
Abstract
During a progressive exercise test, the anaerobic threshold (theoretically) occurs when aerobic metabolism, limited as it is by the amount of O2 delivered by the cardiovascular system, is insufficient to meet the energy requirements of the exercising muscles. The anaerobic threshold is a physiologic phenomenon that is not affected by patient effort or motivation and may be determined on a submaximal exercise test. Consequently, it is an excellent index of the cardiovascular system’s capacity to support the hemodynamic demands of exercise. Because anaerobic metabolism produces CO2 (through the buffering of lactic acid by bicarbonate) but does not consume O2, during a progressive exercise test, the ventilatory anaerobic threshold (VAT) is marked by an increase in V̇CO2 out of proportion to the associated increase in V̇O2. This phenomenon can be detected by expiratory gas analysis. The V̇O2 at this point is termed the “VAT.” Other parameters from submaximal exercise may also shed light upon a patient’s cardiopulmonary function.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Submaximal exercise
- Ventilatory anaerobic threshold
- V̇E/V̇CO2 slope
- End-tidal pCO2
- Oxygen uptake efficiency slope
- Oxygen uptake-work rate relationship
- Exercise electrocardiogram
Ventilatory Anaerobic Threshold (VAT)
As discussed earlier in this book, during a progressive exercise test, the anaerobic threshold (theoretically) occurs when aerobic metabolism, limited as it is by the amount of O2 delivered by the cardiovascular system, is insufficient to meet the energy requirements of the exercising muscles. The anaerobic threshold is a physiologic phenomenon that is not affected by patient effort or motivation and may be determined on a submaximal exercise test. Consequently, it is an excellent index of the cardiovascular system’s capacity to support the hemodynamic demands of exercise. Because anaerobic metabolism produces CO2 (through the buffering of lactic acid by bicarbonate) but does not consume O2, during a progressive exercise test, the ventilatory anaerobic threshold (VAT) is marked by an increase in V̇CO2 out of proportion to the associated increase in V̇O2. This phenomenon can be detected by expiratory gas analysis. The V̇O2 at this point is termed the “VAT” [1,2,3,4].
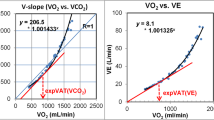
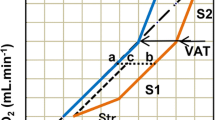
A number of methods may be employed to identify the VAT during a progressive exercise test (Fig. 12.1). If V̇CO2 is plotted vs. V̇O2, an inflection point is observed at the VAT, reflecting the disproportionate increase in V̇CO2 that occurs secondary to the buffering of lactic acid by bicarbonate [5]. Alternatively, the VAT can be identified as the point in time when the ratio of minute ventilation over V̇O2 (the V̇E/V̇O2 ratio) begins to increase, while the V̇E/V̇CO2 ratio is flat or declining. The reason for this phenomenon can be understood from Eq. 4.1 from Chap. 4, which indicates that alveolar ventilation (V̇A, which is the dominant component of V̇E) increases in proportion with V̇CO2. Hence, if V̇CO2 increases due to anaerobic metabolism, V̇E increases in parallel and the V̇E/V̇CO2 ratio does not change. (The V̇E/V̇CO2 ratio may actually decline during this phase of exercise because ventilation/perfusion matching improves and physiologic dead space declines as pulmonary artery pressure increases, allowing more blood to flow to the previously underperfused apices of the lungs (i.e., West Zone 1; see Chap. 3). However, because the V̇O2 does not increase as rapidly as the V̇CO2 beyond the VAT, the V̇E/V̇O2 ratio begins to rise. This method of identifying the VAT is worthwhile because it can readily distinguish between hyperventilation (e.g., due to anxiety), which leads to increases in both the V̇E/V̇O2 and the V̇E/V̇CO2 ratios, and the VAT, which is characterized by an increase in the V̇E/V̇O2 ratio but not the V̇E/V̇CO2 ratio [3]. Finally, the VAT may also be identified as the point in time when the end-tidal pO2 begins to rise. Once again, this phenomenon arises because the increased V̇A associated with the VAT brings more air (and oxygen) per time interval into the alveolus. Since the V̇O2 does not increase as much as the V̇A, there is more O2 left in the alveolus at the end of each breath, and the end-tidal pO2 rises [6].

Methods for determining the ventilator anaerobic threshold (VAT). (a) When V̇CO2 is plotted vs. V̇O2, an inflection point is observed at the VAT. (b) The ratio of minute ventilation over V̇O2 (the V̇E/V̇O2 ratio) begins to increase, while the V̇E/V̇CO2 ratio is flat or declining. (c) End-tidal pO2 reaches a minimum and begins to rise. Figure (c) also depicts a typical end-tidal pCO2 vs. time curve during a progressive exercise test. After some initial anticipatory hyperventilation, when the end-tidal pCO2 is somewhat less than 40 mm Hg, the end-tidal pCO2 rises to ~40 mm Hg (prior to the VAT). It remains at that level until the respiratory compensation point (after the VAT) when the end-tidal pCO2 falls, reflecting the respiratory alkalosis that develops in compensation for the accumulating lactic (metabolic) acidosis. AT ventilatory anaerobic threshold, Exer exercise, HR heart rate, PETCO2 end-tidal pCO2, pETO2 end-tidal pO2, Rec recovery, VE/VCO2 ratio of minute ventilation to carbon dioxide production, VE/VO2 ratio of minute ventilation to oxygen consumption, VCO2 carbon dioxide production, VO2 oxygen consumption
The V̇O2 at the VAT is clinically relevant because it reflects the level of oxygen delivery beyond which the circulatory system is no longer able to completely fulfill the metabolic needs (i.e., ATP requirements) of the exercising muscles. The VAT therefore may convey important information regarding the health and capabilities of the circulatory system. Moreover, the anaerobic threshold is a physiologically determined phenomenon. Unlike peak exercise parameters, it is not affected by a subject’s effort or motivation [1, 2].
One can infer from the aforementioned discussion that some subjectivity exists regarding the determination of the VAT. This introduces a degree of uncertainty into the measurement that should be borne in mind when interpreting VAT data.
Any cardiovascular condition that impairs the delivery of oxygen to the exercising muscles will tend to lower the anaerobic threshold. This includes obstructive lesions, regurgitant lesions, shunt lesions, disorders of systolic or diastolic function, absence of a pulmonary ventricle, pulmonary vascular disease, peripheral vascular disease (e.g., coarctation of the aorta, Takyasu’s arteritis), chronotropic defects, and rhythm disturbances that impair atrioventricular synchrony. Patients with Barth’s syndrome tend to have extremely low anaerobic thresholds because their mitochondria cannot take up oxygen normally, and they therefore rely more heavily on anaerobic metabolism for the generation of ATP [7]. For similar reasons, patients with other rare mitochondrial defects in which the oxidation of fuels is partially uncoupled from the generation of ATP also have low anaerobic thresholds. In contrast, patients with glycogen storage diseases and other conditions that impair the generation of lactate often do not have a detectable VAT, nor does the respiratory exchange ratio (RER) rise to normal levels at peak exercise, even when they expend a maximal effort [8].
Prediction equations exist for the calculation of normal values for the VAT on the basis of age, size, and gender [9]. VAT is also commonly expressed as a percentage of predicted peak V̇O2. In the absence of cardiovascular disease, VAT rarely falls below 40% of the predicted peak V̇O2. However, VAT is often depressed below this value in patients with conditions that significantly impair the ability to increase cardiac output or oxygen delivery appropriately during exercise [3]. In children with congenital heart disease (CHD), the VAT is often depressed in a manner similar to, albeit milder than, the peak V̇O2 [10]. Hence, when reliable peak V̇O2 data are available, VAT data does not often provide significant additional clinical information. However, if a patient does not expend an optimal effort and does not achieve an RER ≥1.09 at peak exercise (and therefore has peak exercise parameters that may be unreliable indicators of a patient’s cardiovascular function), the VAT can provide a valuable window into the patient’s cardiovascular health. Identification of the VAT is also worthwhile because the heart rate (HR) at the VAT has been recommended as the target HR for rehabilitation training [4].
Among patients who cannot augment their forward stroke volume normally during exercise, the V̇O2 at the VAT tends to be less affected than the peak V̇O2 because she/he can compensate for the stroke volume deficit by increasing his/her heart rate more rapidly than normal during submaximal exercise and thereby maintain relatively normal O2 delivery. However, the patient cannot increase the peak HR beyond normal peak values and therefore is unable to compensate for the stroke volume deficit at peak exercise. Similar considerations apply to patients with chronotropic defects.
V̇E/V̇CO2 Slope
Empirically, it has been observed that V̇E rises linearly in proportion with V̇CO2 during a progressive exercise test until a point above the VAT, when the accumulating lactic acidosis engenders a compensatory increase in V̇E out of proportion to the increase in V̇CO2. The V̇E/V̇CO2 slope is the slope of the linear portion of this curve. It may be thought of as an index of gas exchange efficiency during exercise, equivalent to the number of additional liters of air that must be breathed out in order to eliminate one additional liter of CO2 [11]. The point where the V̇E/V̇CO2 slope begins to deviate from linearity has been termed the “respiratory compensation” point [6].
Some physiologists choose to measure the V̇E/V̇CO2 slope from the data throughout exercise, including points beyond the respiratory compensation point. This practice is probably misguided. As an individual exercises beyond the respiratory compensation point, more and more data points are generated that deviate further and further from the linear portion of the V̇E vs. V̇CO2 relationship. The V̇E/V̇CO2 slope therefore becomes an effort-dependent parameter––a property that is particularly undesirable when seeking to apply these V̇E/V̇CO2 slope data to clinical situations.
The normal value for the V̇E/V̇CO2 slope is somewhat age dependent (Fig. 12.2). In older adolescents and young adults, it should be less than 28. Thereafter, gas exchange within the lungs gradually becomes more inefficient (as does almost everything with age) and normal values rise. The V̇E/V̇CO2 slope also tends to be higher in children and younger adolescents [12]. The reason for this observation probably relates to the fact that lung volumes (and hence the tidal volume during exercise) increase rapidly during childhood and adolescence, much more rapidly than the anatomic dead space volume. Hence, the dead space/tidal volume ratio declines during the pediatric years. As indicated by Eq. 4.4 in Chap. 4, this physiologic development results in less ventilation for any given level of V̇CO2. The V̇E/V̇CO2 slope is often elevated in patients with tetralogy of Fallot (TOF), [13], congestive heart failure (CHF) [14, 15], atrial switch procedures [16], and pulmonary hypertension [17, 18]. In these patients, V̇E/V̇CO2 slope elevation has been associated with an increased risk of mortality. Although multiple factors may influence the V̇E/V̇CO2 slope, among patients with pediatric and congenital heart disease, pulmonary blood flow maldistribution and consequent ventilation/perfusion (V/Q) mismatch are probably the most important pathophysiologic processes that underlie these observations and associations [4, 19, 20].

Percentiles for V̇E/V̇CO2 slope in normal male and female children, respectively. The slopes were calculated from the data up to the respiratory compensation point. (Reprinted with permission from [12])
Efficient gas exchange across the alveolar/capillary membrane requires optimal V/Q matching. Patients who have undergone repair of TOF often have residual pulmonary artery stenoses that cause PBF misdistribution, which in turn has been linked to V̇E/V̇CO2 slope elevation and depressed peak V̇O2 [19, 21, 22]. These stenoses can have a particularly deleterious effect upon the physiology of the postoperative tetralogy patient and a strong, negative impact upon a patient’s prognosis. Effective relief of these stenoses has been associated with improvements in peak V̇O2 and the V̇E/V̇CO2 slope (see Chap. 14) [22].
If you ask a radiologist to identify the earliest sign of congestive heart failure detectable on a chest X-ray, she/he would probably say “cephalization of pulmonary blood flow” (which is the result of the elevation of the left atrial and pulmonary capillary wedge pressure). To an exercise physiologist, this radiologic finding implies pulmonary blood flow maldistribution. As the CHF worsens and the pulmonary capillary wedge pressure rises, more fluid enters the alveolar capillary membrane, gas exchange is further impaired, and the V̇E/V̇CO2 slope rises further. As the CHF worsens and the pulmonary capillary wedge pressure rises yet higher, frank pulmonary edema develops, gas exchange is further impaired, and the V̇E/V̇CO2 slope rises still further. Hence, in patients with CHF, a strong link between pulmonary capillary wedge pressure and the V̇E/V̇CO2 slope probably accounts for the prognostic power of the V̇E/V̇CO2 slope that has been observed in this patient population. In a similar manner, for patients who have had an atrial switch procedure for the transposition of the great arteries (TGA), elevation of the V̇E/V̇CO2 slope probably reflects the progressive systemic (right) ventricular dysfunction that often develops in these patients as they age.
In patients with pulmonary hypertension, pulmonary blood flow (PBF) maldistribution results from pulmonary vascular obstructive disease. As the vascular obstruction progresses, the PBF maldistribution worsens, gas exchange within the lungs becomes more and more inefficient, and the V̇E/V̇CO2 slope rises. Hence, for patients with this condition, the V̇E/V̇CO2 slope reflects the extent of disease within the pulmonary vasculature [17, 18]. (This physiology may also be relevant to TGA patients who develop pulmonary vascular obstructive disease after an atrial switch procedure.)
The V̇E/V̇CO2 slope is also almost always elevated in patients with Fontan procedures [23]. Once again, this observation is probably due, to a large extent, to PBF maldistribution (and associated V/Q mismatch) secondary to the absence of a pulmonary ventricle [24, 25]. In Fontan patients, however, the degree of V̇E/V̇CO2 slope elevation is not associated with increased mortality because, in contrast to the conditions enumerated earlier, the elevated slope is intrinsic to the patients’ single ventricle physiology and not necessarily related to the progression/severity of the underlying cardiovascular disease process [4, 26].
Right-to-left intracardiac or intrapulmonary shunting will also cause the V̇E/V̇CO2 slope to be elevated. The shunting allows CO2-rich systemic venous blood to enter the systemic arterial circulation. The consequent increase in arterial pCO2 is sensed by arterial chemoreceptors, inducing central nervous system respiratory centers to increase the patient’s respiratory drive (and V̇E) and causing the V̇E/V̇CO2 slope to rise. The resulting alveolar hyperventilation reduces the pCO2 of the blood returning from the lungs and helps to normalize the patient’s arterial pCO2. Eliminating right-to-left shunting (e.g., by closing a Fontan patient’s fenestration; see Chap. 15) almost always produces a reduction in the V̇E/V̇CO2 slope [4, 27].
The V̇E/V̇CO2 slope will also be elevated in patients with interstitial lung disease––a condition that may occasionally be encountered in patients with pediatric and congenital heart disease. The interstitial lung disease impairs gas exchange across the alveolar capillary membrane. Patients will therefore have to breathe more to excrete any given amount of CO2, and their V̇E/V̇CO2 slope will be elevated.
Patients with obstructive (and to a lesser extent, restrictive) lung disease will tend to have low V̇E/V̇CO2 slopes. The lung disease will limit the patient’s capacity to increase V̇E during exercise. CO2 excretion may, however, be relatively well maintained if there is a concomitant increase in alveolar pCO2 (see Eq. 4.1 in Chap. 4), and the V̇E/V̇CO2 slope will therefore be depressed. In patients with a coexistent lung disease and V/Q mismatch secondary to a cardiovascular problem, the V̇E/V̇CO2 slope depression secondary to the lung disease may offset the elevation that results from V/Q mismatch, and “pseudonormalization” of the V̇E/V̇CO2 slope may be observed.
Some investigators have used the V̇E/V̇CO2 ratio (generally at the VAT) as an index of gas exchange efficiency instead of the V̇E/V̇CO2 slope. Although both parameters reflect more or less the same physiology, there is much more variability in the V̇E/V̇CO2 ratio, because it is determined from a single data point rather than the slope of a linear relationship. Normal values for the V̇E/V̇CO2 ratio are also less well established. The use of this parameter in place of the more standard V̇E/V̇CO2 slope should therefore be avoided, when possible.
End-Tidal pCO2
Because CO2 diffuses easily across the alveolar-capillary membrane, the alveolar pCO2 rapidly comes into equilibrium with the pCO2 of the blood leaving the alveolus. Hence, in the absence of shunts or inhomogeneous ventilation/perfusion (V/Q) matching, end-tidal pCO2 closely matches arterial pCO2 [28, 29]. Consequently, the end-tidal pCO2 should be ~40 mm Hg at rest and during early phases of a progressive exercise test. However, as the exercise intensity increases beyond the VAT and a lactic (metabolic) acidosis develops, homeostatic mechanisms come into play and cause individuals to increase their ventilation, lower their arterial pCO2, and generate compensatory respiratory alkalosis (see Chap. 4). This process will be reflected by a decline in the end-tidal pCO2 (Fig. 12.1c).
Many patients will be anxious or have “anticipatory hyperventilation” prior to and during the early phases of cardiopulmonary exercise testing (CPET). End tidal pCO2 levels below 40 mm Hg are therefore commonly encountered during these phases of a CPET. However, most individuals will cease hyperventilating within 2 or 3 minutes of the initiation of exercise, and their pCO2 levels will rise to ~40 mm Hg until after the VAT, when they begin to decline once again on account of the compensatory respiratory alkalosis that develops in response to the lactic acidosis.
In fit individuals who are able to achieve high exercise intensities and metabolic rates, end-tidal pCO2 will tend to exceed arterial pCO2 at higher levels of exercise [29, 30]. This observation relates to the fact that the arterial pCO2 reflects the average pCO2 of blood leaving the alveolus over the course of a breath. Because CO2 diffusion is so rapid and the pCO2 within the alveolus comes into equilibrium with the blood so quickly, at rest and during lighter exercise intensities, the alveolar pCO2 rapidly plateaus and the alveolar pCO2 toward the end of the breath does not differ significantly from the alveolar pCO2 near the beginning of the breath. Consequently, the blood leaving the alveolus has approximately the same pCO2 throughout the respiratory cycle and the end-tidal pCO2 closely matches the arterial pCO2. However, at higher levels of exercise intensity, the quantity of CO2 delivered to the alveolus is so great that the alveolar pCO2 does not plateau; it rises continually over the course of the breath. Hence, blood leaving the alveolus early in the course of a breath will have a lower pCO2 (generally less than 40 mm Hg) than blood leaving the alveolus toward the end of the breath. The end-tidal pCO2 reflects the blood leaving the alveolus at the end of the breath, and will therefore exceed the arterial pCO2, which reflects the average pCO2 of blood leaving the alveolus. Hence, it is not unusual to observe end-tidal pCO2 levels >40 mm Hg at high levels of exercise intensity in fit individuals. However, even under these circumstances, a decline in end-tidal pCO2 levels at higher levels of exercise, beyond the “respiratory compensation point”, should always be observed. In contrast, patients with severe lung disease may be unable to excrete all of the CO2 their muscles are producing at higher levels of exercise. Consequently, their arterial and end-tidal pCO2 levels will rise and the decline in end-tidal pCO2 that normally occurs beyond the respiratory compensation point will not be observed.
Abnormally low end-tidal pCO2 levels are commonly observed in patients with congenital heart disease. This phenomenon is not due to low arterial pCO2. It is typically due to right-to-left shunting and/or V/Q mismatch. The physiology underlying each of these mechanisms will now be discussed.
As discussed in Chap. 4, venous pCO2 levels rise dramatically during exercise, reaching levels of 60 mm Hg or higher. Hence, when blood shunts right to left during exercise, it is not only low in oxygen content; it is also high in CO2 content. The elevated CO2 levels would be sensed by chemoreceptors in the aorta and arch vessels and trigger homeostatic mechanisms to correct this imbalance by increasing alveolar ventilation and driving down pCO2 levels in the alveolus and the blood leaving the alveolus (i.e., the pulmonary venous blood). The pulmonary venous pCO2 levels will decline to levels somewhat below 40 mm Hg, so that when this blood mixes with the CO2-rich right-to-left shunting blood, the resultant mixture, in the aorta and systemic arteries, would have a pCO2 of 40 mm Hg. Under these circumstances the end-tidal pCO2 will be low (and lower than the arterial pCO2), reflecting the low alveolar pCO2 [4, 27].
In patients with V/Q mismatch, the air leaving alveoli with low perfusion would tend to have low pCO2. For instance, in the extreme case of an alveolus with ventilation but no perfusion, the air leaving the alveolus will have a pCO2 of 0 mm Hg (i.e., equivalent to room air) because the air in that alveolus would not have participated in gas exchange. The low pCO2 air from the underperfused alveoli will dilute out the CO2 in air coming from other alveoli. The end tidal pCO2 will therefore be low, lower than the arterial pCO2, which, of course, in the absence of a right-to-left shunt, reflects the pCO2 of blood returning from perfused alveoli [4, 21, 22].
Oxygen Uptake Efficiency Slope
Empirically it has been observed that there is a linear relationship between V̇O2 and log V̇E. The slope of this relationship is termed the oxygen uptake efficiency slope (OUES) . In patients who expend a good effort, the OUES has been found to correlate closely with the peak V̇O2. Because determination of the OUES does not require a peak effort, it has been advocated as a good submaximal index of exercise function. The OUES is, however, somewhat effort dependent; estimates of the OUES generated from the first 75% of an exercise test are significantly lower than values obtained when data from all of exercise is included [31].
In tests where the peak V̇O2 data is reliable, the OUES probably does not add important clinical information. Its prognostic power has also been found to be inferior to the V̇E/V̇CO2 slope in patients with congestive heart failure [32]. However, in tests where a peak effort is not expended, the OUES can, because of its strong correlation with peak V̇O2, provide worthwhile insights into a patient’s cardiopulmonary function (in some populations). It must also be noted that the OUES has been studied primarily in adults with acquired heart disease. Experience with children is limited. Moreover, as with peak V̇O2, the OUES is strongly dependent upon age, size, and gender [33]. These issues may be partially mitigated by normalizing the OUES for body surface area [34]. However, as discussed previously, in patients with congenital heart disease, one may encounter physiologic anomalies (e.g., right-to-left shunts, pulmonary artery stenoses) uncommon among adults with acquired heart disease that may influence the relationship between V̇O2 and V̇E and disrupt the tight correlation between the OUES and peak V̇O2 [35].
Oxygen Uptake-Work Rate Relationship: ΔV̇O2/ΔWR
In most people, chemical energy is converted into mechanical energy with more or less the same efficiency. Hence, after an initial delay near the onset of work and until the V̇O2 plateaus near V̇O2max, there is a linear relationship between the V̇O2 and the work rate. The slope of this linear portion of the V̇O2-work rate relationship is termed the “ΔV̇O2/ΔWR.” In normal individuals, the value of the ΔV̇O2/ΔWR is 10.3 ± 1.0 ml/min/watt. In patients with impaired oxygen delivery to the muscles, a greater proportion of the energy required by the muscles is derived from anaerobic metabolism, and the ΔV̇O2/ΔWR is low [36]. Hence, cyanotic patients and patients with impairment of the cardiac output response to exercise will have a low ΔV̇O2/ΔWR. Among patients encountered in the field of pediatric cardiology, a disproportionately low ΔV̇O2/ΔWR may also be encountered in patients with coarctation of the aorta [37] or peripheral vascular disease secondary to Takyasu’s arteritis. In contrast, individuals who cannot pedal efficiently (e.g., on account of neurologic or emotional issues) will tend to have an elevated ΔV̇O2/ΔWR. Similarly, obese individuals may have to expend more-than-normal amounts of energy to move their heavy limbs and chest walls during exercise and therefore may also have an elevated ΔV̇O2/ΔWR. Of course, the issue of unmeasured energy expenditure in obese subjects is even more relevant with treadmill exercise, as that modality requires them to carry their entire body weight, rather than resting most of it on the seat of a cycle ergometer.
The Exercise Electrocardiogram
Analysis of exercise electrocardiogram (EKG) data is an integral component of an exercise test. The influence of exercise on the incidence and nature of rhythm disturbances should be assessed. In structurally normal hearts, ectopy that is suppressed by exercise is thought to be benign [38]. In contrast, myocardial ischemia, cardiomyopathies, and conditions such as the prolonged QT syndrome and catecholamine-sensitive ventricular tachycardia are often characterized by an increase in the frequency and complexity of ectopy during exercise [39]. Rhythm disturbances are also commonly encountered in patients who have had surgery for congenital heart disease. In these patients, the absence or suppression of an arrhythmia during an exercise test may have little predictive value. However, patients whose arrhythmias develop or worsen with exercise appear to be at greater risk for future serious arrhythmic events, and exercise testing therefore plays an important role in the assessment and management of this difficult clinical issue [39].
The influence of exercise on conduction abnormalities should also be assessed. Patients with significant AV nodal disease may develop progressively higher grade AV block during exercise. In contrast, patients with AV block secondary to elevated resting vagal tone (e.g., many athletes) typically develop normal AV conduction during exercise [40]. In patients with Wolff-Parkinson-White syndrome (WPW), the sudden loss of pre-excitation during exercise is thought to indicate that the bypass tract has a relatively long anterograde effective refractory period and that the subject is therefore at low risk for sudden cardiac death [41, 42]. Indeed, this finding on an exercise EKG functions as a key decision point within the Pediatric and Congenital Electrophysiology Society’s and the Heart Rhythm Society’s suggested algorithm for the management of asymptomatic patients with WPW [42]. Patients with the prolonged QT syndrome may have normal QT intervals at rest but may be unable to shorten their QT interval appropriately during exercise [43, 44]. In patients with pacemakers, analysis of the exercise EKG may help assess whether the pacemaker is functioning properly and whether the pacemaker settings are optimal [45]. These topics are discussed in greater detail in Part V of this textbook.
The influence of exercise on the ST segment and T-wave morphology should also be analyzed. It must be emphasized, however, that the incidence of coronary artery disease in the pediatric population is quite low, whereas baseline ST-T abnormalities and/or bundle branch blocks (which render the interpretation of ST-T changes less reliable) are common. Consequently, the sensitivity and specificity of ST-T wave changes for the detection of coronary artery anomalies in pediatric subjects are unknown (but probably are not very good) [46,47,48]. Exercise-induced ST-T wave changes are more commonly encountered in pediatric patients with cardiomyopathies and/or myocardial ischemia secondary to excessively high myocardial oxygen demand during exercise (e.g., patients with aortic stenosis). More severe ST-T changes are certainly more suggestive of myocardial ischemia, especially when associated with chest pain and other abnormalities [46, 49, 50]. However, the correlation between ST-T changes and myocardial ischemia in pediatric patients, although not precisely known, is probably no more than moderate [51, 52]. Radionuclide-based myocardial perfusion imaging is often employed to help in the assessment of patients thought to be at increased risk for myocardial ischemia and has been found to be helpful in patients with Kawasaki disease [53] and hypertrophic cardiomyopathy [54]. However, perfusion abnormalities of questionable clinical significance, unassociated with significant detectable coronary artery pathology, are commonly found in patients following the arterial switch operation [48]. The value of myocardial perfusion studies in patients with congenital coronary artery malformations has also not been established.
References
Wasserman K. Determinants and detection of anaerobic threshold and consequences of exercise above it. Circulation. 1987;76(Supp VI)(6:Pt 2):VI-29-VI-39.
Wasserman K. The Dickinson W. Richards lecture. New concepts in assessing cardiovascular function. Circulation. 1988;78(4):1060–71.
Wasserman K. The anaerobic threshold measurement to evaluate exercise performance. Am Rev Respir Dis. 1984;129(2:Pt 2):t-40.
Rhodes J, Ubeda TA, Jenkins KJ. Exercise testing and training in children with congenital heart disease. Circulation. 2010;122(19):1957–67.
Beaver WL, Wasserman K, Whipp BJ. A new method for detecting anaerobic threshold by gas exchange. J Appl Physiol. 1986;60(6):2020–7.
Wasserman K, Hansen DA, Sue DY, Casaburi R, Whipp BJ. Principles of exercise testing and interpretation. 3rd ed. Philadelphia: Lippincott; 1999.
Spencer CT, Byrne BJ, Bryant RM, Margossian R, Maisenbacher M, Breitenger P, et al. Impaired cardiac reserve and severely diminished skeletal muscle O(2) utilization mediate exercise intolerance in Barth syndrome. Am J Physiol Heart Circ Physiol. 2011;301(5):H2122–9.
Tarnopolsky M. Exercise testing in metabolic myopathies. Phys Med Rehabil Clin N Am. 2012;23(1):173–86, xii
Cooper DM, Weiler-Ravell D. Gas exchange response to exercise in children. Am Rev Respir Dis. 1984;129(2:Pt 2):S47–8.
Rhodes J. Exercise testing. In: Keane JF, Lock JE, Fyler DC, editors. Nadas’ pediatric cardiology. Philadelphia: Elsevier; 2006. p. 275–87.
Wasserman K, Hansen JE, Sue DY, Casaburi R, Whipp BJ. Principles of exercise testing and interpretation. Philadelphia: Lippincott; 1999. p. 173–4.
Giardini A, Odendaal D, Khambadkone S, Derrick G. Physiologic decrease of ventilatory response to exercise in the second decade of life in healthy children. Am Heart J. 2011;161(6):1214–9.
Giardini A, Specchia S, Tacy TA, Coutsoumbas G, Gargiulo G, Donti A, et al. Usefulness of cardiopulmonary exercise to predict long-term prognosis in adults with repaired tetralogy of fallot. Am J Cardiol. 2007;99:1462–7.
Chua TP, Ponikowski P, Harrington D, Anker SD, Webb-Peploe K, Clark AL, et al. Clinical correlates and prognostic significance of the ventilatory response to exercise in chronic heart failure. J Am Coll Cardiol. 1997;29(7):1585–90.
Francis DP, Shamim W, Davies LC, Piepoli MF, Ponikowski P, Anker SD, et al. Cardiopulmonary exercise testing for prognosis in chronic heart failure: continuous and independent prognostic value from VE/VCO(2)slope and peak VO(2). Eur Heart J. 2000;21(2):154–61.
Giardini A, Hager A, Lammers AE, Derrick G, Muller J, Diller GP, et al. Ventilatory efficiency and aerobic capacity predict event-free survival in adults with atrial repair for complete transposition of the great arteries. J Am Coll Cardiol. 2009;53(17):1548–55.
Arena R, Lavie CJ, Milani RV, Myers J, Guazzi M. Cardiopulmonary exercise testing in patients with pulmonary arterial hypertension: an evidence-based review. J Heart Lung Transplant. 2010;29:159–73.
Wensel R, Opitz CF, Anker SD, Winkler J, Hoffken G, Kleber FX, et al. Assessment of survival in patients with primary pulmonary hypertension: importance of cardiopulmonary exercise testing. Circulation. 2002;106(3):319–24.
Buller NP, Poole-Wilson PA. Mechanism of the increased ventilatory response to exercise in patients with chronic heart failure. Br Heart J. 1990;63(5):281–3.
Ponikowski P, Francis DP, Piepoli MF, Davies LC, Chua TP, Davos CH, et al. Enhanced ventilatory response to exercise in patients with chronic heart failure and preserved exercise tolerance: marker of abnormal cardiorespiratory reflex control and predictor of poor prognosis. Circulation. 2001;103(7):967–72.
Rhodes J, Dave A, Pulling MC, Geggel RL, Marx GR, Fulton DR, et al. Effect of pulmonary artery stenoses on the cardiopulmonary response to exercise following repair of tetralogy of fallot. Am J Cardiol. 1998;81(10):1217–9.
Sutton NJ, Peng L, Lock JE, Lang P, Marx GR, Curran TJ, et al. Effect of pulmonary artery angioplasty on exercise function after repair of tetralogy of fallot. Am Heart J. 2008;155(1):182–6.
Troutman WB, Barstow TJ, Galindo AJ, Cooper DM. Abnormal dynamic cardiorespiratory responses to exercise in pediatric patients after Fontan procedure. J Am Coll Cardiol. 1998;31(3):668–73.
Grant GP, Mansell AL, Garofano RP, Hayes CJ, Bowman FO Jr, Gersony WM. Cardiorespiratory response to exercise after the fontan procedure for tricuspid atresia. Pediatr Res. 1988;24(1):1–5.
Matsushita T, Matsuda H, Ogawa M, Ohno K, Sano T, Nakano S, et al. Assessment of the intrapulmonary ventilation-perfusion distribution after the fontan procedure for complex cardiac anomalies: relation to pulmonary hemodynamics. J Am Coll Cardiol. 1990;15(4):842–8.
Fernandes SM, Alexander ME, Graham DA, Khairy P, Rodriguez E, Pearson DD, et al. Ability of exercise testing to predict morbidity and mortality in adults with Fontan surgery. Congenit Heart Dis. 2011;6:294–303.
Meadows J, Lang P, Marx G, Rhodes J. Fontan fenestration closure has no acute effect on exercise capacity but improves ventilatory response to exercise. J Am Coll Cardiol. 2008;52(2):108–13.
Jones NL, McHardy GJ, Naimark A, Campbell EJ. Physiological dead space and alveolar-arterial gas pressure differences during exercise. Clin Sci. 1966;31(1):19–29.
Whipp BJ, Wasserman K. Alveolar-arterial gas tension differences during graded exercise. J Appl Physiol. 1969;27(3):361–5.
Wasserman K, Hansen JE, Sue DY, Casaburi R, Whipp BJ. Principles of exercise testing and interpretation. Philadelphia: Lippincott; 1999. p. 82.
Baba R, Nagashima M, Goto M, Nagano Y, Yokota M, Tauchi N, et al. Oxygen uptake efficiency slope: a new index of cardiorespiratory functional reserve derived from the relation between oxygen uptake and minute ventilation during incremental exercise. J Am Coll Cardiol. 1996;28(6):1567–72.
Arena R, Myers J, Hsu L, Peberdy MA, Pinkstaff S, Bensimhon D, et al. The minute ventilation/carbon dioxide production slope is prognostically superior to the oxygen uptake efficiency slope. J Card Fail. 2007;13(6):462–9.
Marinov B, Mandadzhieva S, Kostianev S. Oxygen-uptake efficiency slope in healthy 7- to 18-year-old children. Pediatr Exerc Sci. 2007;19(2):159–70.
Tsai YJ, Li MH, Tsai WJ, Tuan SH, Liao TY, Lin KL. Oxygen uptake efficiency slope and peak oxygen consumption predict prognosis in children with tetralogy of fallot. Eur J Prev Cardiol. 2016;23(10):1045–50.
Giardini A, Specchia S, Gargiulo G, Sangiorgi D, Picchio FM. Accuracy of oxygen uptake efficiency slope in adults with congenital heart disease. Int J Cardiol. 2009;133(1):74–9.
Wasserman K, Hansen JE, Sue DY, Casaburi R, Whipp BJ. Principles of exercise testing and interpretation. 3rd ed. Philadelphia: Lippincott; 1999.
Rhodes J, Geggel RL, Marx GR, Bevilacqua L, Dambach YB, Hijazi ZM. Excessive anaerobic metabolism during exercise after repair of aortic coarctation. J Pediatr. 1997;131(2):210–4.
Jacobsen JR, Garson A Jr, Gillette PC, McNamara DG. Premature ventricular contractions in normal children. J Pediatr. 1978;92:36–8.
Wiles HB. Exercise testing for arrhythmia: children and adolescents. Prog Pediatr Cardiol. 1993;2(2):51–60.
Jones NL. Clinical exercise testing. 4th ed. Philadelphia: W.B. Saunders; 1997.
Bricker JT, Porter CJ, Garson A Jr, Gillette PC, McVey P, Traweek M, et al. Exercise testing in children with Wolff-Parkinson-White syndrome. Am J Cardiol. 1985;55(8):1001–4.
Cohen MI, Triedman JK, Cannon BC, Davis AM, Drago F, Janousek J, et al. PACES/HRS expert consensus statement on the management of the asymptomatic young patient with a Wolff-Parkinson-White (WPW, ventricular preexcitation) electrocardiographic pattern: developed in partnership between the Pediatric and Congenital Electrophysiology Society (PACES) and the Heart Rhythm Society (HRS). Endorsed by the governing bodies of PACES, HRS, the American College of Cardiology Foundation (ACCF), the American Heart Association (AHA), the American Academy of Pediatrics (AAP), and the Canadian Heart Rhythm Society (CHRS). Heart Rhythm. 2012;9(6):1006–24.
Vincent GM, Jaiswal D, Timothy KW. Effects of exercise on heart rate, QT, QTC and QT/QS2 in the Romano-Ward inherited long QT syndrome. Am J Cardiol. 1991;68(5):498–503.
Weintraub RG, Gow RM, Wilkinson JL. The congenital long QT syndromes in childhood. J Am Coll Cardiol. 1990;16(3):674–80.
Bricker JT, Garson A Jr, Traweek MS, Smith RT, Ward KA, Vargo TA, et al. The use of exercise testing in children to evaluate abnormalities of pacemaker function not apparent at rest. Pacing Clin Electrophysiol. 1985;8(5):656–60.
James FW, Kaplan S, Glueck CJ, Tsay JY, Knight MJ, Sarwar CJ. Responses of normal children and young adults to controlled bicycle exercise. Circulation. 1980;61(5):902–12.
Mahle WT, McBride MG, Paridon SM. Exercise performance after the arterial switch operation for d-transposition of the great arteries. Am J Cardiol. 2001;87(6):753–8.
Weindling SN, Wernovsky G, Colan SD, Parker JA, Boutin C, Mone SM, et al. Myocardial perfusion, function and exercise tolerance after the arterial switch operation. J Am Coll Cardiol. 1994;23(2):424–33.
Shimizu M, Ino H, Okeie K, Emoto Y, Yamaguchi M, Yasuda T, et al. Exercise-induced st-segment depression and systolic dysfunction in patients with nonobstructive hypertrophic cardiomyopathy. Am Heart J. 2000;140(1):52–60.
Yetman AT, Hamilton RM, Benson LN, McCrindle BW. Long-term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1998;32(7):1943–50.
Fletcher GF, Ades PA, Kligfield P, Arena R, Balady GJ, Bittner VA, et al. Exercise standards for testing and training: a scientific statement from the American Heart Association. Circulation. 2013;128(8):873–934.
Kane DA, Fulton DR, Saleeb S, Zhou J, Lock JE, Geggel RL. Needles in hay: chest pain as the presenting symptom in children with serious underlying cardiac pathology. Congenit Heart Dis. 2010;5(4):366–73.
Hijazi ZM, Udelson JE, Snapper H, Rhodes J, Marx GR, Schwartz SL, et al. Physiologic significance of chronic coronary aneurysms in patients with Kawasaki disease. J Am Coll Cardiol. 1994;24(7):1633–8.
Dilsizian V, Bonow RO, Epstein SE, Fananapazir L. Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1993;22(3):796–804.
Acknowledgment
Portions of this chapter were based upon the author’s previously published works:
-
Rhodes J, Ubeda TA, Jenkins KJ. Exercise testing and training in children with congenital heart disease. Circulation. 2010;122(19):1957–67.
-
Rhodes J. Exercise testing. In: Keane JF, Lock JE, Fyler DC, editors. Nadas’ pediatric cardiology. 2nd ed. Philadelphia: Elsevier; 2006.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rhodes, J. (2019). Parameters from Submaximal Exercise. In: Rhodes, J., Alexander, M., Opotowsky, A. (eds) Exercise Physiology for the Pediatric and Congenital Cardiologist. Springer, Cham. https://doi.org/10.1007/978-3-030-16818-6_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-16818-6_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16817-9
Online ISBN: 978-3-030-16818-6
eBook Packages: MedicineMedicine (R0)