
Abstract
Myasthenia gravis (MG), Lambert-Eaton myasthenic syndrome (LEMS) and neuromyotonia represent the three autoantibody-mediated disorders at the neuromuscular junction. They give muscle weakness and fatigability as their dominating symptoms. The weakness has usually a preferred localization to some but not all muscles. MG subgroups reflect pathogenesis and direct therapy. Patients should always be classified according to type of antibody, thymus pathology, age at symptom onset and generalized versus pure ocular symptoms. LEMS and neuromyotonia are subgrouped according to paraneoplasia or not. All conditions have well-defined autoantibodies that bind in vivo and directly induce the muscle weakness. Therapy includes symptomatic drugs influencing the acetylcholine receptor activity in the postsynaptic membrane and immunosuppressive treatment influencing the pathogenic autoantibodies. This immunoactive treatment is not yet specific for the disease-inducing antigen-antibody interaction. Treatment is usually effective, and most patients obtain mild symptoms only or a full clinical remission. Comorbidities need to be treated, especially a thymoma in paraneoplastic MG or neuromyotonia and a lung cancer in paraneoplastic LEMS. Supportive therapy is important, and a well-adapted daily training program is recommended. Severe exacerbations (myasthenic crisis) with the need for respiratory support are rare, occur mainly together with infections, and need immediate intensive care.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The neuromuscular junction is a predilection site for disease. The site is crucial for muscle function, and disorders at this junction lead to weakness in the muscle. The disorders can be immune-mediated through the action of autoantibodies. In addition, genetic disorders and toxins can interfere with neuromuscular transmission. More than 100 mutations have been detected in either presynaptic or postsynaptic molecules, most commonly in the postsynaptic acetylcholine receptors (AChR ) [1]. Such mutations usually lead to a stable generalized weakness with symptom debut during the first couple of years after birth. Rarely congenital myasthenia due to genetic disorders can be misdiagnosed as immune-mediated disease, and vice versa. The neuromuscular junction is a predilection site for animal and plant toxins. The induction of muscle paralysis is an excellent strategy both for attacking a potential prey and for defence. Botulinum toxin binds presynaptically, whereas curare and α-bungarotoxin are examples of postsynaptic toxins binding to the AChR.
Action potentials in the motor nerve lead to the release of acetylcholine from the presynaptic terminal. This release acts through the activation of voltage-gated calcium channels in the presynaptic cell membrane, allowing calcium to enter the neuron and triggering vesicles containing acetylcholine to fuse with the cell membrane. The acetylcholine traverses the synaptic cleft and binds to AChR. AChR serve as ligand-gated ion channels, so that binding of acetylcholine opens the central pore, sodium ions flow into the muscle cell, and this generates the muscle depolarization that eventually results in muscle contraction. Autoantibodies specific for immune-mediated disorders interfere with various parts of this cascade, all leading to impaired function and muscle weakness.
There are three main immune-mediated disorders of the neuromuscular junction: myasthenia gravis (MG), Lambert-Eaton myasthenic syndrome (LEMS ) and neuromyotonia. These disorders should be further subgrouped according to clinical and non-clinical biomarkers (Table 1) [2,3,4,5,6]. The three main diseases are characterized by their clinical picture, autoantibodies and neurophysiological characteristics. The MG subgroups are less distinct. Some patients with debut after 50 years can have thymic hyperplasia, a thymoma can be detected some years after MG debut, or a patient can have purely ocular symptoms for many months before progressing into generalized muscle weakness. Such patients challenge the formal subgroup classification. The absence of antibodies in seronegative MG depends on the sensitivity of the applied tests [7]. In ocular MG, the muscle weakness is clinically restricted to the ocular muscles. This is common early in the disease, but applies to only around 10% after 2 years [8].
Both LEMS and neuromyotonia can be paraneoplastic, associated most commonly with small-cell lung carcinoma and thymoma, respectively. The disease with and without cancer is otherwise clinically indistinguishable.
This chapter will give updated information on all aspects of the immune-mediated disorders at the neuromuscular junction, but with focus on therapeutic strategies and aspects that influence therapy. MG as by far the most common of the diseases will be described most detailed.
Epidemiology
MG has a prevalence of approximately 150 per million in most populations, and an annual incidence of around 10 per million [4, 9, 10]. In most Western populations, there is one peak of incidence around age 30 years, and then a gradually increase from age 50 years, at least until age 80 years. In China, there is an additional incidence peak in children around age 5 years. This juvenile MG in the Far East is usually mild and often ocular and otherwise resembles early-onset MG with AChR antibodies [11]. In MG with AChR antibodies and symptom debut before age 50, there is a clear preponderance of females, a two- to three-fold increase compared to males. Late-onset MG is more common in males. This means that in the total MG population the sex ratio is near to one. In countries with a young population, MG is more common in females.
MG prevalence has increased gradually for many decades. This does not necessarily mean that the risk for getting the disease has increased [12]. Prevalence depends on disease prognosis. Today, with the improved treatment, only a slight increase in mortality will lead to a higher prevalence compared to the situation before any effective treatment with perhaps a 50% mortality after 10 years. A second reason for the reported increase in MG prevalence is an improved case-finding. Previously MG was a clinical diagnosis, and thus given only to patients with the typical clinical picture, and recognized by the responsible doctor. Today the diagnosis relies for a large part on highly specific autoantibody analyses. Such antibody tests are performed with increasing frequency, and also in individuals with atypical or mild muscle weakness and fatigue with only a minimal clinical suspicion of MG. The number of neurologists has increased and access to specialists for the whole population has improved in most countries. All this has led to a better case-finding and therefore a higher MG prevalence. Studies using well-organized national patient registries and with a detailed examination of defined cohorts are expected to find more cases than previous and old reports from single or multiple hospital charts only. Finally, population demographics influence MG prevalence. Especially in Western countries, the ageing of the population leads to a higher prevalence of MG since MG has the highest incidence in the older age groups (Fig. 1).

Illustration of the relative prevalence of the various MG subgroups in European and North American populations. The size of the seronegative group without detectable muscle antibodies depends on the sensitivity of the assays used
MG incidence has similarly been reported higher in recent years than previously, and also when adjusted for population demographics. Improved case-finding may explain this increase. A modest and real increase in MG in elderly people has been suggested but not proven. There are no known reasons for a potential increase in MG incidence. The incidence of thymomas has not increased, but again case-finding is better, this being due to more widespread use of thoracal CT or MR. Many thymomas are detected as a coincidence at such examinations, and some few patients turn out to have a mild undiagnosed MG with AChR antibodies.
MuSK MG has a particular geographic pattern. It has a much higher prevalence in the Mediterranean area than in the Scandinavian countries, and with a clear tendency for a south-north divide [13]. However, in China the south-north divide seems to be inverse with the highest frequency in the north [14]. The geographical difference is for a large part, or entirely, explained by genetic population differences, especially HLA gene variation.
Any occurrence of MG clusters in location and time should help in finding etiologic MG factors. No such clusters have been reported in epidemiological studies. Migration studies would help in differentiating between hereditary and environmental factors causing MG. However, MG is a rare disease and good studies with sufficient statistical power are lacking. Studies support the genetic influence, whereas no new potential environmental factors have been put forward [15]. Best estimates have hypothesized that environmental and genetic factors might be equally important in causing MG [16].
LEMS is much rarer than MG. It has been reported with a prevalence of 2–3 per million that is fifty times less common than MG [17]. The annual incidence was 0.5 per million, which was fourteen times less than MG. Approximately one half of new patients with LEMS have a small-cell lung carcinoma. These patients have a poor prognosis for survival, which explains the discrepancy between prevalence and incidence figures when LEMS and MG are compared [18]. LEMS occurs in 0.5–3% of all patients with small cell lung carcinoma and is probably not always recognized as a distinct comorbidity in these patients. The lowest incidence figures reflect what is observed in clinical practice, whereas the highest occur in prospective studies with clinical, neurophysiological and immunological follow-up of all patients. Younger patients with small cell lung carcinoma are more prone to develop LEMS than the older ones, a ten-year age difference in patients with and without LEMS [17, 19]. LEMS can occur in all age groups, but very rarely in children. Mean age at debut in a European cohort was 58 years, definitely higher than for MG. LEMS without carcinoma is equally common in males and females, whereas LEMS with small-cell lung carcinoma reflects smoking habits in the population.
Neuromyotonia is a very rare disease, much rarer than LEMS. No reliable epidemiological data exist, only small series of single patients. In up to one-third of patients, neuromyotonia co-exist with a thymoma and is paraneoplastic.
Clinical Manifestations
MG is characterized by muscle weakness. This muscle weakness is similar for all MG subgroups. Typical for MG is variation over time. The muscles are often strong in the morning and before being used. The weakness increases after repetitions and sustained use, so that fatigue is common. Patients experience this as a chronic muscle weakness, with variation over time, and with a reduced ability for physically demanding tasks. Symptoms can be matched by weakness measured by formal testing [20], but such testing is not always feasible.
The muscle weakness in MG is localized to some but not all muscles and muscle groups. It is confined to skeletal muscle. Most MG patients experience a distinct weakness in extraocular muscles. This leads to two symptoms; diplopia and ptosis. These manifestations can be observed by clinical examination. The ocular muscle weakness is often markedly asymmetrical, with ptosis on one eye only, and divergent eye movements. The asymmetry makes the clinical diagnosis easier. Eye muscle weakness is often a debut symptom of MG. In 15% of patients, the eye muscle weakness persists as the only MG symptom and sign [8]. In 90% of MG patients with eye symptoms only after 2 years, the disease will remain as a pure ocular MG.
Most MG patients have a more generalized weakness. Difficulties with swallowing and chewing and a weak voice are typical (“bulbar symptoms”). Neck and shoulder muscles are often weak, and problems with lifting the arms above the head are common. Trunk muscles are often weak, whereas muscles distally in the extremities, in hands, fingers and feet, usually have a normal strength. Apart from the eye muscles, the weakness is usually symmetrical. Variation over time is the same for all muscle groups.
Respiratory muscle weakness is the life-threatening symptom of MG. The diaphragm is usually not involved in MG. However, this can occur, especially during infections or after other triggering events such as surgery with narcosis. Aspiration due to weak swallowing, infection and respiratory muscle weakness is a feared combination. MG crisis with the need of respiratory support is rare, but a significant proportion of patients experience it, even in a well-treated cohort. Unexplained need for respiratory support, for example, during a pneumonia, can be a manifestation of an undiagnosed MG.
MG patients do not develop muscle atrophy. They do not experience muscle pain. They have no weakness in smooth muscle, and usually no cardiac muscle abnormalities.
MG muscle weakness is always reversible. Even if the paresis has lasted for a long time, one should not give up, but continue and intensify the immunosuppressive treatment to induce an improvement. This is especially important during a myasthenic crisis. Respiratory support should be maintained long term if necessary, and the weakness will improve with optimal treatment.
The clinical manifestations for early-onset MG and late-onset MG with AChR antibodies are similar. Early-onset patients tend to have a milder disease and with a better response to therapy [4, 21]. Juvenile MG with debut age below 15 years is rare in Western countries, and has the same manifestations as early onset MG in general [22]. However, the subgroup with MG onset before age 7 in China and other Far East countries usually have a mild disease, often with ocular manifestations only and with a good prognosis [11].
Thymoma MG constitutes 10% of all MG patients. They tend to have a more severe MG, and hardly ever with a spontaneous remission. Thymoma can in the same patient be associated with other rare autoimmune manifestations including neuromyotonia and the POEMS syndrome [23].
MG with MuSK antibodies has usually pronounced weakness in facial and bulbar muscles. The patients tend to have a more severe disease, with insufficient response to symptomatic treatment and with the need for long-term immunosuppression [13]. MuSK MG sometimes leads to modest muscle atrophy. This disease also tends to have less variation in muscle strength during the day. Limb weakness is uncommon, and some patients do not have any symptoms from eye muscles. Respiratory weakness can occur.
MG with LRP4 antibodies is rare, and appears even rarer because most centres do not test for this antibody. The clinical manifestations are usually mild, often with ocular symptoms being the most prominent [24].
The seronegative MG group is highly heterogeneous. We only include patients with generalized symptoms in this group, as MG patients with pure ocular symptoms and no antibodies should be categorized as ocular MG. One-third to one half of ocular MG patients do not have antibodies by standard tests. The seronegative, generalized patients include several with antibodies against AChR, MuSK or LRP4 when tested with assays that are more sensitive [7, 25]. The clinical manifestations in these patients are similar to those with detectable antibodies in routine tests, although as a group somewhat milder. Atypical clinical manifestations and no detection of muscle antibodies should always lead to a critical re-examination of the MG diagnosis.
Ocular MG is characterized by ptosis and diplopia, often intermittently and with asymmetry. These symptoms occur early, and shortly after debut, ocular MG is common. During the next weeks and months, most patients develop distinct non-ocular manifestations as well. However, if the disease is purely ocular 2 years after onset, it will remain as an ocular MG in 90% of the patients [8].
LEMS has muscle weakness as a hallmark. This weakness is usually most pronounced in the legs, leading to difficulties in walking. The weakness is usually mostly proximal, and up to 80% of the patients experience proximal weakness in both legs and arms [18]. Facial and bulbar muscle weakness is common, as well as eye muscle complaints. Some patients have also distal muscle weakness. There is little variation during the day and no fatigue as in MG. On the contrary, some patients experience an improvement of muscle strength during repetitive activity after an initial weakness. LEMS patients with small cell lung carcinoma tend to have more severe muscle weakness, and often with a gradual progression. LEMS also affects respiratory muscles. Both symptomatic and immunosuppressive treatment has a more variable effect in LEMS compared to MG, and especially so in paraneoplastic LEMS [5, 26]. Absence of tendon reflexes is typical in LEMS. LEMS patients have also autonomic dysfunction. Dry mouth, dry eyes, erectile dysfunction, constipation and reduced sweating represent common symptoms in LEMS. The autonomic symptoms are mild to moderate, and they have less significance for the patients than the muscle weakness.
Neuromyotonia has less distinct muscle weakness, but rather a feeling of fatigue and stiffness in affected muscles. This combines with muscle cramps and muscle twitching, often resembling gross fasciculations. Neuromyotonia implies a reduced capacity for using the muscles, most common being walking difficulties. The symptoms occur most commonly in the legs but can also affect the trunk, arms, face, and neck muscles. A minority of the patients experience mild sensory symptoms [27, 28].
Pathogenesis
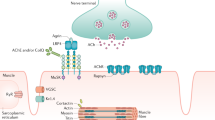
MG, LEMS and neuromyotonia are all caused by antibodies against proteins at the neuromuscular junction (Fig. 2). These antibodies bind in vivo and thereby induce the clinical manifestations of the disorders.

The neuromuscular junction with the key molecules instrumental in the autoimmune disorders MG, LEMS and neuromyotonia. Antibodies against AChR, MuSK and LRP4 postsynaptically and against voltage-gated calcium channels (VGCC) and voltage-gated potassium channels (VGKC) presynaptically cause the muscle weakness and dysfunction
AChR antibodies bind to many epitopes on the extracellular part of the receptor, and to all AChR subunits [3, 16, 29]. There is a major immunogenic region, a predilection site for antibody binding. The IgG antibodies inhibit receptor function by destruction or by blocking for acetylcholine binding. Destruction is more important than blockade and is induced either by cross-linking of AChR or by complement activation. Blockade occurs either directly or through conformational AChR changes. New synthesis of AChR is not inhibited by AChR antibodies and takes place with increased speed in MG. AChR half-life is markedly reduced in MG patients, usually to less than half the normal. This explains the great restorative potential in MG.
MuSK and LRP4 are proteins that functionally and anatomically link to AChR in the postsynaptic membrane. Binding of IgG antibodies to these membrane proteins inhibits their function, and thereby the function of AChR [4, 30, 31]. MuSK antibodies are monovalent so they do not cross-link MuSK molecules, nor do they activate complement. LRP4 antibodies are believed to interfere with the AChR-mediated neuromuscular transmission via an interaction with agrin.
MG patients can have circulating antibodies against other muscle proteins. Whereas AChR, MuSK and LRP4 antibodies very rarely occur together in the same patient, these additional antibodies are present together with AChR antibodies. They are specific or semi-specific for MG. Antibodies against titin are detected in 20–30% of MG patients [16, 32]. In thymoma MG, they appear in nearly 100% of patients; in late-onset MG, they are frequent, whereas they are seen only rarely in other MG subgroups. Ryanodin receptor antibodies are frequent in thymoma MG, rare in late-onset MG, and very rare in the other MG subgroups [33]. Antibodies against titin and ryanodine receptor indicate a more severe disease, with a higher need for long-term immunosuppressive therapy, and in sufficient doses [32]. These antibodies are directed against intracellular antigens, and it is not known if they bind in vivo or if they are merely biomarkers. Antibodies against the membrane molecule agrin have been detected in some MG patients, and in patients both without and with other antibodies [34]. No pathogenetic role has yet been defined. Antibodies against the voltage-gated K+-channel Kv1.4 in skeletal muscle are seen in many AChR MG patients. In Japanese patients, they reflect a more severe disease and often with cardiac complications [35]. This was not found in a North-European cohort [36]. Any pathogenic effect of these antibodies remains to be proven.
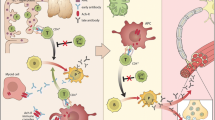
Thymus plays a pathogenic role in some but not all MG patients (Fig. 3). This is most obvious in those 10% of MG patients with a thymoma. One-third of all patients with a thymoma develop MG, and even more have AChR antibodies. Thymoma cells express muscle-like antigens, and they are able to present these antigens for developing thymocytes [37]. T lymphocytes that are capable of inducing antibody production against AChR and other muscle antigens are exported from the thymus with a thymoma [38]. The antibodies are produced in plasma cells/B lymphocytes in activated lymphoid tissue throughout the body. MG with a thymoma is therefore a true paraneoplastic disease. Early-onset MG patients have typically thymus hyperplasia. Thymus is enlarged, and it has a high number of lymphoid follicles. This thymus exports AChR-antibody-inducing T lymphocytes. T cells with this reactivity have been stimulated inside thymus, and they have escaped the normal intrathymic mechanisms to control autoimmunity. Myoid muscle-like cells and epithelial antigen-presenting cells probably both play a role in this AChR sensitization [16, 38]. Most late-onset MG patients with AChR antibodies and some of those with early onset have what appears as a normal thymus. In some of these, pathological biomarkers similar to those of the hyperplastic thymus can be found, and the pathogenesis is probably the same. However, in most such patients, no pathology has been identified [39]. Thus, it is questionable if thymus represents a pathogenic element in all MG patients with AChR antibodies. Ocular MG can have thymus hyperplasia, and this means an increased risk for generalization of symptoms. It is not known what triggers the immunization against AChR in the hyperplastic thymus. Virus infection has been proposed as a potential factor. Although Epstein-Barr virus was claimed to appear in MG thymus some years ago [40], no signs of infection causing MG have been convincingly shown [41]. It is therefore completely unknown why some individuals start to develop thymic hyperplasia, with MG as the consequence. MuSK MG and LRP4 MG do not have any thymus pathology.

MG with AChR antibodies has a pathogenesis that involves the neuromuscular junction, thymus, genetic predisposing factors and unknown triggering or causative factors. In thymoma MG, the thymic tumour represents this causative event
Genetic factors are important in the development of MG. First-degree relatives have a 10–100 times increased risk [42]. Three to seven per cent of MG patients have a first- or second-degree relative with MG [43, 44]. Specific HLA alleles correlate to early-onset MG, late-onset MG, thymoma MG and MuSK MG [45]. Additional genes regulating immune processes increase or decrease the risk for MG [46]. In nearly all such studies, the MG patients have not been defined by subgroup. Some of the risk genes are common for several autoimmune disorders and not specific for MG, particularly in the early-onset MG subgroup [47]. The genetics seem to account for less than fifty per cent of the MG risk.
Neither epidemiological, clinical nor experimental studies have succeeded to identify the external factors that lead to MG. The geographical variation can be explained by genetic influence, and good migration studies are lacking. Those undertaken have failed to come up with potential external factors.
MuSK MG represents a separate disease, with separate genetic and non-genetic causative factors [25]. Thymus is not involved. HLA and non-HLA gene susceptibility is specific for MuSK MG, but explains only a modest part of the total disease risk, similar to the other MG subgroups.
LEMS is caused by antibodies to voltage-gated calcium channels in the presynaptic nerve terminal. These channels are located in the cell membrane, where their calcium transport is necessary for the release of acetylcholine after receiving the triggering nerve signal. The antibodies reduce the number of active channels, they block channel activity, and the calcium influx into the cell is reduced. The consequence is that the quantal release of acetylcholine becomes lower than it should have been [48, 49]. Most antibodies bind to the alpha-1 channel subunit, but the exact pattern of epitope reactivity varies between patients. Voltage-gated calcium channel antibodies are found in at least 85% of all LEMS patients [18]. Whether all the remaining patients have undetectable antibodies against the same channel or there are alternative disease mechanisms is not known.
Small-cell lung carcinoma represents a trigger for the autoantibody production in LEMS through molecular mimicry. Structures antigenically very similar to normal voltage-gated calcium channels appear as tumour-related neoantigens in small-cell lung carcinoma. LEMS usually starts early in tumour development. Most patients with small-cell lung carcinoma and the relevant neoantigens do not develop LEMS. However, some of them have the antibodies without any symptoms. LEMS can rarely be a paraneoplastic manifestation of other cancers [18]. No triggers have been identified for LEMS patients without a cancer. These patients do not have an increased risk for malignancies. LEMS patients without cancer have a proven genetic disposition shown by a linkage to specific HLA-patterns [50]. This is similar to other autoimmune disorders. LEMS patients with small-cell lung carcinoma do not have this pattern, reflecting the difference in etiology. The reason why some but not all patients with small-cell lung carcinoma develop LEMS is unknown. Some differences in the tumours with and without LEMS have been found, but non-tumour aspects are probably more important [19].
Neuromyotonia is caused by antibodies to voltage-gated potassium channels in the presynaptic nerve terminal at the neuromuscular junction, or by antibodies to the channel complex proteins. These antibodies bind to extracellular parts of the channels in vivo and reduce the ionic transport through the channels [51]. There seems to be a correlation between antibody concentration, channel function, and symptom severity [52]. The reduced potassium transport across the neuronal membrane leads to a hyperexcitability. Thymoma is found in 20% of patients with neuromyotonia, and also other neoplasms are associated with neuromyotonia. Antibodies generated against tumour antigens cross-react with the neuronal voltage-gated potassium channels. The same antibodies can bind in the central nervous system and give an autoimmune encephalitis. Some patients may have concurrent autoimmune paraneoplastic manifestations due to a spectrum of autoantibodies [6, 53]. The majority of neuromytonia patients do not have a paraneoplastic condition. The cause of the disease in these patients is not known.
Diagnosis
MG can in most patients be diagnosed clinically. The clue is to consider the disease when relevant. This means to evaluate the possibility in all patients with diplopia, with ptosis, and with otherwise unexplained muscle weakness. In elderly patients, stroke is a common diagnosis at referral, whereas young patients are sometimes believed to have unspecific fatigue conditions. Clinical testing can be normal. One should examine strength in the symptomatic muscles after exercise, for example, as a ptosis test or after continued arm elevation.
AChR antibody testing has a diagnostic sensitivity of 75% and a specificity of near 100% for the best commercial tests [2, 3]. Thus, it is well suited as a screening test, recommended in all patients with a suspicion of MG. The lack of false-positive results is a huge advantage. MuSK antibodies should be tested in samples without AChR antibodies and where MG is still suspected. Sensitivity and specificity for the best commercial MuSK antibody tests are similar to those for AChR antibodies [25]. There are not yet any commercial assays for LRP4 antibodies, so such testing is done mostly for research [24]. More sensitive assays have been developed both for AChR and MuSK antibodies [7]. This shows that a proportion of patients regarded as seronegative indeed belong to one of the other MG subgroups. It is not yet sufficiently clear if these sensitive assays have the same disease specificity. They are not yet commercially available. With a strong suspicion of MG and negative tests, retesting should be done after 6–12 months.
AChR and MuSK antibody concentrations do not reflect MG severity. Some patients with mild disease and a good prognosis have high titres, and patients with low antibody concentrations can have severe MG. There is a tendency for antibody concentrations to fluctuate in parallel with disease development in the same patient [54]. Therefore, repeated AChR and MuSK antibody measurements can be helpful when considering adjustments in ongoing immunosuppressive therapy, and also when considering if a deterioration in function is due to MG or comorbidity.
Titin antibodies are a sensitive marker for thymoma, but with low specificity [32, 55]. Combined with imaging of the mediastinum, it gives an optimal test result. Presence of titin antibodies makes early-onset MG with thymic hyperplasia unlikely. Titin antibodies also indicate a more severe MG, with a long-term need for immunosuppressive treatment. Ryanodine receptor antibodies are in a similar way associated with thymoma, and with a higher specificity, but is not available as a commercial kit.
Imaging of the mediastinum should be performed in all MG patients. It is important to identify the thymoma that is present in 10% of the patients. Both sensitivity and specificity are far from 100%. CT and MR seem to be similar. However, new MR protocols are in development [56, 57]. This should improve thymoma diagnostics, and also lead to a more reliable diagnosis of thymic hyperplasia by imaging. Standard imaging often reveals only an enlarged thymus that could be due to hyperplasia, neoplasia or represent a normal variant. Specialized pathological examination of the removed thymus is important, and for both microtumours, lymphoid follicles and other hyperplasia markers [38]. In most patients with late-onset MG, histological examination of the thymus does not reveal any pathology [39].
Neurophysiological tests can be used to diagnose MG. Repetitive nerve stimulation has a suboptimal sensitivity but a good specificity. Single-fibre EMG has a higher sensitivity but lower specificity. These tests are important diagnostic tools in patients where antibodies cannot be detected or where such tests are unavailable [4]. In patients who already have a clinical and antibody diagnosis, neurophysiological tests are usually unnecessary to perform. However, in MG with purely ocular symptoms, it may be of interest to examine if there are electrophysiological signs of generalization. The selection of muscles for testing is always crucial.
Comorbidity risks should be evaluated both at time of diagnosis and during follow-up. Relevant tests should be performed [58, 59]. The same is true for potential side effects of MG treatment.
LEMS and neuromyotonia are diagnosed based on clinical suspicion, positive tests for the relevant antibody, and typical results at specific neurophysiological tests. Both voltage-gated calcium channel and voltage-gated potassium channel antibody test assays have very high specificity and high sensitivity. Repetitive nerve stimulation at the optimal frequency shows a diagnostic increment in LEMS, reflecting improved channel function and increased release of acetylcholine after multiple stimulations.
Once LEMS or neuromyotonia have been diagnosed, one should search for a small-cell lung carcinoma (LEMS), a thymoma (neuromyotonia) or another cancer (both disorders). Smokers and non-smokers should follow the same examination program, although the risk for lung cancer differs markedly. PET examination should be included if a tumour has not already been detected. In LEMS without a detected small-cell carcinoma at diagnosis, one should follow-up with PET or other sensitive techniques every 6 months for the next 2 years [19].
Treatment
MG responds to symptomatic therapy and to immunosuppression [2, 4, 60, 61] (Table 2). Acetylcholine esterase inhibition leads to symptom relief as long as the drug is active. Pyridostigmine is the favoured drug. Ambenonium chloride and neostigmine are usually less effective but represent alternatives. 3,4 diaminopyridine increases the amount of acetylcholine in the synapse by increasing its presynaptic release. This drug has little or no effect in most MG patients. Pyridostigmine should be given as first-choice drug to all MG subgroups. However, patients with MuSK MG have usually a limited effect of cholinergic treatment [13]. The optimal dose is decided from effect and cholinergic side effects. These are most commonly gastrointestinal, but also from other parts of the autonomic nerve system. Dose can vary from day to day, reflecting variation in patient needs and tasks. Patients can self-administer their optimal dose, regarding both single dose and dose frequency. Some patients become symptom free on pyridostigmine and do not require further drug therapy.
Most MG patients should be treated with immunosuppressive drugs. The combination of prednisolone and azathioprine is regarded as first-choice immunosuppressive treatment. Prednisolone dose should be increased gradually over a few weeks. After obtaining a remission, prednisolone dose should be gradually reduced. It is usually wise to keep a small dose long term, even if a remission seems stable. Prednisolone as MG treatment is given by many centres every second day. This gives a satisfactory effect and may reduce the side effects. Azathioprine takes some months before a clinical effect appears. This drug represents long-term treatment. Most patients tolerate azathioprine without any side effects. Patients can be tested for thiopurine methyl transferase activity before treatment. This is low in 10% of the population, which increases the risk for intolerance to azathioprine. The main reason for immunosuppressive treatment is to control present symptoms. An additional indication should be to prevent deterioration and the development of a more severe MG. This has especially been discussed for ocular MG, if early treatment with prednisolone and azathioprine can prevent generalization [8]. Data indicate that this can be true for some patients.
If the first-choice immunosuppressive drugs fail, there are several options. Failure can be due to lack of effect or side effects. One should be ambitious in the immunosuppressive MG treatment, not accepting symptoms of functional significance or side effects influencing quality of life. Often, second-line immunosuppressive drugs are combined with prednisolone or azathioprine.
Rituximab is recommended as an effective drug in MG. It binds selectively to the CD20 antigen on B lymphocytes and should therefore be well suited for antibody-mediated diseases such as MG. No controlled trials have so far been published, but widespread experience from series of patients with moderate and severe MG shows a therapeutic effect [62]. The optimal treatment schedule has not been defined, but most centres use the same induction schedule as for rheumatic disease and multiple sclerosis. Follow-up treatment depends on the clinical MG development. JC virus-related progressive multifocal leukoencephalopathy is a very rare side effect with rituximab, occurring in perhaps 1 in 30,000 patients [63]. There is no need to check for JC virus before starting with rituximab.
Mycophenolate mofetil is often used for mild to moderate MG. Clinical experience favours the use of this drug, together with uncontrolled study reports. However, two prospective and controlled studies failed to reach the primary end points [64, 65]. This could be due to weakness of the studies, but indicates that this drug is not very potent in MG. Alternative second-line immunosuppressive drugs for MG include methotrexate, cyclosporine, tacrolimus and cyclophosphamide. Neither MG subgroup nor any other MG biomarkers favour one of these immunosuppressive drugs more specifically. However, rituximab seems to be particularly well suited for MuSK MG treatment [66].
Thymectomy should be undertaken early in the course of MG. Patients with early-onset MG have a well-proven effect on MG disease development that comes early and increases during several months after surgery. Thymoma patients should have their thymus removed together with the tumour. It is crucial that the surgeon removes all thymus tissue. This can be done by thoracoscopic, minimally invasive techniques or by traditional sternotomy. The key factor is access and visibility to the mediastinum so that all thymus tissue can be identified and removed. It is not always easy to decide whether a patient should be thymectomized. Patients with generalized MG debut before age 50 and AChR antibodies should definitely have surgery [67]. The same is true for older patients with an enlarged thymus at imaging, being suspected of thymic hyperplasia or even a thymoma. Patients up to the age 60–65 with a normal imaging result are also sometimes thymectomized, but probably not if they have titin antibodies as an indicator of late-onset MG. Patients without any detectable antibodies represent a challenge as we know that some of them in fact have AChR antibodies and thymic hyperplasia. For this group, we recommend specialized imaging of the mediastinum and sensitive antibody tests. For ocular MG, a benefit of thymectomy has not been proven [8]. However, in the presence of AChR antibodies, an enlarged thymus on imaging and neurophysiological signs of generalization, we recommend thymectomy. Even with negative imaging and a pure ocular disease after extensive tests, there are data showing a reduced risk of MG generalization after thymectomy [8]. Thymectomy should not be done in patients with MuSK or LRP4 antibodies, and not in the oldest patients.
Many therapeutic monoclonal antibodies have immunosuppressive actions. Several of them influence autoantibodies: their production, transport and binding, as well as consequences of their binding to the antigen. They might well have a benefit in MG, but the great majority have not been tested properly. Ocrelizumab is a humanized anti-CD20 monoclonal antibody and ofatumumab is a fully humanized antibody against the same antigen. These drugs should be at least as good as rituximab for MG, are very much more expensive, and have not yet been tested. Eculizumab is a humanized monoclonal antibody against the terminal complement protein C5. This drug has a proven but moderate effect in MG [68, 69]. Cost-benefit considerations make it prohibitive for MG patients now as it is extremely expensive, but in the future complement will probably be a target for immunotherapy in MG.
Intravenous immunoglobulin (IvIg) is a well-proven treatment for MG. The effect appears after a few days and is often remarkable. It lasts for approximately 3 months. IvIg is the treatment of choice for MG exacerbations, for severe MG periods, and before surgery or other challenges that could deteriorate their MG. IvIg (or alternatively plasma exchange) should always be given in myasthenic crisis when the patients have a need for respiratory support. The response rate is around 80% [70]. Long-term treatment with IvIg is unusual, but remains an alternative in patients responding well to the other immunosuppressive treatments. IvIg treatment should be combined with immunosuppressive drugs, often in a higher dose than before, or in a combination with new and more potent drugs. IgG can be given subcutaneously. This treatment has not been tested systematically in MG, but it may be an alternative for medium- to long-term treatment [71]. There are ongoing trials using modified IgG molecules or IgG-modifying agents as long-term MG treatment [72].
Plasma exchange has the same indications as IvIg in MG treatment. The therapeutic effect is similar, and is well proven. The frequency of side effects is also similar, but the risk for severe side effects may be higher for plasma exchange. The choice between plasma exchange and IvIg usually depends on local availability, experience and organization. In some patients, one of the treatments is clearly superior. This means that both IvIg and plasma exchange should be available at centres treating patients with severe MG.
For myasthenic crisis, respiratory support and intensive care are crucial. Any infections precipitating or complicating the crisis should be treated vigorously. The patients should be mobilized as soon as possible. A myasthenic crisis is always reversible.
Patients with MG should have a daily physical exercise program. Exercise improves muscle strength also in MG patients. The program should be adapted to their disease, regarding intensity, duration and variation in strength between muscle groups [73]. The exercise program should be combined with sufficient rest. Overweight should be avoided.
MG patients with persisting diplopia and ptosis may benefit from assistive devices, or even local surgery [8]. Most patients should continue to work full time, although physically demanding occupations should be avoided. MG patients tolerate most drugs. However, both patient and doctor should be aware of the possibility of a drug-induced MG exacerbation when initiating a new drug treatment. Muscle relaxants, penicillamine, fluoroquinolones, macrolides and aminoglycosides should be avoided in MG. Statins should be initiated at the same indications with and without MG, but if MG aggravates or is unmasked, the statins should be withdrawn.
LEMS treatment includes symptomatic and immunosuppressive drugs [5, 26]. 3,4 diaminopyridine is the drug preferred to facilitate the cholinergic transmission. Most patients experience a marked and long-lasting effect of such treatment. The effect is better in patients without a small-cell lung carcinoma. Pyridostigmine usually has less effect, adds nothing but side effects in combination with 3,4 diaminopyridine, but can be tried as an alternative in LEMS patients with an inadequate response to 3,4 diaminopyridine. Most LEMS patients need also immunosuppressive drugs. There are no controlled studies, so treatment guidelines rely mostly on clinical practice. The drugs used are the same as for MG: with prednisolone and azathioprine as the first choice, rituximab and mycophenolate mofetil as second choices and several other drugs with an expected effect. IvIg and plasma exchange can be used as in MG but have a usually only a moderate effect. Treatment of the cancer is essential in those with small-cell lung carcinoma. Effective cancer treatment will sometimes improve also the LEMS.
Neuromyotonia treatment includes symptomatic and immunosuppressive measures [28, 52]. Antiepileptic drugs and botulinum toxin can improve muscle stiffness, spasms and pain. The immunosuppressive drugs to be tried are the same as for MG, but experience is limited due to the rarity of this condition. Potential treatment includes IvIg or plasma exchange for exacerbations, severe disease and critical situations.
MG, LEMS and neuromyotonia patients all need optimal treatment of any comorbid conditions. It is important to identify such conditions and to separate them from the neuromuscular disease [58, 59]. Especially in elderly patients, this can be difficult. Specialists tend to care and take responsibility only for one condition. That is a challenge for the patient and even pose a threat for the total care. The neurologist should take responsibility as others usually do not dare to interfere with the treatment for these rare neuromuscular conditions. Cardiovascular disease and respiratory disease are highly relevant, and many patients have additional autoimmune disorders. Insomnia and mild anxiety are common, as in the general population [74].
MG females in reproductive age should get specific information about pregnancy and giving birth [75, 76]. Pyridostigmine, prednisolone and azathioprine are regarded as safe during pregnancy and should be continued if they are needed for MG. IvIg and plasma exchange are also safe and represent effective treatment for exacerbations during pregnancy. Methotrexate, mycophenolate mofetil and cyclophosphamide are teratogenic, whereas rituximab should not be given during the last 6 months before conception because risk of B-cell depletion in the baby. Most patients with MG give birth in an ordinary way, but the percentage with caesarean section is somewhat higher than in women without MG [77]. Neonatal myasthenia due to transfer across placenta of mother’s IgG antibodies occurs in 10–15% of the newborn babies. This can occur for both AChR and MuSK MG, and for LEMS. Neonatal myasthenia lasts for days or a few weeks, until mother’s antibodies disappear. The baby does not produce any muscle antibodies. The risk for neonatal myasthenia means that all females with MG shall give birth at institutions with experience in intensive neonatal care including respiratory support. AChR antibodies induce in rare cases permanent changes in the developing child in utero [78]. Such persistent myopathy can be mild but also severe and with arthrogryposis. This is so uncommon that MG women should be supported in their wish to have children. Breastfeeding is recommended, except in the rare cases where the mother is treated with methotrexate, mycophenolate mofetil or cyclophosphamide.
References
Beeson D. Congenital myasthenic syndromes: recent advances. Curr Opin Neurol. 2016;29:565–71.
Gilhus NE. Myasthenia Gravis. N Engl J Med. 2016;375:2570–81.
Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis – autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12:259–U291.
Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14:1023–36.
Titulaer MJ, Lang B, Verschuuren J. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098–107.
Lang B, Makuch M, Moloney T, et al. Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies. J Neurol Neurosurg Psychiatry. 2017;88:353–61.
Hong Y, Zisimopoulou P, Trakas N, et al. Multiple antibody detection in ’seronegative’ myasthenia gravis patients. Eur J Neurol. 2017;24:844–50.
Kerty E, Elsais A, Argov Z, Evoli A, Gilhus NE. EFNS/ENS Guidelines for the treatment of ocular myasthenia. Eur J Neurol. 2014;21:687–93.
Heldal AT, Owe JF, Gilhus NE, Romi F. SEROPOSITIVE MYASTHENIA GRAVIS: A NATIONWIDE EPIDEMIOLOGIC STUDY. Neurology. 2009;73:150–1.
Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010;10:46.
Hong Y, Skeie GO, Zisimopoulou P, et al. Juvenile-onset myasthenia gravis: autoantibody status, clinical characteristics and genetic polymorphisms. J Neurol. 2017;264:955–62.
Pedersen EG, Hallas J, Hansen K, Jensen PEH, Gaist D. Late-onset myasthenia not on the increase: a nationwide register study in Denmark, 1996-2009. Eur J Neurol. 2013;20:309–14.
Guptill JT, Sanders DB, Evoli A. ANTI-MuSK Antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. 2011;44:36–40.
Hong Y, Li HF, Skeie GO, et al. Autoantibody profile and clinical characteristics in a cohort of Chinese adult myasthenia gravis patients. J Neuroimmunol. 2016;298:51–7.
Boldingh MI, Maniaol A, Brunborg C, et al. Prevalence and clinical aspects of immigrants with myasthenia gravis in northern europe. Muscle Nerve. 2017;55:819–27.
Romi F, Hong Y, Gilhus NE. Pathophysiology and immunological profile of myasthenia gravis and its subgroups. Curr Opin Immunol. 2017;49:9–13.
Wirtz PW, Nijnuis MG, Sotodeh M, et al. The epidemiology of myasthenia gravis, Lambert-Eaton myasthenic syndrome and their associated tumours in the northern part of the province of South Holland. J Neurol. 2003;250:698–701.
Titulaer MJ, Wirtz PW, Kuks JBM, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: A clinical picture in 97 patients. J Neuroimmunol. 2008;201:153–8.
Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association Prediction Score Accurately Predicts Small-Cell Lung Cancer in the LEMS. J Clin Oncol. 2011;29:902–8.
Vinge L, Andersen H. Muscle strength and fatigue in newly diagnosed patients with myasthenia gravis. Muscle Nerve. 2016;54:709–14.
Andersen JB, Gilhus NE, Sanders DB. Factors affecting outcome in myasthenia gravis. Muscle Nerve. 2016;54:1041–9.
Popperud TH, Boldingh MI, Rasmussen M, Kerty E. Juvenile myasthenia gravis in Norway: Clinical characteristics, treatment, and long-term outcome in a nationwide population-based cohort. Eur J Paediatr Neurol. 2017;21:707–14.
Antoine JC, Camdessanche JP. Paraneoplastic neuropathies. Curr Opin Neurol. 2017;30:513–20.
Zisimopoulou P, Evangelakou P, Tzartos J, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun. 2014;52:139–45.
Tsonis AI, Zisimopoulou P, Lazaridis K, et al. MuSK autoantibodies in myasthenia gravis detected by cell based assay - A multinational study. J Neuroimmunol. 2015;284:10–7.
Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database of Syst Rev. 2011.
Kucukali CI, Kurtuncu M, Akcay HI, Tuzun E, Oge AE. Peripheral nerve hyperexcitability syndromes. Rev Neurosci. 2015;26:239–51.
Maddison P. Neuromyotonia. Clin Neurophysiol. 2006;117:2118–27.
Binks S, Vincent A, Palace J. Myasthenia gravis: a clinical-immunological update. J Neurol. 2016;263:826–34.
Koneczny I, Stevens JAA, De Rosa A, et al. IgG4 autoantibodies against muscle-specific kinase undergo Fab-arm exchange in myasthenia gravis patients. J Autoimmun. 2017;77:104–15.
Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. 2012;69:445–51.
Romi F, Skeie GO, Gilhus NE, Aarli JA. Striational antibodies in myasthenia gravis - Reactivity and possible clinical significance. Arch Neurol. 2005;62:442–6.
Skeie GO, Mygland A, Treves S, Gilhus NE, Aarli JA, Zorzato F. Ryanodine receptor antibodies in myasthenia gravis: Epitope mapping and effect on calcium release in vitro. Muscle Nerve. 2003;27:81–9.
Gasperi C, Melms A, Schoser B, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology. 2014;82:1976–83.
Suzuki S, Baba A, Kaida K, et al. Cardiac involvements in myasthenia gravis associated with anti-Kv1.4 antibodies. Eur J Neurol. 2014;21:223–30.
Romi F, Suzuki S, Suzuki N, Petzold A, Plant GT, Gilhus NE. Anti-voltage-gated potassium channel Kv1.4 antibodies in myasthenia gravis. J Neurol. 2012;259:1312–6.
Gilhus NE, Willcox N, Harcourt G, et al. Antigen presentation by thymoma epithelial-cells from myasthenia-gravis patients to potentially pathogenic t-cells. J Neuroimmunol. 1995;56:65–76.
Marx A, Pfister F, Schalke B, Saruhan-Direskeneli G, Melms A, Strobel P. The different roles of the thymus in the pathogenesis of the various myasthenia gravis subtypes. Autoimmun Rev. 2013;12:875–84.
Myking AO, Skeie GO, Varhaug JE, Andersen KS, Gilhus NE, Aarli JA. The histomorphology of the thymus in late onset, non-thymoma myasthenia gravis. Eur J Neurol. 1998;5:401–5.
Cavalcante P, Serafini B, Rosicarelli B, et al. Epstein-Barr Virus persistence and reactivation in myasthenia gravis thymus. Ann Neurol. 2010;67:726–38.
Gilhus NE. Myasthenia and the neuromuscular junction. Curr Opin Neurol. 2012;25:523–9.
Avidan N, Le Panse R, Berrih-Aknin S, Miller A. Genetic basis of myasthenia gravis - A comprehensive review. J Autoimmun. 2014;52:146–53.
Pirskanen R. Genetic aspects in myasthenia-gravis - family study of 264 finnish patients. Acta Neurol Scand. 1977;56:365–88.
Salvado M, Canela M, Maria J, et al. Study of the prevalence of familial autoimmune myasthenia gravis in a Spanish cohort. J Neurol Sci. 2016;360:110–4.
Lisak RP, Barcellos L. New Insights Into the Genetics of Autoimmune Myasthenia Gravis An Evolving Story. JAMA Neurol. 2015;72:386–7.
Renton AE, Pliner HA, Provenzano C, et al. A Genome-Wide Association Study of Myasthenia Gravis. JAMA Neurol. 2015;72:396–404.
Bach JF. The etiology of autoimmune diseases: the case of myasthenia gravis. In: Wolfe GI, Meriggioli MN, Ciafaloni E, Ruff RL, editors. Myasthenia Gravis and Related Disorders I. Boston: Wiley Periodicals; 2012. p. 33–9.
Verschuuren J, Strijbos E, Vincent A. Neuromuscular junction disorders. Handbook of clinical neurology. Amsterdam: Elsevier; 2016;133:447–466..
Takamori M. Lambert-Eaton myasthenic syndrome: Search for alternative autoimmune targets and possible compensatory mechanisms based on presynaptic calcium homeostasis. J Neuroimmunol. 2008;201:145–52.
Titulaer MJ, Verschuuren J. Lambert-Eaton myasthenic syndrome - Tumor versus nontumor forms. In: Kaminski HJ, Barohn RJ, editors. Myasthenia gravis and related disorders: 11th international conference; 2008. p. 129–34.
Fleisher J, Richie M, Price R, Scherer S, Dalmau J, Lancaster E. Acquired neuromyotonia heralding recurrent thymoma in myasthenia gravis. JAMA Neurol. 2013;70:1311–4.
Song J, Jing SS, Quan C, et al. Isaacs syndrome with CASPR2 antibody: A series of three cases. J Clin Neurosci. 2017;41:63–6.
Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72:241–55.
Heldal AT, Eide GE, Romi F, Owe JF, Gilhus NE. Repeated acetylcholine receptor antibody-concentrations and association to clinical myasthenia gravis development. PLoS One. 2014;9:e114060.
Stergiou C, Lazaridis K, Zouvelou V, et al. Titin antibodies in "seronegative" myasthenia gravis - A new role for an old antigen. J Neuroimmunol. 2016;292:108–15.
Priola AM, Priola SM, Giraudo MT, et al. Chemical-shift and diffusion-weighted magnetic resonance imaging of thymus in myasthenia gravis usefulness of quantitative assessment. Investig Radiol. 2015;50:228–38.
Priola AM, Priola SM, Gned D, Giraudo MT, Fornari A, Veltri A. Comparison of CT and chemical-shift MRI for differentiating thymoma from non-thymomatous conditions in myasthenia gravis: value of qualitative and quantitative assessment. Clin Radiol. 2016;71:E157–69.
Gilhus NE, Nacu A, Andersen JB, Owe JF. Myasthenia gravis and risks for comorbidity. Eur J Neurol. 2015;22:17–23.
Nacu A, Andersen JB, Lisnic V, Owe JF, Gilhus NE. Complicating autoimmune diseases in myasthenia gravis: a review. Autoimmunity. 2015;48:362–8.
Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology. 2016;87:419–25.
Skeie GO, Apostolski S, Evoli A, et al. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2010;17:893–902.
Iorio R, Damato V, Alboini PE, Evoli A. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol. 2015;262:1115–9.
Randall KL. Rituximab in autoimmune diseases. Aust Prescr. 2016;39:131–4.
Sanders DB, McDermott M, Thornton C, et al. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71:394–9.
Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71:400–6.
Evoli A, Alboini PE, Damato V, et al. Myasthenia gravis with antibodies to MuSK: an update. Ann N Y Acad Sci. 2018;1412:82–9.
Wolfe GIKH, Aban IB, Minisman G, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375:511–22.
Howard JF, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48:76–84.
Gilhus NE. Eculizumab: a treatment option for mysthenia gravis? Lancet Neurol. 2017;16:947–8.
Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database of Systematic Reviews. 2012.
Beecher G, Anderson D, Siddiqi ZA. Subcutaneous immunoglobulin in myasthenia gravis exacerbation A prospective, open-label trial. Neurology. 2017;89:1135–41.
Kiessling P, Lledo-Garcia R, Watanabe S, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci Transl Med. 2017;9:eaan1208.
Rahbek MA, Mikkelsen EE, Overgaard K, Vinge L, Andersen H, Dalgas U. Exercise in myasthenia gravis: a feasibility study of aerobic and resistance training. Muscle Nerve. 2017;56:700–9.
Andersen JB, Owe JF, Engeland A, Gilhus NE. Total drug treatment and comorbidity in myasthenia gravis: a population-based cohort study. Eur J Neurol. 2014;21:948–55.
Hoff JM, Daltveit AK, Gilhus NE. Myasthenia gravis in pregnancy and birth: identifying risk factors, optimising care. Eur J Neurol. 2007;14:38–43.
Norwood F, Dhanjal M, Hill M, et al. Myasthenia in pregnancy: best practice guidelines from a UK multispecialty working group. J Neurol Neurosurg Psychiatry. 2014;85:538–43.
Hoff JM, Daltveit AK, Gilhus NE. Myasthenia gravis - Consequences for pregnancy, delivery, and the newborn. Neurology. 2003;61:1362–6.
Hacohen Y, Jacobson LW, Byrne S, et al. Fetal acetylcholine receptor inactivation syndrome A myopathy due to maternal antibodies. Neurol Neuroimmunol Neuroinflammation. 2015;2:e57.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gilhus, N.E. (2019). Myasthenia Gravis and Other Immune-Mediated Disorders of the Neuromuscular Junction. In: Mitoma, H., Manto, M. (eds) Neuroimmune Diseases. Contemporary Clinical Neuroscience. Springer, Cham. https://doi.org/10.1007/978-3-030-19515-1_26
Download citation
DOI: https://doi.org/10.1007/978-3-030-19515-1_26
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-19514-4
Online ISBN: 978-3-030-19515-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)