin acetate (Cancidas) approved in 2001 [72], Micafungin (Mycamine) approved in 2005 [73], and Anidulafungin (Eraxis) approved in 2006 [74, 75].
The echinocandin antifungals inhibit the synthesis of β-1,3-D-glucan which is an essential component in the fungal cell wall [72,73,74,75]. The excellent tolerability of this antifungal class is likely due in part to the fact that there is no mammalian analogous structure to β-1,3-D-glucan unlike ergosterol and cholesterol in the fungal and mammalian cell membranes that are involved in the mechanisms of action of both amphotericin B and azole antifungals.
The echinocandins all display fungicidal activity against most Candida species and are considered to be the initial treatment of choice for candidemia [31, 76]. Candida parapsilosis intrinsically exhibits higher echinocandin MICs than other Candida strains and azole antifungals may be preferred over echinocandins for this species [31]. While echinocandins are typically considered to be the initial treatment of choice for invasive Candida glabrata infections [31], the incidence of echinocandin-resistant Candida glabrata appears to be on the rise likely due to over-use of these agents [77]. While echinocandins have been proposed as the preferred treatment for Candida auris, echinocandin-resistant strains have been reported in addition to those with azole-resistance and/or amphotericin B resistance [7].
Unfortunately, echinocandins only exhibit fungistatic activity against Aspergillus [76]. As a result, echinocandin monotherapy is not recommended for the primary treatment of invasive aspergillosis, but only for salvage therapy [32]. Combination therapy of echinocandins with either azole antifungals or amphotericin B formulations have been proposed but are not recommended in the 2016 Aspergillus IDSA guidelines [32]. A randomized clinical trial comparing voriconazole monotherapy to voriconazole and anidulafungin for invasive aspergillosis failed to demonstrate a statistically significant mortality difference between monotherapy and combination therapy [78]. Post-hoc analysis suggested that galactomannan positivity might identify a patient population that could benefit from combination therapy [78]. Asperillus ustus infections have been reported to break through echinocandin prophylaxis in stem cell transplant recipients [42].
The echinocandin antifungals as a class are usually very well tolerated [76]. Hepatotoxicity has been reported but appears to be less common than seen with either amphotericin B formulations or voriconazole [79]. Histamine-mediated infusion reactions have been rarely reported [72,73,74,75]. Case reports in critically ill patients and animal studies have suggested that the echinocandins may impair left ventricular function in critically ill patients, but further research is needed regarding the incidence and clinically significance of this proposed toxicity [80].
The echinocandin antifungals must be administered intravenously due to insignificant oral bioavailability [72,73,74,75,76]. The echinocandins are all highly protein bound mostly to albumin and distribute well in clinically relevant human tissues with the exception of the central nervous system and urinary tract [31, 72,73,74,75,76]. Caspofungin acetate is either metabolized by hydrolysis or N-acetylation or spontaneously degrades to an inactive open-ring formulation [72]. Caspofungin acetate does require a loading dose of 70 mg to rapidly achieve target concentration [72]. Caspofungin acetate dose reduction to 35 mg/day is recommended in patients with moderate liver dysfunction (Child-Pugh score B) and the prescribing information states that inadequate data exists in patients with severe liver dysfunction (Child-Pugh score C) [72]. Micafungin metabolism is metabolized initially by arylsufatase and is not significantly affected by CYP3A oxidative metabolism [73]. Micafungin does not require a loading dose and also does not require dose modification for patients with mild to severe liver dysfunction (Child-Pugh score A-C) [73]. Anidulafungin is not significantly hepatically metabolized and instead demonstrates slow chemical breakdown under physiological conditions to an inactive, open-ring peptide [75]. Anidulafungin does require a loading dose of twice the maintenance dose but does not require dose modification even in the setting of severe liver dysfunction [75]. None of the echinocandins are significantly affected by renal dysfunction [72,73,74,75,76].
Echinocandin drug interactions vary slightly between agents but are typically less problematic than those encounted with the azole antifungals. Despite the lack of a clinically significant pharmacokinetic drug interaction of caspofungin acetate with cyclosporine or tacrolimus, the combination of caspofungin acetate with cyclosporine has been reported to result in increased risk of transaminitis [72]. A recent publication suggests that the caspofungin acetate-cyclosporine interaction is unlikely to cause increased risk of hepatotoxicity [81]. Caspofungin acetate metabolism may be increased by hepatic cytochrome inducers such as rifampin, nevirapine, efavirenz, carbamazepine, dexamethasone, or phenytoin [72]. Micafungin drug interactions are rarely clinically important – the prescribing information notes that patients receiving concurrent sirolimus, nifedipine, or itraconazole should be monitored for signs of toxicity and reduce the dose of the concurrent agent if necessary [73] although it is in fact rarely necessary. When anidulafungin was first released, the recommended initial diluent contained a high percentage of ethanol which created concerns regarding possible drug interactions with metronidazole or use in patients with either a history of substance abuse or religious prohibitions to ethanol ingestion [74]. This is no longer a problem since the current prescribing information recommends reconstitution with either sterile water, D5W, or normal saline [75]. There are no known clinically relevant drug interactions with anidulafungin [75].
Caspofungin acetate dosing [73] | ||
Caspofungin acetate 70 mg IV ×1 then 50 mg IV Q24H for all indications except esophageal candidiasis | ||
For esophageal candidiasis: | 50 mg IV Q24H without loading dose | |
Concurrent CYP inducers: | 70 mg IV Q24H | |
(rifampin, nevirapine, efavirenz, carbamazepine, dexamethasone, or phenytoin) | ||
Higher doses of caspofungin acetate (100–150 mg IV Q24H) have been utilized and tolerated but without evidence for clinical superiority [82]. | ||
Renal dysfunction: | No dose adjustment | |
Hepatic dysfunction: | No dose adjustment required for Child Pugh Class A | |
Child Pugh Class B: | Consider 70 mg IV ×1 then 35 mg IV Q24H | |
Child Pugh Class C: | Insufficient data | |
Micafungin dosing [74] | ||
Candidemia, Acute Disseminated Candidiasis, | 100 mg IV Q24H | |
Candida Peritonitis and Abscesses | ||
Esophageal Candidiasis | 150 mg IV Q24H | |
Prophylaxis of Candida Infections in | 50 mg IV Q24H | |
Stem Cell Transplant Recipients | ||
Antifungal Prophylaxis – Acute Leukemia or | 100 mg IV Q24H [83] | |
Myelodysplastic Syndrome after Chemotherapy (not FDA approved) | ||
Renal dysfunction: | No dose adjustment | |
Hepatic dysfunction: | No dose adjustment | |
Anidulafungin dosing [76] | ||
Candidemia and other Candida | 200 mg IV ×1 then 100 mg IV Q24H | |
Infections (intra-abdominal, peritonitis) | ||
Esophageal Candidiasis | 100 mg IV ×1 then 50 mg IV Q24H | |
Combination therapy for Aspergillosis | 200 mg IV ×1 then 100 mg IV Q24H [78] | |
(not FDA approved) | ||
Renal dysfunction: | No dose adjustment | |
Hepatic dysfunction: | No dose adjustment | |
Hepatic dysfunction: | No dose adjustment required for Child Pugh Class A and B | |
No recommendation for Child Pugh Class C | ||
Summary Table
Antifungal | Notes |
|---|---|
Amphotericin B | Broad fungicidal activity |
Resistance most common in Candida lusitaniae, Aspergillus terreus, Scedosporium species, and Trichosporin beigelii. Also reports of resistant Candida auris and Fusarium species | |
Amphotericin B deoxycholate (Fungizone) | “Amphoterrible” |
Tolerability limited by infusion reactions, nephrotoxicity, and potassium and magnesium wasting. | |
Premedication with hydrocortisone 50 mg PO/IV, acetaminophen 650 mg PO, and/or diphenhydramine 25–50 mg PO/IV may reduce infusion reactions. Rigors can be treated with opioids such as hydromorphone 0.5 mg IV. | |
While NS 500 mL IV before and after each dose may reduce or delay nephrotoxicity, use of lipid formulations are safer from renal standpoint. | |
Extended infusion amphotericin B deoxycholate reported to cause fewer infusion reactions and nephrotoxicity. | |
Monitor patient closely for hypokalemia and hypomagnesemia. Consider scheduled oral potassium and/or magnesium supplementation. Amiloride 5–10 mg PO BID may also be utilized to reduce amphotericin associated hypokalemia. | |
Amphotericin B Colloidal Dispersion (ABCD) or Amphotericin B Cholesteryl Sulfate (Amphotec) | Less nephrotoxic than amphotericin B deoxycholate |
Highest incidence of infusion rates of all amphotericin formulations. | |
Amphotericin B Lipid Complex (ABLC, Abelcet) | Less nephrotoxic than amphotericin B deoxycholate |
Infusion reactions comparable to amphotericin B deoxycholate | |
Liposomal Amphotericin B (Ambisome) | Lowest incidence of infusion reactions and nephrotoxicity of the amphotericin formulations |
Can rarely cause acute, severe atypical infusion reactions which may require change to an alternative agent | |
Nystatin (Mucostatin) | Comparable in vitro activity to amphotericin B but utilized clinically almost exclusively for oral, cutaneous, and vaginal candidiasis. |
Intravenous liposomal nystatin may have a role in the future for the treatment of invasive fungal infections in neutropenic patients, but remains an investigational agent at this time. | |
Flucytosine (Ancobon) | Active against Candida except Candida krusei, Cryptococcus, and Aspergillus |
Used almost exclusively for treatment of Cryptococcal meningitis in combination with amphotericin B | |
Dose limiting toxicity is myelosuppression | |
Therapeutic drug monitoring has been proposed but long turn-around time prevents from this from being clinically useful given usual 2 week duration of therapy | |
Fluconazole (Diflucan) | Active against Candida albicans, Candida keyfr, Candida dublinensis, Candida tropicalis, Candida parapsilosis, Candida guillermondii, and Candida lusitaniae. Reduced activity against Candida glabrata due to efflux pump production – high dose fluconazole may be effective against sensitive dose-dependent Candida glabrata |
Active against Cryptococcus – used for maintenance therapy and secondary prophylaxis in the treatment of cryptococcal meningitis or first line for mild to moderate cryptococcosis | |
Usually well tolerated but may cause hepatotoxicity, QTc prolongation, and rash | |
Excellent bioavailability, can be used oral or IV at same dosing | |
Best azole penetration into urinary tract, central nervous system, and the eye | |
Mild to moderate inhibitor of CYP450 3A4, 2C9, and 2C19 | |
Voriconazole (Vfend) | Activity against yeasts comparable to fluconazole but also usually active against Candida krusei and may be active against fluconazole resistant Candida glabrata. First line treatment of invasive aspergillosis (except for Aspergillus ustus) and scedosporiosis. Increasing reports of resistant Fusarium in clinical isolates. Not active against mucormycosis. |
Monitor patients for hepatotoxicity, QTc prolongation, rash, visual disturbances, and encephalopathy including hallucinations | |
Photosensitizer – avoid prolonged sunlight exposure, cover up, and use high quality sunscreens of SPF30+ | |
Long term use can result in fluorosis and periostitis | |
Excellent bioavailability if taken at least 1 h from food or enteral feedings. | |
Excellent distribution including clinically significant central nervous system and ocular penetration. Not recommended for urinary tract infections. | |
Metabolized primarily by CYP2C19 with substantial generic variance in metabolism | |
CYP2C19 genotyping may prove to be useful in determine initial voriconazole dose | |
Therapeutic drug monitoring is recommended especially in the treatment setting | |
Potent inhibitor of CYP 3A4, CYP2C9, and CYP2C19 leading to many clinically relevant drug interactions | |
Posaconazole (Noxafil) | Activity comparable to voriconazole except for addition of activity against mucormycosis |
FDA approved for antifungal prophylaxis, not treatment other than for esophageal candidiasis | |
Tolerability comparable to fluconazole but monitor for hepatoxicity, QTc prolongation, and rash | |
Available in delayed release tablets and intravenous formulations which achieve more reliable concentrations than the original oral suspension formulation which requires concurrent food intake as well as acidic gastric environment for optimal absorption | |
Potent inhibitor of CYP 3A4 resulting in many clinically relevant drug interactions | |
Isavuconazonium sulfate (Cresemba) | Activity comparable to posaconazole |
FDA approved for treatment of aspergillosis and mucormycosis | |
Tolerability comparable to fluconazole and posaconazole, monitor for hepatotoxicity and rash | |
Does not cause QTc prolongation, modest QTc shortening effect | |
Excellent bioavailability, available in capsules or IV formulation | |
Prolonged half-life, loading dose during first 2 days of therapy is essential otherwise sub-therapeutic for weeks | |
Distributes well into body tissues except urinary tract | |
Data lacking on central nervous system penetration but better brain parenchyma penetration than into cerebrospinal fluid | |
Primarily metabolized by CYP 3A4 – avoid concurrent potent inhibitors or inducers of CYP 3A4 | |
Mild to moderate inhibitor of CYP 3A4 which may result in less significant drug interactions than voriconazole or posaconazole | |
Echinocandins Caspofungin acetate (Cancidas) Micafungin(Mycamine) Anidulafungin (Eraxis) | Fungicidal against most Candida species, first line for Candidemia |
Less potent against Candida parapsilosis | |
Case reports of echinocandin resistance in Candida glabrata and Candida auris | |
Fungistatic against Aspergillus other than Aspergillus ustus, which is resistant to echinocandins | |
Have been used in combination with amphotericin or azole antifungals for salvage therapy of aspergillosis but randomized clinical trial failed to demonstrate mortality benefit except perhaps in galactomannan assay positive cases | |
Usually very well tolerated, monitor for hepatotoxicity | |
Rare histaminic infusion related reactions | |
May impair left ventricular ejection fraction in critically ill patients | |
Limited to intravenous administration | |
Excellent distribution except for central nervous system and urinary tract | |
Caspofungin acetate and anidulafungin require loading dose on day 1 of therapy, but micafungin does not | |
Consider caspofungin acetate dose reduction in patients with moderate liver dysfunction, not required for micafungin or anidulafungin | |
Caspofungin acetate requires dose increase if given with enzyme inducers | |
Caspofungin acetate-cyclosporine drug interaction may result in increased incidence of transaminitis | |
Micafungin drug interactions with sirolimus, nifedipine, or itraconazole are rarely clinically significant | |
Anidulafungin has no known drug interactions |
References
National Center for Biotechnology Information. PubChem Compound Database; CID=71311649. https://pubchem.ncbi.nlm.nih.gov/compound/71311649. Accessed 20 Apr 2018.
Saravolatz LD, Ostrosky-Zeichner LO, Marr KA, Rex JH, Cohen SH. Amphotericin B: time for a new “Gold Standard”. Clin Infect Dis. 2003;37(3):415–25.
Amphotericin B deoxycholate prescribing information. X-Gen Pharmaceuticals, Inc. Big Flats, New York. Revised December 2009.
Ellis D. Amphotericin B: spectrum and resistance. J Antimicrob Chemother. 2002;49(Suppl 1):7–10.
Van Der Linden JWM, Warris A, Verweij PE. Aspergillus species intrinsically resistant to antifungal agents. Med Mycol. 2011;49(Suppl 1):S82–9.
Walsh TJ, Melcher GP, Rinaldi MG, Lecciones J, McGough DA, et al. Trichosporin beigelii, an emerging pathogen resistant to amphotericin B. J Clin Microbiol. 1990;28(7):1616–22.
Sarma S, Upadhyay S. Current perspective on emergence, diagnosis, and drug resistance in Candida auris. Infect Drug Resist. 2017;10:155–65.
Stempel JM, Hammond SP, Sutton DA, Weiser LM, Marty FM. Invasive fusariosis in the voriconazole era: single center 13-year experience. Open Forum Infect Dis. 2015;2(3):ofv099. https://doi.org/10.1093/ofid/ofv099. eCollection 2015 Sep
Patel R. Antifungal agents. Part 1. Amphotericin B preparation and flucytosine. Mayo Clinical Proceedings. 1998;73(12):1205–25.
Eriksson U, Seifert B, Schaffner A. Comparsion of effects of amphotericin B deoxycholate infused over 4 or 24 hours. Br Med J. 2010;322:1–6.
Karimzadeh I, Farsaei S, Khalili H, Dashti-Khavidak S. Are salt loading and prolonging infusion period effective in prevention of amphotericin B induced nephrotoxicity? Expert Opin Drug Safety. 2012;11(6):969–83.
Bearden T, Muncey LA. The effect of amiloride on amphotericin B-induced hypokalemia. J Antimicrob Chemother. 2001;48:109–11.
Hamill RJ. Amphotericin B formulations: a comparative review of efficacy and toxicity. Drugs. 2013;73(9):919–34.
Wingard JR, White MH, Anaissie E, Raffalli JR, Goodman J, et al. A randomized, double-blind comparative trial evaluating the safety of liposomal amphotericin B versus amphotericin B lipid complex in the empirical treatment of febrile neutropenia. Clin Infect Dis. 2000;31:1155–63.
Bowden R, Chandrasekar P, White MH, Pietrelli L, Gurwith M, et al. A double-blind, randomized, controlled trial of amphotericin B colloidal dispersion verus amphotericin B for treatment of invasive aspergillosis in immunocompromised patients. Clin Infect Dis. 2002;35(4):359–66.
Roden MM, Nelson LD, Knudsen TA, Jarosinski PF, Starling JM, et al. Triad of acute infusion-related reactions associated with Liposomal Amphotericin B. Clin Infect Dis. 2003;36:1213–20.
Pham P, Bartlett JG. Amphotericin B. Johns Hopkins ABX Guide. Accessed 4–20-18.
Cornely OA, Maertens J, Bresnik M, Ebrahimi R, Ullmann AJ, et al. Liposomal amphotericin B as initial therapy for invasive mold infections: a randomized trial comparing a high-loading dose regimen with standard dosing (AmBiLoad trial). Clin Infect Dis. 2007;44:1289–97.
Gubbins PO, Amsden JR, McConnell SA, Anaissie EJ. Pharmacokinetics and Buccal Mucosal Concetrations of 15 mg/kg total dose of liposomal amphotericin B as single dose, weekly dose, or daily dose in peripheral stem cell transplant patients. Antimicrob Agents Chemother. 2009;53(9):3664–74.
Cordonnier C, Mohty M, Faucher C, Pautas C, Robin M, et al. Safety of weekly high dose of liposomal amphotericin B for prophylaxis of invasive fungal infection in immunocompromised patients. Int J Antimicrob Agents. 2008;31:135–41.
Annino L, Chierichini A, Anaclerico B, Finolezzi E, Norata M, et al. Prospective phase II single-center study of the safety of a single very high dose of liposomal amphotericin B for antifungal prophylaxis in patients with acute myeloid leukemia. Antimicrob Agents Chemother. 2013;57(6):2596–602.
Rijinders BJ, Cornelissen JJ, Slobbe L, Becker MJ, Doorduign JK, et al. Aerosolized liposomal amphotericin B for the prevention of invasive pulmonary aspergillosis during prolonged neutropenia. Clin Infect Dis. 2008;46:1401–11.
National Center for Biotechnology Information. PubChem Compound Database; CID= 6433272. https://pubchem.ncbi.nlm.nih.gov/compound/6433272. Accessed 27 Aug 2018.
Ng AW, Wasan KM, Lopez-Berestein G. Development of liposomal polyene antibiotics: an historical perspective. J Pharm Pharm Sci. 2003;6(1):67–83.
Offner F, Krcmery V, Boogaerts M, Doyen C, Engelhard D, et al. Liposomal nystatin in patients with invasive aspergillosis refractory to or intolerant of amphotericin B. Antimicrob Agents Chemother. 2004;48(12):4808–12.
Pam PA, Bartlett JG. Nystatin. Johns Hopkins ABX Guide. Accessed 8-27-18.
National Center for Biotechnology Information. PubChem Compound Database; CID=3366. https://pubchem.ncbi.nlm.nih.gov/compound/3366. Accessed 24 Apr 2018.
Ancobon (flucytosine) capsules prescribing information. Valeant Pharmaceuticals International. Costa Mesa, California. Revised May 2006.
Vermes A, Guchelaar H-J, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity, and drug interactions. J Antimicrob Chemother. 2000;46(2):171–9.
Day JN, Chau TTH, Wolbers M, Mai PP, Dung NT, et al. Combination antifungal therapy for cryptococcal meningitis. N Engl J Med. 2013;14:1291–302.
Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr K, et al. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62(4):e1–e50.
Patterson TG, Thompson GR, Denning DW, Fishman JA, Hadley S, Herbrectht R, et al. Practice guidelines for the diagnosis and management of aspergillus: 2016 update. Clin Infect Dis 2–16. 63(4):e1–e60.
Merry M, Boulware DR. Cryptococcal meningitis treatment strategies affected by the explosive cost of fluctyosine in the United States: a cost-effectiveness analysis. Clin Infect Dis. 2016;62(12):1564–8.
Pham P, Bartlett JG. Flucytosine. Johns Hopkins ABX Guide. Accessed 4-24-18.
Ghannoum MA, Rice LB. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin Microbiol Rev. 1999;12(4):501–17.
National Center for Biotechnology Information. PubChem Compound Database; CID=3365. https://pubchem.ncbi.nlm.nih.gov/compound/3365. Accessed 24 Apr 2018.
Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291–322.
Zeuli JD, Wilson JW, Estes LL. Effect of combined fluroquinolone and azole use on QT prolongation in hematology patients 2013; 57(3): 1121–1127.
National Center for Biotechnology Information. PubChem Compound Database; CID=71616. https://pubchem.ncbi.nlm.nih.gov/compound/71616. Accessed 24 Apr 2018.
Pham P, Bartlett JG. Fluconazole. Johns Hopkins ABX Guide. Accessed 4-24-18.
Vfend (Voriconazole) prescribing information. Roerig, division of Pfizer. New York. Revised June 2010.
Pavie J, Lacroix C, Hermoso DG, Robin M, Ferry C, et al. Breakthrough disseminated aspergillus ustus infection in allogeneic hematopoietic stem cell transplant reccipients receiving voriconazole or caspofungin prophylaxis. J Clin Microbiol. 2005;43(9):4902–4.
Goldman C, Akiyama MJ, Torres J, Louie E, Meehan SA. Scedosporium aposperium and role of combination antifungal therapy and GM-CSF. Med Mycol Case Rep. 2016;11:40–3.
Lewis RE, Liao G, Wang W, Prince RA, Kontoyiannis DP. Voriconazole pre-exposure selects for breakthrough mucormycosis in a mixed model of Aspergillus fumigatus-Rhizopus oryzae pulmonary infection. Virulence. 2011;2(4):348–55.
Pham PA. Bartlett JG. Voriconazole. Johns Hopkins ABX Guide. Accessed 4-29-18.
Pascual A, Calandra T, Bolay S, Buclin T, Bille J, Marchetti O. Voriconazole therapeutic drug monitoring in patient with invasive mycoses improves efficacy and safety outcomes. Clin Infect Dis. 2008;46:201–11.
Drinkwater DC, Davidson-Moncada J, Heckendorn E, Myers J, Whitman TJ. A patient with ulcerated nodules on his face. Clin Infect Dis. 2014;58:901–2.
Thompson GR, Bays D, Cohen SH, Pappagianis D. Fluroride excess in coccidioidomycosis patients receiving long term antifungal therapy. Antimicrob Agents Chemother. 2012;56(1):563–4.
Williams D. The effect of enteral nutrition supplements on serum voriconazole levels. J Oncol Pharm Pract. 2011;18(1):128–31.
Obeng O, Egelund EF, Asiltan A, Peloquin CA, Johnson JA. CYP2C19 polymorphisms and therapeutic drug monitoring of voriconazole.
Dickmeyer NJ, Kiel PJ. Dosing voriconazole in an obese patient. Clin Infect Dis. 2011;53:745.
Smith J, Safdar N, Knansinski V, Simmons W, Bhavnani SM, Ambrose PG, Andes D. Antimicrob Agents Chemother. 2006;50:1570–22.
Turner RB, Martello JL, Malhorta A. Worsening renal function in patients with baseline renal impairment treated with intravenous voriconazole. Int J Antimicrob Agents. 2015;46(4):362–6.
National Center for Biotechnology Information. PubChem Compound Database; CID=468595. https://pubchem.ncbi.nlm.nih.gov/compound/468595. Accessed 29 Apr 2018.
Nagappan V, Deresinski S. Posaconazole: A broad-spectrum triazole antifungal agent. Clin Infect Dis. 2007;45:1610–6.
Noxafil (posaconazole) prescribing information. Merck. Whitehouse Station. Revised 2014.
Walish TJ, Raad I, Patterson TF, Chandrasekar P, Donowitz GR, et al. Treatment in invasive aspergillosis with posaconazole in patients who are refractory to or intolerant of conventional therapy. Clin Infect Dis. 2007;44:2–12.
Greenberg RN, Mullane K, van Burik J-AH, Raad I, Abzug MJ, et al. Posconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother. 2006;50(1):126–33.
Pam PA, Bartlett JG. Posconazole. Johns Hopkins ABX Guide. Accessed 4-29-18.
Green MR, Woolery JE. Optimising absorption of posaconazole. Mycoses. 2011;54:e775–9.
National Center for Biotechnology Information. PubChem Compound Database; CID=72196309. https://pubchem.ncbi.nlm.nih.gov/compound/72196309. Accessed 3 May 2018.
National Center for Biotechnology Information. PubChem Compound Database; CID=6918485. https://pubchem.ncbi.nlm.nih.gov/compound/6918485. Accessed 3 May 2018.
Miceli MH, Kauffman CA. Isavuconazole: a new broad-spectrum triazole antifungal agent. Clin Infect Dis. 2015;61(10):1558–65.
Cresemba (isavuconazonium sulfate) prescribing information. Astellas Pharma Inc. Northbrook. Revised June 2015.
Cornely OA, Bohme A, Schmitt-Hoffman A, Ulmann AJ. Safety and pharmacokinetics of isavuconazole as antifungal prophylaxis in acute myeloid leukemia patients with neutropenia. Antimicrob Agents Chemother. 2015;59:2078–85.
Viljoen J, Schmitt-Hoffman A, Ghannoum M. A phase 2, randomized, double-blind, multicenter trial to evaluate the safety and efficacy of three dosing regimens of isavuconazole compared to fluconazole in patients with uncomplicated esophageal candidiasis. Antimicro Agents Chemother. 2015;59(3):1671–9.
Maertens JA, Raad II, Marr KA, Patterson TF, Kontoyiannis DP, et al. Isavuconazole versus voriconazole for primary treatment of invasive mould disease caused by Aspergillus and other filamentous fungi (SECURE). Lancet. 2016;387(10020):760–9. https://doi.org/10.1016/S0140-6736(15)01159-9.. Epub 2015 Dec 10
Pettit NN, Carver PL. Isavuconazole: a new option for the management of invasive fungal infections. Ann Pharmacother. 2015;49(7):825–42.
National Center for Biotechnology Information. PubChem Compound Database; CID=66577041. https://pubchem.ncbi.nlm.nih.gov/compound/66577041. Accessed 3 May 2018.
National Center for Biotechnology Information. PubChem Compound Database; CID=477468. https://pubchem.ncbi.nlm.nih.gov/compound/477468. Accessed 3 May 2018.
National Center for Biotechnology Information. PubChem Compound Database; CID=166548. https://pubchem.ncbi.nlm.nih.gov/compound/166548. Accessed 3 May 2018.
Cancidas (caspofungin acetate) prescribing information. Merck and Company, Inc. Whitehouse Station. Revised Mar 2018.
Mycamine (micafungin) prescribing information. Astellas Pharma US, Inc. Northbrook. Revised Aug 2016.
Eraxis (anidulafungin) prescribing information. Pfizer. New York. Original prescribing information on February 16, 2006.
Eraxis (anidulafungin) prescribing information. Pfizer. New York. Revised January 2018.
Chen SC, Slavin MA, Sorrell TC. Echinocandin antifungal drugs in fungal infections: a comparison. Drugs. 2011;17(1):11–41.
Perlin DS. Echinocandin resistance in Candida. Clin Infect Dis. 2015;61(Suppl 6):S612–7.
Marr KA, Sclamm HT, Herbrect R, Rottinghaus ST, Bow EJ, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2016;162(2):81–9.
Wang J-L, Chang C-H, Young-Xu Y, Chan KA. Systematic review and meta-analysis of the tolerability and hepatotoxicity of antifungals in empirical and definitive therapy for invasive fungal infection. Antimicrob Agents Chemother. 2018;62:2409–19.
Koch C, Uhle F, Wolff M, Arens C, Schulte A, Li L, et al. Cardiac effects of echinocandins after central venous administration in adult rats. Antimicrob Agents Chemother. 2018;62(5):1612–9.
Nishimoto M, Koh H, Tokuwame A, Makuuchi Y, Kuono M, et al. Drug interactions and safety profiles with concomitant use of casofungin and calcineurin inhibitors in allogeneic haemtolopoietic cell transplantation. Br J Clin Pharmacol. 2017;83(9):2000–7.
Betts RF, Nucci M, Talwar D, Gareca M, Queiroz-Telles T, et al. A multi-center double-blind trial of high-dose caspofungin treatment regimen versus standard caspofungin treatment regimen for adjust patients with invasive candidiasis. Clin Infect Dis. 2009;48:1676–84.
Epstein DJ, Seo SK, Huang YT, Park JH, Klimek VM, et al. Micafungin versus posaconazole prophylaxis in acute leukemia or myelodysplastic syndrome: a randomized study. J Infect 2018; pii: S0163-4453(18)30132-4. doi: https://doi.org/10.1016/j.jinf.2018.03.015. [Epub ahead of print].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Quilitz, R. (2019). Antifungal Medications in Neutropenia. In: Velez, A., Lamarche, J., Greene, J. (eds) Infections in Neutropenic Cancer Patients. Springer, Cham. https://doi.org/10.1007/978-3-030-21859-1_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-21859-1_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-21858-4
Online ISBN: 978-3-030-21859-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)