
Abstract
This chapter is focused on skin regeneration with cultured cells and bioengineered skin substitutes. Different kinds of multipotent stem cells can be found in hair and skin tissues. Cell culture is a well-established research tool in biology and medicine. Several cell culture techniques have been developed in order to obtain different cell types. Different culture techniques will be discussed (use of proteolytic enzymes, 3D cultures, and automatic bioprocessing). Several skin regeneration approaches will be described. In the field of aesthetic medicine, the injection of autologous cultured cells seems to be a good method to restore aging skin. Nevertheless its application is far from being widespread. The challenge of current bioengineering efforts is to generate functional organ systems from dissociated cells that have been expanded under defined tissue culture conditions. Tissue engineering is emerging as a significant potential solution for tissue and organ failure. Clinical applications, efficacy and safety, and trends and limitations of the current techniques will also be discussed. Even though there is still a long way to go, the rise of cell culture and tissue engineering is providing powerful tools for regenerative medicine evolution. Further large-scale and rigorous studies with long-term follow-up should be performed to assess the safety of cell culture and skin substitutes. Great improvements have been made in this field, and now the challenge is its application to routine clinical practice.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
Cell culture is a well-established research tool in biology and medicine [1]. The challenge of current bioengineering efforts is to generate functional organ systems from dissociated cells that have been expanded under defined tissue culture conditions [2].
Skin Histology and Physiology
Skin is formed by dermal and epidermal tissues (Fig.1). Keratinocytes are the major component of the epidermal tissue (more than 95%), with melanocytes and Langerhans cells representing a minority population [4]. Human keratinocytes derive from the epidermal stratum basale [4,5,6,7,9] from hair follicles [9,10,11,13] and, as recently suggested, also from eccrine sweat glands [14].

Schematic representation of human skin (Reproduced from Tavakolpour et al. [3])
As cells divide and differentiate through the epidermal layers, their protein expression changes [15]. Cell behavior is governed by chemical messengers. Surface proteins attach to various ligands, such as growth factors, and trigger specific signaling pathways involved in stem cell differentiation. Cells are also influenced by their surrounding ECM (extracellular matrix) [16]. Each type of tissue has its own unique ECM composition. Several studies have demonstrated that ECM greatly influences cell development, migration, proliferation, differentiation, shape, and function [16,17,18,19,21]. ECM and surface proteins perform the so-called mechanotransduction, which transmits mechanical signals to the cell nucleus and alters gene expression [16]. It has been shown that despite the genetically programmed cell expression, phenotype can be changed by modifying the interaction with the ECM. An example of the transformation that the extracellular environment may generate is found in embryonic cells [22].
Dermal tissue comprises a dense connective tissue structure in which the major cellular components are fibroblasts. The ECM and a variety of cytokines are synthesized by fibroblasts to induce epidermal and vascular endothelial cell growth. ECM is made up of collagen, glycoproteins, and proteoglycans (chondroitin sulfate, HA, heparin, etc.). Fibroblasts produce abundant extracellular proteins (especially collagen and elastin). Collagen allows cell adhesion, growth, proliferation, and differentiation [23]. The dermis constantly renews itself via the process of degradation, rebuilding, and regeneration. Theoretically, the dermis cannot regenerate like the liver, bone, and the epidermis after being destroyed. Scarring is hard to avoid during the process of natural healing [24]. The scarless healing of fetal wounds is an ideal healing method [25]. Tissue formation and healing mechanisms are still unclear [26]. A lot can be learned about skin physiology and cell–ECM interactions by studying wound healing which involves cell migration, proliferation, differentiation, apoptosis, and the synthesis and remodeling of the extracellular matrix (ECM) [25]. Injuries involving the epidermis alone or the superficial layer of the dermis will re-epithelialize without surgical intervention, provided there is a sufficient number of keratinocyte stem cells in the remaining epidermis or in the residual dermis. If epidermal keratinocytes are missing, regeneration may be achieved by epithelial stem cells derived from hair follicles and/or sweat glands [26]. During wound healing the cell is fully reprogrammed. They have to de-differentiate and the genes and proteins expressed change. The Wnt pathway leads stem cell function and renewal and reprograms differentiated cells to have stem cell-like properties [15]. Growth factors can be considered the engine of wound healing, but their use as a monotherapy in clinical practice has not worked well. Human serum (a soup of factors) helps keratinocytes but is detrimental to fibroblasts [15, 27]. Aoki et al. [27] demonstrated that dermal fibroblasts, bone marrow stromal cells (BMSCs), and preadipocytes derived from subcutaneous adipose tissue promoted epidermal regeneration [27].
Fibroblasts are a heterogeneous population of mesenchymal origin that can be found in numerous tissues. Fibroblasts from different anatomical sites have their own characteristic phenotypes, synthesizing different extracellular matrix (ECM) proteins and cytokines [28]. Dermal fibroblasts release cytokines and growth factors that have autocrine and paracrine effects [29]. Autocrine activity promotes collagen synthesis and fibroblast proliferation [30]. Paracrine activity affects keratinocyte growth and differentiation [31]. Dermal fibroblasts promote the development of keratinocyte layers in addition to promoting keratinocyte proliferation [32]. Human fibroblasts regulate vascular and lymphatic endothelial cell proliferation [33].
The adult hair follicle (HF) is composed of mesenchymal cells that provide signals to regulate epithelial stem cell function during tissue regeneration [34]. The HF is accessible to experimental modulation and can be easily removed in its entirety. Moreover, the HF is the only mammalian organ that, for the entire lifespan, cyclically undergoes consecutive transformations. The HF cycles between a state of relative “quiescence” (telogen) and rapid and massive growth (anagen); and finally it cycles back toward telogen, via an apoptosis-driven organ involution (catagen) [35].
Skin Stem Cells
Stem cells (SCs) have the unique capacity to self-renew and to differentiate into the cell lineages that constitute their tissue of origin. Hair follicle and skin tissue, apart from bone marrow, are perhaps the only tissues, which hold the niche for diverse kinds of stem cells: melanocyte stem cells, keratinocyte stem cells, and mesenchymal stem cells [36].
Epidermal stem cells (Epi-SCs) reside in the basal layer of interfollicular epidermis and in the hair follicle bulge [37], which is a specialized portion of the outer root sheath epithelium defined as the insertion site of the arrector pili muscle (Fig. 2). Bulge cells contribute not only to the generation of new HFs with each hair cycle but also to the repair of the epidermis during wound healing [39]. Because Epi-SCs in the hair follicle are difficult to acquire, Wang et al. [40] investigated whether Epi-SCs in the epidermis were capable of regenerating epidermal appendages. Among the potential keratinocyte donor sites, the foreskin seems to be a promising source [41].

Depiction of the hair follicle stem cell niche (Reproduced from Lee et al. [38])
Adult dermal SCs have not yet been fully defined [2]. Endogenous dermal stem cells (DSCs) have been demonstrated within the adult mammalian dermis [42], which might serve to regenerate dermis or rejuvenate dermal papilla to restore follicle growth. DSCs reside in the HF mesenchyme. Endogenous DSCs can be grown in vitro as self-renewing multipotent cells named skin-derived precursors (SKPs), which can generate both mesodermal and neural derivatives [2, 43]. In addition, SKPs display all the predicted properties of multipotent dermal SCs including HF morphogenesis demonstrated in rodents but not in humans [42]. Once transplanted into skin, SKPs can generate new dermis and reconstitute the dermal papilla and connective tissue sheath [43]. It has been proved that rodent dermal papilla cells can be removed from HFs and transplanted in their intact state into recipient skin, where they induce de novo HF development and hair growth [44].
Thus, SKPs are attractive tools for regenerating the skin dermis. However, isolating SKPs from human skin requires invasive surgical procedures, and the isolated cells may have limited or variable abilities to proliferate and/or differentiate. Mesenchymal stem cells (MSCs) also have the same problems. Therefore, there have been many studies that generated MSCs from pluripotent stem cells [45].
The amelanotic melanocytes (AMMC) are considered to be melanocyte stem cell population [46].
Cell Lineages Obtained from Skin
Skin is an established tissue source for cell-based therapy. Various cell lineages may be obtained from skin. In addition, the ease of tissue harvest and the multipotent nature of skin and HF stem cells have promoted basic and clinical research in this area [47].
Some neural crest stem cells persist within crest-derived tissues. HFSCs (hair follicle stem cells) and SSCs (skin stem cells) are both originated from neural crest cells. Although both types of stem cells can differentiate into neuronal and melanocyte lineages, HFSCs are a better source for melanocyte differentiation and SSCs are more inclined to neuronal differentiation [47].
The hair follicles have been shown to harbor pluripotent neural crest stem cells [48] which can be differentiated into melanocytes, neuronal cells, adipose cells, and other lineages [47]. Bulge cells can differentiate into all types of cutaneous epithelial cells including sebaceous glands and interfollicular epidermal keratinocytes [2].
DSC and SKP are of neural crest in origin and are capable of differentiating into melanocyte and neural lineages. SKPs are closer to the neuronal cell lineage, while DSCs are closer to the melanocyte progenitors [49]. SKPs can generate both mesodermal and neural derivatives, including adipocytes, skeletogenic cell types, and Schwann cells [42, 48,49,50,51,54], but tend to have spontaneous differentiation toward neuronal lineage. When SKPs are transplanted to full-thickness skin wounds, they originate a variety of fibroblast phenotypes and fill the lesion with new dermal tissue. Transplanted SKPs are also able to integrate into the mesenchyme of existing HFs and initiate formation of new HFs when cotransplanted with epithelial cells [42].
Adult cells can return to the embryonic stage with the possibility of differentiating toward all the specialized cell categories [55]. Skin fibroblasts can be reprogrammed to hiPSCs (human induced pluripotent stem cells) with the potentiality of obtaining all the cellular lineages that can be derived from them. iPSC can be differentiated into specific cells with a wide spectrum of cellular phenotypes. Fibroblasts differentiated from iPSC acquired an augmented biological potency that exceeded those from their parental fibroblasts, characterized by their increased production and assembly of ECM, functional features important for application of these cells in regenerative therapies [24].
Sugiyama et al. provided an induction protocol of SKPs from human iPSCs [2]. The human iPSC-derived SKPs (hiPSC-SKPs) express several genes and proteins that have been previously reported to be expressed by human SKPs [50]. As for their differentiation potential, hiPSC-SKPs can successfully differentiate into adipocytes, osteocytes, and Schwann cells. In addition, hiPSC-SKPs were able to induce hair follicular keratinization when they were co-cultured with epidermal keratinocytes. These observations suggest that hiPSC-SKPs may facilitate the regeneration of human full-thickness skin, including skin appendages [2].
Human epidermal keratinocytes and epidermal SCs have also been developed from induced pluripotent stem cells (iPSCs). Additionally, iPSC-derived epidermal cells have the ability to reconstitute HFs with mouse dermal cells [56].
Background
The first milestones in skin research were the enzymatic separation of the epidermis and dermis [57] and the in vitro culture of human skin epithelial cells [58]. Cell culture appeared with the introduction of trypsinization by Moscona et al. [59]. In 1975, Rheinwald and Green started serial cultivation of keratinocytes (autologous epidermal cultures) and showed that the limitations of epidermal cell cultures were not intrinsic, but due to the relationship between keratinocytes and fibroblasts [5]. An epidermal graft could be expanded to more than 500 times its size within 3–4 weeks [6]. After the first clinical grafting of autologous cultured epithelium prepared from autologous epidermal cells performed by O’Connor et al. [7] in 1981, cultured epidermal autografts (CEAs) were tested in almost all leading burn centers worldwide [26].
In 1981 Bell et al. generated a dermo-epidermal substitute [60]. This technique was transformed into the product Apligraf® (human allogeneic fibroblasts and keratinocytes). On the basis of this development, dermo-epidermal skin substitutes consisting of human autologous keratinocytes and fibroblasts in bovine collagen were also transplanted in severe burn patients [61]. Researchers revealed that the cross-talk between fibroblasts and keratinocytes was essential for the establishment of a functional basement membrane [62]. In 1981, a further significant advance was the development of a bilayered “artificial skin” [63] commonly referred to as Integra® which was commercially launched in the United States in 1996. The appealing idea of combining cultured keratinocytes with Integra® generated a fascinating new field of research. However, reality has shown that simple cultured epidermal autografts do not take well on the neodermis produced by Integra® [26].
Clinical use of injectable autologous skin-derived fibroblasts was first started by Isolagen Technologies in 1995 to repair dermal and subcutaneous contour deformity. Long-term correction and no allergic adverse effects have been reported, which made autologous fibroblasts a promising alternative to the use of other foreign materials [64].
Until 1990, preclinical human hair research had been limited to histological studies or to difficult-to-perform in vivo assays with human skin transplanted onto mice [35], while human HFs could not be maintained and studied ex vivo. The main challenges were not only to maintain the HFs viable, but to keep their function. In 1990 Philpott et al. [65] developed an ex vivo model for the study of isolated human scalp HFs. Not only was the morphology and keratin synthesis of the HFs preserved up to day 4, but also more importantly, the follicles demonstrated rates of de novo hair shaft growth approximating that seen in vivo. This ability to maintain viable human HFs ex vivo constituted a methodological breakthrough in human hair research and raised the possibility of investigating the effects of a wide range of hormones, neurotransmitters, growth factors, cytokines, and drugs on human HF biology, while simultaneously promising new insights into the pathogenesis of a range of hair growth disorders [35].
Skin Cell Culture
Cell culture is the process by which cells are grown under controlled conditions in a favorable artificial environment. When primary cultures reach confluence (they fill all the substrate), the cells are then subcultured (passaged) by transferring them to a new medium (Fig. 3). The stem cells are key pieces in cell cultures. These cells can be maintained in culture for a longer period of time compared to other cell types [47].

Cell culture procedure
Cultures are affected by many factors which influence cell function, proliferation, differentiation, and transcriptional status. Temperature, pH, time, replicative potential, medium composition (growing factors and other cell signals), cell characteristics and origin, passage number, physical and physiological stress (i.e., shear stress), plastic plate adherence, aggregate size, agitation rate, impeller size, and volume of culture medium are decisive factors. Each cell type needs its appropriate culture medium with rigorously controlled characteristics. Stress imposed by inadequate culture conditions induces senescence [4]. Plastic culture flasks are commonly used for cell culture of single cell types [1]. An incubator is used to grow cell cultures maintaining optimal temperature, humidity, and CO2 and oxygen concentrations. The survival rate of a given type of cultured cell depends on the degree of adhesion to the plastic plate, meaning that the adherence of cells is an essential survival factor [66].
Establishment of a novel culture method can sometimes open up huge new fields of cell biology and medicine [66]. One difficulty is the large discrepancy between cell kinetics in vivo and in vitro due to the extreme difficulty to reproduce an anatomical or physiological microenvironment. Several factors, including cytokines, scaffold material, cell–cell interactions, and physical stress, constitute this artificial microenvironment [1].
A variety of studies have been developed to understand and control cell cultures, and certain assumptions have been raised. First, the presence of fetal bovine serum in cell culture medium is questionable since there is a lack of characterization and quantifying of growth factors. Furthermore, the ideal culture media should contain specific nutrients according to the cell type. Second, the in vitro cell culture environment is very different from the in vivo environment. Third, despite widespread use of proteolytic enzymes, it must be taken into account that using these enzymes in the cell passage or tissue digestion promotes the destruction of both the ECM and surface proteins and may, thus, modify signaling and mechanotransduction of signals to the nucleus [16, 17].
Unlike germline and stem cells, somatic cells have a limited lifespan. They stop dividing when cultured in vitro for a certain period of time [67]. Typical human primary keratinocytes possess an in vitro lifespan of around 15–20 population doublings in serum-free and chemically defined media [68]. When cells encounter the so-called Hayflick limit, they enter a state of permanent quiescence, often named cellular senescence [4, 69]. Continuous replication of typical primary human cells is prevented by two events: mortality stage 1 (M1) or “replicative senescence” and mortality stage 2 (M2) or “cellular crisis.” Cells entering senescence first stop responding to exogenous mitogenic stimuli and acquire increased cellular adhesion to the extracellular matrix while losing cell–cell contacts. In addition to prolonged in vitro culture of primary cells, various types of cellular stresses including telomere erosion, DNA damage, overexpression of tumor suppressor genes or oncogenes, oxidative stress, continuous mitogenic stimuli, and a variety of chemicals can also induce senescence [4]. Unrepairable severe terminal telomere shortening eventually leads to cellular crisis, a state characterized by massive cell death [70].
Cell Characterization
Each cellular type holds a particular protein expression profile (Table 1) which can be detected by different methods (i.e., immunofluorescence, flow cytometry, etc.).
Different cell type culture protocols will be summarized. Detailed description of cell culture techniques is out of scope of this chapter.
Fibroblast Culture
Techniques for culturing fibroblasts were long established prior to the discovery made in 1975 by Rheinwald and Green [5] for culturing and expanding keratinocytes, which require fibroblasts to support their proliferation. Dermal fibroblasts can be extracted from skin biopsies either through enzymatic degradation or by explant culture. The medium used for culturing fibroblasts is usually supplemented with fetal calf serum, which previously raised concerns regarding transmission of bovine spongiform encephalopathy (BSE). However, the serum is obtained only from BSE-free countries [29].
Fibroblasts and adult stem cells divide through asymmetric division, which means that the replicating cell gives rise to one adult stem cell and one specialized stem cell (fibroblast), suggesting the continuity of cell division until complete differentiation of the stem cells [74].
Fibroblasts are readily cultured in the laboratory (Fig. 4) and incorporation of fibroblasts into tissue-engineered skin substitutes has produced encouraging results including symptomatic pain relief, rapid healing, less scarring, and better cosmetic results [29]. Growth parameters and the characteristics of fibroblasts in culture will be influenced by passage number, age of the donor, subtype of fibroblast (reticular or papillary dermis), and anatomical site. Older donor skin fibroblasts tend to migrate more slowly, reach cell culture senescence earlier, have a prolonged cell population doubling time, and are less responsive to growth factors. Other factors that influence fibroblast behavior in culture include vitamins, such as vitamin C, and antioxidants, including coenzyme Q10. For example, in the presence of vitamin C, fibroblasts produce twofold more collagen, a response that is independent of the age of the fibroblasts [75]. Likewise, coenzyme Q10 promotes wound healing by increasing cell proliferation and fibroblast mobility [76].

Phase contrast images of minipig keratinocytes and fibroblasts grown in monolayer culture (Reproduced from Dame et al. [77])
Several protocols for culturing fibroblasts have been described. Solakoglu et al. [78] used rat biopsies which provided dermal connective tissue that was treated with collagenase B and DNAase. Fibroblast culture is usually performed in flasks at 37 °C with 5% CO2 in humidified air [74, 78, 79]. Many different culture mediums have been used:
-
Eça et al. 2012 added culture medium containing L-amino acids, Earle’s salts, and sodium bicarbonate, supplemented with human serum from the patients to the culture flasks containing the dermis fragments [74].
-
Solakoglu et al. used DMEM (Dulbecco’s modified Eagle’s medium)-F12 medium and fetal calf serum [78].
-
Weiss utilized Iscove’s modified Dulbecco’s medium (IMDM) with phenol red supplemented with antibiotics and fetal bovine serum (FBS) [79].
-
Zhao et al. used DMEM supplemented with FBS, penicillin, streptomycin, and glutamine [80].
-
Sugiyama et al. cultured human primary fibroblasts in DMEM including 5% FBS [2].
-
Kumar et al. cultured immortalized human foreskin fibroblast (I-HFF) in IMDM supplemented with fetal bovine serum, L-glutamine, non-essential amino acids, and penicillin and streptomycin [47].
When cells reach confluence they are detached from the culture plate with trypsin solution [64, 74, 78, 79]. Solakoglu et al. cultured fibroblasts for 3 weeks by 2 or 3 passages [78]. These cultures expand rapidly resulting in a higher percentage of live cells with the human serum technique than with the use of fetal bovine serum [74]. Culturing fibroblasts at the air–liquid interface (ALI) culture system, which imitates the skin microenvironment, promotes optimal differentiation approaching that of skin in vivo [29].
Autologous fibroblasts can be cultured for their posterior injection [79, 80]. After biopsy collection, skin samples are inspected for quality and transferred to tissue culture plating. After an antibiotic wash, biopsy tissue is subjected to enzymatic dissociation in a collagenase enzyme cocktail at 37 °C. Cells are then seeded into a culture flask with IMDM with phenol red supplemented with antibiotics and fetal bovine serum (FBS) [79].
Keratinocyte Culture
Ex vivo keratinocyte short lifespan has limited many skin-related applications. In order to overcome this difficulty, many attempts to immortalize primary keratinocytes have been made with success. Different kinds of primary cells are able to become immortal through a variety of cellular events including overexpression of telomerase, epigenetic gene silencing, oxidative DNA damage, inactivation of cell cycle regulatory genes, overexpression of cellular or viral oncogenes, and inhibition of a specific host kinase. Nevertheless, immortalized keratinocyte cell lines turn out to have several undesirable genetic abnormalities. In spite of these genetic defects, immortalized keratinocytes seem to maintain some properties of normal keratinocytes, which enable them to be used as a substitute for primary keratinocytes in various skin research fields [4].
Cells that have a lifespan of 20–50 passages under in vitro culture conditions are mostly blast cells, such as fibroblasts. Cells that have a lifespan of less than 10 passages under in vitro culture conditions are typically epithelial cells, such as keratinocytes. In many epithelial cells, epidermal growth factor (EGF) has been shown to be able to increase their lifespan to 10–20 passages before senescence [4]. Human primary keratinocytes can be cultured in keratinocyte medium (J-TEC) [2].
Melanocyte Culture
Kumar et al. [47] induced differentiation of SSCs and HFSCs into melanocytes. For melanocyte differentiation, 70–80% confluent cultures of SSCs and HFSCs are incubated in molecular, cellular, and developmental biology (MCDB) 201 medium and Ham’s F12 nutrient mix, supplemented with fetal calf serum, L-glutamine, L-ascorbic acid, phorbol 12-myristate 13-acetate (PMA), cholera toxin, fibroblast growth factor, and penicillin and streptomycin. PMA is used as an inducer of melanocyte, promoting cell proliferation and helping the formation of multiple dendrites [81]. Geneticin is used to remove the contaminating fibroblasts. The protocol to differentiate the stem cells into melanocytes lasts 21 days. The culture dish becomes homogenously confluent with melanocytes in almost 25–30 days. The authors found no visible difference in the melanocytes differentiated from HFSCs and SSCs after staining with HMB-45 and S-100 antibodies for immunofluorescence. Nevertheless there were a higher percentage of cells functionally active in melanocytes derived from SSCs than in those derived from HFSCs [47].
Adipocyte Culture
Aoki et al. described a unique culture technique for floating adipocytes called “ceiling culture” [66]. Mature adipocytes are mesenchymal cells with abundant lipid droplets within their cytoplasm. As the gravity of mature adipocytes is lower than that of culture medium, mature adipocytes float in medium, and it is quite difficult for floating cells to attach to a plastic culture plate. The authors cultured adipocytes in flasks that were completely filled with medium. Under these conditions, adipocytes became attached to the ceiling of the flask. Then these cells were able to proliferate, form a cell monolayer, and exhibit accumulation of intracytoplasmic lipid droplets after reaching confluency. The authors established an adipose tissue-organotypic culture system in addition to the ceiling culture system, which was able to maintain the proliferative ability and function of mature adipocytes for more than 4 weeks [66].
Wang et al. cultured Epi-SCs derived from the epidermis of neonatal mice or adult human foreskin in CnT-07 PCT epidermal keratinocyte medium containing dexamethasone, insulin, rosiglitazone, and XAV939 for 3 days to induce sebocyte differentiation [40].
Recently, dedifferentiated fat (DFAT) has gained attention in regenerative medicine, because it contains multipotent stem cells [82]. The ceiling culture method is a fundamental technique for the fabrication of DFAT cells, which are able to differentiate into other mesenchymal cell types such as adipocytes, chondrocytes, and osteoblasts [66].
Neuronal Culture
For neuronal differentiation, 70–80% confluent cultures of stem cells are incubated in the neurobasal medium containing penicillin and streptomycin supplemented with basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), B-27 supplement, and L-glutamine. The contaminating fibroblasts are removed with geneticin. The cells start to change their morphology after 4–5 days of culture [47].
Skin-derived precursors (SKPs) are the only neural stem cells which can be isolated from an accessible tissue such as skin. Bayati et al. presented a protocol to enrich neural SKPs by monolayer adherent culture [83]. This culture method helps to increase the number of neural precursor cells. The authors found that serum-free adherent culture reinforced by growth factors was effective on proliferation of skin-derived neural precursor cells (skin-NPCs). The cells of enriched culture possessed a multipotential capacity to differentiate into neurogenic, glial, adipogenic, osteogenic, and skeletal myogenic cell lineages.
Skin Stem Cells (SSCs) and Hair Follicle Stem Cells (HFSCs)
Kumar et al. carried out in vitro expansion of skin stem cells (SSCs) and hair follicle stem cells (HFSCs) by explant culture method [47]. Skin tissue measuring approximately 2 × 2 mm and individual hair follicles were used as explants. Culture was performed according to the modified Rheinwald system [5] consisting of DMEM and Ham’s F12 nutrient mix, supplemented with fetal bovine serum, epidermal growth factor, hydrocortisone, insulin, transferrin, cholera toxin, and penicillin and streptomycin over a fibronectin-coated culture dish. HFSC could be expanded for 10 passages as compared to SSC which could be taken for up to eight passages [47].
Wang et al. demonstrated that a combination of cultured human Epi-SCs and skin-derived precursors (SKPs) was capable of reconstituting functional hair follicles and sebaceous glands (SG) in mice. The Epi-SCs formed de novo epidermis along with hair follicles, and SKPs contributed to dermal papilla in the neogenic hair follicles. Notably, a combination of culture-expanded Epi-SCs and SKPs derived from the adult human scalp could generate hair follicles and hair. In addition, Epi-SCs were able to differentiate into sebocytes and form de novo SGs, which excreted lipids [40]. Rapid attachment to plastic culture dishes has been recognized as a property of Epi-SCs [84]. Therefore Wang et al. selected the cells that rapidly attached to the dish. Epi-SCs were then cultured in the CnT-07 progenitor cell-targeted (PCT) epidermal keratinocyte medium. The dermis was digested with collagenase to isolate SKPs. Their culture was performed in Dulbecco’s modified Eagle’s medium/F12, supplemented with B27, epidermal growth factor, and basal fibroblast growth factor in untreated dishes [40].
Like many stem cell cultures, SKPs are typically grown in static tissue culture flasks as nonadherent, spherical colonies. Recently, Agabalyan et al. presented a new technique consisting of enhanced expansion of SKPs in computer-controlled stirred-suspension bioreactors. Rat SKPs (rSKPs) were isolated from the back skin and were grown in Dulbecco’s modified Eagle’s medium (DMEM) low glucose/F12 with basic fibroblast growth factor, platelet-derived growth factor (PDGF)-BB, B27 supplement, and penicillin/streptomycin. Following primary colony formation, SKPs were dissociated to single cells using collagenase and replated. SKPs were passaged three or four times in static cultures to obtain sufficient numbers of cells to introduce in the bioreactor. Then rSKPs were cultured for three passages in 500-mL computer-controlled DASGIP Parallel Bioreactor Systems (Eppendorf, Hamburg, Germany). The variable bioreactor set points were regulated at 60 rpm, 37 °C, pH 7.4, and a 21% dissolved oxygen concentration [43].
Sugiyama et al. developed a method to induce human SKPs (hSKPs) from human induced pluripotent stem cells (hiPSCs). The induction efficiency of this method is very high (over 95%) in a short period and the hiPSC-SKPs exhibit SKP characteristics. To generate SKPs from hiPSCs, the authors established a differentiation protocol in which hiPSCs were initially differentiated to the multipotent neural crest stage as precursor cells of SKPs. Human iPSCs were treated with human recombinant noggin and SB to promote highly efficient neural induction [2]. A human iPS cell line (201B7) was generated by introducing four transcription factors (Oct3/4, Sox2, Klf4, and c-Myc) into human skin fibroblasts [55]. The hiPSCs were cultured on inactivated SNL feeder cells using hiPSC medium containing DMEM/F12, knockout serum replacement, non-essential amino acids, L-glutamine, β-mercaptoethanol, bFGF, and penicillin and streptomycin. When hiPSC colonies reached 80–90% confluence, they were plated on SNL feeder cells in hiPSC medium without bFGF, including noggin and SB431542. Then, they were cultured in SKPs medium containing DMEM/F12, B27 supplement, penicillin and streptomycin, bFGF, EGF, and CHIR99021 (CHIR). When cells reached 80% confluence, they were dissociated using Accutase cell detachment solution and were subcultured in new dishes in SKPs medium without CHIR. After 5 days, a sufficient number of cells were obtained, which were termed hiPSC-SKPs[2].
Sugiyama et al. described adipogenic, osteogenic, and neurogenic (Schwann cell) differentiation from hiPSC-SKP the same as from traditional SKPs. In addition, hiPSC-SKPs can differentiate into osteogenic cells, unlike SKPs. hiPSC-SKPs can also induce follicular type keratinization. Epidermal keratinocytes and hiPSC-SKPs express trichohyalin, a hair follicle-specific protein [2].
Human Embryonic Stem Cell-Derived Endothelial Precursor Cell (hESC-EPC) Culture
To differentiate hESC effectively into endothelial cells, several approaches have been taken, including altering cytokines in the medium and co-culturing with other cells such as stromal cells [85]. The embryoid bodies (EB) formed spontaneously from hESC have frequently been used to promote differentiation of hESC into endothelial cells [86]. Culture and differentiation of hESC has been described previously [87]. Cells are cultured on human collagen-coated dishes in EGM-2/MV medium. For expansion of hESC-EPC, cells are passaged by trypsinization [85].
Stem cells have been shown to have a therapeutic effect in several ischemic animal models. We examined the wound-healing effect of secretory factors released by hESC-EPC. Conditioned medium (CM) of hESC-EPC was prepared and applied in a mouse excisional wound model. hESC-EPC CM accelerated wound healing, increased the tensile strength of wounds, and caused more rapid re-formation of granulation tissue and re-epithelialization of wounds. In vitro, hESC-EPC CM improved the proliferation and migration of dermal fibroblasts and epidermal keratinocytes. hESC-EPC CM also increased the extracellular matrix synthesis of fibroblasts. hESC-EPCs secrete many growing factors and interleukins important in angiogenesis and wound healing [85].
Human Hair Follicle Organ Culture (HFOC)
Langan et al. described HFOC culture conditions and quality control [35]. Remarkably, even after having been removed from the human body, the HF maintains some of its in vivo characteristics in HFOC. HF growth ex vivo is influenced by the stage of the microdissected follicle, its rate of growth, its intrinsic hair cycle, the rate of matrix keratinocyte proliferation, the differentiation of matrix keratinocytes into the mature hair shaft, and the HF epithelial stem cell proliferation/apoptosis. It is also important that the major stem cell component (bulge) is absent in microdissected and amputated HFs and is only present when full-length HFs are microdissected and cultured [35]. Successful growth of anagen VI terminal HFs ex vivo for up to 2 weeks has been demonstrated [88].
Despite its important role in preclinical hair research, human HFOC clearly has major limitations due to elimination of neural, vascular, and endocrine controls of human HF biology, as well as multiple factors contained in serum. Probably, the rapid HF entry into catagen ex vivo reflects that the HF is significantly stressed by the trauma of microdissection, denervation, and serum and hormone deprivation [35]. It must also be noted that anagen scalp HFs in HFOC operate in the absence of their epithelial and melanocyte stem cell populations in the bulge, even though keratin 15+ or keratin 19+ epithelial progenitor cells and amelanotic melanoblasts are still present in the proximal outer root sheath (ORS) of organ-cultured human HFs [89]. The most important limitation of current human HFOC techniques is that human anagen HFs routinely fail to reach even the telogen phase before they degenerate ex vivo [35].
The original HFOC model [65] has been adapted for a wide range of applications. Microdissected HFs can be cultured in a serum-free medium (Williams’ E), supplemented with L-glutamine, hydrocortisone, insulin, penicillin, and streptomycin, and maintained at 37 °C in 5% CO2 air [35].
It is now possible to knock-down defined genes in human HFOC and to assess the gene expression profile of defined microdissected human HF in situ [12]. These recent developments have greatly enhanced the usefulness and instructiveness of HFOC for preclinical hair research [35].
Dermal Papilla Cell Culture
Human dermal papilla cells can be cultured in dermal papilla cell medium (Cell Applications) [2].
Reproducing a Physiological Microenvironment
Physiological stress is an important factor in both the morphogenesis and homeostasis of various organs. Physical stress has been implicated in the physiologic responses observed in cell behavior [90]. To replicate the tissue architecture, cell–cell interactions, and specific physical microenvironment, Aoki et al. demonstrated the effectiveness of a three-dimensional collagen gel culture system and further established two simple culture systems: air–liquid interface (ALI) and fluid flow stress (FFS) [66]. The microenvironment physical stress (forces of gas and fluid streaming) has a huge effect on the proliferation and differentiation of various cell types [66, 90]. The ALI culture system consists of three components: outer plastic dish, inner cell insert, and collagen gel scaffold. Skin is constantly exposed to air; thus the ALI system closely imitates the skin microenvironment [66].
Alternatives to Proteolytic Enzymes in Cell Culture
Proteolytic enzymes affect the balance between ECM degradation and deposition during cell passage. These enzymes not only destroy the ECM but also interfere in surface proteins; therefore profound changes in stem cell behavior may be produced [16].
Huang et al. studied the proteomic changes caused by the use of proteolytic enzymes (such as trypsin) in cell passage [91]. They found that 36 proteins were differentially expressed in the trypsin-treated cells. Proteins related to the regulation of metabolism, growth, the mitochondria electron transport, and cell adhesion showed less expression, while proteins that regulate apoptosis showed more expression. Cell detachment without proteolytic enzymes may maintain membrane proteins and preserve mesenchymal stem cell properties. Therefore, alternatives to the use of trypsin are being developed, such as cell culture in cell sheets or hydrogel 3D culture [16].
Culture in Cell Sheets
Cell sheets culture was developed to promote cell passage without the use of proteolytic enzymes [16]. Yamada et al. developed the cell sheets technology, in which the cells and their ECM are collected together, without proteolytic enzymes treatment or any tool for extracting cells [92]. The plates are coated with thermo-responsive polymers which change its cell adhesion property as the temperature changes. Several groups have been using the cell sheets technology [91, 92]. It has been demonstrated that, after three passages, cells grown in cell sheets preserve both viability and proliferation properties, and differentiation to some extent [93].
Hydrogel 3D Culture
Hydrogel can mimic the tissue-specific cellular 3D microenvironment by manipulating the ECM physicochemical properties and components, according to tissue and culture requirements. However, it is a challenge to promote appropriate oxygen, soluble factors, and the requirements of cell nutrients transport in hydrogel 3D culture. Hydrogel can be used to culture cells in bioreactors, as a 3D culture which avoids the use of proteolytic enzymes, as a mechanical vehicle to 3D cell/organ printing, and as a biocompatible material to be implanted in vivo. Despite the challenges, hydrogel 3D culture, which avoids proteolytic enzymes, is a good alternative solution for either preservation or manipulation of ECM components [16].
Three-Dimensional (3D) Cultures
Two-dimensional (2D) cell culture systems have routinely been adopted around the world for the past four decades [66]. The 2D cell culture microenvironment affects cellular function, since only one side of the cell is in contact with the ECM and the neighboring cells [16]. Creation of a 3D scaffold provides a better physiologic microenvironment for cultured cells and is expected to more closely develop the cellular function.
Several types of 3D skin culture systems have been developed. A 3D skin culture system was introduced by Ozbun et al. [94] to grow differentiating epithelial tissues that mimicked important morphological and biochemical aspects of skin. This technique is often called an organotypic raft culture due to its apparent floating nature with growing keratinocytes on top of a collagen lattice with fibroblasts [4]. Organotypic raft culture promotes stratification and full differentiation of keratinocytes when placed at the air–liquid interface [4]. Aoki et al. demonstrated the advantages of using a collagen gel-based 3D cell culture system to analyze the effects of adipose tissue on various cell types in vitro [66]. As previously described, hydrogel 3D is an interesting culture alternative [16].
Automatic Bioprocessing
Although static tissue culture is sufficient to generate cells for experimental purposes, it is impractical for generating the large quantities of SKPs that would be required in an autologous cell therapy to repopulate the HF mesenchyme or dermis to enhance wound healing. Static tissue culture methods are time and labor intensive, and manual handling and inherent cellular variation between flasks are influential factors. Therefore, controlled cell culture processes must be developed to efficiently and safely generate sufficient stem cell numbers for clinical use. Computer-controlled stirred-suspension bioreactors can be used for this purpose and generate a large number of DSCs while maintaining their phenotype and at least some of their inherent inductive function [43].
Previous studies using cell types that included murine embryonic stem cells, human embryonic stem cells, multipotent adult progenitor cells from bone marrow, neural precursor cells, mesenchymal stem cells, and induced pluripotent stem cells have all shown that stirred-suspension bioreactors are an effective alternative for culturing stem cells. Stirred-suspension bioreactors offer several advantages over static cultures, including reduced labor and costs, higher yield, more cellular homogeneity, reduced space requirements, and increased cell density per volume. They also allow for precise monitoring and control of key process variables, such as physiochemical environment, thus providing a healthy environment for cells and often leading to increased cell proliferation [43]. The shear stresses produced in stirred-suspension bioreactors can stimulate proliferation and differentiation of stem cells [95]. Moreover, authors have shown that shear stress in stirred suspension can play a role in the expression of stem cell markers [96]. Exposure to shear force might liberate single cells from proliferating aggregates, thereby reducing the average colony size and allowing for formation of new colonies and producing an increase in viable cell number in stirred-suspension bioreactors [43].
Compared with static culture, stirred-suspension bioreactors generated fivefold greater expansion of viable SKPs which were able to reconstitute the HF mesenchyme, to induce de novo hair follicle morphogenesis, and to exhibit bipotency, reconstituting the dermal papilla and connective tissue sheath, although bioreactor-grown SKPs exhibited a significant reduction in hair-forming ability compared with static-expanded SKPs [43]. Little phenotypic differences were found in SKPs exposed to either static or automated bioreactor expansion, and most DSC markers, with the exception of SOX2, were sustained over multiple passages. In static culture, a subset of aggregates consistently adhered to the culture flask, and the frequency of adhesion appeared to increase over passages. All rSKP aggregates grown in the bioreactor remained in suspension, as the reactors were siliconized before use, thus preventing adhesion to the vessel surface [43]. Adhesion is indicative of differentiation and would largely contribute to the limited expansion observed in static culture [42].
Skin Engineering
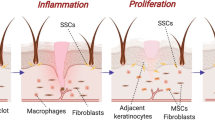
Significant progress has been made over the past 25 years in the development of engineered substitutes that mimic human skin (Fig. 5), either to be used as grafts or for the establishment of in vitro human skin models [98]. Several methods have been described to build skin maintaining its structure and function [24].

Preparation of a bioengineered skin substitute (Reproduced from Hakim et al. [97])
Insights from developmental biology are already pointing to the development of “intelligent materials” that work with nature’s own mechanisms of organogenesis and repair. Biologically active and appropriate matrices and factors in combination with automated (tissue printing) techniques are designed to produce a new generation of complex skin substitutes in a desired number and with a constant quality [26]. Tissue engineering is emerging as a potential solution for tissue and organ failure (Fig. 6) [40]. Tissue engineering has given tools to cover large surface wounds which has been one of the major challenges in clinical research [41]. Successful regeneration of skin with skin substitutes depends on two factors: the presence of self-renewing keratinocyte stem cells for re-epithelialization and a functional dermal substitute consisting of the appropriate cellular and acellular components, which allow no or only limited scarring of the developing skin [26]. To prevent immunological incompatibility, autologous cells may be used. However, the number of cells required for the construction of an organ or tissue is much greater than the number of cells obtained from an autologous donor cell source. In this way, expanding the number of cells in cell culture for a long period is required until the necessary amount of cells is obtained. Nevertheless, maintaining the cell characteristics throughout the expansion process is a challenge as cell processing and cell expansion protocols have not been established yet [16].

Tissue engineering concept (Reproduced from Pandey et al. [99])
Several “commercial” treatment modalities exist along with skin grafting. Various problems are still encountered. A vascularized wound bed is required for prompt graft attachment. If a dermal substitute reaches a threshold thickness [100], vascularization is too slow resulting in epidermal necrosis or graft loss. Therefore, most dermal substitutes thicker than 1 mm (Integra®, Matriderm®) are applied using a two-step approach, giving the dermal substitute sufficient time to vascularize. Transplantation of an epidermal component needs an additional operation. Features of the transplanted epidermal component may be missing elasticity, contraction, lack of pigmentation, and thereby lack of protection against UV radiation [26].
Keratinocytes and fibroblasts in tissue-engineered skin produce the correct concentration and combination of growth factors and cytokines important for efficient and effective wound repair as well as providing the necessary ECM components. In addition, one of the advantages of using skin substitutes is that they can be cryopreserved and that following thawing, the fibroblasts retain the ability to proliferate and produce appreciable amounts of VEGF, hepatocyte growth factor, basic FGF, TGF-β1, and IL-8 [29, 101].
Several commercial products have been developed during the last 30 years (Table 2). They can be permanent or temporary; autologous, allogeneic, or xenogeneic; and made of natural or synthetic materials as scaffolds for cell attachment. They can be classified into three types.
Types of Skin Substitutes
Epidermal Substitutes
Epidermal substitutes contain autologous keratinocytes, often grown in the presence of murine fibroblasts. Most products belong to the category of “cultured epidermal autografts” (Epicel®, EpidexTM, MySkinTM). Developing the final substitute from a skin biopsy takes about 3 weeks [126]. Thus, burn wounds initially need to be treated with temporary wound dressings. Several studies and multicenter trials [126, 127] show a wide range of take rates with an average value of 50% or less [128], and statements about outcomes are inconclusive due to the diversity of methods. Disadvantages are mainly their slow preparation time, variable engraftment rates, difficult handling, and their high production costs [26]. Another approach for epidermal replacement is the use of autologous keratinocytes in suspension (ReCell®) [129].
Dermal Substitutes
Dermal substitutes used as dermal regeneration templates play an important role in skin reconstruction by improving wound healing and scar formation [26]. Engineered dermal substitutes promote new tissue growth and optimize healing conditions by secretion of growth factors and deposition of dermal matrix proteins [29, 130]. There are currently a range of dermal substitutes which may be acellular or cellular. Some of them consist of acellular matrices, which are permanently incorporated into the patient’s wound bed (AlloDerm®, Integra®, Matriderm®) [108, 112, 114]. The four types of commonly used natural materials are collagen, chitosan, hyaluronic acid, and carboxymethyl chitosan. Dermal substitutes need to be covered by a permanent epidermal substitute [130]. These substitutes are colonized and vascularized by the underlying cells usually 3–4 weeks after application [111]. As the autologous neodermis regenerates, the scaffold gradually disappears. Histologic evaluation of biopsies did not show any evidence of immunologic response [131]. More recent approaches are using thinner dermal layers, with the aim of transplanting the dermo-epidermal substitute in a single step [132]. Artificial three-dimensional scaffolds have been used as effective dermal regeneration templates for treating full-thickness skin defects [24].
Incorporation of stromal fibroblasts into dermal substitutes has shown great promise for their application in repairing tissues by fabricating dermal substitutes. In contrast to allogeneic cells, autologous fibroblasts carry no risk of rejection or cross-infection [29]. However, there is often a delay in obtaining sufficient autologous cells, whereas allogeneic cells are cryopreserved and therefore readily available. hiPSCs offer a novel source of autologous cells for dermal regeneration. iPSC-derived fibroblasts may improve efficacy and function for future regenerative therapies [24].
The attempt to incorporate growth factors has, in most cases, been disappointing due to their instability. Loading of functional genes into the scaffolds is a way to produce growth factors, which have drawbacks such as enzymatic degradation of the DNA and low cell transfection efficiencies [24].
Dermo-epidermal Substitutes
More than 10 years have passed since the development of cultured skin substitute (CSS), which consists of cultured autologous epidermis and dermal fibroblasts. However, the CSS does not regenerate the appendages. The incorporation of an epidermal component composed of differentiated keratinocyte layers onto a cellular dermal substrate leads to the formation of a bilayered skin substitute [40].
Few engineered, “off-the-shelf” dermo-epidermal substitutes have been produced. Human allogeneic neonatal keratinocytes and fibroblasts are combined with a scaffold to form a temporary covering (Apligraf®, OrCel®) [29, 100, 130]. Apligraf® was the first bilayered living skin equivalent produced [29]. For autologous cultured dermo-epidermal substitutes [130], keratinocytes and fibroblasts are obtained from a burned patient’s biopsy and added to a collagen–glycosaminoglycan substrate [133]. In terms of graft take and scar appearance, this substitute yields results superior to conventional techniques [130, 134], but further clinical studies need to confirm these results [26]. However it has the disadvantage that it is an expensive method, which requires five weeks of preparation [134].
Keck et al. described the construction of a multilayered skin substitute with human preadipocytes from subcutaneous tissue and cultured keratinocytes seeded onto a scaffold (Matriderm®) [118].
Skin Appendages
Wang et al. identified clinically applicable stem cells for de novo regeneration of the hair follicle and sebaceous glands (SG), suggesting a great potential to develop novel bioengineered skin substitutes with appendage regeneration capacity. The authors demonstrated that a combination of culture-expanded Epi-SCs derived from adult human epidermis and culture-expanded adult human SKPs was sufficient to regenerate de novo hair follicles and hairs. In addition, they evidenced that Epi-SCs from the epidermis differentiated into sebocytes in vitro and formed functional SGs in vivo upon appropriate induction [40].
Stem Cells in Tissue Engineering
Recent research regarding the use of stem cells for skin tissue engineering has mainly concentrated on mesenchymal stem cells (MSCs), adipose-derived stem cells (ASCs), embryonic stem cells, and induced pluripotent stem cells (iPSCs). Meanwhile, a few researchers have begun to focus on dermal-derived stem cells [42, 135, 136]. These stem cells are famous for their ability to perform multi-directional differentiation [24].
MSCs in the bone marrow have the ability to differentiate into a variety of cells and tissues derived from the mesoderm and the neural ectoderm. Under certain conditions, hMSCs can differentiate into epidermal-like cells [137]. Therefore, the bone marrow MSCs are used as seed cells to construct full-thickness skin tissue [24]. MSCs have also been used in the induction of vascularization of tissue engineering scaffolds [138].
Research investigating differentiation from ASCs into epidermal cells is very minimal but may result in breakthroughs in the treatment of severe trauma and extensive burns [24]. Therefore, ASCs may be ideal seed cells for skin tissue engineering research [139].
Clinical Applications
Skin Substitutes
Many clinical indications for treatment with skin substitutes have been described:
Chronic Ulcers
Dermal equivalents and bilayered skin substitutes have been used to treat chronic nonhealing wounds, such as venous, diabetic, and pressure ulcers [29].
Burns
Burn injuries may be divided into partial-thickness burns, involving loss of epidermis and papillary dermis, and full-thickness burns where damage is deeper. Superficial partial-thickness burns may result in full regeneration by re-epithelialization without scar formation, in comparison with full-thickness burns where scarring inevitably occurs. Nevertheless, all burn injuries can lead to loss of fluid and proteins, and increase susceptibility to infection, thus requiring immediate attention. Nonbiological topical treatments and biological dressings may be used. Tissue-engineered skin substitutes as temporary biological dressings (i.e., AlloDerm®) are also effective and promote wound healing [140]. Alternatively, the use of a bilayered skin substitute such as Apligraf® or OrCel® requires just one step for skin replacement. The advantage of using Apligraf® as opposed to cadaver skin as a biological dressing is that Apligraf® is readily available, of reproducible quality, and does not predispose the patient to infectious disease transmission. Furthermore, Apligraf® incorporates neonatal fibroblasts which have higher proliferative rates and offer the possibility of producing near normal dermis [141]. On the other hand, treatment of burns may involve two steps [29]. First a dermal template with artificial epidermis initially allows autogenous neovascularization and autologous fibroblast migration into the dermal scaffold. Second, following formation of the neodermis, the temporary epidermis is removed and replaced by an epidermal autograft. An example of this type of product is Integra®[29].
Genodermatoses and Other Dermatological Conditions
Tissue-engineered skin substitutes have been used with variable success in epidermolysis bullosa (EB), pyoderma gangrenosum, hydroxyurea-induced leg ulcers, bullous morphea ulcers, and ulcerative sarcoidosis. Fibroblasts for cell-based therapy and gene therapy have been used for the treatment of recessive dystrophic EB [29].
Cosmetic and Reconstructive Surgery
Tissue-engineered skin substitutes have also been used for the treatment of wounds following cancer excision [29]. They have the advantage of not inducing donor-site defects as well as allowing monitoring for local tumor recurrence. Dermagraft® has been successfully used for covering intraoral defects following oral squamous cell carcinoma [142]. In addition, the use of Apligraf® produced better cosmetic results in wounds following Mohs or excisional surgery [143].
Cultured Fibroblasts
Cultured fibroblasts may be utilized to promote tissue repair in a variety of conditions ranging from acute and chronic wounds through to their application in aesthetic and reconstructive surgery. For permanent engraftment, autologous fibroblasts are necessary. However, allogeneic fibroblasts may be used as a biological dressing or for preconditioning of the wound bed prior to graft application, especially when wounds are very large. In addition, using autologous fibroblasts in dermal substitutes has led to better restoration of dermal tissue and minimal scar formation compared with allogeneic dermal substitutes [29].
Skin Stem Cells
The skin stem cells (SSCs) are in clinical setup for a long period of time and have been used for the management of vitiligo, burn, and other pigmentary disorders. Hair follicle stem cells (HFSCs) have also been used for cell-based clinical needs, especially in vitiligo [47].
hiPSC-SKPs can provide an unlimited number of dermal SCs and could contribute to skin dermal regeneration that was lost due to injury or disease [2]. Enhanced expansion of SKPs in computer-controlled stirred-suspension bioreactors might provide a safe and efficient method to generate large numbers of DSCs, thereby permitting drug screening for compounds that might influence HF growth or cell-based strategies to repopulate the skin and hair follicle after injury or disease [43].
Secretory Factors
Secretory factors released from stem cells could be an important mediator of stem cell therapy in ischemic tissue diseases [85].
Clinical Applications in Aesthetic Medicine
Currently the use of cell culture techniques and tissue engineering is not widespread in aesthetic clinical practice.
Fibroblast Injections
Fibroblasts and ECM decrease during skin aging resulting in the formation of wrinkles; therefore a therapy for rhytids consisting of autologous cell injection has been proposed. Cell injection is neither a dermal filler, nor stem cells (but may contain stem cells, which is under investigation), nor growth factors [74]. Cultured autologous fibroblasts seem to be the first successful implementation of cell therapy for the treatment of wrinkles. This treatment offers the promise of maintained growth of cells, which may persist longer than other fillers [79].
Autologous fibroblasts are the first and only autologous fibroblast cell therapy approved by the US Food and Drug Administration (FDA) for aesthetic use that is grown from patient biopsies and injected back into facial skin (Fig. 7). Fibroblast cell injections appear to be best suited for fine lines with the current on-label indication of nasolabial wrinkle correction [79]. There is evidence that autologous fibroblast injections can improve the appearance of facial wrinkles and depressed scars [145, 146]. Other indications are wounds [147] and subcutaneous atrophy [80]. Some physicians are currently using the product for off-label indications such as glabella folds, periocular rhytids, tear troughs, upper lip rhytids, marionette lines, chest wrinkles, necklace lines, and atrophic skin of the dorsal hands. Long-term results are expected, but not proved. Anecdotal evidence suggests that some patients treated in clinical trials 8 years ago still show clinical benefit for NLF [79].

Correcting nasojugal groove with autologous cultured fibroblast injection (Reproduced from Moon et al. [144])
Fibroblasts secrete different kinds of ECM proteins, of which collagen is the most likely involved in correcting dermal and subcutaneous defects. Type I and type III collagens are the most abundant types of collagens in the skin. In adults, type I collagen constitutes approximately 80% of dermal collagen, whereas type III collagen is abundant in healing tissue, and then it is gradually replaced by the stronger and tougher type I collagen [148]. In Zeng et al. study [64], both types of collagens were secreted by the transplanted human fibroblasts and accumulated gradually during the 3 months.
Unlike traditional fillers, cultured autologous fibroblast cells are injected more superficially and treatment may require months to show improvement. Therefore patients must be informed that this treatment does not work immediately. Autologous cells give gradual improvement after three consecutive treatments over several weeks. Autologous fibroblast cell injection may be a good alternative for patients who do not want foreign materials injected. Side effects are minimal and comparable with other injected agents. Compared with other fillers, additional costs for harvesting and culturing before injection are incurred. Autologous fibroblast treatment may be synergistic to volume fillers. Autologous fibroblasts may provide a long-term solution to the increase of dermal collagen bundles [79]. In addition cultured fibroblasts can extend the longevity of bovine collagen [78].
Clinical trials for autologous fibroblast therapy have been conducted since 2001 [79]. A major trial (N = 215) with living autologous fibroblast cells for the treatment of facial contour defects was reported in 2007 [149], showing initial hopeful results. Live fibroblasts (20 million/mL) were given in three doses administered at 1-week to 2-week intervals. Efficacy evaluations were performed 1, 2, 4, 6, 9, and 12 months after the first injection. Results indicated that living fibroblasts produced greater improvements in dermal deformities and acne scars. At a 12-month follow-up, patients treated with live fibroblasts continued to show benefit from treatment. No serious adverse events were reported. This finding led to the initial conclusion that autologous fibroblast injections could safely and effectively produce improvements in rhytids, acne scars, and other dermal defects continuing for at least 12 months after injection [79].
Eça et al. performed a study to assess the safety and efficacy of the injection of autologous fibroblasts cultivated in the patient’s own serum for dermal repair of skin flaccidity and wrinkles [74]. A skin biopsy was performed in the groin region. Next the dermis was mechanically separated from the epidermis and the hair follicles and then fragmented and transferred into culture flasks. After the primary culture reached 70% confluence, the cells were treated with trypsin solution, centrifuged, and resuspended in PBS (phosphate-buffered saline). Then two aliquots were separated: 1 mL for expansion and 1 mL for injection. The aliquot for expansion was cultured constituting the first cell passage (first population doubling). When confluence of 70% was reached, the cells at first population doubling were once again submitted to trypsinization, with 50% of the cells being used for injection and the remainder for expansion until completion of the second population doubling, when the entire cell content was injected. Injections into the superficial dermis in forehead wrinkles, perioral wrinkles, nasolabial fold, chin, and periorbital skin were performed using a retrograde linear threading technique. Injections were given over four sessions, with a minimal interval of 15 days between each session. The first injection was performed after the first passage (first population doubling). Injections were given every 15 days at the second, third, and fourth population doublings. The cell population increased progressively. The cells resulted in 98% viable cells at the fourth population doubling. Sixty days after completing the four intradermal injections, significant improvement was found in periorbital flaccidity in two cases, with slight improvement in surface lines in one case. No improvement was found in deeper wrinkles. Six months after completion of treatment, no further changes were found. A total of 6.4 × 106 fibroblasts/mL was injected, resulting in a good response in the periorbital region, although surface wrinkles and deeper wrinkles may require a greater number of fibroblasts, as shown in the Weiss study [149] previously described.
The current autologous fibroblast therapy product called Isolagen Therapy™ (Laviv™) is the first cell therapy cleared by the FDA for aesthetic improvement and the first to show statistically significant benefits in large blinded controlled trials [150]. A personalized biopsy kit/shipper is sent to the practice location. The biopsies are performed and processed for culture the next morning. Three 3-mm punch biopsies are performed in the retroauricular area with just enough depth to obtain cells from the dermis, but not as deep as adipose tissue, and placed in the transport media vial. After fibroblast culture, fibroblasts are harvested and cryopreserved. Before use, the cells are thawed, washed with PBS and Dulbecco’s modified Eagle’s medium, resuspended at a concentration of 1.0 to 2.0 × 107 cells/mL, and shipped overnight at 2–8 °C to the treatment center for administration the next day. Efficacy and safety tests are performed. Before use at the treatment center, the cell suspension is stored at 2–8 °C and then allowed to warm to room temperature for 30 min before use. Only topical anesthetic is used. The area of treatment is cleaned using alcohol with time allowed for the alcohol to evaporate. After gentle inversion of the vial to dislodge clumped cells, aspiration into a 1-mL or 3-mL syringe is performed using a 22-gauge to 25-gauge needle. The cell suspension is injected using 30-gauge needle in a retrograde threading technique or in small aliquots of 0.05–0.1 mL directly into a wrinkle. Serial puncture is most commonly used. No lidocaine or epinephrine is added to the cell suspension before injection because it could be harmful to cells. The injection is into the superficial papillary dermis and confirmed by the appearance of blanching and wheal formation. No massage or other manipulations of the areas are performed to avoid risk of damaging the cells. Subjects should avoid the use of soaps or any other products to the face for 72 h after each injection session, although mild washing is permitted. Indirect application of ice to the treatment area is not recommended. The treatment consists of three sessions, each 5 weeks apart, with a dose of 0.1 mL of a suspension of 1.0 to 2.0 × 107 cells/mL [79]. Clinical improvement in NLF wrinkles was seen 2 months after the start of treatment, with continuous improvement in the follow-up months 2–6 after a series of three injections [150]. Unlike most dermal filler products, autologous fibroblast therapy benefit is not expected to degrade over 6 months [151, 152]. Prior studies of autologous fibroblasts showed continued benefit 1 year after treatment [149]. Zhao et al. proved that cultured autologous skin fibroblasts survive for at least 5 months after injection [80].
Cultured fibroblasts can be injected combined with hyaluronic acid (HA) to obtain longer-lasting results [78, 153]. Solakoglu et al. [78] used cross-linked HA as a biodegradable polymer scaffold for cultured human fibroblasts. Dermal fibroblasts obtained from rat skin biopsies were cultured and injected. The density of the cells in mixture was approximately 30 × 106/mL. At the end of the fourth and eighth months, the injected fibroblasts, elastin, and collagen production were found to be stable and well tolerated. Syntheses of collagen and elastin were demonstrated. HA bulks surrounded by fibroblasts suggest an interaction between HA and fibroblasts. HA also promoted vascular angiogenesis. There were no signs of apoptosis, inflammation, or necrosis, which was expected because the injected cells were autologous. Therefore, cultured human dermal fibroblasts combined with hyaluronic acid can provide a long-lasting material and should be regarded as a new method in dermal renovation [78].
Scar formation and contraction should be avoided when using fibroblast treatment. In case of correction of dermal and subcutaneous depression, where the skin remains intact, the possibility of wound contraction is very low. Thus fibrosis and scar formation are our primary concern, which may be caused by excessive growth or secretion of cultured fibroblasts after transplantation. The extracts of the dermis (extract D) could inhibit the proliferation of fibroblasts [154].
Stem Cell-Conditioned Medium
Microneedle fractional radiofrequency is a safe and effective skin rejuvenation method, and better results may be expected when combined with stem cell-conditioned medium [155]. The stem cell-conditioned medium (hESC-EPC CM) [85] is composed of a large number of growth factors and cytokines. In vitro, hESC-EPC CM significantly improved the proliferation and migration of dermal fibroblasts and epidermal keratinocytes and also increased collagen synthesis of fibroblasts. hESC-EPCs secrete cytokines and chemokines which are important in angiogenesis and wound healing [156]. Patients received three sessions at 4-week intervals. Histologic examination revealed marked increase in dermal thickness and dermal collagen content. Side effects were minimal [155].
Efficacy and Safety
Since the first transplants of adult stem cells from bone marrow in 1959 [157], there is no record in the scientific literature of any case of tumor formation resulting from the injection of these cells [74]. The technique is considered safe at an expansion of up to the fourth population doubling[158].
Immunological Impact of Allogeneic Cells
There have also been a number of studies investigating the immunological impact of allogeneic cells [29]. It has been suggested that allogeneic cells are replaced by host cells. In addition, large trials involving grafting of allogeneic skin equivalents onto venous ulcers did not reveal evidence of rejection clinically or immunologically in the patients [159]. There was no demonstration of antibodies specific for human leukocyte class I antigens expressed on allogeneic cells and no proliferation of T cells in patients. One of the reasons for the perceived lack of acute rejection in immunocompetent hosts is that dermal fibroblasts lack major histocompatibility complex class II antigens necessary for antigen presentation [160]. It has also been proposed that during in vitro culture, the antigen-presenting cells, such as Langerhans cells, are gradually lost following serial passages [161]. One study assessed the persistence of allogeneic fibroblasts in an acute wound (porcine model) and found that after 1 week, allogeneic fibroblasts were not detectable by polymerase chain reaction [162].
Malignancies Development
It is essential that clinical safety be ensured as these cells return to patients. It is also important to investigate and understand the changes in cell culture to be certain that the cells do not carry mutations or unwanted differentiations that may cause any pathology in the medium- to long-term horizon [16].
Hayflick et al. [69] have shown that human fibroblasts could maintain their genomic stability after 40 generations, although cells at the tenth passage were too senescent for injection. Clinically, cells at passages 3–4 are most suitable for injection in terms of cell quantity and proliferative and secretory activity [64]. Eça et al. [74] ensured that no genetic alterations occurred in fibroblast expansion up to the fourth passage. Zeng et al. [64] noted that the proliferative behavior of the cultured cell population remained stable from passages 5 to 10 without overactive division or apoptosis. Cells at passages 5 and 10 maintained their normal somatic cell diploid karyotype, and no mutations or other translocations were discovered. No chromosomal abnormalities were found in in vitro expanded human fibroblasts. It was also noted that the proliferation was active at the first month and returned to normal later, indicating that the proliferation of the injected cells was under certain regulations, which prevented the cells from hyperplasia. The fact that no macrophages were found suggested that there was no abnormal apoptosis or necrosis of the injected cells. Moreover, normal cell morphology without dysplasia was observed from histological sections, and no tumors were detected from gross inspection together, suggesting that no oncogenic transformation or fibrosis formation had occurred at least 3 months after transplantation [64]. However, small segment mutations or other subtle molecular events were not completely excluded. The in vivo section of the study has limitations since the injected cells were not labeled. Second, the viability and stability of the injected cells were only demonstrated by microscope observation. Telomerase activity and carcinogenicity should be measured in a further study [64].
A theoretic risk of the enhancement of malignancies with autologous cell therapy has been raised [163]. Although one basal cell carcinoma was reported near a treatment site in the pivotal trial, it is unrelated to the autologous fibroblast injection for two reasons:
-
1.
Given the number of subjects and duration of the trial, the incidence of cutaneous malignancy is consistent with background rates.
-
2.
Basal cell carcinoma is not a fibroblast-derived tumor. To date, no dermatofibrosarcomas have been reported in thousands of injections [79].
Animal Disease Transmission
The use of fetal bovine serum (FBS) in fibroblast culture medium may increase the risk of infection from bovine diseases or of a reaction to foreign proteins. In addition, cell division and consequently the number of fibroblasts have been shown to be greater in autologous dermal fibroblasts cultivated in human serum than in FBS [164].
Autologous Cultured fibroblasts
The advantage of injecting a live autologous filler is obvious as it leads to longer-term correction and eliminates the problem of hypersensitivity and foreign body granulomatous reactions [29]. Many authors have studied the safety and efficacy of the injection of cultured autologous fibroblasts [29, 64, 74, 78, 79, 149]. The preclinical safety and efficacy of autologous cultured human skin fibroblasts has been studied and their proliferation and secretion activity has been demonstrated. The implanted fibroblasts can survive in vivo for more than 5 months and actively secrete new collagen [64, 80]. The use of cultured human skin-derived fibroblasts has been proven to be safe, providing a basic support for clinical use of autologous fibroblast transplantation [64].
Side effects of cultured autologous fibroblasts are minimal and temporary and comparable with other injected agents [79]. A major trial reported in 2007 [149] found no serious treatment-related adverse events. No hypersensitivity reactions have been noted, and no long-term nodules or other local problems have been seen [150]. Solakoglu et al. found no complications after injection of cultured fibroblasts combined with stabilized hyaluronic acid (HA) [78]. Furthermore, there have been no reports of hypertrophic or keloid scarring following the injection of autologous fibroblasts, suggesting that fibroblast proliferation and collagen synthesis are naturally regulated by cell–cell and cell–ECM contact and negative feedback [145]. Therefore autologous fibroblast therapy is as safe as many of the existing filler therapies [79].
Skin Substitutes
A systematic review which included 20 randomized controlled trials assessed the safety and efficacy of bioengineered skin substitutes in comparison with standard methods in the management of burns. Nevertheless the numerous subgroup analyses and the diversity of skin substitutes limited the ability to draw conclusions [41, 130]. Bioengineered skin substitutes, as Biobrane®, TransCyte®, Dermagraft®, Apligraf®, Integra®, and CEA (cultured epithelial autograft), have proved to be at least as safe as the classical skin replacements or topical agents/wound dressings [130]. Apligraf® is composed of allogeneic keratinocytes and fibroblasts which are not detectable beyond 6 weeks [165].
Present and Future of Cell Cultures
Stem Cells
The possible therapeutic use of somatic cells derived from embryonic stem cells is currently a hot topic in regenerative medicine [41]. Although skin biopsies are a regular source of keratinocytes and skin stem cells, the generation of keratinocytes from human embryonic stem cells could be a useful technique [166]. Nevertheless obtaining skin stem cells from the own patient’s skin might simplify the procedure.
Work on embryonic stem cells [167] and the discovery of Yamanaka et al. [168], which demonstrated that adult cells can return to the embryonic stage with the possibility of producing all the specialized cell categories, give hope in the near future to cellular genetic therapy, which might replace tissue and organ transplants [134].
The usage of stem cells may help to overcome the limitations of current technologies (i.e., the lack of vascular networks, sensory receptors) [24]. Though using stem cells has been partially effective, the potential risks of malignant teratoma formation and long-term adverse effects of the stem cells should be taken into account, and therefore more extensive studies are required [169].
Tissue Engineering
Tissue engineering of skin is based on 25 years of research and rests on a strong background of technologies and cell and molecular biology. Despite initial unrealistic expectations, tissue-engineered skin has already delivered considerable benefits to patients with burns, accidents, infections, and chronic wounds. In this regard, “skingineering” has a huge potential that has just begun to be realized [26]. The challenge that still remains is the generation of a complex dermo-epidermal substitute that can be safely and effectively transplanted with minimal scarring in one single-step procedure. Skin regeneration aims to achieve structural and functional reconstruction, reducing scar formation and improving the quality of wound healing [24]. Regeneration rather than scar formation is key [15].
The invention of Integra® was a major step in skin engineering. Controlled release of angiogenic factors from matrices, seeding endothelial cells directly into the matrix, and engineering the vasculature directly into the tissue would largely contribute to speeding up vascularization in these complex skin grafts [26]. The combination of tissue-engineered skin substitutes with cytokines and growth factors may in the future be used to enhance wound healing as well as the possibility of incorporating defensins for antimicrobial benefit [29]. The use of cells with its preserved matrix could exempt the need for a scaffold [16].
Stem cell biology also has to be integrated in this future concept. Human adult stem cells can be a source to generate skin in vitro [26]. iPSCs provide a novel source of bioactivity for scaffolds to promote cell proliferation and differentiation via different signal transduction pathways. However, the currently available dermal substitutes have certain deficiencies to overcome, such as inconvenient usage processes, missing hair follicles, and poor resistance to infection, and most of them require secondary transplantation in autologous skin. These include further research into the differences between fetal scarless wound healing and adult wound healing and improving vascularization of scaffold materials and seed cells, especially stem cells [24, 100].
Skin Three-Dimensional Bioprinting
In recent years, skin three-dimensional bioprinting is a potential technology which can generate stratified constructs to simulate function and morphology of the natural skin [98, 135, 169]. Until now, relatively simple skin architectures composed of keratinocytes and fibroblasts have been successfully generated through bioprinting techniques (Fig. 8). These skin architectures have demonstrated similarities to native skin and have performed some skin functions in in vivo studies [24, 98, 165,166,173].

Skin bioprinting (Reproduced from Augustine [170])
Cubo et al. generated a human bilayered skin using bioinks containing human plasma as well as primary human fibroblasts and keratinocytes that were obtained from skin biopsies in less than 35 min. The generated skin was very similar to human skin and indistinguishable from handmade bilayered dermo-epidermal equivalents [98]. The barrier function of the skin is closely relative to the maturation and formation of the stratum corneum. Therefore the bioprinted skin constructs cannot achieve complete function due to the absence of a fully differentiated epidermal region [165,166,173]. The density of ECM and cellular component can be controlled in bioprinting and the cell viability can be maintained during the whole printing process [165,166,167,174]. Furthermore, the thicknesses of printed constructs can be customized and controlled according to the wound depth [175]. Therefore 3D bioprinting is a suitable technology to generate bioengineered skin for therapeutical and industrial applications in an automatized manner [98].
Cell Cultures
The future application of fibroblasts for gene therapy also offers a huge potential in providing new strategies for treating skin genodermatoses [29]. In the future, autologous fibroblast cell therapy might replace superficial synthetic fillers, but currently that is doubtful [79].
HFOC is a versatile and accessible assay system which has only just begun being employed for research into human genodermatoses and miRNA function, chronobiology, cutaneous neuroendocrinology, HF-associated progenitor cell biology, melanocyte biology, epithelial stem cell immunopathology, and mitochondrial biology [35].
In postpartum humans, skin appendages lost in injury are not regenerated. Wang et al. demonstrated that transplantation of culture-expanded epidermal stem cells and skin-derived progenitors led to de novo regeneration of functional hair follicles and sebaceous glands. This finding could be the basis for the development of novel bioengineered skin substitutes with epidermal appendage regeneration capacity [40].
Future studies will identify the inductive signals that might explain the limited inductive capacity after bioreactor expansion. It is promising that inductive function is partially retained after extensive cell expansion [43].
Cell Reprogrammation and Immortalization
The reprogrammation of differentiated somatic cells to iPSCs offers an opportunity to generate pluripotent patient-specific cell lines [41]. These iPSC lines could help in model human diseases, drug discovery, and cellular transplantation therapies. Nevertheless, there are lots of factors regarding safety that should be resolved [24, 170,177,178]. Better understanding of cellular senescence will allow the immortalization of various kinds of primary cells which will be essential not only for regenerative medicine but also for the economic development of a three-dimensional skin culture system [4].
Conclusions
-
Even though there is still much work to be done, the rise of cell culture and tissue engineering is providing powerful tools for regenerative medicine evolution.
-
It is important to maintain not only the tissue structure but also to preserve its function.
-
Efforts should be ideally directed to efficiently and safely obtain large amounts of high-quality product reducing costs, and to decrease the use of allogeneic products, thus diminishing potential risks.
-
Further progress in automatic processing may improve reproducibility and reliability and may contribute to speeding up the process, reducing costs, increasing efficiency, homogenizing protocols, and decreasing manual procedures variability. Computer-controlled bioreactors and skin 3D bioprinting can contribute in this regard.
-
The advance of cell therapy will possibly make it more accessible and affordable, and in the future rejuvenation treatments might not be as we know them today but would mostly consist of autologous treatments with a decrease of secondary effects.
-
Further large and rigorous studies with long-term follow-up should be performed to assess the safety of cell culture and skin substitutes.
-
The use of stem cells may help to overcome some of the limitations of current tissue engineering techniques (i.e., angiogenesis, sensory receptors generation).
-
Great improvements have been made in this field, and now the challenge is its application to routine clinical practice.
References
Freshney RI. Culture of animal cells: a manual of basic technique and specialized applications. Wiley-Blackwell: Hoboken, NJ; 2016.
Sugiyama-Nakagiri Y, Fujimura T, Moriwaki S. Induction of skin-derived precursor cells from human induced pluripotent stem cells. PLoS One. 2016;11:e0168451. https://doi.org/10.1371/journal.pone.0168451.
Tavakolpour S, Daneshpazhooh M, Mahmoudi H. Skin cancer: genetics, immunology, treatments, and psychological care. In: Mehdipour P, editor. Cancer genetics and psychotherapy. Cham: Springer; 2017. https://doi.org/10.1007/978-3-319-64550-6_18.
Choi M, Lee C. Immortalization of primary keratinocytes and its application to skin research. Biomol Ther (Seoul). 2015;23:391–9. https://doi.org/10.4062/biomolther.2015.038.
Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–44.
Rheinwald JG, Green H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature. 1977;265:421–4.
O’Connor N, Mulliken JB, Banks-Schlegel S, Kehinde O, Green H. Grafting of burns with cultured epithelium prepared from autologous epidermal cells. Lancet. 1981;1:75–8.
Li A, Simmons PJ, Kaur P. Identification and isolation of candidate human keratinocyte stem cells based on cell surface phenotype. Proc Natl Acad Sci U S A. 1998;95:3902–7.
Webb A, Li A, Kaur P. Location and phenotype of human adult keratinocyte stem cells of the skin. Differentiation. 2004;72:387–95. https://doi.org/10.1111/j.1432-0436.2004.07208005.x.
Limat A, Mauri D, Hunziker T. Successful treatment of chronic leg ulcers with epidermal equivalents generated from cultured autologous outer root sheath cells. J Invest Dermatol. 1996;107:128–35.
Limat A, French LE, Blal L, Saurat JH, Hunziker T, Salomon D. Organotypic cultures of autologous hair follicle keratinocytes for the treatment of recurrent leg ulcers. J Am Acad Dermatol. 2003;48:207–14. https://doi.org/10.1067/mjd.2003.69.
Ohyama M, Terunuma A, Tock CL, Radonovich MF, Pise-Masison CA, Hopping SB, et al. Characterization and isolation of stem cell-enriched human hair follicle bulge cells. J Clin Invest. 2006;116:249–60. https://doi.org/10.1172/JCI26043.
Ohyama M, William J. Cunliffe scientific awards. Advances in the study of stem-cell-enriched hair follicle bulge cells: a review featuring characterization and isolation of human bulge cells. Dermatology. 2007;214:342–51. https://doi.org/10.1159/000100889.
Biedermann T, Pontiggia L, Böttcher-Haberzeth S, Tharakan S, Braziulis E, Schiestl C, et al. Human eccrine sweat gland cells can reconstitute a stratified epidermis. J Invest Dermatol. 2010;130(8):1996–2009. https://doi.org/10.1038/jid.2010.83.
Hill R. Skin regeneration symposium Cambridge. Regen Med. 2016;11:443–57. https://doi.org/10.2217/rme-2016-0062.
Penna V, Lipay MV, Duailibi M, Duailibi SE. The likely role of proteolytic enzymes in unwanted differentiation of stem cells in culture. Future Sci OA. 2015;1:FSO28. https://doi.org/10.4155/fso.15.26.
Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A. 1997;94:849–54.
Lelièvre SA, Bissell MJ. Communication between the cell membrane and the nucleus: role of protein compartmentalization. J Cell Biochem Suppl. 1998;30-31:250–63.
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. In: Ltda AMS, editor. Cell junctions, cell adhesion, and the extracellular matrix. 3rd ed. Brazil: Rio Grande do Sul; 2002. p. 949–1010.
DeMali KA, Sun X, Bui GA. Force transmission at cell–cell and cell–matrix adhesions. Biochemistry. 2014;53:7706–17. https://doi.org/10.1021/bi501181p.
Faulk DM, Johnson SA, Zhang L, Badylak SF. Role of the extracellular matrix in whole organ engineering. J Cell Physiol. 2014;229:984–9. https://doi.org/10.1002/jcp.24532.
Spencer VA, Xu R, Bissell MJ. Gene expression in the third dimension: the ECM-nucleus connection. J Mammary Gland Biol Neoplasia. 2010;15:65–71. https://doi.org/10.1007/s10911-010-9163-3.
Ruszczak Z. Effect of collagen matrices on dermal wound healing. Adv Drug Deliv Rev. 2003;55:1595–611.
Zhou H, You C, Wang X, Jin R, Wu P, Li Q, et al. The progress and challenges for dermal regeneration in tissue engineering. J Biomed Mater Res A. 2017;105:1208–18. https://doi.org/10.1002/jbm.a.35996.
Rolfe KJ, Grobbelaar AO. A review of fetal scarless healing. ISRN Dermatol. 2012;2012:698034. https://doi.org/10.5402/2012/698034.
Böttcher-Haberzeth S, Biedermann T, Reichmann E. Tissue engineering of skin. Burns. 2010;36:450–60. https://doi.org/10.1016/j.burns.2009.08.016.
Aoki S, Toda S, Ando T, Sugihara H. Bone marrow stromal cells, pre-adipocytes, and dermal fibroblasts promote epidermal regeneration in their distinctive fashions. Mol Biol Cell. 2004;15:4647–57.
Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–82. https://doi.org/10.1073/pnas.162488599.
Wong T, McGrath JA, Navsaria H. The role of fibroblasts in tissue engineering and regeneration. Br J Dermatol. 2007;156:1149–55. https://doi.org/10.1111/j.1365-2133.2007.07914.x.
Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell. 1993;4:637–45.
Werner S, Smola H. Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell Biol. 2001;11:143–6.
El-Ghalbzouri A, Gibbs S, Lamme E, Van Blitterswijk CA, Ponec M. Effect of fibroblasts on epidermal regeneration. Br J Dermatol. 2002;147:230–43.
Trompezinski S, Berthier-Vergnes O, Denis A, Schmitt D, Viac J. Comparative expression of vascular endothelial growth factor family members, VEGF-B, -C and -D by normal human keratinocytes and fibroblasts. Exp Dermatol. 2004;13:98–105. https://doi.org/10.1111/j.0906-6705.2004.00137.x.
Rendl M, Polak L, Fuchs E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev. 2008;22:543–57.
Langan EA, Philpott MP, Kloepper JE, Paus R. Human hair follicle organ culture: theory, application and perspectives. Exp Dermatol. 2015;24:903–11. https://doi.org/10.1111/exd.12836.
Kumar A, Mohanty S, Nandy SB, Gupta S, Khaitan BK, Sharma S, et al. Hair & skin derived progenitor cells: in search of a candidate cell for regenerative medicine. Indian J Med Res. 2016;143:175–83. https://doi.org/10.4103/0971-5916.180205.
Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339–73.
Lee B, Dai X. Transcriptional control of epidermal stem cells. In: Hime G, Abud H, editors. Transcriptional and translational regulation of stem cells. Advances in experimental medicine and biology, vol. vol. 786. Dordrecht: Springer; 2013. https://doi.org/10.1007/978-94-007-6621-1_9.
Ito M, Yang Z, Andl T, Cui C, Kim N, Millar SE, et al. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature. 2007;447:316–20. https://doi.org/10.1038/nature05766.
Wang X, Wang X, Liu J, Cai T, Guo L, Wang S, et al. Hair follicle and sebaceous gland de novo regeneration with cultured epidermal stem cells and skin-derived precursors. Stem Cells Transl Med. 2016;5:1695–706. https://doi.org/10.5966/sctm.2015-0397.
Mcheik JN, Barrault C, Levard G, Morel F, Bernard FX, Lecron JC. Epidermal healing in burns: autologous keratinocyte transplantation as a standard procedure: update and perspective. Plast Reconstr Surg Glob Open. 2014;2:e218. https://doi.org/10.1097/GOX.0000000000000176.
Biernaskie J, Paris M, Morozova O, Fagan BM, Marra M, Pevny L, et al. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell. 2009;5:610–23. https://doi.org/10.1016/j.stem.2009.10.019.
Agabalyan NA, Borys BS, Sparks HD, Boon K, Raharjo EW, Abbasi S, et al. Enhanced expansion and sustained inductive function of skin-derived precursor cells in computer-controlled stirred suspension bioreactors. Stem Cells Transl Med. 2017;6:434–43. https://doi.org/10.5966/sctm.2016-0133.
Cohen J. The transplantation of individual rat and guinea pig whisker papillae. J Embryol Exp Morphol. 1961;9:117–27.
Sabapathy V, Kumar S. hiPSC-derived iMSCs: NextGen MSCs as an advanced therapeutically active cell resource for regenerative medicine. J Cell Mol Med. 2016;20:1571–88. https://doi.org/10.1111/jcmm.12839.
Zhu WY, Zhang RZ, Ma HJ, Wang DG. Isolation and culture of amelanotic melanocytes from human hair follicles. Pigment Cell Res. 2004;17:668–73. https://doi.org/10.1111/j.1600-0749.2004.00190.x.
Kumar A, Mohanty S, Sahni K, Kumar R, Gupta S. Extracted hair follicle outer root sheath cell suspension for pigment cell restoration in vitiligo. J Cutan Aesthet Surg. 2013;6:121–5. https://doi.org/10.4103/0974-2077.112679.
Sieber-Blum M, Grim M, Hu YF, Szeder V. Pluripotent neural crest stem cells in the adult hair follicle. Dev Dyn. 2004;231:258–69. https://doi.org/10.1002/dvdy.20129.
Zabierowski SE, Fukunaga-Kalabis M, Li L, Herlyn M. Dermis derived stem cells: a source of epidermal melanocytes and melanoma? Pigment Cell Melanoma Res. 2011;24:422–9. https://doi.org/10.1111/j.1755-148X.2011.00847.x.
Toma JG, Akhavan M, Fernandes KJ, Barnabé-Heider F, Sadikot A, Kaplan DR, et al. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3:778–84. https://doi.org/10.1038/ncb0901-778.
Fernandes KJ, McKenzie IA, Mill P, Smith KM, Akhavan M, Barnabé-Heider F, et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6:1082–93. https://doi.org/10.1038/ncb1181.
McKenzie IA, Biernaskie J, Toma JG, Midha R, Miller FD. Skin-derived precursors generate myelinating Schwann cells for the injured and dysmyelinated nervous system. J Neurosci. 2006;26:6651–60. https://doi.org/10.1523/JNEUROSCI.1007-06.2006.
Hunt DP, Morris PN, Sterling J, Anderson JA, Joannides A, Jahoda C, et al. A highly enriched niche of precursor cells with neuronal and glial potential within the hair follicle dermal papilla of adult skin. Stem Cells. 2008;26:163–72.
Krause MP, Dworski S, Feinberg K, Jones K, Johnston AP, Paul S, et al. Direct genesis of functional rodent and human Schwann cells from skin mesenchymal precursors. Stem Cell Rep. 2014;3:85–100. https://doi.org/10.1016/j.stemcr.2014.05.011.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Introduction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. https://doi.org/10.1016/j.cell.2007.11.019.
Yang R, Zheng Y, Burrows M, Liu S, Wei Z, Nace A, et al. Generation of folliculogenic human epithelial stem cells from induced pluripotent stem cells. Nat Commun. 2014;5:3071. https://doi.org/10.1038/ncomms4071.
Billingham RE, Medawar P. Technique of free skin grafting in mammals. J Exp Biol. 1950;28:385–402.
Karasek M. In vitro culture of human skin epithelial cell. J Invest Dermatol. 1966;47:533–40.
Moscona A, Moscona H. The dissociation and aggregation of cells from organ rudiments of the early chick embryo. J Anat. 1952;86:287–301.
Bell E, Ehrlich HP, Buttle DJ, Nakatsuji T. Living tissue formed in vitro and accepted as skin-equivalent tissue of full thickness. Science. 1981;211:1052–4.
Boyce ST, Goretsky MJ, Greenhalgh DG, Kagan RJ, Rieman MT, Warden GD. Comparative assessment of cultured skin substitutes and native skin autograft for treatment of full thickness burns. Ann Surg. 1995;222:743–52.
Delvoye P, Pierard D, Noel A, Nusgens B, Foidart JM, Lapiere CM. Fibroblasts induce the assembly of the macromolecules of the basement membrane. J Invest Dermatol. 1988;90:276–82.
Burke JF, Yannas IV, Quinby WC Jr, Bondoc CC, Jung WK. Successful use of a physiologically acceptable artificial skin in the treatment of extensive burn injury. Ann Surg. 1981;194:413–28.
Zeng W, Zhang S, Liu D, Chai M, Wang J, Zhao Y. Preclinical safety studies on autologous cultured human skin fibroblast transplantation. Cell Transplant. 2014;23:39–49. https://doi.org/10.3727/096368912X659844.
Philpott MP, Green MR, Kealey T. Human hair growth in vitro. J Cell Sci. 1990;97:463–71.
Aoki S, Takezawa T, Sugihara H, Toda S. Progress in cell culture systems for pathological research. Pathol Int. 2016;66:554–62. https://doi.org/10.1111/pin.12443.
Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36.
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–8. https://doi.org/10.1038/23962.
Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66:407–25.
Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–17. https://doi.org/10.1038/nrm2636.
Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells. 2005;23:727–37. https://doi.org/10.1634/stemcells.2004-0134.
Malik N, Rao MS. A review of the methods for human iPSC derivation. Methods Mol Biol. 2013;997:23–33. https://doi.org/10.1007/978-1-62703-348-0_3.
Eça LP, Pinto DG, de Pinho AM, Mazzetti MP, Odo ME. Autologous fibroblast culture in the repair of aging skin. Dermatol Surg. 2012;38:180–4. https://doi.org/10.1111/j.1524-4725.2011.02192.x.
Phillips CL, Combs SB, Pinnell SR. Effects of ascorbic acid on proliferation and collagen synthesis in relation to the donor age of human dermal fibroblasts. J Invest Dermatol. 1994;103:228–32.
Woan KV, Narain NR, Persaud I, et al. Coenzyme Q10 enhances the proliferation and migration of fibroblasts and keratinocytes: a possible implication for wound healing. J Invest Dermatol. 2005;124s:A57.
Dame MK, Spahlinger DM, DaSilva M, Perone P, Dunstan R, Varani J. Establishment and characteristics of Gottingen minipig skin in organ culture and monolayer cell culture: relevance to drug safety testing. In Vitro Cell Dev Biol Anim. 2008;44(7):245–52. https://doi.org/10.1007/s11626-008-9091-3.
Solakoglu S, Tiryaki T, Ciloglu SE. The effect of cultured autologous fibroblasts on longevity of cross-linked hyaluronic acid used as a filler. Aesthet Surg J. 2008;28:412–6. https://doi.org/10.1016/j.asj.2008.04.008.
Weiss RA. Autologous cell therapy: will it replace dermal fillers? Facial Plast Surg Clin North Am. 2013;21:299–304. https://doi.org/10.1016/j.fsc.2013.02.008.
Zhao Y, Wang J, Yan X, Li D, Xu J. Preliminary survival studies on autologous cultured skin fibroblasts transplantation by injection. Cell Transplant. 2008;17:775–83.
Eves PC, Beck AJ, Shard AG, Mac NS. A chemically defined surface for the co-culture of melanocytes and keratinocytes. Biomaterials. 2005;26:7068–81. https://doi.org/10.1016/j.biomaterials.2005.05.015.
Matsumoto T, Kano K, Kondo D, Fukuda N, Iribe Y, Tanaka N, et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J Cell Physiol. 2008;215:210–22. https://doi.org/10.1002/jcp.21304.
Bayati V, Gazor R, Nejatbakhsh R, Negad DF. Enrichment of skin-derived neural precursor cells from dermal cell populations by altering culture conditions. Stem Cell Invest. 2016;3:83–92. https://doi.org/10.21037/sci.2016.10.10.
Bickenbach JR. Isolation, characterization, and culture of epithelial stem cells. Methods Mol Biol. 2005;289:97–102.
Lee MJ, Kim J, Lee KI, Shin JM, Chae JI, Chung HM. Enhancement of wound healing by secretory factors of endothelial precursor cells derived from human embryonic stem cells. Cytotherapy. 2011;13:165–78. https://doi.org/10.3109/14653249.2010.512632.
Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Endothelial cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2002;99:4391–6. https://doi.org/10.1073/pnas.032074999.
Monaco JL, Lawrence WT. Acute wound healing an overview. Clin Plast Surg. 2003;30:1–12.
Westgate GE, Gibson WT, Kealey T, Philpott MP. Prolonged maintenance of human hair follicles in vitro in a serum-free medium. Br J Dermatol. 1993;129:372–9.
Purba TS, Haslam IS, Poblet E, Jiménez F, Gandarillas A, Izeta A, et al. Human epithelial hair follicle stem cells and their progeny: current state of knowledge, the widening gap in translational research and future challenges. BioEssays. 2014;36:513–25. https://doi.org/10.1002/bies.201300166.
Brown TD. Techniques for mechanical stimulation of cells in vitro: a review. J Biomech. 2000;33:3–14.
Huang HL, Hsing HW, Lai TC, Chen YW, Lee TR, Chan HT. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci. 2010;17:36. https://doi.org/10.1186/1423-0127-17-36.
Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y, Sakurai Y. Thermoresponsive polymeric surfaces: control of attachment and detachment of cultured cells. Makromol Chem Rapid Commun. 1990;11:571–6. https://doi.org/10.1002/marc.1990.030111109.
Yang L, Cheng F, Liu T, Lu JR, Song K, Jiang L, et al. Comparison of mesenchymal stem cells released from poly(N-isopropylacrylamide) copolymer film and by trypsinization. Biomed Mater. 2012;7:035003. https://doi.org/10.1088/1748-6041/7/3/035003.
Ozbun MA, Patterson NA. Using organotypic (raft) epithelial tissue cultures for the biosynthesis and isolation of infectious human papillomaviruses. Curr Protoc Microbiol. 2014;34:14B.3.1–18. https://doi.org/10.1002/9780471729259.mc14b03s34.
Brindley D, Moorthy K, Lee JH, Mason C, Kim HW, Wall I. Bioprocess forces and their impact on cell behavior: implications for bone regeneration therapy. J Tissue Eng. 2011;2011:620247. https://doi.org/10.4061/2011/620247.
Gareau T, Lara GG, Shepherd RD, Krawetz R, Rancourt DE, Rinker KD, et al. Shear stress influences the pluripotency of murine embryonic stem cells in stirred suspension bioreactors. J Tissue Eng Regen Med. 2014;8:268–78. https://doi.org/10.1002/term.1518.
Hakim N, editor. Artificial organs, new techniques in surgery series 4. Springer-Verlag London Limited; 2009. https://doi.org/10.1007/978-1-84882-283-2_6.
Cubo N, Garcia M, Del Cañizo JF, Velasco D, Jorcano JL. 3D bioprinting of functional human skin: production and in vivo analysis. Biofabrication. 2016;9:015006. https://doi.org/10.1088/1758-5090/9/1/015006.
Pandey AR, Singh US, Momin M, et al. J Polym Res. 2017;24:125. https://doi.org/10.1007/s10965-017-1286-4.
MacNeil S. Progress and opportunities for tissue-engineered skin. Nature. 2007;445:874–80. https://doi.org/10.1038/nature05664.
Kubo K, Kuroyanagi Y. A study of cytokines released from fibroblasts in cultured dermal substitute. Artif Organs. 2005;29:845–9. https://doi.org/10.1111/j.1525-1594.2005.00138.x.
Moustafa M, Simpson C, Glover M, Dawson RA, Tesfaye S, Creagh FM, et al. A new autologous keratinocyte dressing treatment for non-healing diabetic neuropathic foot ulcers. Diabet Med. 2004;21:786–9.
Zhu N, Warner RM, Simpson C, Glover M, Hernon CA, Kelly J, et al. Treatment of burns and chronic wounds using a new cell transfer dressing for delivery of autologous keratinocytes. Eur J Plast Surg. 2005;28:319–30.
Wright KA, Nadire KB, Busto P, Tubo R, McPherson JM, Wentworth BM. Alternative delivery of keratinocytes using a polyurethane membrane and the implications for its use in the treatment of full-thickness burn injury. Burns. 1998;24:7–17.
Carsin H, Ainaud P, Le Bever H, Rives J, Lakhel A, Stephanazzi J, et al. Cultured epithelial autografts in extensive burn coverage of severely traumatized patients: a five year single-center experience with 30 patients. Burns. 2000;26:379–87.
Tausche AK, Skaria M, Böhlen L, Liebold K, Hafner J, Friedlein H, et al. An autologous epidermal equivalent tissue-engineered from follicular outer root sheath keratinocytes is as effective as split-thickness skin autograft in recalcitrant vascular leg ulcers. Wound Repair Regen. 2003;11:248–52.
Renner R, Harth W, Simon JC. Transplantation of chronic wounds with epidermal sheets derived from autologous hair follicle--the Leipzig experience. Int Wound J. 2009;6:226–32. https://doi.org/10.1111/j.1742-481X.2009.00609.x.
Wainwright DJ. Use of an acellular allograft dermal matrix (AlloDerm) in the management of full-thickness burns. Burns. 1995;21:243–8.
Gordley K, Cole P, Hicks J, Hollier L. A comparative, long term assessment of soft tissue substitutes: AlloDerm, Enduragen, and Dermamatrix. J Plast Reconstr Aesthet Surg. 2009;62:849–50. https://doi.org/10.1016/j.bjps.2008.05.006.
Cooper ML, Hansbrough JF, Spielvogel RL, Cohen R, Bartel RL, Naughton G. In vivo optimization of a living dermal substitute employing cultured human fibroblasts on a biodegradable polyglycolic acid or polyglactin mesh. Biomaterials. 1991;12:243–8.
Kearney JN. Clinical evaluation of skin substitutes. Burns. 2001;27:545–51.
Branski LK, Herndon DN, Pereira C, Mlcak RP, Celis MM, Lee JO, et al. Longitudinal assessment of Integra in primary burn management: a randomized pediatric clinical trial. Crit Care Med. 2007;35:2615–23. https://doi.org/10.1097/01.CCM.0000285991.36698.E2.
Stiefel D, Schiestl C, Meuli M. Integra artificial skin for burn scar revision in adolescents and children. Burns. 2010;36:114–20. https://doi.org/10.1016/j.burns.2009.02.023.
Haslik W, Kamolz LP, Nathschläger G, Andel H, Meissl G, Frey M. First experiences with the collagen-elastin matrix Matriderm as a dermal substitute in severe burn injuries of the hand. Burns. 2007;33:364–8. https://doi.org/10.1016/j.burns.2006.07.021.
Schneider J, Biedermann T, Widmer D, Montano I, Meuli M, Reichmann E, et al. Matriderm versus Integra: a comparative experimental study. Burns. 2009;35:51–7. https://doi.org/10.1016/j.burns.2008.07.018.
Shevchenko RV, James SL, James SE. A review of tissue engineered skin bioconstructs available for skin reconstruction. J R Soc Interface. 2010;7:229–58. https://doi.org/10.1098/rsif.2009.0403.
Sheridan RL, Morgan JR, Cusick JL, Petras LM, Lydon MM, Tompkins RG. Initial experience with a composite autologous skin substitute. Burns. 2001;27:421–4.
Keck M, Haluza D, Lumenta DB, Burjak S, Eisenbock B, Kamolz LP, et al. Construction of a multi-layer skin substitute: simultaneous cultivation of keratinocytes and preadipocytes on a dermal template. Burns. 2011;37:626–30. https://doi.org/10.1016/j.burns.2010.07.016.
Gómez C, Galán JM, Torrero V, Ferreiro I, Pérez D, Palao R, et al. Use of an autologous bioengineered composite skin in extensive burns: clinical and functional outcomes. A multicentric study. Burns. 2011;37:580–9. https://doi.org/10.1016/j.burns.2010.10.005.
Kirsner RS. The use of Apligraf in acute wounds. J Dermatol. 1998;25:805–11.
Edmonds M, European and Australian Apligraf Diabetic Foot Ulcer Study Group. Apligraf in the treatment of neuropathic diabetic foot ulcers. Int J Low Extrem Wounds. 2009;8:11–8. https://doi.org/10.1177/1534734609331597.
Eisenberg M, Llewellyn DM, Moran K, Kerr A. Successful engraftment of cultured human epidermal allograft in a child with recessive dystrophic epidermolysis bullosa. Med J Aust. 1987;147:520–1.
Windsor ML, Eisenberg M, Gordon-Thomson C, Moore GP. A novel model of wound healing in the SCID mouse using a cultured human skin substitute. Australas J Dermatol. 2009;50:29–35. https://doi.org/10.1111/j.1440-0960.2008.00512.x.
Tavis MJ, Thornton JW, Bartlett RH, et al. A new composite skin prosthesis. Burns. 1979;8:123–30.
Demling RH, DeSanti L. Management of partial thickness facial burns (comparison of topical antibiotics and bio-engineered skin substitutes). Burns. 1999;25:256–61.
Wood FM, Kolybaba ML, Allen P. The use of cultured epithelial autograft in the treatment of major burn injuries: a critical review of the literature. Burns. 2006;32:395–401. https://doi.org/10.1016/j.burns.2006.01.008.
Meuli M, Raghunath M. Burns (Part 2). Tops and flops using cultured epithelial autografts in children. Pediatr Surg Int. 1997;12:471–7.
Horch RE, Kopp J, Kneser U, Beier J, Bach AD. Tissue engineering of cultured skin substitutes. J Cell Mol Med. 2005;9:592–608.
Wood FM. Clinical potential of cellular autologous epithelial suspension. Wounds. 2002;15:16–22.
Pham C, Greenwood J, Cleland H, Woodruff P, Maddern G. Bioengineered skin substitutes for the management of burns: a systematic review. Burns. 2007;33:946–57. https://doi.org/10.1016/j.burns.2007.03.020.
Mansbridge J. Commercial considerations in tissue engineering. J Anat. 2006;209:527–32. https://doi.org/10.1111/j.1469-7580.2006.00631.x.
Wood FM, Stoner ML, Fowler BV, Fear MW. The use of a non-cultured autologous cell suspension and Integra dermal regeneration template to repair full-thickness skin wounds in a porcine model: a one-step process. Burns. 2007;33:693–700. https://doi.org/10.1016/j.burns.2006.10.388.
Boyce ST. Design principles for composition and performance of cultured skin substitutes. Burns. 2001;27:523–33.
Sabeh G, Sabé M, Ishak S, Sweid R, Ayoubi M, Chahal AM. Greffes séquentielles de cellules cutanées: premiers résultats d’un nouveau procédé et revue de la littérature. J Med Liban. 2015;63:47–58.
Driskell RR, Clavel C, Rendl M, Watt FM. Hair follicle dermal papilla cells at a glance. J Cell Sci. 2011;124:1179–82. https://doi.org/10.1242/jcs.082446.
Hu MS, Rennert RC, McArdle A, Chung MT, Walmsley GG, Longaker MT, Lorenz HP. The role of stem cells during scarless skin wound healing. Adv Wound Care (New Rochelle). 2014;3:304–14. https://doi.org/10.1089/wound.2013.0471.
Chun-mao H, Su-yi W, Ping-ping L, Hang-hui C. Human bone marrow-derived mesenchymal stem cells differentiate into epidermal-like cells in vitro. Differentiation. 2007;75:292–8. https://doi.org/10.1111/j.1432-0436.2006.00140.x.
Wang C, Lin K, Chang J, Sun J. Osteogenesis and angiogenesis induced by porous beta-CaSiO(3)/PDLGA composite scaffold via activation of AMPK/ERK1/2 and PI3K/Akt pathways. Biomaterials. 2013;34(1):64–77. https://doi.org/10.1016/j.biomaterials.2012.09.021.
Kim HJ, Park SS, Oh SY, Kim H, Kweon OK, Woo HM, et al. Effect of acellular dermal matrix as a delivery carrier of adipose-derived mesenchymal stem cells on bone regeneration. J Biomed Mater Res B Appl Biomater. 2012;100:1645–53. https://doi.org/10.1002/jbm.b.32733.
Burd A, Chiu T. Allogeneic skin in the treatment of burns. Clin Dermatol. 2005;23:376–87. https://doi.org/10.1016/j.clindermatol.2004.07.019.
Waymack P, Duff RG, Sabolinski M. The effect of a tissue engineered bilayered living skin analog, over meshed split-thickness autografts on the healing of excised burn wounds. The Apligraf Burn Study Group. Burns. 2000;26:609–19.
Gath HJ, Hell B, Zarrinbal R, Bier J, Raguse JD. Regeneration of intraoral defects after tumour resection with a bioengineered human dermal replacement (Dermagraft). Plast Reconstr Surg. 2002;109:889–93.
Gohari S, Gambla C, Healey M, Spaulding G, Gordon KB, Swan J, et al. Evaluation of tissue-engineered skin (human skin substitute) and secondary intention healing in the treatment of full thickness wounds after Mohs micrographic or excisional surgery. Dermatol Surg. 2002;28:1107–14.
Moon KC, Lee HS, Han SK, et al. Aesth Plast Surg. 2018;42:815. https://doi.org/10.1007/s00266-017-1044-3.
Watson D, Keller GS, Lacombe V, Fodor PB, Rawnsley J, Lask GP. Autologous fibroblasts for treatment of facial rhytids and dermal depressions: a pilot study. Arch Facial Plast Surg. 1999;1:165–70.
Gonzalez MJ, Sturgill WH, Ross EV, Uebelhoer NS. Treatment of acne scars using the plasma skin regeneration (PSR) system. Lasers Surg Med. 2008;40:124–7. https://doi.org/10.1002/lsm.20617.
Velander P, Theopold C, Bleiziffer O, Bergmann J, Svensson H, Feng Y, et al. Cell suspensions of autologous keratinocytes or autologous fibroblasts accelerate the healing of full thickness skin wounds in a diabetic porcine wound healing model. J Surg Res. 2009;157:14–20. https://doi.org/10.1016/j.jss.2008.10.001.
Li J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol. 2007;25:9–18. https://doi.org/10.1016/j.clindermatol.2006.09.007.
Weiss RA, Weiss MA, Beasley KL, Munavalli G. Autologous cultured fibroblast injection for facial contour deformities: a prospective, placebo-controlled, phase III clinical trial. Dermatol Surg. 2007;33:263–8. https://doi.org/10.1111/j.1524-4725.2007.33060.x.
Smith SR, Munavalli G, Weiss R, Maslowski JM, Hennegan KP, Novak JM. A multicenter, double-blind, placebo-controlled trial of autologous fibroblast therapy for the treatment of nasolabial fold wrinkles. Dermatol Surg. 2012;38:1234–43. https://doi.org/10.1111/j.1524-4725.2012.02349.x.
Narins RS, Brandt FS, Lorenc ZP, Maas CS, Monheit GD, Smith SR. Twelve-month persistency of a novel ribose-cross-linked collagen dermal filler. Dermatol Surg. 2008;3:S31–9. https://doi.org/10.1111/j.1524-4725.2008.34240.x.
Smith SR, Jones D, Thomas JA, Murphy DK, Beddingfield FC 3rd. Duration of wrinkle correction following repeat treatment with Juvederm hyaluronic acid fillers. Arch Dermatol Res. 2010;302:757–62. https://doi.org/10.1007/s00403-010-1086-8.
Yoon ES, Han SK, Kim WK. Advantages of the presence of living dermal fibroblasts within restylane for soft tissue augmentation. Ann Plast Surg. 2003;51:587–92. https://doi.org/10.1097/01.sap.0000096424.23397.2a.
Muir I, Padilla-Lamb A, Stewart JE, Wheatley DN. Growth inhibition of culture fibroblast by extracts from human dermis. Br J Plast Surg. 1997;50:186–93.
Lee HJ, Lee EG, Kang S, Sung JH, Chung HM, Kim DH. Efficacy of microneedling plus human stem cell conditioned medium for skin rejuvenation: a randomized, controlled, blinded split-face study. Ann Dermatol. 2014;26:584–91. https://doi.org/10.5021/ad.2014.26.5.584.
Wu Y, Wang J, Scott PG, Tredget EE. Bone marrow-derived stem cells in wound healing: a review. Wound Repair Regen. 2007;15:S18–26. https://doi.org/10.1111/j.1524-475X.2007.00221.x.
Thomas ED, Lochte HL Jr, Cannon JH, Sahler OD, Ferrebee JW. Supralethal whole body irradiation and isologous marrow transplantation in man. J Clin Invest. 1959;38:1709–16. https://doi.org/10.1172/JCI103949.
Gragnani A, Giannoccaro FB, Sobral CS, Moraes AA, França JP, Ferreira AT, et al. Dimethylaminoethanol affects the viability of human cultured fibroblasts. Aesthet Plast Surg. 2007;31:711–8. https://doi.org/10.1007/s00266-006-0208-3.
Falanga V, Margolis D, Alvarez O, Auletta M, Maggiacomo F, Altman M, et al. Rapid healing of venous ulcers and lack of clinical rejection with an allogeneic cultured human skin equivalent. Human skin equivalent investigators group. Arch Dermatol. 1998;134:293–300.
Theobald VA, Lauer JD, Kaplan FA, Baker KB, Rosenberg M. ‘Neutral allografts’—lack of allogeneic stimulation by cultured human cells expressing MHC class I and class II antigens. Transplantation. 1993;55:128–33.
Phillips TJ, Manzoor J, Rojas A, Isaacs C, Carson P, Sabolinski M, et al. The longevity of a bilayered skin substitute after application to venous ulcers. Arch Dermatol. 2002;138:1079–81.
Price RD, Das-Gupta V, Harris PA, Leigh IM, Navsaria HA. The role of allogenic fibroblasts in an acute wound healing model. Plast Reconstr Surg. 2004;113:1719–29.
Barnas JL, Simpson-Abelson MR, Brooks SP, Kelleher RJ Jr, Bankert RB. Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J Immunol. 2010;185(5):2681–92. https://doi.org/10.4049/jimmunol.1000896.
Mazlyzam AL, Aminuddin BS, Saim L, Ruszymah BH. Human serum is an advantageous supplement for human dermal fibroblast expansion: clinical implications for tissue engineering of skin. Arch Med Res. 2008;39:743–52. https://doi.org/10.1016/j.arcmed.2008.09.001.
Griffiths M, Ojeh N, Livingstone R, Price R, Navsaria H. Survival of Apligraf in acute human wounds. Tissue Eng. 2004;10:1180–95. https://doi.org/10.1089/ten.2004.10.1180.
Guenou H, Nissan X, Larcher F, Feteira J, Lemaitre G, Saidani M, et al. Human embryonic stem cell derivatives for full reconstruction of the pluristratified epidermis: a preclinical study. Lancet. 2009;374:1745–53. https://doi.org/10.1016/S0140-6736(09)61496-3.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cells lines derived from human blastocysts. Science. 1998;282:1145–7.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. https://doi.org/10.1016/j.cell.2006.07.024.
Ng WL, Wang S, Yeong WY, Naing MW. Skin bioprinting: impending reality or fantasy? Trends Biotechnol. 2017;35:278. https://doi.org/10.1016/j.tibtech.2016.08.009.
Augustine R. Prog Biomater. 2018;7:77. https://doi.org/10.1007/s40204-018-0087-0.
Koch L, Deiwick A, Schlie S, Michael S, Gruene M, Coger V, et al. Skin tissue generation by laser cell printing. Biotechnol Bioeng. 2012;109:1855–63. https://doi.org/10.1002/bit.24455. Epub 2012 Feb 13.
Michael S, Sorg H, Peck CT, Koch L, Deiwick A, Chichkov B, et al. Tissue engineered skin substitutes created by laser-assisted bioprinting form skin-like structures in the dorsal skin fold chamber in mice. PLoS One. 2013;8:e57741. https://doi.org/10.1371/journal.pone.0057741.
Lee V, Singh G, Trasatti JP, Bjornsson C, Xu X, Tran TN, et al. Design and fabrication of human skin by three-dimensional bioprinting. Tissue Eng Part C Methods. 2014;20:473–84. https://doi.org/10.1089/ten.TEC.2013.0335.
Lee W, Lee V, Polio S, Keegan P, Lee JH, Fischer K, et al. On-demand three-dimensional freeform fabrication of multi-layered hydrogel scaffold with fluidic channels. Biotechnol Bioeng. 2010;105:1178–86. https://doi.org/10.1002/bit.22613.
Lee W, Debasitis JC, Lee VK, Lee JH, Fischer K, Edminster K, et al. Multi-layered culture of human skin fibroblasts and keratinocytes through three-dimensional freeform fabrication. Biomaterials. 2009;30:1587–95. https://doi.org/10.1016/j.biomaterials.2008.12.009.
Hussein SM, Nagy K, Nagy A. Human induced pluripotent stem cells: the past, present, and future. Clin Pharmacol Ther. 2011;89:741–5. https://doi.org/10.1038/clpt.2011.37.
Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. 2012;10:678–84. https://doi.org/10.1016/j.stem.2012.05.005.
Turksen K. Revisiting the bulge. Dev Cell. 2004;6:454–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jáñez, L. (2019). Skin Cell Cultures and Skin Engineering. In: Pinto, H., Fontdevila, J. (eds) Regenerative Medicine Procedures for Aesthetic Physicians. Springer, Cham. https://doi.org/10.1007/978-3-030-15458-5_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-15458-5_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-15457-8
Online ISBN: 978-3-030-15458-5
eBook Packages: MedicineMedicine (R0)