
Abstract
The periplasm of Gram-negative bacteria contains a specialized chaperone network that facilitates the transport of unfolded membrane proteins to the outer membrane as its primary functional role. The network, involving the chaperones Skp and SurA as key players and potentially additional chaperones, is indispensable for the survival of the cell. Structural descriptions of the apo forms of these molecular chaperones were initially provided by X-ray crystallography. Subsequently, a combination of experimental biophysical methods including solution NMR spectroscopy provided a detailed understanding of full-length chaperone–client complexes . The data showed that conformational changes and dynamic re-organization of the chaperones upon client binding, as well as client dynamics on the chaperone surface are crucial for function. This chapter gives an overview of the structure-function relationship of the dynamic conformational rearrangements that regulate the functional cycles of the periplasmic molecular chaperones Skp and SurA.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
For Gram-negative bacteria, such as E. coli, around 35% of the proteome is directed to the bacterial cell envelope (Krogh et al. 2001; Orfanoudaki and Economou 2014; De Geyter et al. 2016; Tsirigotaki et al. 2017). The cell envelope is composed of two membranes: the inner membrane and the outer membrane, which are separated by the aqueous periplasm (Van Wielink and Duine 1990). The periplasm is a viscous and oxidizing compartment that contains a thin layer of peptidoglycan playing a structural role as a bacterial cell wall. Periplasmic proteins are synthesized as pre-proteins in the cytoplasm and are subsequently translocated across the inner membrane by either of two secretory pathways, depending on the signal sequence they carry, the Sec machinery and the twin arginine translocation (TAT) system (Wickner et al. 1991; Driessen et al. 2001; Natale et al. 2008; Lycklama et al. 2012; Tsirigotaki et al. 2017).
Thereby, the majority of proteins are processed by the Sec machinery, which translocates its clients in an unfolded conformation. As proteins exit the Sec machinery and enter the periplasm, a complex chaperone network in charge of protein quality control ensures their folding and integrity. The network comprises two main pathways for clients, the protection of protein folding under stress conditions and the transport of client proteins towards the outer membrane. The first pathway includes the chaperones HdeA/HdeB , which are activated by a pH-controlled mechanism that helps to prevent protein aggregation in acidic environment (Hong et al. 2005; Kern et al. 2007; Ding et al. 2015; Zhang et al. 2016; Yu et al. 2017; Salmon et al. 2018; Yu et al. 2018), and the chaperone Spy (Quan et al. 2011; Stull et al. 2016; He et al. 2016). The second pathway, the transport of outer membrane proteins (OMPs) through the periplasm is controlled by the network of two chaperones SurA and Skp (Bitto and McKay 2002; Walton and Sousa 2004; Korndörfer et al. 2004). Additionally, the protease DegP is responsible for the degradation of off-pathway OMPs (Jiang et al. 2008; Clausen et al. 2011; Ge et al. 2014a, b) and has also been shown to display chaperone activity under low temperature conditions (Spiess et al. 1999). Under heat shock conditions, in turn, the chaperone FkpA has been demonstrated to play a key role in OMP biogenesis (Ge et al. 2014a, b). In addition, the biogenesis of OM lipoproteins requires the dedicated chaperone LolA (Tajima et al. 1998; Miyamoto et al. 2001; Taniguchi et al. 2005; Okuda and Tokuda 2009).
Besides these two pathways, the periplasm contains several folding catalysts that are speeding up two slow reaction steps, the cis–trans isomerization of peptidyl-prolyl bonds and the formation of disulfide bonds. Cis–trans isomerization is generally a slow process that can often be rate-limiting for protein folding and that can be accelerated by enzymes with peptidyl-prolyl cis-trans isomerases (PPIase) activity (Liu and Walsh 1990; Bardwell et al. 1991; Hayano et al. 1991; Scholz et al. 1998). Four proteins with PPIase activity have been identified in the E. coli periplasm, FkpA (Arie et al. 2001; Saul et al. 2004; Helbig et al. 2011), PpiA (Liu and Walsh 1990; Hayano et al. 1991), PpiD (Antonoaea et al. 2008; Matern et al. 2010) and SurA (Bitto and McKay 2002), among which SurA and FkpA are the main players. A second slow process in protein folding is the formation of disulfide bonds. In the periplasm, the formation of disulfide bond is catalyzed by the Dsb redox system that comprises the oxidation system DsbA and DsbB and the isomerization system DsbC and DsbD (Joly and Swartz 1997; Messens and Collet 2006; Vertommen et al. 2008; Ito and Inaba 2008; Heras et al. 2009; Denoncin and Collet 2013). In this chapter, we focus on the recent advances of the structural characterization of the chaperones Skp and SurA that transport the OMPs through the periplasm and promote their insertion into the outer membrane by the BAM complex.
The biogenesis of OMPs is a challenging task for the cell, since the proteins have to cross two biophysical barriers to reach the outer membrane: The inner membrane and the aqueous periplasm. On this pathway, the translocation of the OMPs across the inner membrane, from the cytoplasm into the periplasm, is facilitated by the SEC translocase (Driessen et al. 2001; van den Berg et al. 2004; Chatzi et al. 2014; Tsirigotaki et al. 2017). The second biophysical barrier for the insoluble OMPs, the aqueous periplasm, is then overcome by a unique network of molecular chaperones. This network is organized around the chaperones Skp (Seventeen Kilodalton Protein) and SurA (survival factor A), which work on two parallel pathways to target the OMP clients to the β-barrel assembly machinery (BAM) complex, which eventually facilitates their folding and insertion (Fig. 6.1) (Rizzitello et al. 2001; De Geyter et al. 2016).

The periplasmic chaperone network in outer membrane biogenesis. After their synthesis in the cytoplasm, unfolded native OMPs are translocated by the SecYEG machinery in a linear fashion across the inner membrane. Subsequently, they are transported by the chaperones Skp or SurA on parallel pathways to the BAM complex for subsequent insertion into the outer membrane
Understanding the structural biology of these two chaperones is of key scientific interest due to their fundamental role in polypeptide transport and OMP biogenesis. A first milestone has been the determination of the crystal structures of the client-free (apo) forms of SurA and Skp (Bitto and McKay 2002; Walton and Sousa 2004; Korndörfer et al. 2004). These structures provided valuable information about the functional mechanisms of the individual components of the chaperone system in single, individually stabilized states. Subsequently, new developments of biophysical techniques including nuclear magnetic resonance (NMR) spectroscopy allowed the observation of these chaperone systems in aqueous solution and in complex with client proteins, thus providing complete descriptions of the chaperones dynamic functional cycles. These findings have established how structural plasticity allows these chaperones to undergo conformational rearrangements and populate conformational ensemble states that overall regulate their functional cycles. Such conformational flexibility enables the chaperones to adapt to their highly dynamic client proteins as well as to fulfil their roles in a complex interaction network in the periplasm.
The Periplasmic Chaperone Skp
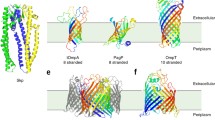
The crystal structure of Skp shows a homo-trimeric assembly where the three subunits are connected by a β-barrel trimerization domain that extends into three α-helical “tentacles” or “arms” that give rise to Skp’s characteristic jelly fish-like shape (Fig. 6.2a, b) (Walton and Sousa 2004; Korndörfer et al. 2004). Each monomer contributes three β-strands to the trimerization interface. Importantly, these strands are however not connected within one protomer subunit but form a small β-barrel across the trimer by connecting β-strands intermolecularly from two adjacent subunits (Fig. 6.2a). Each arm is constituted by two long α-helixes in coiled-coil arrangement, extending from the oligomerization domain and forming a central cavity. The presence of a central cavity bears some resemblance to the well-characterized Hsp60 (GroEL, CTT) chaperone, where a central cavity serves as a protective environment for the client protein (Braig et al. 1995; Xu et al. 1997; Ditzel et al. 1998; Horwich et al. 2007). The overall organization and shape of Skp bears further similarity to the ATP-independent holdase chaperone prefoldin, with the difference that the N and C-termini of Skp are located at the β-barrel trimerization domain, while for prefoldin they are located at the extremity of the arms (Siegert et al. 2000; Martín-Benito et al. 2002; Ohtaki et al. 2008). Just like prefoldin, Skp is classified as an ATP-independent holdase chaperone.

Dynamic adaptation of the periplasmic chaperone Skp and its client protein. a Structure of Skp trimer [PDB code 1SG2 (Korndörfer et al. 2004)] with the individual protomers coloured in green, pink and yellow. The position of phenylalanine 30 is highlighted by red arrows. b Skp amino acid sequence and secondary structure. The β-sheets and α-helixes are represented by grey arrows and rectangular shapes, respectively. Phenylalanine 30 is highlighted as in (a). c Close-up on Skp arms highlighting the pivot element in helices α2 and α3. The flexible helix α2.A (red) is linked by the hinge at phenylalanine 30 to the more rigid helix α2.B (yellow). The helicity of segment Ser89–Arg93 (purple) of helix α3.A (orange) is stabilized upon client binding [adapted from (Burmann et al. 2013)]. d Structural model of a single conformation from the dynamic Skp(blue)–tOmpA (purple) ensemble. The residues highlighted in green correspond to the alanine identified as interacting with the unfolded tOmpA [adapted from (Callon et al. 2014)]. e Model of the multivalent binding of OMPs that are too large to fit in the cavity of one trimeric Skp. The arms are in an open conformation to expand the size of the cavity [adapted from (Holdbrook et al. 2017)]
The protein Skp was functionally identified as a binder of OMPs in a pull-down experiment with unfolded OmpF covalently linked to Sepharose beads (Chen and Henning 1996). OmpF is however not the only client protein of Skp, as shown in a subsequent study where the Skp clientome was mapped by a combined strep-tag affinity and proteomic approach (Jarchow et al. 2008). Overall, more than 30 proteins were identified in the Skp clientome, including the OMPs OmpA and LamB, but also several soluble periplasmic proteins, such as MalE and OppA, demonstrating Skp’s functional importance for a broad spectrum of clients (Table 6.1). The functional relevance of the interaction of Skp with soluble proteins was additionally highlighted by studies showing that Skp chaperone activity can improve the expression yields of recombinant proteins not only in the periplasm, but also in the cytosol when a Skp variant devoid of its signal-sequence is used (Bothmann and Plückthun 1998; Levy et al. 2001).
The complex of an OMP client protein and Skp was initially characterized by NMR spectroscopy and fluorescence spectroscopy (Walton et al. 2009; Qu et al. 2009). A full understanding of the dynamic nature of the complexes at atomic resolution was however only obtained subsequently with a complete description of structure and dynamics of Skp–OmpX and Skp–tOmpA (transmembrane domain of OmpA) complexes determined by solution NMR spectroscopy (Burmann et al. 2013; Callon et al. 2014). The OMP polypeptide was found to adopt a disordered conformation that interacts within Skp’s central cavity by both hydrophobic contacts and interactions between the charged loops of the OMP and charged parts of the cavity. Notably, these works exploited state-of-the-art NMR experiments to determine for the first time the dynamic properties and structure of a chaperone–client complex with a full-length native client protein with atomic resolution. Moreover, these studies demonstrated the conformational flexibility of Skp in its apo and in its client-bound holo state in solution. In the apo form, a hinge element located at the conserved residue phenylalanine 30 allows the Skp arms to explore a large degree of conformational flexibility (Fig. 6.2a–c). Upon OMP binding, the flexibility of the arms is decreased, leading to a stabilization of the cavity around the bound client protein. While bound, the OMP client protein explores a dynamic landscape of disordered states within the cavity of Skp with no polypeptide segment of OMP stably bound to the chaperone. Thereby, the OMP encapsulated in the cavity has a reduced compactness by approximately a factor of two in comparison to the disordered OMP in denaturant solution. The local lifetime of the individual conformations of at most 1 ms is in stark contrast to the long global lifetime of the complex in the range of hours. This discrepancy arises by avidity from a combination of multiple weak local interactions with a short lifetime. A detailed analysis of a combination of distance-based NMR experiments (NOE and PRE) confirmed the contact interface between Skp and the bound OMP in the cavity (Fig. 6.2d).
The observed position of the OMP client protein in the Skp cavity also raised the question by which mechanism Skp adapts to a wide range of different clients ranging in size from 150 residues (OmpX) to more than 700 residues (FhuA). A study combining molecular modelling, small angle X-ray scattering (SAXS) and NMR spectroscopy confirmed that the hinges on the arms permit to dramatically extend the size of the cavity in an ATP-independent manner to adapt to the size of client proteins (Fig. 6.2e) (Holdbrook et al. 2017). A complementary model was established by systematically analysing the binding capacity of a large variety of OMPs with a size range from 8 to 16 strands to Skp (Schiffrin et al. 2017). These data demonstrated that a higher Skp:OMP ratio is required to bind larger OMPs (16-stranded), while for smaller OMPs the Skp cavity can be expanded to adapt to the size of the client proteins. One common characteristic of Skp is that it exists only as a multiple of the trimeric state, indicating that this stoichiometry corresponds to the active form of the chaperone. Thereby, a recent study established that at physiological concentration Skp exists in an equilibrium between a trimeric and a monomeric state (Sandlin et al. 2015). However, the conformation of the monomeric state is still controversial as Sandlin et al. hypothesized a folded conformation by the mean of a van’t Hoff analysis, while Burmann et al. (2013) observed a disordered conformation . To date no study has been able to shed light on characteristics and functionality of the Skp monomeric state.
While structure and dynamics of Skp in apo and holo states have been extensively characterized, the mechanism allowing the release of OMPs from Skp into the outer membrane is less clear. Burmann et al. (2013) have proposed a mechanism hypothesizing that in a ternary Skp–OMP–BAM transition complex the weak local affinity and the rapid structural interconversion of the OMP within the Skp cavity would allow client release by dynamic rearrangements of the polypeptide while it remains bound to Skp. According to this model, the OMP bound to Skp might be inserted sequentially one β-hairpin at a time into the lipid membrane. Furthermore, the BAM complex could guide the unfolded OMP, by transient contacts with BamA POTRA domains to the membrane entry point.
Taken together, the key element regulating Skp’s functional cycle is its dynamic nature , which allows both to operate large motion of its arms and to adapt to highly dynamic client proteins . The fast dynamics of the encapsulated OMP client permit its release in an ATP-independent manner. The recent demonstration of the biological relevance of a second non-characterized monomeric state could lead to additional critical insights into the functional cycle of Skp.
The Periplasmic Chaperone SurA
The periplasmic chaperone SurA is generally considered the main pathway for the transport of OMPs to the BAM complex (Sklar et al. 2007; Lazar and Kolter 1996; Rouvière and Gross 1996). Deletion of surA results in a lower OMP density and accumulation in the periplasm of the major OMPs OmpA, OmpF, and LamB in an unfolded form (Rouvière and Gross 1996). The expression of these proteins is down-regulated upon induction of the σE-dependent cytoplasmic stress response, which is activated when mis- or unfolded proteins are accumulating in the periplasm (Lazar and Kolter 1996; Missiakas et al. 1996; Rouvière and Gross 1996). The drastically decreased levels of outer membrane-inserted OMPs in SurA mutant strains result in phenotypes characterized by a defective cell envelope resulting in an increased sensitivity to hydrophobic agents such as SDS-EDTA and to antibiotics such as novobiocin (Lazar and Kolter 1996).
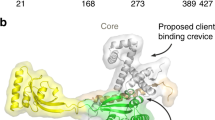
SurA is composed of four distinct domains: a large N-terminal domain, two parvulin-like peptidyl-prolyl isomerase (PPIase) domains and a short C-terminal helix followed by a short β-strand (Fig. 6.3a, b) (Bitto and McKay 2002). The combination of the PPIase 1 domain (P1) with the N and C-terminal domains form the core of the protein and the PPIase 2 domain (P2) is flexibly connected to this core by two linkers. The C-terminal 10 last residues form a short β-strand that forms an anti-parallel β-sheet with an N-terminal β-hairpin. While the exact amino acid sequence of the C-terminus is not essential, its shortening drastically affects chaperone activity (Chai et al. 2014). A minimal construct of SurA, composed of the N-terminal domain directly linked to the C-terminal domain, displays activity in vitro and can complement the SurA deletion phenotype in vivo (Behrens et al. 2001; Webb et al. 2001). This suggests that the N/C-domain pair is responsible for the chaperone activity and corroborates the initial observation of a large cavity on the N-terminal domain that could be a binding site for unfolded clients (Fig. 6.3a, b) (Bitto and McKay 2002).

Conformational rearrangements of the periplasmic chaperone SurA regulate its function. a Crystal structure of apo SurA (PDB 1M5Y). The core of the protein is formed by the N-terminal domain (green) together with the PPIase domain I (P1) (blue) and the C-terminal helix (magenta). The PPIase domain II (P2) (red) is connected by two flexible linkers and is thus located 30 Å away from the core. The hypothesized client binding site on the N-terminal domain is highlighted in yellow. b SurA domain composition coloured as in (a). c Schematic illustration of the regulation of SurA functional cycle by conformational dynamics of the two PPIase domains relatively to the core, coloured are as in (a) [adapted from (Soltes et al. 2016)]. d Structure of the dimeric complex of SurAΔP2 (SurA lacking the domain P2) bound to a peptide mimicking a client protein (PDB 2PV3), coloured as in (a). The peptide is coloured in orange. e Different conformations of the P1 domain in the apo state (left) and in the monomer extracted from the dimeric complex with a peptide (right). The formation of the complex triggers a major conformational change that leads to the dissociation of the P1 domain from the core domain
How the PPIase domains are relevant for the function of SurA is still unclear, especially as several SurA homologues have only one or no PPIase domain (Alcock et al. 2008). In general, parvulin-like domains feature enzymatic activity to catalyze the cis–trans conversion of peptide bonds preceding prolyl residues (Rahfeld et al. 1994; Rudd et al. 1995). For SurA only the P2 domain exhibits significant PPIase activity and both PPIase domains can simultaneously be deleted without significant loss of function (Rouvière and Gross 1996; Behrens et al. 2001). Isolated PPIase domains 1 and 2 do not exhibit chaperone activity and do not complement activity in a surA depletion mutant (Behrens et al. 2001). However, it cannot be excluded that their PPIase activity is important for specific client proteins and/or under certain conditions. In this context, it has recently been demonstrated that while the deletion of the P2 domain does not completely abolish the chaperoning activity it is significantly decreasing it (Soltes et al. 2016). This discovery could potentially be explained by a large dynamic conformational rearrangement of the P2 domain that allows closure of the N-terminal domain by a clamp-like mechanism. Soltes et al. also showed that the binding of the P1 domain to the core stabilizes the protein but inhibits the access to the binding site, while the flexible unbound state increases the chaperone activity at the cost of a decreased stability (Fig. 6.3c). The combination of the studies above suggests that the N/C-domain could represent a platform providing the basal chaperone activity that then has been evolutionarily diversified by addition of accessory domains.
The involvement of SurA in the assembly and transport of β-barrel OMPs to the outer membrane is clearly demonstrated by multiple studies (Rizzitello et al. 2001; Sklar et al. 2007). A specific set of OMPs that require SurA for their proper maturation has been revealed by a differential proteomics experiment including the formerly known SurA client proteins OmpF, OmpA, FhuA, and LamB as well as newly discovered client proteins such as LptD (Vertommen et al. 2009). Some motifs determining client specificity of SurA were identified using phage display experiments (Bitto and McKay 2003; Xu et al. 2007). These studies have shown that SurA recognizes peptide sequences rich in aromatic amino acids and arranged in the specific pattern Ar–X–Ar, where Ar is an aromatic residue and X can be any residue. The Ar–X–Ar sequence is found with increased abundance at the C-terminus of transmembrane β-barrel OMPs, indicating a possible role as recognition signal for SurA. This study lead to the development of a high affinity peptide mimicking the C-terminus of OMPs and the crystal structure of SurA with this peptide bound could be solved (Xu et al. 2007) (Fig. 6.3d). The structure shows a hydrophobic binding site on the P1 domain, however, not only one but two P1 domains are binding to one peptide, thus triggering a 2:1 SurA:peptide stoichiometry. Thereby, the P1 domain undergoes large conformational changes, which result in its separation from the core domain, while the structures of the N- and C-terminal domains remain unaffected compared to the apo form (Fig. 6.3e). Overall, the biological relevance of this structure remains unclear, since the binding of a short peptide is not necessarily representative for a full-length client protein. Furthermore, the position of the binding site contradicts in vivo data showing that the chaperoning activity is not affected by P1 deletion. This discrepancy could perhaps be explained by a decoupling of the sites of substrate recognition and chaperone activity. In the structure by Xu et al. the peptide bound to the P1 domain is oriented with its N-terminus towards the SurA N/C—domain. A full-length client protein could then bind the N/C-domain via a non-specific interaction.
Overall, the structural properties of SurA–OMP complexes are by far not as well characterized as the Skp–OMP complex, as no atomic resolution information is available. To obtain a complete picture it will be necessary to determine, among others, which functional role the conformational rearrangements of the domains have. A complete characterization of the structure and dynamics of SurA in its apo and holo states, is highly desired in order to draw a complete the picture of the molecular mechanism underlying the function of SurA.
Skp- and SurA-Mediated Stabilization and Folding of Client Proteins
The increasingly detailed structural knowledge of the periplasmic molecular chaperone network is accompanied by several studies characterizing how these chaperones interact with and influence the folding behaviour of client OMPs in vitro. Trimeric Skp has been found to form stable 1:1 (Skp3:OMP) complexes with small OMP clients such as unfolded OmpA, but also complexes in 2:1 (Skp3:OMP) ratio with larger OMPs such as unfolded BamA and FhuA (Schiffrin et al. 2016; Thoma et al. 2015). Thereby, Skp can bind unfolded client OMPs with high affinities, corresponding to dissociation constants in the low nM range, as determined for multiple unfolded OMP clients, such as OmpA, OmpG and BamA as well as OmpLA, OmpW and PagP (Qu et al. 2007; Moon et al. 2013). While the stoichiometry of SurA:OMP complexes has not been studied in detail, it has been proposed to bind OmpC as well as FhuA in a 2:1 ratio (Thoma et al. 2015; Li et al. 2018). The affinity of SurA to OMP clients is generally much weaker than of Skp. While SurA’s affinity to full-length nascent client proteins has been determined to a lesser extent, it has been found to bind unfolded OmpG and OmpF with affinities in the low µM range (Bitto and McKay 2004). Similar values were found for its affinity to heptapeptide WEYIPNV, which contains the Ar–X–Ar motif frequently found in OMPs (Bitto and McKay 2003).
A higher client affinity of Skp compared to SurA is also consistent with experiments showing Skp binding to unfolded OmpC at faster rates than SurA as well as 6-fold longer lifetimes of chaperone-client complexes formed by Skp with unfolded FhuA compared to SurA-FhuA complexes (Wu et al. 2011; Thoma et al. 2015). Moreover, Skp has been shown to outcompete SurA in equimolar mixtures of both chaperones when binding unfolded FhuA or OmpA (Thoma et al. 2015; Schiffrin et al. 2017). The client OMP either remains bound to Skp when preassembled Skp–client complexes are added to apo SurA, or transfers to Skp when preassembled SurA–client complexes are added to apo Skp. Differences in client binding were further elucidated by a recent study reporting a mild disaggregase function for Skp, which could break down scant aggregates containing up to 5 OmpC monomers, whereas such behavior could not be observed for SurA (Li et al. 2018). These data are also in full agreement with the notion that Skp binds its clients in a tighter and more compact conformation than SurA.
While chaperones can maintain client OMPs dynamically unfolded in solution over prolonged time periods, proper folding of the client requires the presence of a lipid membrane which provides the thermodynamic stabilization of the native form (Moon et al. 2013; Burmann et al. 2013; Thoma et al. 2015). Thereby, the transition from a chaperone-stabilized state to a folded, membrane-embedded state depends on the biophysical properties of the lipid bilayer. For example, OmpA and PagP fold into negatively charged lipid bilayers from solutions containing Skp, but not into neutral bilayers (Bulieris et al. 2003; Patel et al. 2009; Patel and Kleinschmidt 2013; McMorran et al. 2013). Moreover, the presence of additional factors such as LPS, transmembrane proteins in general and in particular the presence of BamA appears to influence the transition from a chaperone-stabilized state into the lipid bilayer (Bulieris et al. 2003; Patel et al. 2009; Patel and Kleinschmidt 2013; Schiffrin et al. 2017). Especially client OMPs folding from complexes with SurA appear to profit greatly from the presence of membrane-embedded BAM, but show relatively little folding into empty lipid bilayers (Schiffrin et al. 2017). Despite the influence of different membrane composition, it evolves as a common theme from these studies that elevated concentrations of Skp prevent folding and membrane insertion of client OMPs (Bulieris et al. 2003; Schiffrin et al. 2016). The inhibitory effect of Skp was also observed when looking at chaperone assisted OMP folding on the single molecule level. The large OMP FhuA has a strong tendency to misfold in the absence of periplasmic chaperones, whereas in the presence of either of the chaperones Skp or SurA, misfolding is prevented. In agreement with the bulk measurements, folding of FhuA is largely abolished in the presence of Skp, however, FhuA can reinsert into the lipid bilayer in a series of folding steps shaped by individual beta-hairpins in the presence of SurA (Fig. 6.4a) (Thoma et al. 2012, 2015). The sequence in which individual β-hairpins insert into the membrane can be random, while a certain tendency is observed to progress from the C-terminal toward the N-terminal end of the protein. Thereby, the yet unfolded segments of the nascent FhuA polypeptide remain protected from misfolding over the entire time of the folding process. In agreement with the higher binding affinity of Skp, in an equimolar mixture of both chaperones, the inhibitory effect Skp exerted on the folding behaviour dominates over the folding promoting effect of SurA (Thoma et al. 2015). Consequentially, these experiments indicate that both chaperones could interact effectively with partially inserted client OMPs, strengthening the concept of the lipid membrane acting not only as a free-energy sink but also as a physical barrier separating partially folded segments from the chaperones (Fig. 6.4b).

[adapted from (Thoma et al. 2015)]
Folding and membrane insertion model of client OMP FhuA from a SurA-FhuA complex. a In the absence of chaperones, the folding pathway of unfolded FhuA (cyan) is directed towards a misfolded state. The presence of SurA (orange) stabilizes the unfolded state of FhuA and promotes stepwise insertion and folding of β-hairpins into the lipid membrane until folding is completed, upon which SurA is released. b The hypothetical folding free-energy landscape of FhuA in the presence of SurA is characterized by a series of free-energy wells corresponding to β-hairpins stably inserted in the lipid membrane. SurA (orange) prevents misfolding of FhuA in solution but is spatially excluded from the lipid membrane (blue) and thus cannot interact with folded intermediates
In summary, these in vitro findings indicate that Skp binds clients with high affinity and a strong tendency to prevent their folding whereas SurA binds unfolded OMPs with lower affinity while promoting their folding. Together with in vivo findings, which indicate severely reduced OMP levels in SurA-depleted strains, but only mild phenotypes upon Skp depletion, the available data indicate a role of SurA acting as the main periplasmic pathway for OMP folding, while Skp forms a secondary backup pathway (Silhavy et al. 2010; Sklar et al. 2007; Wu et al. 2011). The concept of SurA acting as the primary pathway is further substantiated by the observation that the delivery of major OMPs, as well as certain large OMPs such as LptD and FhuA seem to rely particularly on the presence of SurA (Vertommen et al. 2009). However, it remains to be elucidated precisely how the differential effects of both chaperones observed in vitro are modulated in vivo to promote efficient delivery of client OMPs to the OM.
Concluding Remarks
During the last decade, ground-breaking works have drastically improved our understanding of the molecular mechanism regulating the periplasmic chaperones Skp and SurA and highlighted the major importance of conformational flexibility in their mechanisms of action. These latest advancements have been made possible by the technical development of SAXS, molecular dynamics and NMR spectroscopy methods, in addition to the ground-laying crystal structures, highlighting the critical importance of the integration of the different methods. The structure and dynamics of the periplasmic chaperone Skp in its apo and holo states have shown that large conformational rearrangements of its cavity are the key factors of its functional cycle, giving Skp the capacity to adapt in response to the properties of its client proteins. Similarly, preliminary results on the chaperone SurA highlight the role played by the dynamic rearrangements of its domains to regulate its functional cycle.
Abbreviations
- ATP:
-
Adenosine 5′-triphosphate
- BAM:
-
β-barrel assembly machinery
- Hsp60:
-
Heat shock protein 60
- NMR:
-
Nuclear magnetic resonance
- NOE:
-
Nuclear Overhauser effect
- OMP:
-
Outer membrane protein
- PPIase:
-
Parvulin-like peptidyl-prolyl isomerase
- PRE:
-
Paramagnetic relaxation enhancement
- SAXS:
-
Small angle X-ray scattering
- Skp:
-
Seventeen Kilodalton Protein
- SurA:
-
Survival factor A
References
Alcock FH, Grossmann JG, Gentle IE, Likić VA, Lithgow T, Tokatlidis K (2008) Conserved substrate binding by chaperones in the bacterial periplasm and the mitochondrial intermembrane space. Biochem J 409:377–387. https://doi.org/10.1042/BJ20070877
Antonoaea R, Furst M, Nishiyama K, Muller M (2008) The periplasmic chaperone PpiD interacts with secretory proteins exiting from the SecYEG translocon. Biochemistry 47:5649–5656. https://doi.org/10.1021/bi800233w
Arie JP, Sassoon N, Betton JM (2001) Chaperone function of FkpA, a heat shock prolyl isomerase, in the periplasm of Escherichia coli. Mol Microbiol 39:199–210. https://doi.org/10.1046/j.1365-2958.2001.02250.x
Bardwell JC, McGovern K, Beckwith J (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. https://doi.org/10.1016/0092-8674(91)90532-4
Behrens S, Maier R, de Cock H, Schmid FX, Gross CA (2001) The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J 20:285–294. https://doi.org/10.1093/emboj/20.1.285
Bitto E, McKay DB (2002) Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10:1489–1498. https://doi.org/10.1016/s0969-2126(02)00877-8
Bitto E, McKay DB (2003) The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J Biol Chem 278:49316–49322. https://doi.org/10.1074/jbc.M308853200
Bitto E, McKay DB (2004) Binding of phage-display-selected peptides to the periplasmic chaperone protein SurA mimics binding of unfolded outer membrane proteins. FEBS Lett 568:94–98. https://doi.org/10.1016/j.febslet.2004.05.014
Bothmann H, Plückthun A (1998) Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nat Biotechnol 16:376–380. https://doi.org/10.1038/nbt0498-376
Braig K, Adams PD, Brünger AT (1995) Conformational variability in the refined structure of the chaperonin GroEL at 2.8 Å resolution. Nat Struct Mol Biol 2:1083. https://doi.org/10.1038/nsb1295-1083
Bulieris PV, Behrens S, Holst O, Kleinschmidt JH (2003) Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J Biol Chem 278:9092–9099. https://doi.org/10.1074/jbc.M211177200
Burmann BM, Wang C, Hiller S (2013) Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat Struct Mol Biol 20:1265–1272. https://doi.org/10.1038/nsmb.2677
Callon M, Burmann BM, Hiller S (2014) Structural mapping of a chaperone-substrate interaction surface. Angew Chem Int Edit 53:5069–5072. https://doi.org/10.1002/anie.201310963
Chai Q, Ferrell B, Zhong M, Zhang X, Ye C, Wei Y (2014) Diverse sequences are functional at the C-terminus of the E. coli periplasmic chaperone SurA. Protein Eng Des Sel 27:111–116. https://doi.org/10.1093/protein/gzu003
Chatzi KE, Sardis MF, Economou A, Karamanou S (2014) SecA-mediated targeting and translocation of secretory proteins. Biochim et Biophys Acta (BBA) 1843:1466–1474. https://doi.org/10.1016/j.bbamcr.2014.02.014
Chen R, Henning U (1996) A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol Microbiol 19:1287–1294. https://doi.org/10.1111/j.1365-2958.1996.tb02473.x
Clausen T, Kaiser M, Huber R, Ehrmann M (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12:152–162. https://doi.org/10.1038/nrm3065
De Geyter J, Tsirigotaki A, Orfanoudaki G, Zorzini V, Economou A, Karamanou S (2016) Protein folding in the cell envelope of Escherichia coli. Nat Microbiol 1:16107. https://doi.org/10.1038/nmicrobiol.2016.107
Denoncin K, Collet J-F (2013) Disulfide bond formation in the bacterial periplasm: major achievements and challenges ahead. Antioxid Redox Signal 19:63–71. https://doi.org/10.1089/ars.2012.4864
Ding J, Yang C, Niu X, Hu Y, Jin C (2015) HdeB chaperone activity is coupled to its intrinsic dynamic properties. Sci Rep 5:1292. https://doi.org/10.1038/srep16856
Ditzel L, Löwe J, Stock D, Stetter K-O, Huber H, Huber R, Steinbacher S (1998) Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell 93:125–138. https://doi.org/10.1016/S0092-8674(00)81152-6
Driessen AJM, Manting EH, van der Does C (2001) The structural basis of protein targeting and translocation in bacteria. Nat Struct Mol Biol 8:492–498. https://doi.org/10.1038/88549
Ge X, Wang R, Ma J, Liu Y, Ezemaduka AN, Chen PR, Fu X, Chang Z (2014a) DegP primarily functions as a protease for the biogenesis of β-barrel outer membrane proteins in the Gram-negative bacterium Escherichia coli. FEBS J 281:1226–1240. https://doi.org/10.1111/febs.12701
Ge X, Lyu Z-X, Liu Y, Wang R, Zhao XS, Fu X, Chang Z (2014b) Identification of FkpA as a key quality control factor for the biogenesis of outer membrane proteins under heat shock conditions. J Bacteriol 196:672–680. https://doi.org/10.1128/jb.01069-13
Hayano T, Takahashi N, Kato S, Maki N, Suzuki M (1991) Two distinct forms of peptidylprolyl-cis-trans-isomerase are expressed separately in periplasmic and cytoplasmic compartments of Escherichia coli cells. Biochem 30:3041–3048. https://doi.org/10.1021/bi00226a009
He L, Sharpe T, Mazur A, Hiller S (2016) A molecular mechanism of chaperone-client recognition. Sci Adv 2–11:e1601625. https://doi.org/10.1126/sciadv.1601625
Helbig S, Patzer SI, Schiene-Fischer C, Zeth K, Braun V (2011) Activation of colicin M by the FkpA prolyl cis-trans isomerase/chaperone. J Biol Chem 286:6280–6290. https://doi.org/10.1074/jbc.m110.165274
Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL (2009) DSB proteins and bacterial pathogenicity. Nat Rev Microbiol 7:215–225. https://doi.org/10.1038/nrmicro2087
Holdbrook DA, Burmann BM, Huber RG, Petoukhov MV, Svergun DI, Hiller S, Bond PJ (2017) A spring-loaded mechanism governs the clamp-like dynamics of the Skp chaperone. Structure 25:1079–1088.e3. https://doi.org/10.1016/j.str.2017.05.018
Hong W, Jiao W, Hu J, Zhang J, Liu C, Fu X, Shen D, Xia B, Chang Z (2005) Periplasmic protein HdeA exhibits chaperone-like activity exclusively within stomach pH range by transforming into disordered conformation. J Biol Chem 280:27029–27034. https://doi.org/10.1074/jbc.m503934200
Horwich AL, Fenton WA, Chapman E, Farr GW (2007) Two families of chaperonin: physiology and mechanism. Annu Rev Cell Dev Bi 23:115–145. https://doi.org/10.1146/annurev.cellbio.23.090506.123555
Ito K, Inaba K (2008) The disulfide bond formation (Dsb) system. Curr Opin Struct Biol 18:450–458. https://doi.org/10.1016/j.sbi.2008.02.002
Jarchow S, Lück C, Görg A, Skerra A (2008) Identification of potential substrate proteins for the periplasmic Escherichia coli chaperone Skp. Proteomics 8:4987–4994. https://doi.org/10.1002/pmic.200800288
Jiang J, Zhang X, Chen Y, Wu Y, Zhou ZH, Chang Z, Sui S-F (2008) Activation of DegP chaperone-protease via formation of large cage-like oligomers upon binding to substrate proteins. Proc Natl Acad Sci USA 105:11939–11944. https://doi.org/10.1073/pnas.0805464105
Joly JC, Swartz JR (1997) In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry 36:10067–10072. https://doi.org/10.1021/bi9707739
Kern R, Malki A, Abdallah J, Tagourti J, Richarme G (2007) Escherichia coli HdeB is an acid stress chaperone. J Bacteriol 189:603–610. https://doi.org/10.1128/jb.01522-06
Korndörfer IP, Dommel MK, Skerra A (2004) Structure of the periplasmic chaperone Skp suggests functional similarity with cytosolic chaperones despite differing architecture. Nat Struct Mol Biol 11:1015–1020. https://doi.org/10.1038/nsmb828
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol 305:567–580. https://doi.org/10.1006/jmbi.2000.4315
Lazar SW, Kolter R (1996) SurA assists the folding of Escherichia coli outer membrane proteins. J Bacteriol 178:1770–1773. https://doi.org/10.1128/jb.178.6.1770-1773.1996
Levy R, Weiss R, Chen G, Iverson BL, Georgiou G (2001) Production of correctly folded Fab antibody fragment in the cytoplasm of Escherichia coli trxB gor mutants via the coexpression of molecular chaperones. Prot Exp Purif 23:338–347. https://doi.org/10.1006/prep.2001.1520
Li G, He C, Bu P, Bi H, Pan S, Sun R, Zhao XS (2018) Single–molecule detection reveals different roles of Skp and SurA as chaperones. ACS Chem Biol. https://doi.org/10.1021/acschembio.8b00097
Liu J, Walsh CT (1990) Peptidyl-prolyl cis-trans-isomerase from Escherichia coli: a periplasmic homolog of cyclophilin that is not inhibited by cyclosporin A. Proc Natl Acad Sci USA 87:4028–4032. https://doi.org/10.1073/pnas.87.11.4028
Lycklama A, Nijeholt JA, Driessen AJ (2012) The bacterial Sec-translocase: structure and mechanism. Philos Trans R Soc B Biol Sci 367:1016–1028. https://doi.org/10.1098/rstb.2011.0201
Martín-Benito J, Boskovic J, Gómez-Puertas P, Carrascosa JL, Simons CT, Lewis SA, Bartolini F, Cowan NJ, Valpuesta JM (2002) Structure of eukaryotic prefoldin and of its complexes with unfolded actin and the cytosolic chaperonin CCT. EMBO J 21:6377–6386. https://doi.org/10.1093/emboj/cdf640
Matern Y, Barion B, Behrens-Kneip S (2010) PpiD is a player in the network of periplasmic chaperones in Escherichia coli. BMC Microbiol 10:251. https://doi.org/10.1186/1471-2180-10-251
McMorran LM, Bartlett AI, Huysmans GHM, Radford SE, Brockwell DJ (2013) Dissecting the effects of periplasmic chaperones on the in vitro folding of the outer membrane protein PagP. J Mol Biol 425:3178–3191. https://doi.org/10.1016/j.jmb.2013.06.017
Messens J, Collet JF (2006) Pathways of disulfide bond formation in Escherichia coli. Int J Biochem Cell Biol 38:1050–1062. https://doi.org/10.1016/j.biocel.2005.12.011
Missiakas D, Betton JM, Raina S (1996) New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol Microbiol 21:871–884. https://doi.org/10.1046/j.1365-2958.1996.561412.x
Miyamoto A, Matsuyama S, Tokuda H (2001) Mutant of LolA, a lipoprotein-specific molecular chaperone of Escherichia coli, defective in the transfer of lipoproteins to LolB. Biochem Biophys Res Commun 287:1125–1128. https://doi.org/10.1006/bbrc.2001.5705
Moon CP, Zaccai NR, Fleming PJ, Gessmann D, Fleming KG (2013) Membrane protein thermodynamic stability may serve as the energy sink for sorting in the periplasm. Proc Natl Acad Sci USA 110:4285–4290. https://doi.org/10.1073/pnas.1212527110
Natale P, Bruser T, Driessen AJ (2008) Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane-distinct translocases and mechanisms. Biochim Biophys Acta 1778:1735–1756. https://doi.org/10.1016/j.bbamem.2007.07.015
Okuda S, Tokuda H (2009) Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proc Natl Acad Sci USA 106:5877–5882. https://doi.org/10.1073/pnas.0900896106
Ohtaki A, Kida H, Miyata Y, Ide N, Yonezawa A, Arakawa T, Iizuka R, Noguchi K, Kita A, Odaka M, Miki K, Yohda M (2008) Structure and molecular dynamics simulation of archaeal prefoldin: the molecular mechanism for binding and recognition of nonnative substrate proteins. J Mol Biol 376:1130–1141. https://doi.org/10.1016/j.jmb.2007.12.010
Orfanoudaki G, Economou A (2014) Proteome-wide subcellular topologies of E. coli polypeptides database (STEPdb). Mol Cell Proteomics 13:3674–3687. https://doi.org/10.1074/mcp.O114.041137
Patel GJ, Behrens-Kneip S, Holst O, Kleinschmidt JH (2009) The periplasmic chaperone Skp facilitates targeting, insertion, and folding of OmpA into lipid membranes with a negative membrane surface potential. Biochemistry 48:10235–10245. https://doi.org/10.1021/bi901403c
Patel GJ, Kleinschmidt JH (2013) The lipid bilayer-inserted membrane protein BamA of Escherichia coli facilitates insertion and folding of outer membrane protein A from its complex with Skp. Biochemistry 52:3974–3986. https://doi.org/10.1021/bi400103t
Qu J, Behrens S, Holst O, Kleinschmidt JH (2009) Binding sites of outer membrane protein A (OmpA) in the complex with the periplasmic chaperone Skp from E. Coli. A site-directed fluorescence study. Biophys J 96:449a–450a. https://doi.org/10.1016/j.bpj.2008.12.2310
Qu J, Mayer C, Behrens S, Holst O, Kleinschmidt JH (2007) The trimeric periplasmic chaperone Skp of Escherichia coli forms 1:1 complexes with outer membrane proteins via hydrophobic and electrostatic interactions. J Mol Biol 374:91–105. https://doi.org/10.1016/j.jmb.2007.09.020
Quan S, Koldewey P, Tapley T, Kirsch N, Ruane KM, Pfizenmaier J, Shi R, Hofmann S, Foit L, Ren G, Jakob U, Xu Z, Cygler M, Bardwell JC (2011) Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat Struct Mol Biol 18:262–269. https://doi.org/10.1038/nsmb.2016
Rahfeld J-U, Schierhorn A, Mann K, Fischer G (1994) A novel peptidyl-prolyl cisltrans isomerase from Escherichia coli. FEBS Lett 343:65–69. https://doi.org/10.1016/0014-5793(94)80608-X
Rizzitello AE, Harper JR, Silhavy TJ (2001) Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J Bacteriol 183:6794–6800. https://doi.org/10.1128/JB.183.23.6794-6800.2001
Rouvière PE, Gross CA (1996) SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev 10:3170–3182. https://doi.org/10.1101/gad.10.24.3170
Rudd KE, Sofia HJ, Koonin EV, Plunkett G, Lazar S, Rouviere PE (1995) A new family of peptidyl-prolyl isomerases. Trends Biochem Sci 20:12–14. https://doi.org/10.1016/S0968-0004(00)88940-9
Salmon L, Stull F, Sayle S, Cato C, Akgül Ş, Foit L, Ahlstrom LS, Eisenmesser EZ, Al-Hashimi HM, Bardwell JCA, Horowitz S (2018) The mechanism of HdeA unfolding and chaperone activation. J Mol Biol 430:33–40. https://doi.org/10.1016/j.jmb.2017.11.002
Sandlin CW, Zaccai NR, Fleming KG (2015) Skp trimer formation is insensitive to salts in the physiological range. Biochemistry 54:7059–7062. https://doi.org/10.1021/acs.biochem.5b00806
Saul FA, Arie JP, Vulliez-le Normand B, Kahn R, Betton JM, Bentley GA (2004) Structural and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J Mol Biol 335:595–608. https://doi.org/10.1016/j.jmb.2003.10.056
Schiffrin B, Calabrese AN, Devine PWA, Harris SA, Ashcroft AE, Brockwell DJ, Radford SE (2016) Skp is a multivalent chaperone of outer-membrane proteins. Nat Struct Mol Biol 1–11. https://doi.org/10.1038/nsmb.3266
Schiffrin B, Calabrese AN, Higgins AJ, Humes JR, Ashcroft AE, Kalli AC, Brockwell DJ, Radford SE (2017) Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane protein folding. J Mol Biol 429:3776–3792. https://doi.org/10.1016/j.jmb.2017.09.008
Scholz C, Scherer G, Mayr LM, Schindler T, Fischer G, Schmid FX (1998) Prolyl isomerases do not catalyze isomerization of non-prolyl peptide bonds. Biol Chem 379:361–365. https://doi.org/10.1016/s0014-5793(98)00871-0
Siegert R, Leroux MR, Scheufler C, Hartl FU, Moarefi I (2000) Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103:621–632. https://doi.org/10.1016/S0092-8674(00)00165-3
Silhavy TJ, Kahne D, Walker S (2010) The bacterial cell envelope. CSH Perspect Biol 2:a000414–a000414. https://doi.org/10.1101/cshperspect.a000414
Sklar JG, Wu T, Kahne D, Silhavy TJ (2007) Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484. https://doi.org/10.1101/gad.1581007
Spiess C, Beil A, Ehrmann M (1999) A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97:339–347. https://doi.org/10.1016/s0092-8674(00)80743-6
Soltes GR, Schwalm J, Ricci DP, Silhavy TJ (2016) The activity of Escherichia coli chaperone SurA is regulated by conformational changes involving a parvulin domain. J Bacteriol 198:921–929. https://doi.org/10.1128/JB.00889-15
Stull F, Koldewey P, Humes JR, Radford SE, Bardwell JCA (2016) Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat Struct Mol Biol 23:53–58. https://doi.org/10.1038/nsmb.3133
Tajima T, Yokota N, Matsuyama S, Tokuda H (1998) Genetic analyses of the in vivo function of LolA, a periplasmic chaperone involved in the outer membrane localization of Escherichia coli lipoproteins. FEBS Lett 439:51–54. https://doi.org/10.1016/s0014-5793(98)01334-9
Taniguchi N, Matsuyama S, Tokuda H (2005) Mechanisms underlying energy- independent transfer of lipoproteins from LolA to LolB, which have similar unclosed beta-barrel structures. J Biol Chem 280:34481–34488. https://doi.org/10.1074/jbc.m507388200
Thoma J, Bosshart P, Pfreundschuh M, Müller DJ (2012) Out but not in: the large transmembrane β-barrel protein FhuA unfolds but cannot refold via β-hairpins. Structure 20:2185–2190
Thoma J, Burmann BM, Hiller S, Müller DJ (2015) Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nat Struct Mol Biol 22:795–802. https://doi.org/10.1038/nsmb.3087
Tsirigotaki A, De Geyter J, Šoštaric N, Economou A, Karamanou S (2017) Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15:4, 15:21–36. https://doi.org/10.1038/nrmicro.2016.161
Van den Berg B, Clemons WM Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA (2004) X-ray structure of a protein-conducting channel. Nature 427:36–44. https://doi.org/10.1038/nature02218
Van Wielink JE, Duine JA (1990) How big is the periplasmic space? Trends Biochem Sci 15:136–137. https://doi.org/10.1016/0968-0004(90)90208-S
Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, Messens J, Bardwell JC, Collet J-F (2008) The disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner. Mol Microbiol 67:336–349. https://doi.org/10.1111/j.1365-2958.2007.06030.x
Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet J-F (2009) Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443. https://doi.org/10.1002/pmic.200800794
Walton TA, Sandoval CM, Fowler CA, Pardi A, Sousa MC (2009) The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc Natl Acad Sci USA 106:1772–1777. https://doi.org/10.1073/pnas.0809275106
Walton TA, Sousa MC (2004) Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell 15:367–374. https://doi.org/10.1016/j.molcel.2004.07.023
Webb HM, Ruddock LW, Marchant RJ, Jonas K, Klappa P (2001) Interaction of the periplasmic peptidylprolyl cis-transisomerase SurA with model peptides. J Biol Chem 276:45622–45627. https://doi.org/10.1074/jbc.M107508200
Wickner W, Driessen AJ, Hartl FU (1991) The enzymology of protein translocation across the Escherichia coli plasma membrane. Annu Rev Plant Physio. Plant Mol Biol 60:101–124. https://doi.org/10.1146/annurev.bi.60.070191.000533
Wu S, Ge X, Lv Z, Zhi Z, Chang Z, Zhao XS (2011) Interaction between bacterial outer membrane proteins and periplasmic quality control factors: a kinetic partitioning mechanism. Biochem J 438:505–511. https://doi.org/10.1042/BJ20110264
Xu X, Wang S, Hu Y-X, McKay DB (2007) The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J Mol Biol 373:367–381. https://doi.org/10.1016/j.jmb.2007.07.069
Xu Z, Horwich AL, Sigler PB (1997) The crystal structure of the asymmetric GroEL–GroES–(ADP)7 chaperonin complex. Nature 388:741–750. https://doi.org/10.1038/41944
Yu X-C, Yang C, Ding J, Niu X, Hu Y, Jin C (2017) Characterizations of the interactions between Escherichia coli periplasmic chaperone HdeA and its native substrates during acid stress. Biochem 56:5748–5757. https://doi.org/10.1021/acs.biochem.7b00724
Yu X-C, Hu Y, Ding J, Li H, Jin C (2018) Structural basis and mechanism of the unfolding-induced activation of HdeA, a bacterial acid response chaperone. J Biol Chem. https://doi.org/10.1101/390104
Zhang S, He D, Yang Y, Lin S, Zhang M, Dai S, Chen PR (2016) Comparative proteomics reveal distinct chaperone–client interactions in supporting bacterial acid resistance. Proc Natl Acad Sci USA 113:10872–10877. https://doi.org/10.1073/pnas.1606360113
Acknowledgements
This work was supported by the Swiss National Science Foundation and the NFP 72 (grants 31003A_166426 and 407240_167125 to S.H.) and by the Swiss Nanoscience institute. The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mas, G., Thoma, J., Hiller, S. (2019). The Periplasmic Chaperones Skp and SurA. In: Kuhn, A. (eds) Bacterial Cell Walls and Membranes . Subcellular Biochemistry, vol 92. Springer, Cham. https://doi.org/10.1007/978-3-030-18768-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-18768-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-18767-5
Online ISBN: 978-3-030-18768-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)