
Abstract
Calcium exchanges and homeostasis are finely regulated between cellular organelles and in response to physiological signals. Besides ionophores, including voltage-gated Ca2+ channels, ionotropic neurotransmitter receptors, or Store-operated Ca2+ entry, activity of regulatory intracellular proteins finely tune Calcium homeostasis. One of the most intriguing, by its unique nature but also most promising by the therapeutic opportunities it bears, is the sigma-1 receptor (Sig-1R). The Sig-1R is a chaperone protein residing at mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs), where it interacts with several partners involved in ER stress response, or in Ca2+ exchange between the ER and mitochondria. Small molecules have been identified that specifically and selectively activate Sig-1R (Sig-1R agonists or positive modulators) at the cellular level and that also allow effective pharmacological actions in several pre-clinical models of pathologies. The present review will summarize the recent data on the mechanism of action of Sig-1R in regulating Ca2+ exchanges and protein interactions at MAMs and the ER. As MAMs alterations and ER stress now appear as a common track in most neurodegenerative diseases, the intracellular action of Sig-1R will be discussed in the context of the recently reported efficacy of Sig-1R drugs in pathologies like Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, or amyotrophic lateral sclerosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
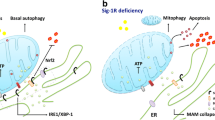
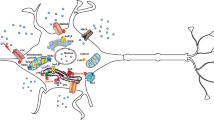
Keywords
- Sigma-1 receptor
- Calcium
- Mitochondria
- ER stress
- UPR
- MAMs
- Neurodegenerative disease
- Alzheimer’s disease
- Amyotrophic lateral sclerosis
- Addiction
- Pain
28.1 Introduction: Physiopathology of Sigma-1 Receptor (Sig-1R)
The Sigma-1 receptor (Sig-1R), discovered in the mid 1970s [1] was identified as a 223-amino acid protein only in the mid-1990s [2, 3]. Although its involvement in physiopathology started to be documented earlier, its cellular role was precised only 10 years ago [4] and cellular biology studies continue to precise its intracellular partners and functions. It shares no homology with any other known protein, except some steroid related/emopamyl-binding enzymes [2, 5, 6]. The protein was initially viewed as a receptor since, very early, specific and selective small molecules have been identified binding to Sig-1Rs and triggering (for so-called agonists) or preventing (for so-called antagonists) biological responses. However, its activation by physiological triggers, including ER stress or oxidative stress [4, 7, 8], and its mode of action, relying on modifications of protein-protein interactions rather than coupling to second messenger systems, suggested more a chaperone-like identity than a classical receptor nature [4]. Indeed, the present review will detail the effects of Sig-1R at intracellular organelles and show that it offers a unique opportunity to finely tune its activity, thereby impacting numerous physiopathological pathways, through a very classical pharmacological approach involving agonists, positive modulators or antagonists.
The Sig-1Rs are expressed in numerous organs, including liver, heart, lung, gonads and the nervous system, and numerous cell types, including, in the latter, neurons and glial cells (astrocytes, microglia, oligodendrocytes, Schwann cells) and vascular cells [9,10,11]. The particular density of Sig-1R in the nervous system is coherent with its importance in numerous psychiatric and neurological conditions. Sig1-Rs have indeed been involved in epilepsy [12, 13], stroke [14,15,16], drug abuse [17,18,19], pain [20, 21], and neurodegenerative pathologies. Interestingly, the last field of research is currently very active and recent evidence show both that Sig-1Rs play a role in the physiopathology of several neurodegenerative disease and that Sig-1R agonists have effective neuroprotective effects in preclinical models that deserve translation in clinical trials and better understanding of the mechanism of action of Sig-1R drugs against neurodegeneration. In parallel to the accumulating evidences that small molecules acting as Sig-1R agonist have pharmacological action in preclinical models of neurodegenerative diseases, and thus therapeutic potential, arguments are also brought confirming that Sig-1R exerts its cell homeostatic and cytoprotective activities mainly by directly targeting ER/mitochondria communication. We will here detail these arguments.
28.2 Sig-1R at the MAM
The ER of a cell spread almost all over a cell either in close proximity or in direct contacts with other subcellular components including the Golgi, mitochondria, nucleus, and plasma membrane. Through those close encounters, the ER plays many critical functions in the cell. One such important contact site for the ER is the mitochondria-associated ER membrane, termed the MAM [22], which harbors not only the lipid exchange [22], mitochondrial DNA exchange, Ca2+ signaling between the ER and mitochondria [4, 23,24,25,26,27] but also plays a role in the ER-nucleus signaling for cellular survival [28]. Recent evidences also indicate that the MAM is the origin of the isolated membrane for autophagy [29]. In addition, the MAM is critical in the formation of inflammasome [30, 31].
The MAM contains a plethora of functional proteins [24, 32]. Among those is the Sig-1R which is an ER molecular chaperone with two transmembrane regions from cellular biology studies [4, 33] but only one from the X-ray crystallographic study [34]. At the MAM, the Sig-1R chaperones the inositol-1,4,5 trisphosphate receptor type 3 (IP3R3) which would otherwise degrade after the stimulation of IP3, ensuring thus proper Ca2+ signaling from the ER into mitochondria [4, 35]. At the MAM, the Sig-1R also chaperones inositol-requiring enzyme 1 (IRE-1), one of the ER stress sensors, to facilitate the signaling of the unfolded protein response from the ER into nucleus to call for the transcriptional activation of antioxidant proteins and chaperones [28]. The Sig-1R was also found to attenuate free radical formation around the MAM area to reduce the activation of caspase that would have degraded the guanine nucleotide exchange factor to inactivate Rac GTPase that is essential for dendritic spine formation [36]. The Sig-1R also plays a role, likely at the MAM, in binding and transferring myristic acid to p35 to facilitate the p35 degradation by proteasome at the plasma membrane, thereby diverting p35 from forming p25 that would otherwise stun the axon elongation [37]. However, it remains to be totally clarified how those molecular actions of the Sig-1R at the MAM may contribute to the overall cellular and physiological functions of the MAM in general in a cell.
Upon the stimulation of Sig-1R agonists, Sig-1Rs dissociate from innate co-chaperone binding immunoglobulin protein (BiP) and translocate to other parts of cell to interact with and regulate the function of receptors, ion channels, and other functional proteins at the plasma membrane, mitochondria, ER reticular network, and nucleus [38, 39]. Thus, due to the nature and dynamics of Sig-1Rs, the receptor plays multiple physiological roles in living systems.
One of the important physiological roles of the Sig-1R is to regulate Ca2+ signaling not only at the MAM but also at the ER reticular network and plasma membrane. Sig-1Rs at the MAM facilitate Ca2+ influx from the ER into mitochondria by chaperoning IP3R3 at the MAM [4]. At the ER reticular network, the supranormal release of Ca2+ from the ER in medium spiny neurons of the YAC128 transgenic Huntington’s disease mice was attenuated by a Sig-1R agonist [40]. The release of Ca2+ from the ER reticular network is mainly controlled by the IP3R type 1 and the ryanodine receptor. Thus, this report suggests an inhibitory effect of Sig-1Rs on the IP3R1 or the ryanodine receptor, which is in contrast to the facilitative effect of Sig-1Rs on IP3R3. More experiments are needed as such.
The following three studies showed inconsistent effects of Sig-1Rs on the [Ca2+]i in nevertheless different systems. Also, the site of action of Sig-1Rs were not identified. By using cultured cortical neurons, Sig-1R agonists were found to attenuate the ischemia-induced increase of [Ca2+]i [41]. A recent study showed an increased [Ca2+]i when Sig-1Rs were activated by agonists in cultured embryonic mouse spinal neurons from ALS-causing mutants [42]. As well, methamphetamine-induced increase of [Ca2+]i was shown to be attenuated by Sig-1R agonists in dopaminergic neurons [43]. Again, sites of action of Sig-1Rs in those three studies were not identified. It remains to be seen if those actions of Sig-1Rs were at the MAM, the ER reticular network, or the plasma membrane. Possibility exists that results were manifestation of concerted actions at all of those sites.
At the plasma membrane, Sig-1Rs showed a presynaptic action in inhibiting N-type Ca2+ channels in cholinergic interneurons in rat striatum, resulting in a decrease in presynaptic [Ca2+]i [44]. The Sig-1R co-immunoprecipitated and co-localized with the N-type Ca2+ channel [24]. In rat brain microvascular endothelial cells, the store-operated calcium entry (SOCE) was attenuated by a Sig-1R agonist cocaine [45]. The mechanism of this interesting action of Sig-1R was reported in an elegant study in the same year. The Sig-1R was shown to bind stromal interaction molecule 1 (STIM1) at the ER when extracellular Ca2+ was depleted, and, as a result slowed down the recruitment of STIM1 to the ER-plasma membrane junction where STIM1 binds Orai1 [46]. The resultant inhibition of SOCE was seen when Sig-1Rs were overexpressed or when cells were treated with Sig-1R agonists. The Sig-1R antagonists or shSig-1R treatment enhanced the SOCE [46].
The calcium channels on the plasma membrane were examined in autonomous neurons taken from neonatal rat intracardiac and superior cervical ganglia (SCG). It was found that Sig-1Rs depressed high-voltage activated calcium currents from all calcium channel subtypes found on the cell body of these neurons, which includes N-, L-, P/Q-, and R-type calcium channels [47]. This study suggests that the activation of sigma receptors on sympathetic and parasympathetic neurons may modulate cell-to-cell signaling in autonomic ganglia and thus the regulation of cardiac function by the peripheral nervous system [47]. No direct interaction between Sig-1Rs and those calcium channels were demonstrated in this study however. The effect of Sig-1Rs on the potassium chloride-induced Ca2+ influx was examined by using retinal ganglion cell line (RGC)-5 and rat primary RGCs by the whole-cell patch clamp technique [48]. Sig-1R agonists inhibited the calcium influx and the Sig-1R antagonist reversed the inhibitory effect of Sig-1R agonist [48]. The Sig-1R was found to co-immunoprecipitate with the L-type calcium channels in this study.
Calcium homeostasis is critical to cellular physiology and plays an important role in many central nervous system (CNS) diseases, in particular the neurodegenerative diseases [49,50,51,52,53,54]. Since Sig-1Rs play critical role in calcium signaling at several loci of a cell, Sig-1Rs may be related to neurodegenerative diseases which show dysfunctional calcium homeostasis. However, a direct link between Sig-1R-regulated calcium signaling and a neurodegenerative disease has only been recently demonstrated in Huntington’s disease as mentioned above [40].
Nevertheless, because Sig-1Rs reside mainly at the MAM which is increasingly recognized as an important loci related to many neurodegenerative diseases [25, 49, 52, 53, 55, 56], it is possible that the Sig-1R at the MAM may participate in those diseases in a manner either directly related to calcium signaling or via other yet-to-be-revealed mechanisms at the MAM.
A study has specifically examined the role of Sig-1Rs at the MAM on ALS. Using primary motor neuron cultures, the study found that the pharmacological or genetic inactivation of Sig-1Rs led to motor neuron axonal degeneration [57]. They also found that the disruption of Sig-1R function in motor neurons disturbed ER-mitochondria contacts and affected intracellular calcium signaling, and was accompanied by activation of ER stress and defects in mitochondrial dynamics and transport (direct quotes) [57]. It is interesting to note that several other studies have implicated Sig-1Rs in ALS although they did not directly examine if the action of Sig-1Rs was at the MAM [58,59,60,61].
Sig-1Rs have been related to Alzheimer’s disease (AD). Several Sig-1R agonists, including PRE-084, MR-22, afobasole, ANAVEX1-41, ANAVEX2-73 or dehydroepiandrosterone, prevented amyloid-β25-35 (Aβ25-35)-induced toxicity in rat neuronal cultures [62, 63] and/or Aβ25-35-induced toxicity and learning impairments in mice in vivo [64,65,66,67,68,69]. Among the biochemical markers of toxicity in both in vitro and in vivo models, the Sig-1R drugs appear particularly effective in alleviating oxidative stress. Similar data were obtained in transgenic animal models of AD by Fisher et al. [70] who reported that AF710B, a mixed M1 mAChR/Sig-1R agonist, administered for 2 months in female 3xTg-AD mice, attenuated memory impairments and neurotoxicity. The drug also diminished soluble and insoluble Aβ species accumulation, the number of plaques and Tau hyperphosphorylation [70], thus confirming the neuroprotection and potentially disease-modifying effects of the drug. Moreover, invalidation of the Sig-1R expression, using Sig-1R knockout mice or a repeated treatment with the Sig-1R antagonist NE-100, increased learning deficits and neurotoxicity in Aβ25–35-injected mice or after cross-breeding with APPSwe,Ldn mice [71]. Therefore, it appeared that the absence of Sig-1R could worsen Aβ toxicity and behavioral deficits while it activation by therapeutic drugs showed neuroprotection.
The role of the MAM in the pathogenesis of AD remains an important area of research. Interestingly, a study has related the toxicity of Aβ to the increase of [Ca2+]i and the overload of mitochondrial calcium [72]. Nanomolar concentrations of Aβ was shown to increase MAM-associated proteins and caused an increase of the MAM [55]. Importantly, knockdown of Sig-1Rs resulted in neurodegeneration [55, 71]. Moreover, a direct examination of the effects of Sig-1R drugs in isolated mitochondria exposed to β-amyloid peptide showed that agonists decreased Aβ1–42-induced increase in reactive oxygen species (ROS) and attenuated Aβ1–42-induced alterations in mitochondrial respiration related to decreases in complex I and IV activity [73]. The Sig-1R agonists increased complex I activity, in a Ca2+-dependent and Sig-1R antagonist-sensitive manner in physiological conditions. These observations identified direct consequences on mitochondria of Sig-1R activity. However, further research on the involvement of the MAM in Alzheimer’s disease is certainly warranted.
Moreover, Sig-1R agonists, and particularly PRE-084, have been shown to be neuroprotective in mouse models of Parkinson’s disease [74], Huntington’s disease [75], amyotrophic lateral sclerosis (ALS) [76, 77], multiple sclerosis [78] or retinal neurodegeneration [79, 80], notably. Several recent reviews addressed the different progresses made so far [17, 51, 81,82,83,84,85,86]. For instance, a study examined the role of Sig-1Rs in Parkinsonism in a mouse model of intrastriatal lesion by 6-hydroxydopamine and found that the Sig-1R agonist significantly improved the fore-limb use [74]. At the molecular level, the study found that the agonist increased the density of dopaminergic fibers at the most denervated striatal regions and also caused an increase of neurotrophic factor brain-derived neurotrophic factor (BDNF) [74]. Interestingly, the agonist treatment induced a wider intracellular distribution of Sig-1Rs [74]. Because Sig-1Rs can translocate to other parts of neuron upon the stimulation by an agonist, it is tempting to speculate that the action of Sig-1Rs in the improvement of Parkinsonism may occur at the MAM as well other parts of neuron.
The Sig-1R has been shown to relate to Huntington’s disease. Most of the evidence cam from studies using a drug called pridopidine which was effective against Huntington’s disease in preclinical models and in phase two clinical trial at the secondary end-point level [87]. Originally thought to be a dopamine D2 ligand, pridopidine was nevertheless found to have a 100-fold higher affinity at the Sig-1R than at the dopamine D2 receptor [88]. In an in vivo radioligand binding assay, behaviorally relevant doses of pridopidine blocked about 57–85% of radiotracer binding to Sig-1Rs while blocked only negligible fraction of D2 receptor [89]. Recently, pridopidine was found to attenuate the phencyclidine-induced memory impairment through the Sig-1R-mediated mechanism as the effect of pridopidine was blocked by a Sig-1R antagonist NE-100 [90].
The exact mechanism and therefore the cellular site of action of Sig-1Rs underlying the action of pridopidine against Huntington’s disease are however not fully clarified. However, couple of studies provide some interesting results. In Q175 knock-in (Q175 KI) vs Q25 WT mouse models, the effect of pridopidine versus sham treatment on genome-wide expression profiling in the rat striatum was analyzed and compared to the pathological expression profile. Then a broad, unbiased pathway analysis was conducted, followed by testing the enrichment of relevant pathways [87]. Results showed that pridopidine upregulated the BDNF pathway (P = 1.73E-10), and its effect on BDNF secretion was Sig-1R-dependent [87]. It remains to be investigated how Sig-1Rs may upregulate BDNF at the molecular level. As mentioned before, the action of pridopidine was examined in a mouse model of Huntington’s disease with a specific focus on intracellular calcium signaling [40]. Results showed that pridopidine attenuates spine loss of medium spiny neurons and the effect was absent with the neuronal deletion of Sig-1Rs [40]. Pridopidine suppressed supranormal ER Ca2+ release, restored ER calcium levels and reduced excessive SOCE entry into spines. Interestingly, normalization of ER Ca2+ levels by pridopidine was prevented by Sig-1R deletion [40]. Whether those effects of Sig-1Rs originate at the MAM or beyond are not clear at present.
28.3 Sig-1R and ER Stress
The ER is an essential organelle of the cell that plays important role in protein folding and quality control [91, 92], lipid synthesis [93] and Ca2+ homeostasis [94]. During the life of the cell, different factors may perturb these functions, leading to a cellular state referred to as ‘ER stress’. These stressors may be intrinsic, i.e., cancer [95,96,97,98], neurodegenerative disease [99, 100], or diabetes [101, 102], or extrinsic, i.e., micro-environmental stress [103], exposure to ER stressors [104], temperature [105] or reactive oxygen species production [106, 107]. Nevertheless, every time the ER is stressed, it triggers an adaptive response. This adaptive response is called the unfolded protein response (UPR). This UPR will help the cells to counter the stress by attenuating protein synthesis, clearing the unfolded proteins and enhancing the ability of the ER to fold proteins.
The UPR is an intracellular signal transduction mechanism that protects cells from ER stress. Three ER-resident transmembrane proteins function as stress sensors: RNA-activated protein kinase (PKR)-like endoplasmic reticular kinase (PERK); activating transcription factor 6 (ATF6); and IRE1. In basal state, these three transmembrane proteins are bound to BiP, an ER resident chaperone and are inactive [108, 109]. Upon a stress, the folding capacity of the ER is surpassed, leading to the dissociation of BiP from PERK, ATF6 and IRE1. This dissociation allows the activation of the three sensors [110]. Their activations transduce the unfolded protein stress signal across ER membrane and lead to UPR activation [111]. PERK is transmembrane ER resident protein of 1116 amino acids with two functional domains, a luminal and a cytosolic Ser/Thr kinase domain [112]. The dissociation of BiP from the luminal domain leads to oligomerization [108] and trans-autophosphorylation [113]. Activation of the PERK pathway leads to attenuation of general protein translation by phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) [114]. Phosphorylated eIF2α inhibits eukaryotic translation initiation 2B activity, thus leading to a decrease of protein synthesis [115]. The blockage of the translation during ER stress diminishes the protein load on the ER folding machinery and is a prerequisite to a reestablishment of the ER homeostasis. In contrast to its attenuation of translation, eIF2α phosphorylation can selectively enhance the translation of mRNAs containing inhibitory upstream open reading frames in their 5′ untranslated region, such as activating transcription factor 4 (ATF4) [116]. The production of ATF4 induces the expression of a plethora of adaptive genes involved in amino acid transport, metabolism, protection from oxidative stress, protein homeostasis and autophagy [117]. Finally, ATF4 favors the expression of CAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), which will result in the expression of genes that are involved in protein synthesis and the UPR. If the expression of CHOP is sustained, the increased protein synthesis will lead to oxidative stress and cell death [118].
The second ER stress sensor is ATF6. ATF6 is also an ER resident transmembrane protein of 670 amino acids and two functional domains, an N-terminal cytosolic containing basic leucine zipper and a C-terminal luminal domain. When BiP dissociates from ATF6, this one is exported to the Golgi where it will be cleaved by site-1 and site-2 proteases [119]. This cleavage releases a fragment of 400 amino acids corresponding to ATF6 cytosolic N-terminal domain. This released fragment of ATF6 will then translocate to the nucleus in order to act as a transcription factor. ATF6 will bind to the promoter of UPR-inducible genes, resulting in an upregulations of proteins, which role is to adjust ER protein folding, including ER chaperones and X-box-binding protein-1 (XBP-1) [120, 121].
The last ER stress sensor is IRE1. IRE1 is, like the two other sensors, an ER resident transmembrane proteins of 977 amino acids with three functional domains, an N-terminal luminal domain, a C-terminal Ser/Thr kinase domain and a C-terminal RNase L domain. The dissociation with BiP triggers oligomerization and activation of its cytosolic kinase domain. This activation facilitates the unconventional splicing of XBP-1 mRNA and subsequent translation of an active transcription factor, XBP1s [111, 121]. XBP1s is a basic leucine zipper transcription factor [122]. XBP1s controls the expression of several targets including chaperones, foldases and components of the ER-associated degradation (ERAD) pathway, in order to stop the ER stress and restore homeostasis [123]. The ERAD system destroys unfolded proteins through degradation in the cytosol [124]. Indeed, unfolded ER proteins are retro-translocated across the ER membrane into the cytosol in order to be degraded by the proteasome, following ubiquitination by ubiquitin-conjugating enzymes [125, 126]. Finally, the RNase activity of IRE1 may also target other genes via a mechanism named regulated IRE1-dependent decay (RIDD) [127]. RIDD is a conserved mechanism in eukaryotes by which IRE1 cleaves its target substrates [128]. The cleaved transcripts are degraded by exoribonucleases [129]. Therefore, RIDD seems to be required for the maintenance of ER homeostasis by diminishing ER protein load via mRNA degradation. Notably, it has been recently suggested that the physiological activity of RIDD may increase with the severity of the ER stress [130].
Interestingly, a substantial number of proteins involved in UPR are localized in MAMs [23]. Indeed, two of the three major proteins involved in UPR, PERK [131] and IRE1 [28], are enriched in MAMs. Intriguingly, some ER chaperones involved in UPR are also enriched in MAMs. For example, Calnexin, a type I integral membrane protein which helps in folding newly synthetize proteins which is essential in mitigating ER stress, is expressed in MAMs [132]. Another chaperone expressed in MAMs is the Sig-1R. The first evidence of the role of Sig-1R in ER stress came from the observation that under Ca2+ depletion or when stimulated by its ligand, Sig-1R dissociates from BiP, thus allowing a sustained Ca2+ efflux from the ER via IP3R [4]. In addition, under ER stress following treatment with tunicamycin or thapsigargin, Sig-1R is upregulated, suggesting that it is protective against ER stress. Interestingly, overexpression on Sig-1R suppressed ER stress-induced activation of PERK and ATF6. IRE1 is expressed in MAMs and Sig-1R regulates the stability of IRE1 [28]. This enhanced stability favors the phosphorylation level of IRE1 under ER stress. Notably, the Sig-1R knock down potentiates the apoptosis of cells under ER stress. They showed that increased apoptosis was due to a diminution of the Xbp1 splicing [28]. In addition, activation of Sig-1R increases Bcl-2 expression, allowing Bcl-2/IP3R interaction, leading to increased mitochondrial Ca2+ uptake and ATP production [133] (see Penke et al. for review [134]).
It is well known that Sig-1R plays an important protective role in retinal disease [85]. Using in situ hybridization, Ola et al. [135] detected the Sig-1R mRNA in retinal ganglion cells, cells of the inner nuclear layer, photoreceptor and retinal pigment epithelium. The mRNA expression was confirmed by immunohistochemistry. Ha et al. [136] described that in Müller cells of the retina from Sig-1R KO mice, the expression of PERK, IRE1 and ATF4 was decreased, whereas the expression of BiP, CHOP and ATF6 was increased. Intriguingly, no difference was detected in whole brain or whole retina. Similar to what was described by Yang et al. [133], Ha et al. saw a decrease expression of Bcl-2 associated with decrease in NFkB and pERK1/2 [136]. Moreover, Wang et al. [137] demonstrated that loss of Sig-1R in a model of retinitis pigmentosa (rd10), aggravates the degeneration of the photoreceptors. They revealed that at P28, the expression level of Xbp1 and CHOP is increased in the rd10 mice without Sig-1R expression.
Since Sig-1R is a receptor that can be activated or inhibited, different groups determined the effect of its activation or inhibition in following ER stress. Ha et al. [138] treated RGC-5 cells with (+)-pentazocine, a potent Sig-1R agonist. RGC-5 cells are a rat retinal ganglion cell line [139]. They showed that whereas the protein level of PERK, ATF4, ATF6 IRE1 and CHOP was upregulated during oxidative stress, in the presence of (+)-pentazocine, their expression level decreased, suggesting that Sig-1R plays a pivotal role in the UPR response. These results confirmed the initial observation of Wang et al. [140] that stimulation of Sig-1R protects against oxidative stress. Indeed, in human cell line FHL124, H2O2 treatment induces apoptosis, associated to an increase level of BiP, ATF6 and p-eIF2α. Application of (+)-pentazocine suppressed the induction of BiP and p-eIF2α. Another agonist, fluvoxamine, alleviates induction of CHOP, cleaved caspase 3 and 4 in cancer neuronal cell SK-N-SH [141]. In another experiment, Omi et al. [142] showed that treatment of neuronal cell line Neuro2a induces overexpression of Sig-1R. This expression is mediated by ATF4, a downstream element of PERK activation. Interestingly, this overexpression is achieved without activating UPR. Intriguingly, the increased translation of ATF4 is dependent of the presence/function of Sig-1R since if the concomitant treatment of Neuro2a cells with Fluvoxamine and NE-100, a Sig-1R antagonist, abolished the ATF4 translation. This result was confirmed by the use of mouse embryonic fibroblasts (MEF) from Sig-1R KO mice. Indeed, fluvoxamine treatment of these MEF did not increase ATF4 expression. Morihara et al. [143] treated mice with Sig-1R agonist aniline derivative compound (Comp-AD) following ischemic stroke, since it is well known that Sig-1R protects against ischemic stroke but the role of ER stress was unknown. So, treatment of mice after 90 min of transient middle cerebral artery, diminished the expression level of p-PERK and p-IRE1, suggesting that activation of Sig-1R protects against ischemic stroke via the attenuation of ER stress.
If Sig-1R activation suppresses effectively ER stress, it should be expected that inhibition of Sig-1R should do the contrary. This was demonstrated by Ono et al. [144] using Sig-1R antagonist, by Hong et al. [145] using Sig-1R KO, and Alam et al. [146] using siRNA to knock down Sig-1R. Ono et al. [144] showed that NE-100 protects ER stress induced cell death in hippocampal HT22 cells after tunicamycin treatment. Indeed, NE-100 application attenuated the upregulation of CHOP. Interestingly, NE-100 treatment alone was capable of upregulate the expression of both ATF6 and BiP. Total ablation of Sig-1R in dopaminergic neurons of substantia nigra in mice led to an elevation of the expression level of p-eIF2α and CHOP [145]. In cardiomyocytes treated with tunicamycin, the downregulation of Sig-1R by siRNA led to an increase of CHOP expression. They also showed that Sig-1R downregulation diminished IRE1 phosphorylation and Xbp1 splicing [146].
Mutations of Sig-1R in human may lead to Juvenile [58] and classic ALS [147] or distal hereditary neuropathy (dHMN) [148,149,150,151]. Interestingly, E102Q mutation, which induces juvenile ALS, leads to ER stress [59, 152]. Indeed, over-expression of Sig-1R mutant in MCF7 cells induced an aggregation of the mutant protein into the ER in contrast to the overexpression of the wild-type Sig-1R, which is localized in the ER, the nuclear envelope end ER-Golgi intermediate compartment. Using ER stress response element (ERSE) reporter assay, they detected an increase in ER stress in MCF7 cells. There was an increase expression level of p-eIF2α, BiP, HSP70, GADD. Moreover, they showed a co-localization of ubiquitin-positive Sig-1R mutant aggregates with 20S proteasome subunit, suggesting possible interference with the ubiquitin proteasome system machinery [59]. In parallel, they demonstrated that the proteasome activity was greatly reduced. In order to confirm the results observed in transfected cells, they generated immortalized primary lymphoblastoid cells (PLCs) from blood samples of ALS patients. In PLCs, they also showed an aggregation of mutant Sig-1R in the ER associated with an increase level of BiP and p-eIF2α together with an increase of HSP70, GADD and ubiquitin conjugates [59].
28.4 Sig-1R in the Nucleus
Although Sig-1R is known to be particularly enriched in MAMs, observation of Sig-1Rs at the nuclear envelope (NE) and within nucleoplasms have also been reported. First, after stimulation by agonists such as cocaine, Sig-1Rs were found to translocate from ER to the NE, where they bind NE protein emerin and recruit chromatin-remodeling molecules [37]. These partners include lamin A/C, barrier-to-autointegration factor, and histone deacetylase (HDAC), to form a complex with the gene repressor specific protein 3 (Sp3). The dynamics of the interaction was confirmed when knockdown of Sig-1Rs attenuated the complex formation [37]. These observations were confirmed and developed by Mavlyutov et al. [153] who expressed APEX2 peroxidase fused to Sig1R-GFP in a Sig1R-null NSC34 neuronal cell line generated with CRISPR-Cas9. They observed that Sig1R actually resides in the nucleoplasmic reticulum, a specialized nuclear compartment formed via NE invagination into the nucleoplasm. A major consequence for this localization appears to be related to neurodegenerative pathologies since accumulation of Sig-1R may be common to neuronal nuclear inclusions in various proteinopathies [154]. Sig-1R immunoreactivity was shown to be co-localized with neuronal nuclear inclusions in TDP-43 proteinopathy, five polyglutamine diseases and intranuclear inclusion body disease, as well as in intranuclear Marinesco bodies in aged normal controls [154]. These authors interestingly proposed that Sig-1Rs might shuttle between the nucleus and the cytoplasm and likely play an important role in neurodegenerative diseases, characterized by neuronal nuclear inclusions, and known to particularly rely on ER-related degradation machinery as a common pathway for the degradation of aberrant proteins [154].
28.5 Conclusion
The Sig-1R protein is not specifically a MAM or ER protein, and direct interactions have been described at or close to the plasma membrane with potassium or sodium ion channels, ether-a-gogo-related gene (ERG) ionophores and metabotropic neurotransmitter receptors [39]. One of the complexity seen with Sig-1R is the multiplicity of its intracellular partners and consequent target pathways affected by its activation, as summarized in Fig. 28.1. This multiplicity of actions within different types of cells, in the brain as well as in other tissues, explain its involvement in numerous physiopathological processes and its value as a potential therapeutic target. Moreover, the well-known observation that bearing a relatively simple pharmacophore, Sig-1R binds small molecules, and even steroids or peptides, of diverse nature with high affinity, contributed to poorly considered it as a pertinent pharmacological target for therapeutic intervention. The data we discussed in this review allow to realize that we are now accumulating evidence on the mechanisms of action of Sig-1R, on its major role at MAMs and the ER, on its efficacy to maintain, and putatively restore cellular integrity and Ca2+ homeostasis (Fig. 28.1). The recent progression of several molecules in clinical phases in Alzheimer’s disease or Huntington’s disease strengthened the validity of Sig-1R as a pharmacological target. Moreover, their efficacies in preclinical models of different pathologies outlined the importance of MAM and ER alterations in neurodegenerative processes.

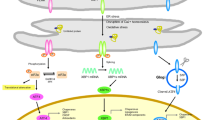
Schematic diagram of the role of 1R in different compartment of the cell and putative major impacts in neurodegenerative diseases (AD, PD, HD, ALS)
In the nucleus, S1R interacts with Emerin [37]. This interaction leads to the recruitment of chromatin-remodeling molecules such as Lamin A/C and HDAC. This interaction is necessary for the creation of a supercomplex with Sp3. In the MAM, S1R interacts with P35 [37]. This will leads to the myristoylation of P25 and axon elongation. S1R interacts with RacGTP in order to foster dendritic spine formation [155]. S1R interacts with IP3R3 in order to allow the proper Ca2+ efflux from the ER to the mitochondria [4]. In the ER, S1R interacts with BiP [4]. Upon stimulation, the dissociation of S1R with BiP induces activation of IRE1 [28] and ATF6 [136, 138, 144]. This will induce the splicing of XBP1 and the transcription of chaperones. The activation of PERK [144, 156] will induce the phosphorylation of eIF2α, to stop protein translation. Finally, S1R interacts with STIM1 and this interaction will regulate Ca2+ fluxes into the ER thought the STIM1/Orai1 axe [46]. In the plasma membrane, S1R interacts with voltage-gated calcium channels (for review, [157]) in order to modulate Ca2+ homeostasis. S1R favors BDNF secretion [74, 87]. BDNF will activate the PI3K/Akt pathway in order to improve cell survival
References
Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE (1976) The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther 197(3):517–532
Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E et al (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A 93(15):8072–8077
Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V (1996) Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem Biophys Res Commun 229(2):553–558
Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131(3):596–610
Maurice T, Gregoire C, Espallergues J (2006) Neuro(active)steroids actions at the neuromodulatory sigma1 (σ1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol Biochem Behav 84(4):581–597
Moebius FF, Reiter RJ, Hanner M, Glossmann H (1997) High affinity of sigma 1-binding sites for sterol isomerization inhibitors: evidence for a pharmacological relationship with the yeast sterol C8-C7 isomerase. Br J Pharmacol 121(1):1–6
Meunier J, Hayashi T (2010) Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J Pharmacol Exp Ther 332(2):388–397
Pal A, Fontanilla D, Gopalakrishnan A, Chae YK, Markley JL, Ruoho AE (2012) The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur J Pharmacol 682(1-3):12–20
Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M et al (2000) Immunocytochemical localization of the sigma1 receptor in the adult rat central nervous system. Neuroscience 97(1):155–170
Palacios G, Muro A, Vela JM, Molina-Holgado E, Guitart X, Ovalle S et al (2003) Immunohistochemical localization of the sigma1-receptor in oligodendrocytes in the rat central nervous system. Brain Res 961(1):92–99
Tagashira H, Bhuiyan S, Shioda N, Hasegawa H, Kanai H, Fukunaga K (2010) Sigma1-receptor stimulation with fluvoxamine ameliorates transverse aortic constriction-induced myocardial hypertrophy and dysfunction in mice. Am J Physiol Heart Circ Physiol 299(5):H1535–H1545
Meurs A, Clinckers R, Ebinger G, Michotte Y, Smolders I (2007) Sigma 1 receptor-mediated increase in hippocampal extracellular dopamine contributes to the mechanism of the anticonvulsant action of neuropeptide Y. Eur J Neurosci 26(11):3079–3092
Vavers E, Svalbe B, Lauberte L, Stonans I, Misane I, Dambrova M et al (2017) The activity of selective sigma-1 receptor ligands in seizure models in vivo. Behav Brain Res 328:13–18
Harukuni I, Bhardwaj A, Shaivitz AB, DeVries AC, London ED, Hurn PD et al (2000) sigma1-receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine affords neuroprotection from focal ischemia with prolonged reperfusion. Stroke 31(4):976–982
Lesage AS, De Loore KL, Peeters L, Leysen JE (1995) Neuroprotective sigma ligands interfere with the glutamate-activated NOS pathway in hippocampal cell culture. Synapse 20(2):156–164
Shen YC, Wang YH, Chou YC, Liou KT, Yen JC, Wang WY et al (2008) Dimemorfan protects rats against ischemic stroke through activation of sigma-1 receptor-mediated mechanisms by decreasing glutamate accumulation. J Neurochem 104(2):558–572
Cai Y, Yang L, Niu F, Liao K, Buch S (2017) Role of sigma-1 receptor in cocaine abuse and neurodegenerative disease. Adv Exp Med Biol 964:163–175
Maurice T, Martin-Fardon R, Romieu P, Matsumoto RR (2002) Sigma1 (σ1) receptor antagonists represent a new strategy against cocaine addiction and toxicity. Neurosci Biobehav Rev 26(4):499–527
Su TP, Hayashi T (2001) Cocaine affects the dynamics of cytoskeletal proteins via sigma1 receptors. Trends Pharmacol Sci 22(9):456–458
Diaz JL, Zamanillo D, Corbera J, Baeyens JM, Maldonado R, Pericas MA et al (2009) Selective sigma-1 (σ1) receptor antagonists: emerging target for the treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem 9(3):172–183
Zamanillo D, Romero L, Merlos M, Vela JM (2013) Sigma 1 receptor: a new therapeutic target for pain. Eur J Pharmacol 716(1-3):78–93
Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265(13):7248–7256
Carreras-Sureda A, Pihan P, Hetz C (2017) The unfolded protein response: at the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front Oncol 7:55
Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19(2):81–88
Raturi A, Simmen T (2013) Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta 1833(1): 213–224
Rizzuto R, Duchen MR, Pozzan T (2004) Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE 2004(215):re1
Walter L, Hajnoczky G (2005) Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. J Bioenerg Biomembr 37(3):191–206
Mori T, Hayashi T, Hayashi E, Su TP (2013) Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8(10):e76941
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N et al (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495(7441):389–393
Sutterwala FS, Haasken S, Cassel SL (2014) Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319:82–95
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225
Poston CN, Krishnan SC, Bazemore-Walker CR (2013) In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteome 79:219–230
Fontanilla D, Hajipour AR, Pal A, Chu UB, Arbabian M, Ruoho AE (2008) Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry 47(27):7205–7217
Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC (2016) Crystal structure of the human sigma1 receptor. Nature 532(7600):527–530
Wu Z, Bowen WD (2008) Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem 283(42):28198–28215
Tsai SY, Hayashi T, Harvey BK, Wang Y, Wu WW, Shen RF et al (2009) Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc Natl Acad Sci U S A 106(52):22468–22473
Tsai SY, Pokrass MJ, Klauer NR, Nohara H, Su TP (2015) Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc Natl Acad Sci U S A 112(21):6742–6747
Maurice T, Su TP (2009) The pharmacology of sigma-1 receptors. Pharmacol Ther 124(2):195–206
Su TP, Su TC, Nakamura Y, Tsai SY (2016) The sigma-1 receptor as a puripotent modulator in living systems. Trends Pharmacol Sci 37(4):262–278
Ryskamp D, Wu J, Geva M, Kusko R, Grossman I, Hayden M et al (2017) The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol Dis 97(Pt A):46–59
Katnik C, Guerrero WR, Pennypacker KR, Herrera Y, Cuevas J (2006) Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J Pharmacol Exp Ther 319(3):1355–1365
Tadic V, Malci A, Goldhammer N, Stubendorff B, Sengupta S, Prell T et al (2017) Sigma 1 receptor activation modifies intracellular calcium exchange in the G93A(hSOD1) ALS model. Neuroscience 359:105–118
Sambo DO, Lin M, Owens A, Lebowitz JJ, Richardson B, Jagnarine DA et al (2017) The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat Commun 8(1):2228
Zhang K, Zhao Z, Lan L, Wei X, Wang L, Liu X et al (2017) Sigma-1 receptor plays a negative modulation on N-type calcium channel. Front Pharmacol 8:302
Brailoiu GC, Deliu E, Console-Bram LM, Soboloff J, Abood ME, Unterwald EM et al (2016) Cocaine inhibits store-operated Ca2+ entry in brain microvascular endothelial cells: critical role for sigma-1 receptors. Biochem J 473(1):1–5
Srivats S, Balasuriya D, Pasche M, Vistal G, Edwardson JM, Taylor CW et al (2016) Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J Cell Biol 213(1):65–79
Zhang H, Cuevas J (2002) Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J Neurophysiol 87(6):2867–2879
Tchedre KT, Huang RQ, Dibas A, Krishnamoorthy RR, Dillon GH, Yorio T (2008) Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction. Invest Ophthalmol Vis Sci 49(11):4993–5002
Erpapazoglou Z, Mouton-Liger F, Corti O (2017) From dysfunctional endoplasmic reticulum-mitochondria coupling to neurodegeneration. Neurochem Int 109:171–183
Joshi AU, Kornfeld OS, Mochly-Rosen D (2016) The entangled ER-mitochondrial axis as a potential therapeutic strategy in neurodegeneration: a tangled duo unchained. Cell Calcium 60(3):218–234
Nguyen L, Lucke-Wold BP, Mookerjee S, Kaushal N, Matsumoto RR (2017) Sigma-1 receptors and neurodegenerative diseases: towards a hypothesis of sigma-1 receptors as amplifiers of neurodegeneration and neuroprotection. Adv Exp Med Biol 964:133–152
Ottolini D, Cali T, Negro A, Brini M (2013) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet 22(11):2152–2168
Ouyang YB, Giffard RG (2012) ER-mitochondria crosstalk during cerebral ischemia: molecular chaperones and ER-mitochondrial calcium transfer. Int J Cell Biol 2012:493934
Zundorf G, Reiser G (2011) Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal 14(7):1275–1288
Hedskog L, Pinho CM, Filadi R, Ronnback A, Hertwig L, Wiehager B et al (2013) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci U S A 110(19):7916–7921
Prudent J, McBride HM (2017) The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr Opin Cell Biol 47:52–63
Bernard-Marissal N, Medard JJ, Azzedine H, Chrast R (2015) Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138(Pt 4):875–890
Al-Saif A, Al-Mohanna F, Bohlega S (2011) A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol 70(6):913–919
Dreser A, Vollrath JT, Sechi A, Johann S, Roos A, Yamoah A et al (2017) The ALS-linked E102Q mutation in sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ 24(10):1655–1671
Luty AA, Kwok JB, Dobson-Stone C, Loy CT, Coupland KG, Karlstrom H et al (2010) Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann Neurol 68(5):639–649
Prause J, Goswami A, Katona I, Roos A, Schnizler M, Bushuven E et al (2013) Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum Mol Genet 22(8):1581–1600
Behensky AA, Yasny IE, Shuster AM, Seredenin SB, Petrov AV, Cuevas J (2013) Afobazole activation of sigma-1 receptors modulates neuronal responses to amyloid-β25–35. J Pharmacol Exp Ther 347(2):468–477
Marrazzo A, Caraci F, Salinaro ET, Su TP, Copani A, Ronsisvalle G (2005) Neuroprotective effects of sigma-1 receptor agonists against beta-amyloid-induced toxicity. Neuroreport 16(11):1223–1226
Antonini V, Marrazzo A, Kleiner G, Coradazzi M, Ronsisvalle S, Prezzavento O et al (2011) Anti-amnesic and neuroprotective actions of the sigma-1 receptor agonist (−)-MR22 in rats with selective cholinergic lesion and amyloid infusion. J Alzheimers Dis 24(3):569–586
Lahmy V, Meunier J, Malmstrom S, Naert G, Givalois L, Kim SH et al (2013) Blockade of Tau hyperphosphorylation and Aβ1–42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and sigma1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 38(9):1706–1723
Meunier J, Ieni J, Maurice T (2006) The anti-amnesic and neuroprotective effects of donepezil against amyloid β25–35 peptide-induced toxicity in mice involve an interaction with the σ1 receptor. Br J Pharmacol 149(8):998–1012
Villard V, Espallergues J, Keller E, Alkam T, Nitta A, Yamada K et al (2009) Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1-41 against amyloid β25–35-induced toxicity in mice. Neuropsychopharmacology 34(6):1552–1566
Villard V, Espallergues J, Keller E, Vamvakides A, Maurice T (2011) Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J Psychopharmacol 25(8):1101–1117
Yang R, Chen L, Wang H, Xu B, Tomimoto H, Chen L (2012) Anti-amnesic effect of neurosteroid PREGS in Aβ25–35-injected mice through σ1 receptor- and α7 nAChR-mediated neuroprotection. Neuropharmacology 63(6):1042–1050
Fisher A, Bezprozvanny I, Wu L, Ryskamp DA, Bar-Ner N, Natan N et al (2016) AF710B, a Novel M1/σ1 agonist with therapeutic efficacy in animal models of Alzheimer’s disease. Neurodegener Dis 16(1-2):95–110
Maurice T, Strehaiano M, Duhr F, Chevallier N (2018) Amyloid toxicity is enhanced after pharmacological or genetic invalidation of the sigma1 receptor. Behav Brain Res 339:1–10
Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L (2008) Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3(7):e2718
Goguadze N, Zhuravliova E, Morin D, Mikeladze D, Maurice T (2019) Sigma-1 receptor agonists induce oxidative stress in mitochondria and enhance complex I activity in physiological condition but protect against pathological oxidative stress. Neurotox Res 35(1):1–18
Francardo V, Bez F, Wieloch T, Nissbrandt H, Ruscher K, Cenci MA (2014) Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 137(Pt 7):1998–2014
Hyrskyluoto A, Pulli I, Tornqvist K, Ho TH, Korhonen L, Lindholm D (2013) Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-κB pathway. Cell Death Dis 4:e646
Mancuso R, Olivan S, Rando A, Casas C, Osta R, Navarro X (2012) Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9(4): 814–826
Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D et al (2014) Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis 62:218–232
Oxombre B, Lee-Chang C, Duhamel A, Toussaint M, Giroux M, Donnier-Marechal M et al (2015) High-affinity sigma1 protein agonist reduces clinical and pathological signs of experimental autoimmune encephalomyelitis. Br J Pharmacol 172(7):1769–1782
Smith SB, Duplantier J, Dun Y, Mysona B, Roon P, Martin PM et al (2008) In vivo protection against retinal neurodegeneration by sigma receptor 1 ligand (+)-pentazocine. Invest Ophthalmol Vis Sci 49(9):4154–4161
Zhao L, Chen G, Li J, Fu Y, Mavlyutov TA, Yao A et al (2017) An intraocular drug delivery system using targeted nanocarriers attenuates retinal ganglion cell degeneration. J Control Release 247:153–166
Francardo V, Schmitz Y, Sulzer D, Cenci MA (2017) Neuroprotection and neurorestoration as experimental therapeutics for Parkinson’s disease. Exp Neurol 298(Pt B):137–147
Mancuso R, Navarro X (2017) Sigma-1 receptor in motoneuron disease. Adv Exp Med Biol 964:235–254
Maurice T, Goguadze N (2017) Role of σ1 receptors in learning and memory and Alzheimer’s disease-type dementia. Adv Exp Med Biol 964:213–233
Maurice T, Goguadze N (2017) Sigma-1 (σ1) receptor in memory and neurodegenerative diseases. Handb Exp Pharmacol 244:81–108
Smith SB, Wang J, Cui X, Mysona BA, Zhao J, Bollinger KE (2018) Sigma 1 receptor: a novel therapeutic target in retinal disease. Prog Retin Eye Res 67:130–149
Weng TY, Tsai SA, Su TP (2017) Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J Biomed Sci 24(1):74
Geva M, Kusko R, Soares H, Fowler KD, Birnberg T, Barash S et al (2016) Pridopidine activates neuroprotective pathways impaired in Huntington disease. Hum Mol Genet 25(18):3975–3987
Sahlholm K, Arhem P, Fuxe K, Marcellino D (2013) The dopamine stabilizers ACR16 and (−)-OSU6162 display nanomolar affinities at the sigma-1 receptor. Mol Psychiatry 18(1): 12–14
Sahlholm K, Sijbesma JW, Maas B, Kwizera C, Marcellino D, Ramakrishnan NK et al (2015) Pridopidine selectively occupies sigma-1 rather than dopamine D2 receptors at behaviorally active doses. Psychopharmacology 232(18):3443–3453
Sahlholm K, Valle-Leon M, Fernandez-Duenas V, Ciruela F (2018) Pridopidine reverses phencyclidine-induced memory impairment. Front Pharmacol 9:338
Braakman I, Bulleid NJ (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem 80:71–99
Hebert DN, Molinari M (2007) In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev 87(4):1377–1408
Fagone P, Jackowski S (2009) Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res 50(Suppl):S311–S316
Meldolesi J, Pozzan T (1998) The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci 23(1):10–14
Corazzari M, Gagliardi M, Fimia GM, Piacentini M (2017) Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol 7:78
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5): 646–674
Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69(2):169–181
Jain BP (2017) An overview of unfolded protein response signaling and its role in cancer. Cancer Biother Radiopharm 32(8):275–281
Hetz C, Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13(8):477–491
Xiang C, Wang Y, Zhang H, Han F (2017) The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 22(1):1–26
Ariyasu D, Yoshida H, Hasegawa Y (2017) Endoplasmic reticulum (ER) stress and endocrine disorders. Int J Mol Sci 18(2):382
Harding HP, Ron D (2002) Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51(Suppl 3):S455–S461
Giampietri C, Petrungaro S, Conti S, Facchiano A, Filippini A, Ziparo E (2015) Cancer microenvironment and endoplasmic reticulum stress response. Mediat Inflamm 2015:417281
Foufelle F, Fromenty B (2016) Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect 4(1):e00211
Liu Y, Sakamoto H, Adachi M, Zhao S, Ukai W, Hashimoto E et al (2012) Heat stress activates ER stress signals which suppress the heat shock response, an effect occurring preferentially in the cortex in rats. Mol Biol Rep 39(4):3987–3993
Bhandary B, Marahatta A, Kim HR, Chae HJ (2012) An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 14(1):434–456
Cao SS, Kaufman RJ (2014) Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21(3):396–413
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2(6):326–332
Shen J, Chen X, Hendershot L, Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3(1):99–111
Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B et al (2018) Endoplasmic reticulum stress signalling – from basic mechanisms to clinical applications. FEBS J 286:241–278
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716):271–274
McQuiston A, Diehl JA (2017) Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Res 6:1897
Schroder M (2006) The unfolded protein response. Mol Biotechnol 34(2):279–290
Rowlands AG, Panniers R, Henshaw EC (1988) The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biol Chem 263(12):5526–5533
Lu PD, Harding HP, Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167(1):27–33
Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D et al (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216(7):2027–2045
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J et al (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15(5):481–490
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R et al (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6(6):1355–1364
Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H et al (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell 13(3):365–376
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7):881–891
Liou HC, Boothby MR, Finn PW, Davidon R, Nabavi N, Zeleznik-Le NJ et al (1990) A new member of the leucine zipper class of proteins that binds to the HLA DR alpha promoter. Science 247(4950):1581–1584
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101(3):249–258
Bonifacino JS, Weissman AM (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol 14:19–57
Hwang J, Qi L (2018) Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci 43(8):593–605
Wu X, Rapoport TA (2018) Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol 53:22–28
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186(3):323–331
Oikawa D, Tokuda M, Hosoda A, Iwawaki T (2010) Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res 38(18):6265–6273
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783):104–107
Maurel M, Chevet E, Tavernier J, Gerlo S (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci 39(5):245–254
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP et al (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19(11):1880–1891
Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L et al (2012) Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J 31(2):457–470
Yang S, Bhardwaj A, Cheng J, Alkayed NJ, Hurn PD, Kirsch JR (2007) Sigma receptor agonists provide neuroprotection in vitro by preserving bcl-2. Anesth Analg 104(5):1179–1184
Penke B, Fulop L, Szucs M, Frecska E (2018) The role of sigma-1 receptor, an intracellular chaperone in neurodegenerative diseases. Curr Neuropharmacol 16(1):97–116
Ola MS, Moore P, El-Sherbeny A, Roon P, Agarwal N, Sarthy VP et al (2001) Expression pattern of sigma receptor 1 mRNA and protein in mammalian retina. Brain Res Mol Brain Res 95(1-2):86–95
Ha Y, Shanmugam AK, Markand S, Zorrilla E, Ganapathy V, Smith SB (2014) Sigma receptor 1 modulates ER stress and Bcl2 in murine retina. Cell Tissue Res 356(1):15–27
Wang J, Saul A, Cui X, Roon P, Smith SB (2017) Absence of sigma 1 receptor accelerates photoreceptor cell death in a Murine model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 58(11):4545–4558
Ha Y, Dun Y, Thangaraju M, Duplantier J, Dong Z, Liu K et al (2011) Sigma receptor 1 modulates endoplasmic reticulum stress in retinal neurons. Invest Ophthalmol Vis Sci 52(1):527–540
Krishnamoorthy RR, Agarwal P, Prasanna G, Vopat K, Lambert W, Sheedlo HJ et al (2001) Characterization of a transformed rat retinal ganglion cell line. Brain Res Mol Brain Res 86(1-2):1–12
Wang L, Eldred JA, Sidaway P, Sanderson J, Smith AJ, Bowater RP et al (2012) Sigma 1 receptor stimulation protects against oxidative damage through suppression of the ER stress responses in the human lens. Mech Ageing Dev 133(11-12):665–674
Tanimukai H, Kudo T (2015) Fluvoxamine alleviates paclitaxel-induced neurotoxicity. Biochem Biophys Rep 4:202–206
Omi T, Tanimukai H, Kanayama D, Sakagami Y, Tagami S, Okochi M et al (2014) Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis 5:e1332
Morihara R, Yamashita T, Liu X, Nakano Y, Fukui Y, Sato K et al (2018) Protective effect of a novel sigma-1 receptor agonist is associated with reduced endoplasmic reticulum stress in stroke male mice. J Neurosci Res 96:1707–1716
Ono Y, Tanaka H, Tsuruma K, Shimazawa M, Hara H (2013) A sigma-1 receptor antagonist (NE-100) prevents tunicamycin-induced cell death via GRP78 induction in hippocampal cells. Biochem Biophys Res Commun 434(4):904–909
Hong J, Wang L, Zhang T, Zhang B, Chen L (2017) Sigma-1 receptor knockout increases alpha-synuclein aggregation and phosphorylation with loss of dopaminergic neurons in substantia nigra. Neurobiol Aging 59:171–183
Alam S, Abdullah CS, Aishwarya R, Orr AW, Traylor J, Miriyala S, et al. (2017) SigmaR1 regulates endoplasmic reticulum stress-induced C/EBP-homologous protein expression in cardiomyocytes. Biosci Rep 37(4)
Watanabe S, Ilieva H, Tamada H, Nomura H, Komine O, Endo F et al (2016) Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol Med 8(12):1421–1437
Gregianin E, Pallafacchina G, Zanin S, Crippa V, Rusmini P, Poletti A et al (2016) Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum Mol Genet 25(17):3741–3753
Horga A, Tomaselli PJ, Gonzalez MA, Laura M, Muntoni F, Manzur AY et al (2016) SIGMAR1 mutation associated with autosomal recessive Silver-like syndrome. Neurology 87(15):1607–1612
Li X, Hu Z, Liu L, Xie Y, Zhan Y, Zi X et al (2015) A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 84(24):2430–2437
Nandhagopal R, Meftah D, Al-Kalbani S, Scott P (2018) Recessive distal motor neuropathy with pyramidal signs in an Omani kindred: underlying novel mutation in the SIGMAR1 gene. Eur J Neurol 25(2):395–403
Fukunaga K, Shinoda Y, Tagashira H (2015) The role of SIGMAR1 gene mutation and mitochondrial dysfunction in amyotrophic lateral sclerosis. J Pharmacol Sci 127(1):36–41
Mavlyutov TA, Yang H, Epstein ML, Ruoho AE, Yang J, Guo LW (2017) APEX2-enhanced electron microscopy distinguishes sigma-1 receptor localization in the nucleoplasmic reticulum. Oncotarget 8(31):51317–51330
Miki Y, Mori F, Kon T, Tanji K, Toyoshima Y, Yoshida M et al (2014) Accumulation of the sigma-1 receptor is common to neuronal nuclear inclusions in various neurodegenerative diseases. Neuropathology 34(2):148–158
Natsvlishvili N, Goguadze N, Zhuravliova E, Mikeladze D (2015 Apr 30) Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria. BMC Biochem 16:11
Mitsuda T, Omi T, Tanimukai H, Sakagami Y, Tagami S, Okochi M, Kudo T, Takeda M (2011 Nov 25) Sigma-1Rs are upregulated via PERK/eIF2α/ATF4 pathway and execute protective function in ER stress. Biochem Biophys Res Commun 415(3):519–525
Kourrich S (2017) Sigma-1 receptor and neuronal excitability. Handb Exp Pharmacol 244:109–130. https://doi.org/10.1007/164_2017_8
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Delprat, B., Crouzier, L., Su, TP., Maurice, T. (2020). At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor?. In: Islam, M. (eds) Calcium Signaling. Advances in Experimental Medicine and Biology, vol 1131. Springer, Cham. https://doi.org/10.1007/978-3-030-12457-1_28
Download citation
DOI: https://doi.org/10.1007/978-3-030-12457-1_28
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-12456-4
Online ISBN: 978-3-030-12457-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)