
Abstract
Dexmedetomidine (4-[(1S)-1-(2,3-dimethylphenyl)ethyl]-1H-imidazole, is a highly potent, selective α2-adrenergic receptor agonist with sedative, analgesic and anxiolytic properties. Although primarily used for sedation in adult intensive care unit patients, novel applications have emerged in the literature, with favorable results in the prevention of delirium, acute postoperative pain, and opioid induced hyperalgesia, amongst others. Dexmedetomidine exerts its action by binding to α2A-, α2B-, and α2C-adrenoceptors, producing plasma concentration-dependent hypotension and bradycardia without respiratory depression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
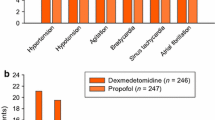


Keywords
- Dexmedetomidine
- Infusion
- Acute postoperative pain
- Complex regional pain syndrome
- Opioid-induced hyperalgesia
- Sedation in the ICU
- Delirium in ICU
- Drug withdrawal and weaning
- Awake fiberoptic intubation
- Procedural sedation
- Awake fiberoptic intubation
Introduction
Dexmedetomidine, or 4-[(1S)-1-(2,3-dimethylphenyl)ethyl]-1H-imidazole, is a potent and selective α2-adrenoceptor agonist used for its sedative, analgesic and anxiolytic properties [1, 2]. It was initially approved in the United States in 1999 (Precedex®; Hospira, Lake Forrest, IL, USA) for intravenous (IV) sedation of mechanically ventilated adult patients in the intensive care unit (ICU), at doses ranging from 0.2 to 0.7 mcg/kg/h for up to 24 h [3]. In 2008, it received additional approval for procedural sedation of non-intubated adult patients in perioperative and non-surgical settings [2]. In Europe it has been approved since 2011 (Dexdor®; Orion Corporation, Espoo, Finland), for sedation in adult ICU patients who are required to remain easily arousable to verbal stimuli [4, 5].
Mechanism of Action
As the pharmacologically active dextro-isomer of medetomidine (a commonly used agent in veterinary medicine) [2], dexmedetomidine binds to transmembrane Gi-coupled α2-adrenergic receptors in the periphery (α2A subtype) and centrally within the brain and spinal cord (α2B and α2C subtypes) [6, 7]. It does not have action at gamma-aminobutyric acid (GABA), opioid or N-methyl-D-aspartate (NMDA) receptors as do benzodiazepines, opioids and propofol. Sedative and antinociceptive actions are mediated through α2A stimulation [8, 9], vasoconstrictive effects are produced by α2B activation, and α2C stimulation modulates dopaminergic neurotransmission, hypothermia and numerous behavioral changes [9]. The suppression of neurotransmission seen with α2-agonists is due to activation of pre- and post-synaptic potassium ion channels. Subsequent potassium efflux and membrane hyperpolarization inhibits norepinephrine release and leads to decreased excitation, particularly in the medulla within the locus coeruleus [6, 9]. This area of the brain is the key site of noradrenergic functions including arousal, sleep, anxiety, and withdrawal from CNS depressants including opioids [6, 9, 10]. Additionally, dexmedetomidine interacts with imidazoline type 1 (I1) receptors, which are not G-coupled, and have roles in memory [11], neuroprotection [12], central hypotension and display antiarrhythmogenic qualities [13, 14]. These receptors are found centrally in the medulla, and peripherally within the cardiac tissue [15]. Overall, α2-agonists have sedative, sympatholytic, and opioid sparing properties [6, 16]. Importantly, dexmedetomidine-based sedation resembles natural sleep, with patients remaining quickly and easily arousable [17,18,19,20].
As compared to the prototypic α2-agonist clonidine, dexmedetomidine has a dose-dependent seven to eight-fold higher α2-receptor selectivity (α2:α1 ratio of 1620:1 vs. 220:1) [2, 16, 21]. Both α1- and α2-receptor activity was found in early preclinical studies with slow high dose infusions (>1000 mcg/kg) or following rapid IV bolus administration [3]. This proclivity for α1-activity at very high plasma concentrations, in addition to the expected α2-receptor actions at recommended plasma concentrations, explains the main hemodynamic side effects of dexmedetomidine. Main among them are transient hypertension, bradycardia and hypotension. The onset of action of dexmedetomidine infusion is 5–8 min when given with a loading dose, with a peak effect after 10–20 min [1]. It is 94% albumin-bound in plasma and has a weight-dependent volume of distribution of 1.3–2.5 L/kg [2]; thus, reduced doses are not required in pediatric patients. Dexmedetomidine undergoes hepatic biotransformation via direct glucuronidation and hydroxylation by cytochrome P450 enzymes (mainly CYP2A6) and is excreted unchanged in the urine (95%) and feces (5%) [2, 3, 5]. There are no active or toxic metabolites, and the elimination half-life ranges between 2–4 h [2]. As with clonidine, a dependence potential has been demonstrated in preclinical studies, however, this has not been researched in clinical trials [22].
Despite the seemingly narrow list of approved indications, numerous off-label uses and routes of administration, including intra-auricular [23], intranasal [24], buccal [25], and epidural [26] have been described in the literature. Indications which include therapeutic IV infusion for pain associated conditions will be the focus of this chapter.
Indications
Perioperative and Intensive Care Unit
Overall, dexmedetomidine is primarily used in perioperative and ICU settings. Its use for sedation in the ICU has been thoroughly researched and is well established [3, 6]. The approved dosing regimen in adults is 1 mcg/kg over 10 min as a loading dose, followed by a 0.2–0.7 mcg/kg/h maintenance infusion for a maximum of 24 h [3]. Studies have described safe infusions in mechanically ventilated ICU patients for upwards of a week in duration [27, 28]. The most recent RCT from 2018 by Skrobik et al. demonstrated that low-dose nocturnal dexmedetomidine infusion can prevent delirium in adult ICU patients [29]. Furthermore, in light of the current opioid epidemic [30], as increasing numbers of patients are admitted to the ICU for the management of withdrawal or acute detoxification [31], dexmedetomidine has shown value in both adult and pediatric patients [32,33,34,35]. It has been approved for awake fiberoptic intubation (AFOI) since 2008 [2], and as per the most recent meta-analysis, remains a suitable agent alongside remifentanil [36]. In a recent trial comparing these two agents, Hu and colleagues found that dexmedetomidine (loading: 1.5 mcg/kg, infusion: 0.7 mcg/kg/h) reduced the incidence of recall to 40%, in comparison to 70% in the remifentanil group, indicating that it may be preferable [37]. The easily arousable nature of dexmedetomidine sedation renders it a common agent for procedures where conscious sedation is required. A large RCT with 326 patients undergoing varied procedures found that dexmedetomidine at 0.5–1.0 mcg/kg loading dose, followed by continuous IV infusion at 0.2–1.0 mcg/kg/h, reduced the need for supplemental midazolam and overall dose required to achieve adequate depth of sedation [38]. However, where dexmedetomidine has been successful in orthopedic, vascular, diagnostic and dental procedures, it is inappropriate for outpatient colonoscopy [39]. This is due to possible vagal stimulation during colonoscopy, which may potentiate the bradycardic side effects of dexmedetomidine. Indeed, due to its shorter duration of action and less hemodynamic disturbance, propofol remains the drug of choice in such scenarios [39]. Furthermore, recent meta-analyses have concluded that, dexmedetomidine-infused patients experience significantly lower rates of postoperative nausea and vomiting (PONV) and shivering, compared to placebo controls [40, 41]. Alone and in combination with other agents, dexmedetomidine has been shown to attenuate the incidence of emergence delirium in children [42,43,44]. The agent may in fact, be the drug of choice to achieve opioid-free total IV anesthesia [45], as studies have demonstrated its effectiveness when combined with propofol and/or ketamine in bariatric [46] and spinal surgery [47]; however, more RCTs are needed in this field and further discussion is outside the scope of this chapter.
Opioid-Sparing and Acute Postoperative Pain
Alleviating the acute postoperative pain seen in up to 80% of patients after surgery is paramount to increasing patient satisfaction, reducing time in recovery and curbing the incidence of chronic postsurgical pain [48]. A multimodal analgesic approach is recommended by the American Society of Anesthesiologists [49]. Due to its inherent antinociceptive qualities, dexmedetomidine has demonstrated promising results in the perioperative period [50, 51].
Initial studies evaluated dexmedetomidine for its opioid-sparing properties [52, 53]. Lin et al. randomized 100 post-hysterectomy patients to receive either morphine or morphine plus dexmedetomidine as part of patient-controlled analgesia (PCA) over the first 24 h after surgery [52]. The investigators found that patients in the combined group required one-third less morphine compared to those who received morphine alone, without any higher incidence of bradycardia, hypotension or respiratory depression [52]. Similarly, Wahlander and colleagues investigated dexmedetomidine as an adjuvant IV infusion (loading dose: 0.5 mcg/kg over 20 min, maintenance infusion: 0.4 mcg/kg/h) in patients following thoracic surgery [53]. Twenty-eight patients with epidural bupivacaine catheters for regional anesthesia at T4 were randomized to receive dexmedetomidine or placebo in the 24 h after surgery [53]. The pain scores were similar in both study arms, however, the placebo group required more epidural rescue fentanyl to maintain adequate analgesia.
A more recent RCT included patients after laparoscopic gynecologic surgery and compared postoperative dexmedetomidine PCA versus fentanyl PCA [54]. Again, no difference in pain scores was seen; however, patient satisfaction was significantly higher in the dexmedetomidine group [54]. Indeed, Peng et al. who performed a meta-analysis of 18 RCTs including 1284 heterogeneous surgical patients, and determined that combined opioid-dexmedetomidine PCA decreased postoperative pain, opioid consumption and related adverse events [55]. Doses of dexmedetomidine were generally low, and ranged from 0.045 to 0.2 mcg/kg/h [55].
Following from these and other studies, the intraoperative use of dexmedetomidine for postoperative pain was investigated. In a study from 2017, Fan et al. analyzed intraoperative dexmedetomidine infusion versus placebo in 45 patients undergoing radical mastectomy [56]. General anesthesia was maintained with propofol, remifentanil and Ringer’s lactate or propofol, as well as remifentanil and dexmedetomidine, followed by morphine-based PCA for 24 h after surgery [56]. The dexmedetomidine group exhibited lower pain scores, a longer time to initial morphine dose postoperatively, and decreased morphine consumption overall [56]. Similar findings were documented in several studies in patients undergoing abdominal surgery [57,58,59]. Ge et al. delivered intraoperative dexmedetomidine at 0.4 mcg/kg/h in hysterectomy [58] and colectomy [59] patients, and found reduced opioid consumption within the first 24 h following surgery. These results were comparative to those seen by Tufanogullari et al. after bariatric surgery; however, in these patients, opioid consumption was not decreased on postoperative days 2 and 7 [57]. Furthermore, the authors concluded that the optimal dose to achieve pain control and reduce the incidence of adverse cardiovascular events was 0.2 mcg/kg/h [57].
Analogous to its use in AFOI, remifentanil is a common agent used in neurosurgical cases where rapid emergence is required. In two trials investigating intraoperative remifentanil versus dexmedetomidine, Rajan et al. [60] and Hwang et al. [47] determined that dexmedetomidine provided superior pain relief and decreased postoperative opioid consumption for up to 48 h. Such results were not seen in a trial by Naik et al. where intraoperative dexmedetomidine (1 mcg/kg loading dose, followed by 0.5 mcg/kg/h infusion) was compared to placebo (normal saline) in patients undergoing major spinal surgery [61]. No differences were seen in postoperative opioid consumption or pain scores; however, dexmedetomidine did reduce intraoperative opioid consumption [61].
Such discrepancies were examined in a 2018 meta-analysis of 11 RCTs involving 674 neurosurgical patients [62]. The investigators concluded that dexmedetomidine reduces perioperative and post-surgical opioid consumption, in addition to reducing postoperative pain intensity [62]; optimal intraoperative doses were, however, not given. Meta-analyses including nasal [63] and general surgery [64] found comparative results, however, further trials for clarifying dosing regimens are required. Additionally, studies in pediatric surgery are lacking.
Chronic Pain Conditions
Studies investigating dexmedetomidine infusion in the treatment of chronic pain conditions are limited. Clinical evidence comes from a case report by Nama et al. [65]. The patient was a 47-year-old female with complex regional pain syndrome-1 (CRPS-1) of the upper left extremity, refractory to conventional therapy [65]. At admission, her pain was 10/10 on the pain scale and a sub-anesthetic IV infusion of ketamine (100 mcg/kg/h) was initiated. After 6 h, with pain at 7/10, a single dexmedetomidine dose of 8 mcg was given as a one-time IV bolus [65]. This led to a decrease of pain to 3/10. At the end of the 19-h infusion the patient reported pain 0/10 and was discharged [65]. The authors concluded that ketamine with adjunct dexmedetomidine is a promising treatment of acute exacerbation of CRPS [65]. Although the extract pathophysiology of CRPS remains unknown, activation and upregulation of NMDA receptors in the spinal cord leading to chronic pain has been implicated through a mechanism termed central sensitization [66]. Ketamine with its NMDA receptor antagonism is a rational agent for decreasing central sensitization. Dexmedetomidine has analgesic activity through activation of α2A- and α2C-adrenoceptors, located on C-fiber primary afferent nerve terminals and the superficial dorsal horn of the spinal cord [8]. Furthermore, dexmedetomidine has been shown to attenuate neuropathic pain by inhibiting purinergic receptor 7 and extracellular signal-regulated kinase signaling in a recent animal model [67]. Chronic constriction injury of the sciatic nerve created neuropathic pain which resolved following 2.5 mcg of intrathecal dexmedetomidine for 3 days [67]. This antinociceptive and sympatholytic effect is distinct from opioids. Indeed, this distinctive mechanism of action has proven effective in cases where opioids or ketamine are unsuccessful. Sheehy and colleagues presented a case series which included 11 adolescent patients with severe vaso-occlusive episodes due to sickle cell disease [68]. All patients were receiving morphine or hydromorphone PCA at rates of 0.02 mg/kg/h (0.02 mg/kg boluses up to every 8 min) or 0.003 mg/kg/h (0.003 mg/kg additional boluses up to 10 min apart), respectively [68]. Furthermore, the patients were receiving ketamine infusions at sub-anesthetic doses less than 1.0 mg/kg/h, in addition to non-steroidal anti-inflammatory drugs as needed. In this report, the authors identified 3 patients who required escalating doses of opioids and ketamine without sufficient pain relief. The patients, aged between 15- and 18-years, reported pain 8/10 or greater despite morphine equivalents up to 9 mg/kg/day. The first patient received a continuous dexmedetomidine infusion at 0.2–0.4 mcg/kg/h over 5 days and subsequently discontinued opioids and ketamine with a pain score 6–7/10. After a 3-day dexmedetomidine infusion at 0.5 mcg/kg/h, the second patient discontinued ketamine, reduced their opioid requirement to 2.1 mg/kg/day and reported a pain score of 2/10 [68]. The third patient reported a pain score of “14” (pain scale 0–10) on the 5th day of hospitalization and was initiated on a 1.0 mcg/kg/h continuous dexmedetomidine infusion over 5 h; this dose was decreased to 0.5 mcg/kg/h for the next 12 h and then maintained at 0.2–0.4 mcg/kg/h over the subsequent 6 days [68]. After the infusion, the patient had a pain score of 6–7/10, with an oral morphine-equivalent of 0.17 mg/kg/day. Important to note, all three patients received a transdermal clonidine patch (0.1–0.2 mg) upon completion of the dexmedetomidine infusion, which was continued for 14, 7 and 7 days, respectively. The authors suspected that these patients may have developed opioid-induced hyperalgesia (OIH) and thus dexmedetomidine may have attenuated their response to opioids. In comparison to those patients who did not receive dexmedetomidine, the marked reduction in opioid requirement and improved pain scores, nevertheless, warrants further research before solid conclusions may be reached. Indeed, as of late 2018, there are very few active or completed trials examining dexmedetomidine in chronic pain conditions including chronic postoperative and cancer pain [69].
Opioid-Induced Hyperalgesia
Patients receiving high dose short-term [70] or long-term opioids are prone to developing opioid-induced hyperalgesia (OIH). Whether through pharmacologic tachyphylaxis and tolerance, or central sensitization by NMDA receptor activation, OIH is characterized by a paradoxical increase in pain intensity and distribution [71]. Where propofol infusion has shown benefit by way of NMDA antagonism [72], the exact mechanism how α2- receptors act synergistically with opioids remains unclear. Nevertheless, limited studies have emerged that show the benefit of dexmedetomidine infusion in the treatment of OIH. The first report, a case series of 11 patients by Belgrade and Hall, demonstrated a decrease in the average pain intensity from 6/10 to 1/10 following dexmedetomidine infusion [73]. The doses ranged from 0.1–0.2 mcg/kg/h over 3 days, to 0.2 mcg/kg/h titrated up to a maximum of 0.7 mcg/kg/h over 24 h [73]. The majority of the patients continued their pre-OIH opioid therapy after infusion, however, at reduced doses. Indeed, the mean oral morphine equivalent of the 11 patients prior to infusion was reduced from 648 to 122 mg following discharge; two of the patients ceased opioids altogether. The authors concluded that dexmedetomidine “rebooted” the opioid sensitivity of these patients [73]. In a similar fashion Patch III et al. presented a case report where dexmedetomidine infusion was successfully utilized as part of a controlled multimodal analgesic plan to treat OIH [74]. The patient was a 55-year-old female with a long history of chronic pain syndrome receiving up 19,702 mcg per day of fentanyl via an intrathecal drug delivery device. Following unremarkable exploratory laparotomy, she was diagnosed with acute pain crisis and OIH secondary to uncontrolled and disproportionate abdominal pain [74]. After consultation with the pain service, a continuous dexmedetomidine infusion was initiated at 0.8 mcg/kg/h, in addition to ketamine at 0.7 mg/kg/h with 10 mg IV boluses every 30 min as required, alongside extra ketorolac, and acetaminophen doses, and lidocaine patches around the incision [74]. Over 48 h both infusions were weaned, as hydromorphone (1 mg) and ketamine IV boluses were increased to 4-h intervals. Her pain subsided on the third postoperative day and she was transferred to the ward, where oral hydromorphone (4 mg) was prescribed every 6 h as needed until discharge; at 3-month follow-up her intrathecal fentanyl dose had reduced to 11,000 mcg per day [74]. Although dexmedetomidine was successfully used in this case, it remains only the second report published on OIH in patients with chronic pain syndromes.
High-dose remifentanil has been increasingly associated with the development of OIH in the acute postoperative setting [75]. To date however, few clinical trials have examined the antihyperalgesic effect of dexmedetomidine. In the first RCT from 2013, Lee and colleagues recruited 90 patients aged 20–65 years who were scheduled for laparoscopically assisted vaginal hysterectomy, to investigate whether dexmedetomidine could attenuate remifentanil-induced secondary hyperalgesia in the initial 24 h postoperative period [76]. The patients were randomized to three groups: placebo (normal saline) and 0.05 mcg/kg/min remifentanil, placebo and 0.3 mcg/kg/min remifentanil, and dexmedetomidine (1.0 mcg/kg over 10 min loading dose, followed by 0.7 mcg/kg/h) and 0.3 mcg/kg/min remifentanil [76]. Patients in the second group (with high dose remifentanil and placebo) were suspected to have developed OIH due to higher postoperative VAS scores, reduced mechanical hyperalgesia thresholds, and greater utilization of morphine PCA [76]; on the contrary, these findings were not seen in the third group who had received dexmedetomidine in addition to high dose remifentanil [76]. The authors concluded that intraoperative dexmedetomidine alleviated OIH symptoms in these patients, adding that it may have preventative properties [76]. Following on from this in 2016, Yu et al. conducted a similar RCT in the same subset of patients, to investigate whether dexmedetomidine combined with flubiprofen axetil (cyclooxygenase inhibitor) had comparable results [77]. Ninety-five female patients were included and randomized to similar groups as the aforementioned RCT; first group only high-dose remifentanil, second group high-dose remifentanil and dexmedetomidine, with the third group receiving high-dose remifentanil, dexmedetomidine (0.5 mcg/kg over 10 min loading dose, followed by 0.6 mcg/kg/h continuous infusion) and flubiprofen axetil [77]. The primary outcome was mechanical pain threshold in the first 24 h after surgery. In this study the patients who received dexmedetomidine, with or without flubiprofen axetil experienced higher pain thresholds (p < 0.05) [77]. Furthermore, the postoperative VAS scores were significantly lower in the two dexmedetomidine groups (p < 0.05). This RCT concluded that when used in combination with flubiprofen axetil, dexmedetomidine provided the most significant attenuation of remifentanil-induced hyperalgesia [77]. Whether gender differences exist between patients was investigated in the most recent RCT from 2018 [78]. Forty-eight patients undergoing thyroidectomy were included and randomized into six male and female groups: control (0.2 mcg/kg normal saline), low-dose and high-dose dexmedetomidine injection (0.2 mcg/kg and 0.6 mcg/kg, respectively) [78]. Dexmedetomidine was given preoperatively and all patients received intraoperative remifentanil at 0.2 mcg/kg/min [78]. No significant differences were seen amongst the genders, and expectedly the dexmedetomidine groups demonstrated less PONV and shivering, in addition to lower pain scores by VAS and significantly higher mechanical hyperalgesia thresholds (p < 0.05) [78]. The underlying mechanism behind the preventative nature of dexmedetomidine on remifentanil-induced hyperalgesia has been attributed to regulation of the NMDA receptor-protein kinase C-calmodulin-dependent protein kinase II pathway in a recent rat model, however, human studies have not been performed [79].
Nevertheless, these few RCTs show that dexmedetomidine is a promising agent for remifentanil-induced hyperalgesia in the postoperative period. However, more studies are required to further our knowledge of its effect in OIH in patients with chronic pain conditions not undergoing surgical procedures.
Contraindications
There are no absolute contraindications to dexmedetomidine infusion. Nevertheless, it is an in vitro inhibitor of CYP2D6, CYP2C9, CYP1A, CYP3A and CYP3A4 [80]. In vivo, however, it does not inhibit the pharmacokinetics of midazolam, a CYP3A4 substrate [81]. Inhibition of CYP2D6 may affect the metabolism of opioids including oxycodone and tramadol, diminishing their analgesic effect [82]; however, this remains unproven. In contrast, dexmedetomidine has been shown to reduce the requirements of other anesthetics including sevoflurane [83], isoflurane [84], propofol [85], and thiopental [86], in addition to having opioid-sparing properties as discussed earlier. Perhaps the most well-known interactions were investigated in pre-registration studies. Antihypertensive agents such as β-blockers may increase the hypotensive and bradycardic effects of dexmedetomidine [5]. These effects may be further exaggerated with concomitant vagal stimulation as seen during sternal separation, colonoscopy and laparoscopic insufflation [39, 87, 88].
Renal impairment does not warrant dose reduction [3]. Hepatic impairment and/or hypoalbuminemia, however, necessitate dose reductions due to diminished clearance and an increase in unbound plasma concentration favoring toxicity [3, 89].
Side Effects
The side effect profile of dexmedetomidine is largely predictable and based on its receptor binding pharmacology. The most common adverse events are hemodynamic in nature, namely transient hypertension, bradycardia, and hypotension [5, 38, 90, 91]. It demonstrates a biphasic effect on blood pressure, producing hypertension at high plasma concentrations and hypotension at lower plasma concentrations [92, 93]. Rapid IV bolus or fast loading dose administration has been associated with progressive increases in mean arterial pressure [2, 93]. These effects are thought to occur due to α2B-receptor activation in vascular smooth muscle, resulting in peripheral vasoconstriction accompanied by a baroreceptor-mediated reduction in heart rate [92, 93]. This increase in systemic and pulmonary vascular resistance may lead to systemic and pulmonary hypertension, which can limit its use in patients with serious cardiac morbidity. At plasma concentrations between 1.9 and 3.2 ng/ml these hypertensive effects prevail, and may be mitigated by decreasing and slowing loading doses and avoiding IV bolus administration [5, 93]. The hypotensive phase occurs at lower plasma concentrations and for a prolonged time after the initial dose, with an average decrease in mean arterial pressure of 13–27%, mediated by presynaptic α2-receptor inhibition of catecholamine release and increased vagal activity [92, 93].
These side effects are to be expected, however, the most serious and life-threatening adverse events are difficult to predict. Numerous case reports and series describing severe bradycardia and/or atrioventricular block precipitating cardiac arrest have been reported in the literature [87, 94,95,96,97,98]. Ingersoll-Weng et al. were the first to report an episode of asystole during intraoperative dexmedetomidine infusion in a 52-year-old patient with myasthenia gravis undergoing thymectomy via median sternotomy [94]. Dexmedetomidine infusion was started prior to induction of general anesthesia, with a loading dose of 1 mcg/kg over 10 min followed by a rate of 0.2 mcg/kg/h. Asystole occurred immediately after sternotomy, lasted for 2 min and resolved following internal cardiac massage and 300 mcg/kg IV epinephrine [94]. The authors concluded that caution should be taken when dexmedetomidine is used in conjunction with other cardiac depressants [94]. Vagal stimulation caused by sternotomy may have also potentiated the bradycardia, which preceded cardiac arrest. Shah et al. reported cardiac arrest and subsequent death during procedural sedation with dexmedetomidine in a 76-year-old patient undergoing exchange of an infected permanent pacemaker [87]. An infusion of 1 mcg/kg was initiated and intended for 20 min duration (total dose 95 mcg). After 15 min, however, having received 71.25 mcg of dexmedetomidine, the patient began to cough, became dyspneic and lost consciousness; pacing spikes without capture were seen on the electrocardiogram [87]. Although these doses are acceptable loading doses, the plasma concentration immediately after the infusion began would have been 1.7 ng/ml, with cardiac depression induced at concentrations exceeding 1.2 ng/ml [92]. Takata et al. reported dexmedetomidine-induced atrioventricular (AV) block and cardiac arrest in a 56-year-old patient under sedation in the ICU on the first postoperative day following a Bentall procedure [95]. As part of the postsurgical regimen the patient was receiving atrial pacing at a rate of 90 bpm [95]. In the ICU a dexmedetomidine infusion was initiated at a rate of 0.3 mcg/kg/h without a loading dose. Five hours and 30 min into the infusion, the interval between a pacing spike and a Q wave increased to 340 ms, and the patient subsequently developed complete AV block without an escape rhythm (asystole) [95]. The authors concluded that even low-dose dexmedetomidine may disturb AV conduction in addition to having an inhibitory effect on cardiac pacing autoregulation, above all, in patients with conduction abnormalities [95]. They added that ventricular pacing alongside standard atrial pacing may be prudent in similar patients [95].
Bharati et al. reported a series of six instances of intraoperative bradycardia leading to asystole [96]. Most were given loading doses of 1 mcg/kg, however, over less than 10 min, which led to plasma concentrations ranging from 0.77 to 1.8 ng/ml [95, 96]. The authors concluded that dexmedetomidine should be used with extreme caution when combined with negative chronotropes and inotropes, furthermore stating that dexmedetomidine should be avoided in elderly patients and those with cardiac disease.
Similar adverse reactions have been seen in pediatric patients. Shepard et al. presented a case of a 3-year-old patient with a history of repaired congenital heart disease, and a permanent pacemaker. On the second day following a mitral valve replacement surgery, the patient developed atrial standstill with a loss of capture after 21 h of dexmedetomidine infusion at a rate of 0.6 mcg/kg/h [98]. Immediate cessation of dexmedetomidine led to resolution without sequela. Similarly, Zhang et al. reported 10 s bouts of asystole in an 18-year-old patient with cystic fibrosis and a history of double-lung transplant in the ICU during mechanical ventilation [97]. On the 22nd postoperative day following laparoscopic fundoplication for gastroesophageal reflux disease, the patient, already on a propofol (0.5–2.5 mg/kg/h) and fentanyl infusions (up to 4 mcg/kg/h), was started on dexmedetomidine with a loading dose of 1 mcg/kg over 10 min followed by an infusion of 0.4–0.7 mcg/kg/h with the intention of weaning from propofol [97]. Over the following hours, propofol and fentanyl infusions were reduced but not stopped. After 8 h of dexmedetomidine (infused at a maximum rate of 0.7–0.8 mcg/kg/h), the patient developed a bradycardia which progressed to a 10-s period of asystole [97]. Over the subsequent 24 h, four additional episodes occurred, three during and one after the dexmedetomidine infusion. These instances of asystole were not pharmacologically treated, and the patient was uneventfully discharged on the 32nd postoperative day [97]. The authors concluded that changes to cardiac autonomic innervation that can occur after double-lung transplant place such patients at increased risk for these phenomena [97].
The consensus of the aforementioned cases is that dexmedetomidine, at any dose, should be used with extreme caution when combined with other common anesthetic agents, and in patients with cardiac abnormalities. Nevertheless, these severe adverse events are exceedingly rare.
The specific α2-andrenoceptor antagonist atipamezole has been shown to rapidly reverse the sedative and hemodynamic effects of dexmedetomidine [99,100,101]. Although widely used in veterinary medicine, to date this agent has not been approved for use in humans [102].
Monitoring
The use of dexmedetomidine requires continuous monitoring of the cardiovascular and respiratory systems. Non-invasive or continuous blood pressure monitoring, electrocardiography and pulse oximetry are the minimal recommendations. BIS targeted between 60 and 80 may be beneficial in neonates and infants. As hepatic impairment may necessitate dose adjustment, liver function tests should be ordered before initiating the infusion. Please review your local monitoring and support guidelines regarding the use of dexmedetomidine.
Algorithm for Dexmedetomidine Infusion Regimens
The following algorithm summarizes the IV infusion dosing regimens published in the literature (Table 10.1). Differing settings and patients may require different doses.
Summary
Dexmedetomidine has numerous uses outside of the approved indications. Doses vary; however, most include a 1 mcg/kg/hr. IV loading dose over 10 min followed by a maintenance infusion of 0.2–0.7 mcg/kg/hr. Duration may safely extend beyond 24 h. Due to the risk of hemodynamic adverse events, careful monitoring of the cardiovascular and respiratory systems with non-invasive blood pressure measurement, electrocardiography, and pulse oximetry are the minimal recommendations. For the management of acute postoperative pain, optimal intraoperative doses range from 0.1–0.7 mcg/kg/h, and 0.045–0.2 mcg/kg/h when combined with an opioid for PCA. When given in the context of an acute CRPS exacerbation, dexmedetomidine as a single 8 mcg IV bolus has shown benefit when used as an adjunct to ketamine infusion. Infusion at 0.1–0.2 mcg/kg/h over 3 days, or 0.2 mcg/kg/h titrated to 0.7 mcg/kg/h over 24 h has shown benefit in alleviating OIH. Dosing for the prevention of delirium in adult non-surgical ICU patients includes a low-dose nocturnal infusion at 0.2 mcg/kg/h titrated by 0.1 mcg/kg/min every 15 min until the desired level of sedation is reached. The doses for alleviating emergence delirium in children vary greatly from intraoperative infusions of 0.2 mcg/kg/h to single IV doses of 0.5–1.0 mcg/kg. Slower loading doses over 10–20 min and longer infusions (up to 10 days) may be necessary when used for mitigating drug withdrawal. For AFOI and procedural sedation doses are also varied, however, 0.5–1.0 mcg/kg over 10 min loading dose followed by 0.2–1.0 mcg/kg/h maintenance infusion is optimal. For preventing PONV a continuous intraoperative infusion at 0.1–0.7 mcg/kg/h is advised.
Most dosing regimens do not differ greatly from those recommended upon registration. However, the data on numerous indications remains scarce and further studies are required.
References
Bhana N, Goa KL, McClellan KJ. Dexmedetomidine. Drugs. 2000;59(2):263–8. discussion 9–70.
Weerink MAS, Struys M, Hannivoort LN, Barends CRM, Absalom AR, Colin P. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893–913.
US Food and Drug Administration. Precedex label. 1999. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021038s017lbl.pdf.
Weatherall M, Aantaa R, Conti G, Garratt C, Pohjanjousi P, Lewis MA, et al. A multinational, drug utilization study to investigate the use of dexmedetomidine (Dexdor(R)) in clinical practice in the EU. Br J Clin Pharmacol. 2017;83(9):2066–76.
European Medicines Agency. Dexdor: EPAR – Product Information. 2018. Available from: https://www.ema.europa.eu/documents/product-information/dexdor-epar-product-information_en.pdf.
Hoy SM, Keating GM. Dexmedetomidine: a review of its use for sedation in mechanically ventilated patients in an intensive care setting and for procedural sedation. Drugs. 2011;71(11):1481–501.
Tan JA, Ho KM. Use of dexmedetomidine as a sedative and analgesic agent in critically ill adult patients: a meta-analysis. Intensive Care Med. 2010;36(6):926–39.
Nazarian A, Christianson CA, Hua XY, Yaksh TL. Dexmedetomidine and ST-91 analgesia in the formalin model is mediated by alpha2A-adrenoceptors: a mechanism of action distinct from morphine. Br J Pharmacol. 2008;155(7):1117–26.
Arcangeli A, D’Alo C, Gaspari R. Dexmedetomidine use in general anaesthesia. Curr Drug Targets. 2009;10(8):687–95.
Correa-Sales C, Rabin BC, Maze M. A hypnotic response to dexmedetomidine, an alpha 2 agonist, is mediated in the locus coeruleus in rats. Anesthesiology. 1992;76(6):948–52.
McKay BE, Lado WE, Martin LJ, Galic MA, Fournier NM. Learning and memory in agmatine-treated rats. Pharmacol Biochem Behav. 2002;72(3):551–7.
Reis DJ, Regunathan S, Golanov EV, Feinstein DL. Protection of focal ischemic infarction by rilmenidine in the animal: evidence that interactions with central imidazoline receptors may be neuroprotective. Am J Cardiol. 1994;74(13):25A–30A.
Khan ZPFC, Jones RM. Alpha-2 and imidazoline receptor agonists. Their pharmacology and therapeutic role. Anaesthesia. 2002;54(2):146–65.
Sullivan AF, Kalso EA, McQuay HJ, Dickenson AH. The antinociceptive actions of dexmedetomidine on dorsal horn neuronal responses in the anaesthetized rat. Eur J Pharmacol. 1992;215(1):127–33.
Mukaddam-Daher S, Gutkowska J. Imidazoline receptors in the heart: a novel target and a novel mechanism of action that involves atrial natriuretic peptides. Braz J Med Biol Res. 2004;37(8):1239–45.
Haselman MA. Dexmedetomidine: a useful adjunct to consider in some high-risk situations. AANA J. 2008;76(5):335–9.
Hall JE, Uhrich TD, Barney JA, Arain SR, Ebert TJ. Sedative, amnestic, and analgesic properties of small-dose dexmedetomidine infusions. Anesth Analg. 2000;90(3):699–705.
Venn RM, Bradshaw CJ, Spencer R, Brealey D, Caudwell E, Naughton C, et al. Preliminary UK experience of dexmedetomidine, a novel agent for postoperative sedation in the intensive care unit. Anaesthesia. 1999;54(12):1136–42.
Venn RM, Grounds RM. Comparison between dexmedetomidine and propofol for sedation in the intensive care unit: patient and clinician perceptions. Br J Anaesth. 2001;87(5):684–90.
Hsu YW, Cortinez LI, Robertson KM, Keifer JC, Sum-Ping ST, Moretti EW, et al. Dexmedetomidine pharmacodynamics: part I: crossover comparison of the respiratory effects of dexmedetomidine and remifentanil in healthy volunteers. Anesthesiology. 2004;101(5):1066–76.
Virtanen R, Savola JM, Saano V, Nyman L. Characterization of the selectivity, specificity and potency of medetomidine as an alpha 2-adrenoceptor agonist. Eur J Pharmacol. 1988;150(1–2):9–14.
Knezevic NN, Yekkirala A, Yaksh TL. Basic/translational development of forthcoming opioid- and nonopioid-targeted pain therapeutics. Anesth Analg. 2017;125(5):1714–32.
Al-Metwalli RR, Mowafi HA, Ismail SA, Siddiqui AK, Al-Ghamdi AM, Shafi MA, et al. Effect of intra-articular dexmedetomidine on postoperative analgesia after arthroscopic knee surgery. Br J Anaesth. 2008;101(3):395–9.
Yuen VM, Irwin MG, Hui TW, Yuen MK, Lee LH. A double-blind, crossover assessment of the sedative and analgesic effects of intranasal dexmedetomidine. Anesth Analg. 2007;105(2):374–80.
Anttila M, Penttila J, Helminen A, Vuorilehto L, Scheinin H. Bioavailability of dexmedetomidine after extravascular doses in healthy subjects. Br J Clin Pharmacol. 2003;56(6):691–3.
Abdallah FW, Brull R. Facilitatory effects of perineural dexmedetomidine on neuraxial and peripheral nerve block: a systematic review and meta-analysis. Br J Anaesth. 2013;110(6):915–25.
Venn M, Newman J, Grounds M. A phase II study to evaluate the efficacy of dexmedetomidine for sedation in the medical intensive care unit. Intensive Care Med. 2003;29(2):201–7.
Shehabi Y, Ruettimann U, Adamson H, Innes R, Ickeringill M. Dexmedetomidine infusion for more than 24 hours in critically ill patients: sedative and cardiovascular effects. Intensive Care Med. 2004;30(12):2188–96.
Skrobik Y, Duprey MS, Hill NS, Devlin JW. Low-dose nocturnal dexmedetomidine prevents ICU delirium. A randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2018;197(9):1147–56.
Manchikanti L, Helm S 2nd, Fellows B, Janata JW, Pampati V, Grider JS, et al. Opioid epidemic in the United States. Pain Physician. 2012;15(3 Suppl):ES9–38.
Donroe JH, Tetrault JM. Substance use, intoxication, and withdrawal in the critical care setting. Crit Care Clin. 2017;33(3):543–58.
Maccioli GA. Dexmedetomidine to facilitate drug withdrawal. Anesthesiology. 2003;98(2):575–7.
Finkel JC, Elrefai A. The use of dexmedetomidine to facilitate opioid and benzodiazepine detoxification in an infant. Anesth Analg. 2004;98(6):1658–9, table of contents
Finkel JC, Johnson YJ, Quezado ZM. The use of dexmedetomidine to facilitate acute discontinuation of opioids after cardiac transplantation in children. Crit Care Med. 2005;33(9):2110–2.
Baddigam K, Russo P, Russo J, Tobias JD. Dexmedetomidine in the treatment of withdrawal syndromes in cardiothoracic surgery patients. J Intensive Care Med. 2005;20(2):118–23.
Zhou LJ, Fang XZ, Gao J, Zhangm Y, Tao LJ. Safety and efficacy of dexmedetomidine as a sedative agent for performing awake intubation: a meta-analysis. Am J Ther. 2016;23(6):e1788–e800.
Hu R, Liu JX, Jiang H. Dexmedetomidine versus remifentanil sedation during awake fiberoptic nasotracheal intubation: a double-blinded randomized controlled trial. J Anesth. 2013;27(2):211–7.
Candiotti KA, Bergese SD, Bokesch PM, Feldman MA, Wisemandle W, Bekker AY, et al. Monitored anesthesia care with dexmedetomidine: a prospective, randomized, double-blind, multicenter trial. Anesth Analg. 2010;110(1):47–56.
Jalowiecki P, Rudner R, Gonciarz M, Kawecki P, Petelenz M, Dziurdzik P. Sole use of dexmedetomidine has limited utility for conscious sedation during outpatient colonoscopy. Anesthesiology. 2005;103(2):269–73.
Jin S, Liang DD, Chen C, Zhang M, Wang J. Dexmedetomidine prevent postoperative nausea and vomiting on patients during general anesthesia: a PRISMA-compliant meta analysis of randomized controlled trials. Medicine (Baltimore). 2017;96(1):e5770.
Wang G, Zhang L, Lou S, Chen Y, Cao Y, Wang R, et al. Effect of dexmedetomidine in preventing postoperative side effects for laparoscopic surgery: a Meta-analysis of randomized controlled trials and trial sequential analysis (PRISMA). Medicine (Baltimore). 2016;95(10):e2927.
Sottas CE, Anderson BJ. Dexmedetomidine: the new all-in-one drug in paediatric anaesthesia? Curr Opin Anaesthesiol. 2017;30(4):441–51.
Wang X, Deng Q, Liu B, Yu X. Preventing emergence agitation using ancillary drugs with sevoflurane for pediatric anesthesia: a network meta-analysis. Mol Neurobiol. 2017;54(9):7312–26.
Makkar JK, Bhatia N, Bala I, Dwivedi D, Singh PM. A comparison of single dose dexmedetomidine with propofol for the prevention of emergence delirium after desflurane anaesthesia in children. Anaesthesia. 2016;71(1):50–7.
Davy A, Fessler J, Fischler M, LE Guen M. Dexmedetomidine and general anesthesia: a narrative literature review of its major indications for use in adults undergoing non-cardiac surgery. Minerva Anestesiol. 2017;83(12):1294–308.
Ziemann-Gimmel P, Goldfarb AA, Koppman J, Marema RT. Opioid-free total intravenous anaesthesia reduces postoperative nausea and vomiting in bariatric surgery beyond triple prophylaxis. Br J Anaesth. 2014;112(5):906–11.
Hwang W, Lee J, Park J, Joo J. Dexmedetomidine versus remifentanil in postoperative pain control after spinal surgery: a randomized controlled study. BMC Anesthesiol. 2015;15:21.
Helander EM, Menard BL, Harmon CM, Homra BK, Allain AV, Bordelon GJ, et al. Multimodal analgesia, current concepts, and acute pain considerations. Curr Pain Headache Rep. 2017;21(1):3.
American Society of Anesthesiologists Task Force on Acute Pain M. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology. 2012;116(2):248–73.
Luo J, Min S. Postoperative pain management in the postanesthesia care unit: an update. J Pain Res. 2017;10:2687–98.
Tang C, Xia Z. Dexmedetomidine in perioperative acute pain management: a non-opioid adjuvant analgesic. J Pain Res. 2017;10:1899–904.
Lin TF, Yeh YC, Lin FS, Wang YP, Lin CJ, Sun WZ, et al. Effect of combining dexmedetomidine and morphine for intravenous patient-controlled analgesia. Br J Anaesth. 2009;102(1):117–22.
Wahlander S, Frumento RJ, Wagener G, Saldana-Ferretti B, Joshi RR, Playford HR, et al. A prospective, double-blind, randomized, placebo-controlled study of dexmedetomidine as an adjunct to epidural analgesia after thoracic surgery. J Cardiothorac Vasc Anesth. 2005;19(5):630–5.
Wang X, Liu W, Xu Z, Wang F, Zhang C, Wang B, et al. Effect of dexmedetomidine alone for intravenous patient-controlled analgesia after gynecological laparoscopic surgery: a consort-prospective, randomized, controlled trial. Medicine (Baltimore). 2016;95(19):e3639.
Peng K, Zhang J, Meng XW, Liu HY, Ji FH. Optimization of postoperative intravenous patient-controlled analgesia with opioid-dexmedetomidine combinations: an updated meta-analysis with trial sequential analysis of randomized controlled trials. Pain Physician. 2017;20(7):569–96.
Fan W, Xue H, Sun Y, Yang H, Zhang J, Li G, et al. Dexmedetomidine improves postoperative patient-controlled analgesia following radical mastectomy. Front Pharmacol. 2017;8:250.
Tufanogullari B, White PF, Peixoto MP, Kianpour D, Lacour T, Griffin J, et al. Dexmedetomidine infusion during laparoscopic bariatric surgery: the effect on recovery outcome variables. Anesth Analg. 2008;106(6):1741–8.
Ge DJ, Qi B, Tang G, Li JY. Intraoperative dexmedetomidine promotes postoperative analgesia and recovery in patients after abdominal hysterectomy: a double-blind. Randomized Clinical Trial Sci Rep. 2016;6:21514.
Ge DJ, Qi B, Tang G, Li JY. Intraoperative dexmedetomidine promotes postoperative analgesia and recovery in patients after abdominal colectomy: a CONSORT-prospective, randomized, controlled clinical trial. Medicine (Baltimore). 2015;94(43):e1727.
Rajan S, Hutcherson MT, Sessler DI, Kurz A, Yang D, Ghobrial M, et al. The effects of dexmedetomidine and remifentanil on hemodynamic stability and analgesic requirement after craniotomy: a randomized controlled trial. J Neurosurg Anesthesiol. 2016;28(4):282–90.
Naik BI, Nemergut EC, Kazemi A, Fernandez L, Cederholm SK, McMurry TL, et al. The effect of dexmedetomidine on postoperative opioid consumption and pain after major spine surgery. Anesth Analg. 2016;122(5):1646–53.
Liu Y, Liang F, Liu X, Shao X, Jiang N, Gan X. Dexmedetomidine reduces perioperative opioid consumption and postoperative pain intensity in neurosurgery: a Meta-analysis. J Neurosurg Anesthesiol. 2018;30(2):146–55.
Lee HS, Yoon HY, Jin HJ, Hwang SH. Can dexmedetomidine influence recovery profiles from general anesthesia in nasal surgery? Otolaryngol Head Neck Surg. 2018;158(1):43–53.
Le Bot A, Michelet D, Hilly J, Maesani M, Dilly MP, Brasher C, et al. Efficacy of intraoperative dexmedetomidine compared with placebo for surgery in adults: a meta-analysis of published studies. Minerva Anestesiol. 2015;81(10):1105–17.
Nama S, Meenan DR, Fritz WT. The use of sub-anesthetic intravenous ketamine and adjuvant dexmedetomidine when treating acute pain from CRPS. Pain Physician. 2010;13(4):365–8.
Zhou QQ, Imbe H, Zou S, Dubner R, Ren K. Selective upregulation of the flip-flop splice variants of AMPA receptor subunits in the rat spinal cord after hindpaw inflammation. Brain Res Mol Brain Res. 2001;88(1–2):186–93.
Lin JP, Chen CQ, Huang LE, Li NN, Yang Y, Zhu SM, et al. Dexmedetomidine attenuates neuropathic pain by inhibiting P2X7R expression and ERK phosphorylation in rats. Exp Neurobiol. 2018;27(4):267–76.
Sheehy KA, Finkel JC, Darbari DS, Guerrera MF, Quezado ZM. Dexmedetomidine as an adjuvant to analgesic strategy during vaso-occlusive episodes in adolescents with sickle-cell disease. Pain Pract. 2015;15(8):E90–7.
clinicaltrials.gov. 2018. Available from: https://clinicaltrials.gov/ct2/results?cond=Chronic+Pain&term=dexmedetomidine&cntry=&state=&city=&dist=.
Guignard B, Bossard AE, Coste C, Sessler DI, Lebrault C, Alfonsi P, et al. Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology. 2000;93(2):409–17.
Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24(6):479–96.
Kaye AD, Chung KS, Vadivelu N, Cantemir C, Urman RD, Manchikanti L. Opioid induced hyperalgesia altered with propofol infusion. Pain Physician. 2014;17(2):E225–8.
Belgrade M, Hall S. Dexmedetomidine infusion for the management of opioid-induced hyperalgesia. Pain Med. 2010;11(12):1819–26.
Patch Iii RK, Eldrige JS, Moeschler SM, Pingree MJ. Dexmedetomidine as part of a multimodal analgesic treatment regimen for opioid induced hyperalgesia in a patient with significant opioid tolerance. Case Rep Anesthesiol. 2017;2017:9876306.
Santonocito C, Noto A, Crimi C, Sanfilippo F. Remifentanil-induced postoperative hyperalgesia: current perspectives on mechanisms and therapeutic strategies. Local Reg Anesth. 2018;11:15–23.
Lee C, Kim YD, Kim JN. Antihyperalgesic effects of dexmedetomidine on high-dose remifentanil-induced hyperalgesia. Korean J Anesthesiol. 2013;64(4):301–7.
Yu Z, Wu W, Wu X, Lei H, Gong C, Xu S. Protective effects of dexmedetomidine combined with flurbiprofen axetil on remifentanil-induced hyperalgesia: a randomized controlled trial. Exp Ther Med. 2016;12(4):2622–8.
Qiu H, Sun Z, Shadhiya F, Arulthas R, Priya GV, Christopher P, et al. The influence of dexmedetomidine on remifentanil-induced hyperalgesia and the sex differences. Exp Ther Med. 2018;16(4):3596–602.
Yuan Y, Sun Z, Chen Y, Zheng Y, Xie KL, He Y, et al. Prevention of remifentanil induced postoperative hyperalgesia by dexmedetomidine via regulating the trafficking and function of spinal NMDA receptors as well as PKC and CaMKII level in vivo and in vitro. PLoS One. 2017;12(2):e0171348.
Rodrigues AD, Roberts EM. The in vitro interaction of dexmedetomidine with human liver microsomal cytochrome P4502D6 (CYP2D6). Drug Metab Dispos. 1997;25(5):651–5.
Karol MD, Maze M. Pharmacokinetics and interaction pharmacodynamics of dexmedetomidine in humans. Best Pract Res Clin Anaesthesiol. 2000;14(2):261–9.
Prommer E. Review article: dexmedetomidine: does it have potential in palliative medicine? Am J Hosp Palliat Care. 2011;28(4):276–83.
Savla JR, Ghai B, Bansal D, Wig J. Effect of intranasal dexmedetomidine or oral midazolam premedication on sevoflurane EC50 for successful laryngeal mask airway placement in children: a randomized, double-blind, placebo-controlled trial. Paediatr Anaesth. 2014;24(4):433–9.
Aantaa R, Jaakola ML, Kallio A, Kanto J. Reduction of the minimum alveolar concentration of isoflurane by dexmedetomidine. Anesthesiology. 1997;86(5):1055–60.
Jang YE, Kim YC, Yoon HK, Jeon YT, Hwang JW, Kim E, et al. A randomized controlled trial of the effect of preoperative dexmedetomidine on the half maximal effective concentration of propofol for successful i-gel insertion without muscle relaxants. J Anesth. 2015;29(3):338–45.
Buhrer M, Mappes A, Lauber R, Stanski DR, Maitre PO. Dexmedetomidine decreases thiopental dose requirement and alters distribution pharmacokinetics. Anesthesiology. 1994;80(6):1216–27.
Shah AN, Koneru J, Nicoara A, Goldfeder LB, Thomas K, Ehlert FA. Dexmedetomidine related cardiac arrest in a patient with permanent pacemaker; a cautionary tale. Pacing Clin Electrophysiol. 2007;30(9):1158–60.
Muntazar M, Kumar FC. Cardiac arrest, a preventable yet a possible risk of dexmedetomidine: fact or fiction? Anesthesiology. 2004;101(6):1478–9. author reply 9–80.
Panzer O, Moitra V, Sladen RN. Pharmacology of sedative-analgesic agents: dexmedetomidine, remifentanil, ketamine, volatile anesthetics, and the role of peripheral mu antagonists. Crit Care Clin. 2009;25(3):451–69. vii.
Martin E, Ramsay G, Mantz J, Sum-Ping ST. The role of the alpha2-adrenoceptor agonist dexmedetomidine in postsurgical sedation in the intensive care unit. J Intensive Care Med. 2003;18(1):29–41.
Bergese SD, Candiotti KA, Bokesch PM, Zura A, Wisemandle W, Bekker AY, et al. A phase IIIb, randomized, double-blind, placebo-controlled, multicenter study evaluating the safety and efficacy of dexmedetomidine for sedation during awake fiberoptic intubation. Am J Ther. 2010;17(6):586–95.
Bloor BC, Ward DS, Belleville JP, Maze M. Effects of intravenous dexmedetomidine in humans. II Hemodynamic changes. Anesthesiology. 1992;77(6):1134–42.
Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology. 2000;93(2):382–94.
Ingersoll-Weng E, Manecke GR Jr, Thistlethwaite PA. Dexmedetomidine and cardiac arrest. Anesthesiology. 2004;100(3):738–9.
Takata K, Adachi YU, Suzuki K, Obata Y, Sato S, Nishiwaki K. Dexmedetomidine-induced atrioventricular block followed by cardiac arrest during atrial pacing: a case report and review of the literature. J Anesth. 2014;28(1):116–20.
Bharati S, Pal A, Biswas C, Biswas R. Incidence of cardiac arrest increases with the indiscriminate use of dexmedetomidine: a case series and review of published case reports. Acta Anaesthesiol Taiwanica. 2011;49(4):165–7.
Zhang X, Schmidt U, Wain JC, Bigatello L. Bradycardia leading to asystole during dexmedetomidine infusion in an 18 year-old double-lung transplant recipient. J Clin Anesth. 2010;22(1):45–9.
Shepard SM, Tejman-Yarden S, Khanna S, Davis CK, Batra AS. Dexmedetomidine-related atrial standstill and loss of capture in a pediatric patient after congenital heart surgery. Crit Care Med. 2011;39(1):187–9.
Karhuvaara S, Kallio A, Scheinin M, Anttila M, Salonen JS, Scheinin H. Pharmacological effects and pharmacokinetics of atipamezole, a novel alpha 2-adrenoceptor antagonist--a randomized, double-blind cross-over study in healthy male volunteers. Br J Clin Pharmacol. 1990;30(1):97–106.
Karhuvaara S, Kallio A, Salonen M, Tuominen J, Scheinin M. Rapid reversal of alpha 2-adrenoceptor agonist effects by atipamezole in human volunteers. Br J Clin Pharmacol. 1991;31(2):160–5.
Scheinin H, Aantaa R, Anttila M, Hakola P, Helminen A, Karhuvaara S. Reversal of the sedative and sympatholytic effects of dexmedetomidine with a specific alpha2-adrenoceptor antagonist atipamezole: a pharmacodynamic and kinetic study in healthy volunteers. Anesthesiology. 1998;89(3):574–84.
Paris A, Tonner PH. Dexmedetomidine in anaesthesia. Curr Opin Anaesthesiol. 2005;18(4):412–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zec, S., Tharian, A., Candido, K.D., Knezevic, N.N. (2019). Dexmedetomidine Infusion Therapy. In: Abd-Elsayed, A. (eds) Infusion Therapy. Springer, Cham. https://doi.org/10.1007/978-3-030-17478-1_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-17478-1_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-17477-4
Online ISBN: 978-3-030-17478-1
eBook Packages: MedicineMedicine (R0)