
Abstract
Hepatocellular carcinoma (HCC) is considered an inflammation-induced cancer as it often develops in the setting of chronic hepatitis. Therefore, the application of immune-based therapies may provide an ideal approach to treatment. As a matter of fact, hepatitis B vaccination can be seen as the first preventive cancer vaccine for HCC. The recent approval of nivolumab as second-line therapy for patients with advanced HCC has been the pinnacle of years of research and clinical trials in the application of immunotherapies to patients with HCC. The liver, unlike most organs, has an immune-tolerant microenvironment due to its high antigen exposure from the gut which is often coupled with chronic immune stimulation from confounding liver disease. As a result, efforts to understand HCC antigens as well as the tumor microenvironment has enabled advances in the application of immunotherapies to HCC. Immune-based therapies can be categorized as checkpoint inhibitors, adoptive cell transfer, cytokine-based therapy, and vaccines. In this chapter, we discuss the application of different immunotherapies to HCC.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Immunotherapy
- Immune checkpoint inhibitors
- Adoptive cell transfer
- Cytokines
- Vaccine
- Antigen
- Tumor microenvironment
Introduction
Hepatocellular carcinoma (HCC) often develops in the setting of chronic liver disease and as such is often considered an inflammation-induced cancer where this inflammation aids to drive carcinogenesis [1]. As an inflammation-induced cancer, patients with a greater lymphocyte density in HCC tumors often correlate with a better prognosis [2]. As a result, immunotherapy may provide an ideal approach of treatment [3]. As of 2017, nivolumab, a monoclonal antibody targeting programmed cell death-1 (PD-1), has been approved for patients with advanced HCC in the second-line setting for those patients who have progressed on sorafenib [4]. In order to have an effective immunotherapy, a tumor must present antigens which are recognized by the immune system, and then the immune system must be able to mount a response against that tumor-associated antigen. However, the tumor microenvironment has adapted ways to evade immune recognition as well as escape from immune therapies. In this chapter, we describe immune-based treatment of HCC first focusing on HCC tumor antigens and liver tumor microenvironment. We will then discuss the application of immune-based therapies in the treatment of patients with advanced HCC as well as future directions of where the field of immunotherapy may be heading in patients with HCC.
Tumor Antigens
The identification of HCC tumor antigens is an important step for potential targeted immune-based therapies. Solid tumors may contain 30–70 mutations that alter the amino acid sequences of proteins. Each of these alterations is foreign to the immune system and is therefore a potential target for immune recognition [5]. However, the immune system cannot recognize all mutations for an effective immune response. The mutation must result in a protein that is expressed by the tumor cell, and this protein must be able to be presented to the immune system on a human leukocyte antigen (HLA) protein [5]. If either of these conditions are not met, the mutation may not be immunogenic.
Tumor antigens can be categorized into three broad categories: tumor-specific antigens, which arise in cancer cells and are completely absent from normal cells; tumor-associated antigens (TAA) which are expressed mainly on tumor cells but also at low levels on normal cells; and cancer/testis antigens which are expressed by tumor cells and expressed in reproductive tissue but otherwise absent in normal adult cells [6]. TAA-specific CD8+ T cell responses have been found to be more detectable in patients with early-stage HCCs as compared to later stages. In addition, patients with TAA-specific responses were found to have a significantly greater median progression-free survival (PFS) compared to patients without TAA-specific responses [7]. Therefore, we can deduce that tumors, which contain more immunogenic antigens tend to have a more favorable prognosis and outcome.
Perhaps the most well-known HCC antigen is alpha-fetoprotein (AFP). AFP is widely considered the most useful biomarker for HCC evaluation with high levels correlating with the development and progression of HCC [8]. AFP is present during fetal development but is largely absent in healthy adults. Other widely studied tumor antigens associated with HCC include glypican-3 (GPC-3), melanoma-associated gene-A1 (MAGE-A1), New York-esophageal squamous cell carcinoma-1 (NY-ESO-1), sarcoma X breakpoint 2 (SSX2), and telomerase reverse transcriptase (hTERT) [1, 7]. Targeting tumor antigens may be an efficient way to control tumor growth, but immune-based approaches focusing on vaccines, cytokines, and nonspecific T cell activation have resulted in largely disappointing results [1, 9]. These immune-based strategies may produce disappointing results as the HCC tissue microenvironment may have a large impact in response to therapy.
Tumor Environment
The tumor microenvironment (TME) is becoming increasingly recognized as having a role in tumor growth promotion and immune evasion. Under normal conditions, the liver experiences a tremendous antigen exposure as a result of portal-venous blood flow with physiologic filtration of environmental and bacterial agents from the gastrointestinal tract. Therefore, to prevent autoimmune damage from constant immune stimulation and antigen exposure, the liver has developed intrinsic tolerogenic mechanisms in the innate and adaptive immune responses. This tolerance to the large flux of antigens can be harmless in respect to the large majority of antigens but may prove to be detrimental with immune tolerance to tumor-associated antigens and HCC progression [10].
In addition to the physiologic immune tolerance of the liver, chronic inflammation promotes immune suppression through continuous cytokine production and recruitment of immune cells [10]. In order to have T cell activation, CD4+ T cells are presented an antigen on major histocompatibility complex (MHC) class II molecules by antigen-presenting cells that recognize a specific T cell receptor (TCR). A costimulatory binding of CD28 on the T cell with CD80 or CD86 on the antigen presenting cell (APC) is then required to propagate the signal for T cell activation. Activated CD4+ T cells are then polarized toward a TH1 phenotype enhancing CD8+ T cell cytotoxicity toward the target antigen (Fig. 12.1). However, once CD4+ T cells are activated, they upregulate the immunosuppressive receptor, cytotoxic T lymphocyte-associated protein (CTLA)-4, to act as a break on the adaptive immune response by competing with CD28 for the binding of CD80/CD86 and therefore taking away the necessary costimulatory signal. CTLA-4 is present on activated T cells, dendritic cells (DCs), and is constitutively expressed on regulatory T cells (Tregs). Under physiologic conditions, immune checkpoints prevent over activation of T cells, thereby limit unwanted collateral tissue damage [11]. However, the TME has usurped this physiological function to produce an immunosuppressive milieu.

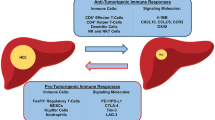
Overview of tumor immune response. Under physiological conditions, antigens are recognized and presented to CD4+ T cells, which further activate CD8+ T cells. T cell activation causes upregulation of CTLA-4 and PD-1 to prevent overactivation of the immune response. The tumor microenvironment creates an immunosuppressive milieu through recruitment of regulatory T cells (Tres), myeloid-derived suppressor cells (MDSCs), and upregulation of immune checkpoints. These immunosuppressive factors lead toward carcinogenesis and tumor progression
In addition to CTLA-4, PD-1 also acts as a check on the immune response. Much like CTLA-4, PD-1 is upregulated in the setting of chronic antigen exposure. PD-1 is expressed by activated CD4+ and CD8+ T cells, natural killer (NK) cells, B cells, as well as Tregs, myeloid-derived suppressor cells (MDSCs), monocytes, and DCs. Programmed death-ligand 1 (PD-L1) and PD-L2 are the ligands of PD-1. PD-L1 is found on APCs, MDSCs, macrophages, various parenchymal cells, as well as tumor cells, while PD-L2 is only expressed by hematopoietic stem cells. The binding of PD-1 with its ligand blocks TCR signaling, inhibits T cell proliferation, and leads to dysfunctional exhausted T cells [1]. Additionally, PD-L1 overexpression in HCC is associated with more aggressive tumors and an increase in postoperative recurrence of HCC [12].
Different immune cells have been reported to suppress antitumor immunity in HCC. Tregs have been shown to accumulate in patients with HCC where an increase in Tregs has been linked to a worse outcome [13]. Tregs inhibit the immune response through competitively binding CTLA-4 to CD28 as well as downregulation of CD80 and CD86, secretion of transforming growth factor (TGF)-β and IL-10, and depletion of IL-2. MDSCs are also found to be increased in patients with HCC and elevated counts often correlate to tumor progression [14,15,16]. MDSCs are a heterogeneous group of immature and immunosuppressive myeloid cells which promote tumor formation by facilitating angiogenesis through VEGF production, impair CD4+ and CD8+ T cells with increased arginase activity, impair NK cell activity via TGF-β, disrupt TCR signaling through reactive oxygen and nitrogen species, and promote Treg expansion. Additionally, MDSCs promote formation of immunosuppressive M2 macrophages which creates a vicious cycle further propagating an immunosuppressive microenvironment through production of suppressive cytokines and promoting T cells to a TH2 phenotype which further induces M2 macrophages and MDSCs [1].
In addition to the aforementioned checkpoint inhibitors, other inhibitory membrane proteins are being recognized as creating an immunosuppressive TME and therefore may represent a potential therapeutic target. TIM-3 is expressed by cells of the innate and adaptive immune system. Galactin-9 is a ligand for TIM-3 and is expressed by multiple tissues, including the liver, and regulates cell differentiation, adhesion, and cell death. Galactin-9 and TIM-3 have been shown to inhibit the T cell response. In addition, TIM-3-expressing T cells often co-express PD-1, indicating the two inhibitory pathways may cooperate to produce a severely exhausted T cell phenotype [9]. Preclinical studies have shown that blocking TIM-3 and PD-1 may produce a greater tumor response in non-small cell lung cancer models [17].
LAG-3 has also been shown to act synergistically with PD-1 to promote tumor immune evasion. LAG-3 is expressed by activated T cells and binds to MHC class II molecules on DCs, thereby decreasing the costimulatory function of DCs and is a hallmark of exhausted T cells [1, 9]. Other immune-inhibitory factors include arginase-1 which is expressed at high levels by MDSCs and tumor-associated machrophages (TAMs) as well as indoleamine 2,3-dioxygenase (IDO) which is expressed by multiple cell types in the TME [1]. Both arginase-1 and IDO metabolize amine acids, arginine and tryptophan, respectively, which deprive immune cells of vital nutrients and generate immunosuppressive by-products [1].
Immunotherapies
As we discussed, the tumor microenvironment creates an immunosuppressive milieu promoting tumor formation and limits the capacity of the host to mount a proper immune response. Investigations are ongoing to create immune-based therapies to promote tumor recognition and ultimately tumor eradication. Here, we discuss various approaches to immune-based therapies, including the application of immune checkpoint inhibitors, cell-based therapies, and cytokine-based therapies. We will then discuss future directions with genetically engineered therapies including chimeric antigen receptors (CAR) T cell therapies and TCR therapies.
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors have gained interest for patients with advanced HCC after success in treating patients with melanoma and non-small cell lung cancer with these agents. Between 2013 and 2017, results of three clinical trials have been reported for patients with advanced HCC treated with immune checkpoint inhibitors; two trials using tremelimumab (anti-CTLA-4), and one trial utilizing nivolumab (anti-PD1) (Table 12.1). Checkpoint inhibitors have several advantages over other types of immunotherapies such as cell-based therapies with regard to commercial availability, wider applicability to a range of pathologic conditions, and no restriction by HLA status.
Sangro et al. reported the first clinical use of immune checkpoint inhibitor, tremelimumab, in a phase II trial in patients with chronic hepatitis C and advanced HCC not amenable to surgical or locoregional therapies [18]. Fifty-seven percent of patients enrolled progressed on previous therapies, and 43% had severer liver disease, i.e., Child-Pugh class B advanced fibrosis/cirrhosis. This is significant given that previously evaluated drugs such as sorafenib have been tested in mostly patients with Child-Pugh class A disease and better natural history. Although the patients received what is now considered to be a suboptimal dose of tremelimumab (15 mg/kg every 90 days to a maximum of four doses), partial response (PR) in 3 patients and stable disease (SD) in 10 patients were observed among 17 evaluable patients [9]. In addition, tremelimumab was generally well tolerated, and a reduction in HCV viral load was also detected.
The next trial used tremelimumab in combination with noncurative tumor ablation utilizing radiofrequency ablation (RFA) or transarterial chemoembolization (TACE) [19]. The hypothesis of adding tumor ablation was the assumption that RFA or TACE may cause immunogenic cell death. This type of cell death could then lead to a systemic release of antigens and a global immune response which may be enhanced by the checkpoint inhibitor [9, 19]. This was a phase I/II study with optimal therapeutic dosing of tremelimumab consisting of 78% of patients who advanced on previous therapies. Fourteen percent of patients were Child-Pugh class B and 75% had viral hepatitis. Of the 19 evaluable patients, 5 patients (26%) had a PR and 12 patients (63%) were deemed to have SD. Again, this trial showed an acceptable safety profile, reduction in viral load, and promising antitumor effects.
The encouraging results of the tremelimumab studies followed into utilizing nivolumab in CheckMate 040 trial [4]. This study consisted of dose-escalation and dose-expansion phases in patients with intermediate or advanced HCC who had progressed, were intolerant, or refused sorafenib. As a result of the dose-escalation study, 3 mg/kg every 2 weeks was chosen for the expansion cohort. Most patients had advanced HCC with extrahepatic metastases and received sorafenib. Of the 212 evaluable patients, they observed complete response (CR) in 3 patients (1%) and PR in 39 patients (18%). Furthermore, response rates were similar in both patients with or without prior sorafenib therapy. These findings resulted in nivolumab being approved in the second-line setting for patients with advanced HCC. Another anti-PD1, pembrolizumab, has shown similar objective response rate of 17% in 104 enrolled patients in a phase II trial (KEYNOTE-224) [20]. A follow-up phase III study however did not meet the primary end points for OS and PFS despite favorable trends, suggesting the need for predictive biomarkers of response (KEYNOTE-240, NCT02702401). Another phase III trial of pembrolizumab is ongoing in Asian patients (KEYNOTE-394, NCT03062358). A phase II trial is underway to evaluate the agent as neoadjuvant therapy following surgical resection or ablation (AURORA, NCT03337841).
Although these aforementioned trials showed promising results for the use of immune checkpoint inhibitors in patients with advanced HCC, additional trails are needed and underway to show efficacy as first-line therapy and in combination with other immune or cytotoxic therapies. In patients with melanoma combining nivolumab and ipilimumab (anti-CTLA-4) produced a greater objective-response rate and progression-free survival as compared to single-agent therapy [21]. Currently, phase III clinical trials are underway with nivolumab in the first-line setting against sorafenib (CheckMate 459) and combination therapies of tremelimumab and durvalumab (anti-PD-L1) along with ablative therapies (NCT02821754) and nivolumab plus sorafenib (NCT03439891).
Cell Bases Therapies
Adoptive cell transfer (ACT) is a highly personalized form of cancer immunotherapy that involves the transfer of host-derived expanded immune cells [22]. Adoptive transfer of autologous tumor-infiltrating lymphocytes (TIL) has been shown to produce a complete and durable tumor regression in patients with metastatic melanoma [23]. There is limited data in the treatment of HCC patients with metastatic or unresectable disease via the transfer of ex vivo expanded autologous TIL. However, ACT has been tried in the adjuvant setting. Takayama et al. studied ACT in the adjuvant setting in 150 patients who had undergone curative resection for HCC with 76 patients receiving adoptive immunotherapy and 74 patients receiving no adjuvant treatment [24]. The ACT treatment consisted of adjuvant activated autologous lymphocyte infusions. ACT treatment increased the recurrence-free survival (RFS) but had no impact on overall survival (OS). Activated T cell transfer has also been applied with adjunctive treatments such as an autologous tumor lysate-pulsed DC vaccine [25]. In this study, patients who underwent a curative HCC liver resection received an adjuvant DC vaccine which was made from a patient’s isolated DCs pulsing with tumor lysate created from the resected tumor along with CD3+ activated T cells. There was a significant difference in both RFS and OS in favor of combination DC vaccine and ACT vs. no adjuvant therapy. The difference between these two studies may highlight the need for combination therapy strategies in the future.
Another strategy of ACT that has been tried in the adjuvant setting for HCC is through the use of cytokine-induced killer (CIK) cells. CIK cells are autologous cells that are expanded ex vivo from a patient’s peripheral blood mononuclear cells (PBMCs) which are cultured with a cytokine cocktail and anti-CD3 antibodies. CIK cells consist of a variety of subpopulations: CD3+/CD56+ cells, CD3−/CD56+ NK cells, and CD3+/CD56− cytotoxic T cells. Therefore, CIK cells have potent antitumor effects with the dual-functional capability of T cells and NK cells. The result indicated its substantial specificity toward tumor cells and the capability of acting independent of TCR [26, 27]. Lee et al. studied the use of CIK cells in the adjuvant setting in patients with resected HCC. The primary end point of this study is RFS and secondary end points include OS and cancer-specific survival. They found that CIK cell immunotherapy was associated with improved RFS, OS, and cancer-specific survival [27].
CIK cell therapy has also been evaluated in the setting of inoperable advanced disease. In a nonrandomized evaluation of patients receiving RFA and TACE, CIK cell therapy was shown to possibly improve OS when given with these locoregional therapies [28]. Additionally, in a phase II randomized trial, it was found that CIK cell therapy can improve OS and PFS as compared to standard treatment [29]. Additional data support the use of tadalafil, a phosphodiesterase type 5 (PDE5) inhibitor, in combination with CIK cell therapy, as it improves efficacy through the suppression of MDSC activity (unpublished work from Greten lab). These studies demonstrate promise in adoptive cell-transfer techniques for patients with advanced nonoperable HCC as well as in the adjuvant setting. However more research and clinical trials are needed in the application of these treatment methods. A major potential drawback of ACT is the need of specialized centers and the difficultly in making these treatments widely and commercially available.
Cytokines
Dr. Steven Rosenberg pioneered one of the first successful immune-based treatments with the application of interleukin-2 (IL-2) for the treatment of metastatic melanoma [30]. However, for patients with HCC, cytokines-based treatments have met with limited success. The use of interferon (IFN) appeared as a logical first choice for the treatment of HCC, and it may show both antiviral and antitumor functions. However, for patients with advanced disease, the tumor response rates to IFN therapy was poor with a partial response rate of 6% (2 of 30 patients) and no benefit in OS [31]. Additionally, IFN therapy was not well tolerated in patients with cirrhosis and HCC resulting in nearly half of the patients discontinuing treatment due to intolerance or adverse events [31]. IFN has also been studied in the adjuvant setting for patients with viral hepatitis-related HCC with conflicting results. Chen et al. investigated the use of adjuvant IFN and failed to find a statistical difference in either RFS or OS [32]. Sun et al. investigated a similar cohort with IFN and found no difference in disease-free survival but found a significant difference in OS in favor of patients receiving adjuvant IFN [33]. Of note, more patients in the IFN-group received a second liver resection than patients in the control group. Although this was not statistically significant, this may influence OS in these patients.
Additional trials with cytokines involve phase I trials with intratumoral delivery of IL-12 [34, 35]. Both these trials included patients with advanced GI tumors, displaying feasibility and safety of the therapy, but did not show promising antitumor response rates, although the studies were underpowered. Further investigations are underway with promising results involving a small molecule inhibitor of TGF-ß receptor 1, LY2157299 (galunisertib) in patients with advanced HCC either alone or in combination with nivolumab or stereotactic body radiotherapy [10].
Vaccine Therapy
The introduction of the hepatitis B vaccine in the 1980s may have virtually restructured the landscape of HCC by preventing assumedly numerous cases of HCC. Similarly, immunization against human papilloma virus has greatly decreased the risk of cervical cancer in a similar fashion. Utilizing similar principles of immune recognition and promoting an adaptive immune response against specific antigens, i.e., vaccination, can be applied not only for cancer prevention but also for cancer treatment.
The basic element underlying cancer vaccination is increased immune recognition of tumor-specific neoantigens that result from either driver or passenger genomic DNA mutations, producing altered proteins to create neoepitopes. Multineoepitope vaccines have been shown to activate both neoantigen-specific CD4+ and CD8+ T cells. In the first priming phase of the immune response, cross presentation between DCs and TH1 cells can induce potent neoepitope-specific cytotoxic T lymphocytes (CTLs) with improved tumor penetration and generating memory CD8+ T cells. In the tumor, the vaccine-induced TH1 CD4+ T cells can further promote an inflammatory TME through increased IFN-γ, thus upregulating MHC class I molecules on tumor cells, which improves killing by the CD8+ T cells. Additionally, the IFN-γ upregulates MHC class II molecules, further sensitizing tumors to recognition by CD4+ T cells [36].
AFP is expressed by a fraction of HCC tumors and during the process of fetal development but not in healthy adult tissues. AFP was the first TAA targeted for vaccine-based therapies in HCC despite limited success. In early studies utilizing AFP peptides or AFP-pulsed DCs, a T cell response was induced, but no clinical benefit of the therapy was observed [37, 38]. In a more recent study, administration of AFP-derived peptides to 15 HCC patients produced T cells that reacted to the peptides and resulted in CR in 1 patient and SD in 8 patients with no adverse events [39].
Other trials have been conducted utilizing peptide vaccine against a carcinoembryonic antigen, GPC3, which is an appealing target for HCC vaccines because of its specificity to HCC and association with a poor prognosis of the patients. Early studies utilizing a GPC3 peptide vaccine found the treatment to be safe and able to induce tumor infiltration of CD8+ T cells. However, the therapy produced only 1 PR out of 33 treated patients with a median time to tumor progression of 3.4 months [40]. Preclinical studies have shown that utilizing anti-PD1 therapy may result in increased response to GPC3 peptide vaccines [41]. The GPC3 vaccine was also tested in the adjuvant setting, demonstrating a significantly improved recurrence rate in patients treated with surgery plus vaccine compared to surgery alone at 1 year but was found to be no longer statistically significant at 2 years [42]. Besides a vaccine, targeting GPC3 through an anti-GPC3 antibody, GC33, has shown to be tolerated and may have promise in further phase II trials [43].
Clinical trials have also been performed using the targeted oncolytic poxvirus, JX-594, which was designed to replicate in and destroy cancer cells. JX-594 was found to be safe in a phase I study and displayed some promising results in a phase II trial with a intrahepatic disease control rate of 46% [44, 45]. Currently, there is an ongoing phase III clinical trial evaluating JX-594 (Pexa Vec) followed by sorafenib versus sorafenib alone in patients with advanced HCC (NCT02562755). Other trials utilizing a vaccine-based strategy with DCs pulsed with antigens have failed to demonstrate a significant clinical benefit [10]. Additionally, a phase II trial of low-dose cyclophosphamide in combination with the telomerase peptide GV1001 in patients with advanced HCC showed no radiologically detectable tumor responses [46]. The above studies demonstrate that although vaccine-based strategies in HCC can mount an immune response as shown by antigen-specific T cells in the blood of treated patients, local factors in the tumor likely prevent tumor eradication. Therefore, further studies are needed to progress vaccine-based immunotherapies, perhaps in combination with other immune therapies, in patients with advanced HCC.
Sensitivity and Resistance to Immune Therapies
Although there has been recent success with immunotherapy in HCC, namely, immune checkpoint inhibitors, a subset of patients do not respond to therapy. Unfortunately, there is little known regarding the characteristics of HCC tumors that may predict response to immunotherapies. Experimental evidence suggests high levels of TIL, IFN signaling, presence of immune checkpoints, or a high tumor mutation burden may favor a positive clinical response to immune-based therapies. Furthermore, gene expression profiles of tumor, stromal, and immune cells indicate approximately 25% of evaluated HCC samples express markers of an inflammatory response which may indicate better sensitivity to immunotherapy [47].
Recent studies have suggested potential mechanisms underlying resistance to and escape from immunotherapies. Resistance to immunotherapy can be classified as primary resistance or adaptive and acquired resistance. Primary resistance occurs when the tumor does not respond to an immunotherapy from the initiation of therapy likely through lack of tumor antigen recognition by T cells. Adaptive or acquired resistance may occur when a tumor is recognized by the immune system, but the tumor protects itself from immune attack. Additionally, resistance can be due to intrinsic tumor properties such as a low mutation burden and high PD-L1 expression or extrinsic tumor properties such as a highly immunosuppressive TME and lack of T cells with antigen-specific TCRs [48].
Further proposed mechanisms of resistance to immunotherapy include tumor cells, which have lost beta2-microglobulin (ß2-m) in tumor cells and can therefore no longer be recognized by CD8+ T cells [49]. Others observed that nonresponders to anti-CLTA-4 with metastatic melanoma have tumors with genomic defects in IFN-γ pathway genes [50]. Additionally, upregulation of alternative immune checkpoints, notably TIM-3, have also been found in lung adenocarcinoma. This suggests the upregulation of alternative immune checkpoints may be associated with adaptive resistance to anti-PD-1 therapy and therefore TIM-3 may be a potential target for combination therapy with anti-PD-1 [17]. Further understanding of the underlying resistance mechanisms to immunotherapies is needed to develop proper combination strategies to improve efficacy of checkpoint blockade for HCC treatment.
Future Therapies
Genetically modified T cells have been applied to cancer therapy, and this technique is likely to be applied to HCC in the near future. T cells can be manipulated to express a high-affinity TCR or a chimeric antigen receptor (CAR). TCRs have a limitation of being restricted to recognize specific MHC molecules and therefore can only be used in patients who possess those specific HLAs. CAR T cells express a genetically engineered fusion molecule that act independently from an MHC molecule and are therefore able to circumvent HLA restrictions. The use of CAR T cells has been thrust into mainstream treatment for patients with CD19+ hematologic malignancies. The use of TCR and CAR T cells for patients with HCC remains in its infancy. Preclinical studies utilizing GPC3-specific T cells showed promising results in HCC-bearing mice [51]. A phase I study is currently underway to investigate its safety and antitumor activity of autologous T cells expressing TCRs for AFP in patients with advanced HCC (NCT03132792).
As alluded to earlier in this chapter, combining multiple immunotherapy modalities may improve HCC response rates. The combination of checkpoint inhibitors, ACT, cytokines, and vaccines have been studied in other malignancies with varying degrees of success. The application of these approaches is likely soon to be applied to HCC. The use of immune-based therapies also raises concern about safety of the agents in HCC patients with underlying liver dysfunction caused by viral hepatitis or other etiologies. An early concern was the possible hepatocyte damage due to an overwhelming immune response against viruses in infected hepatocytes. However, the earlier application of immunotherapy, namely, checkpoint inhibitors, appeared to be generally safe and well-tolerated in patients with advanced HCC and therefore has led to the pursuit of combination with other treatment modalities [52]. Additionally, effective adjuvant therapy is lacking in HCC, and the use of immunotherapies may provide a benefit to this patient population and requires further evaluation. Phase I and II clinical trials are planned to test nivolumab as neoadjuvant therapy after surgical resection of HCC tumors (NCT03510871, NCT03299946).
Conclusion
The application of immunotherapies has restructured the treatment approach to numerous malignancies such as melanoma and non-small cell lung cancer and is now being adopted for the treatment of HCC patients. Although the liver microenvironment is immunosuppressive and chronic liver disease contributes to further immune tolerance, the early application of immune-based therapies has demonstrated promising results in clinical trials. Further basic, translational, and clinical studies are required to better understand the complex interactions between tumor cells, immune cells, and immunotherapies in the tumor microenvironment to develop a treatment strategy to eliminate HCC cells.
References
Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12(12):681–700. https://doi.org/10.1038/nrgastro.2015.173.
Wada Y, Nakashima O, Kutami R, Yamamoto O, Kojiro M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology. 1998;27(2):407–14. https://doi.org/10.1002/hep.510270214.
Greten TF, Duffy AG, Korangy F. Hepatocellular carcinoma from an immunologic perspective. Clin Cancer Res. 2013;19(24):6678–85. https://doi.org/10.1158/1078-0432.ccr-13-1721.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502. https://doi.org/10.1016/s0140-6736(17)31046-2.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. https://doi.org/10.1126/science.1235122.
Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–22. https://doi.org/10.1038/nrc.2016.154.
Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59(4):1415–26. https://doi.org/10.1002/hep.26731.
Tsuchiya N, Sawada Y, Endo I, Saito K, Uemura Y, Nakatsura T. Biomarkers for the early diagnosis of hepatocellular carcinoma. World J Gastroenterol. 2015;21(37):10573–83. https://doi.org/10.3748/wjg.v21.i37.10573.
Greten TF, Sangro B. Targets for immunotherapy of liver cancer. J Hepatol. 2017; https://doi.org/10.1016/j.jhep.2017.09.007.
Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62(6):1420–9. https://doi.org/10.1016/j.jhep.2015.02.038.
Inarrairaegui M, Melero I, Sangro B. Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin Cancer Res. 2018;24(7):1518–24. https://doi.org/10.1158/1078-0432.Ccr-17-0289.
Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):971–9. https://doi.org/10.1158/1078-0432.Ccr-08-1608.
Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65(6):2457–64. https://doi.org/10.1158/0008-5472.Can-04-3232.
Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015;64(5):842–8. https://doi.org/10.1136/gutjnl-2014-307990.
Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR −/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62(8):1421–30. https://doi.org/10.1007/s00262-013-1447-1.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–43. https://doi.org/10.1053/j.gastro.2008.03.020.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. https://doi.org/10.1038/ncomms10501.
Sangro B, Gomez-Martin C, de la Mata M, Inarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8. https://doi.org/10.1016/j.jhep.2013.02.022.
Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. 2017;66(3):545–51. https://doi.org/10.1016/j.jhep.2016.10.029.
Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19(7):940–52. https://doi.org/10.1016/S1470-2045(18)30351-6.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17. https://doi.org/10.1056/NEJMoa1414428.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8. https://doi.org/10.1126/science.aaa4967.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–7. https://doi.org/10.1158/1078-0432.Ccr-11-0116.
Takayama T, Sekine T, Makuuchi M, Yamasaki S, Kosuge T, Yamamoto J, et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356(9232):802–7. https://doi.org/10.1016/S0140-6736(00)02654-4.
Shimizu K, Kotera Y, Aruga A, Takeshita N, Katagiri S, Ariizumi S, et al. Postoperative dendritic cell vaccine plus activated T-cell transfer improves the survival of patients with invasive hepatocellular carcinoma. Hum Vaccin Immunother. 2014;10(4):970–6.
Schmidt-Wolf IG, Lefterova P, Mehta BA, Fernandez LP, Huhn D, Blume KG, et al. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp Hematol. 1993;21(13):1673–9.
Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148(7):1383–91 e6. https://doi.org/10.1053/j.gastro.2015.02.055.
Huang ZM, Li W, Li S, Gao F, Zhou QM, Wu FM, et al. Cytokine-induced killer cells in combination with transcatheter arterial chemoembolization and radiofrequency ablation for hepatocellular carcinoma patients. J Immunother (Hagerstown, MD: 1997). 2013;36(5):287–93. https://doi.org/10.1097/CJI.0b013e3182948452.
Yu X, Zhao H, Liu L, Cao S, Ren B, Zhang N, et al. A randomized phase II study of autologous cytokine-induced killer cells in treatment of hepatocellular carcinoma. J Clin Immunol. 2014;34(2):194–203. https://doi.org/10.1007/s10875-013-9976-0.
Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–8. https://doi.org/10.4049/jimmunol.1490019.
Llovet JM, Sala M, Castells L, Suarez Y, Vilana R, Bianchi L, et al. Randomized controlled trial of interferon treatment for advanced hepatocellular carcinoma. Hepatology. 2000;31(1):54–8. https://doi.org/10.1002/hep.510310111.
Chen LT, Chen MF, Li LA, Lee PH, Jeng LB, Lin DY, et al. Long-term results of a randomized, observation-controlled, phase III trial of adjuvant interferon Alfa-2b in hepatocellular carcinoma after curative resection. Ann Surg. 2012;255(1):8–17. https://doi.org/10.1097/SLA.0b013e3182363ff9.
Sun HC, Tang ZY, Wang L, Qin LX, Ma ZC, Ye QH, et al. Postoperative interferon alpha treatment postponed recurrence and improved overall survival in patients after curative resection of HBV-related hepatocellular carcinoma: a randomized clinical trial. J Cancer Res Clin Oncol. 2006;132(7):458–65. https://doi.org/10.1007/s00432-006-0091-y.
Mazzolini G, Alfaro C, Sangro B, Feijoo E, Ruiz J, Benito A, et al. Intratumoral injection of dendritic cells engineered to secrete interleukin-12 by recombinant adenovirus in patients with metastatic gastrointestinal carcinomas. J Clin Oncol. 2005;23(5):999–1010. https://doi.org/10.1200/jco.2005.00.463.
Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, Herrero I, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol. 2004;22(8):1389–97. https://doi.org/10.1200/jco.2004.04.059.
Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–60. https://doi.org/10.1126/science.aar7112.
Butterfield LH, Economou JS, Gamblin TC, Geller DA. Alpha fetoprotein DNA prime and adenovirus boost immunization of two hepatocellular cancer patients. J Transl Med. 2014;12:86. https://doi.org/10.1186/1479-5876-12-86.
Butterfield LH, Ribas A, Meng WS, Dissette VB, Amarnani S, Vu HT, et al. T-cell responses to HLA-A∗0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin Cancer Res. 2003;9(16. Pt 1):5902–8.
Nakagawa H, Mizukoshi E, Kobayashi E, Tamai T, Hamana H, Ozawa T, et al. Association between high-avidity T-cell receptors, induced by alpha-fetoprotein-derived peptides, and anti-tumor effects in patients with hepatocellular carcinoma. Gastroenterology. 2017;152(6):1395–406.e10. https://doi.org/10.1053/j.gastro.2017.02.001.
Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18(13):3686–96. https://doi.org/10.1158/1078-0432.Ccr-11-3044.
Sawada Y, Yoshikawa T, Shimomura M, Iwama T, Endo I, Nakatsura T. Programmed death-1 blockade enhances the antitumor effects of peptide vaccine-induced peptide-specific cytotoxic T lymphocytes. Int J Oncol. 2015;46(1):28–36. https://doi.org/10.3892/ijo.2014.2737.
Sawada Y, Yoshikawa T, Ofuji K, Yoshimura M, Tsuchiya N, Takahashi M, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology. 2016;5(5):e1129483. https://doi.org/10.1080/2162402x.2015.1129483.
Zhu AX, Gold PJ, El-Khoueiry AB, Abrams TA, Morikawa H, Ohishi N, et al. First-in-man phase I study of GC33, a novel recombinant humanized antibody against glypican-3, in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2013;19(4):920–8. https://doi.org/10.1158/1078-0432.CCR-12-2616.
Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–36. https://doi.org/10.1038/nm.3089.
Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–42. https://doi.org/10.1016/s1470-2045(08)70107-4.
Greten TF, Forner A, Korangy F, N'Kontchou G, Barget N, Ayuso C, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209. https://doi.org/10.1186/1471-2407-10-209.
Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology. 2017;153(3):812–26. https://doi.org/10.1053/j.gastro.2017.06.007.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23. https://doi.org/10.1016/j.cell.2017.01.017.
Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88(2):100–8.
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e9. https://doi.org/10.1016/j.cell.2016.08.069.
Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–28. https://doi.org/10.1158/1078-0432.Ccr-14-1170.
Brown ZJ, Heinrich B, Steinberg SM, Yu SJ, Greten TF. Safety in treatment of hepatocellular carcinoma with immune checkpoint inhibitors as compared to melanoma and non-small cell lung cancer. J Immunother Cancer. 2017;5(1):93. https://doi.org/10.1186/s40425-017-0298-2.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Brown, Z.J., Greten, T.F. (2019). Immune Therapies. In: Hoshida, Y. (eds) Hepatocellular Carcinoma. Molecular and Translational Medicine. Humana, Cham. https://doi.org/10.1007/978-3-030-21540-8_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-21540-8_12
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-21539-2
Online ISBN: 978-3-030-21540-8
eBook Packages: MedicineMedicine (R0)