
Abstract
Chemical bonding in transition metal complexes is typically described by Dewar–Chatt–Duncanson model which separates donation (ligand → metal) and back-donation (metal → ligand) charge transfer processes—these are with no doubt crucial factors which determine a number of properties of metal complexes. This contribution highlights the importance of various non-covalent interactions including untypical homopolar dihydrogen contacts C–H•••H–C in metal complexes. The selected systems are: (1) Zn(II) species containing NTA (nitrotriacetic acid ), NTPA (nitrotri-3-propanoic), BPy (2,2′-bipyridyl) ligands, (2) cis-NiL2–hexane (L–thiourea -based ligand) complex, and (3) hydrogen storage materials LiNMe2BH3 and KNMe2BH3. It is shown consistently by various methods and bonding descriptors including for example the charge and energy decomposition scheme ETS-NOCV , Interacting Quantum Atoms (IQA) , Reduced Density Gradient (NCI) , Quantum Theory of Atoms in Molecules (QTAIM) and NMR spin-spin 1J(C–H) coupling constants, that London dispersion dominated C–H•••H–C interactions and other more typical hydrogen bonds (e.g. C–H•••N, C–H•••O) driven mostly by electrostatics, are crucial for determination of the structures and stability of the selected metal complexes. Although London dispersion forces are the fundamental factor (~70% of the overall stabilization) contributing to C–H•••H–C interactions, the charge delocalization (outflow of electrons from the σ(C–H) bonds engaged in C–H•••H–C and the accumulation in the interatomic H•••H region) as well as electrostatic terms are also non-negligible (~30%). Remarkably, hydride–hydride interactions B–H•••H–B in LiNMe2BH3 are found to be repulsive due to dominant destabilizing electrostatic contribution as opposed to stabilizing C–H•••H–C.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Homopolar dihydrogen interactions
- Transition metal complexes
- Non-covalent interactions
- Hydrogen storage materials
- ETS-NOCV
- London dispersion forces
1 Introduction
Chemical bonding in transition metal complexes is usually described in terms of Dewar–Chatt–Duncanson model which accounts for donation (ligand → metal) and back-donation (metal → ligand) charge transfer processes [1, 2]. They are crucial for the molecular stability as well as for the determination of various catalytic and spectroscopic properties [1,2,3,4,5]. Very recently, however, more and more attention is paid to London dispersion forces and different types of non-covalent interactions which can, additionally to typical donor/acceptor phenomena, influence the chemistry of transition metal complexes [6, 7]. One shall emphasize that the DFT method together with the recent breakthrough developments of semi-empirical dispersion corrections by Grimme [8, 9] allows for identification and better understanding a number of very important physical phenomena in real materials [6, 7].
Transition metal complexes often contain sterically demanding ligands which traditionally are associated with the source of repulsion—however, the recent topical review by Schreiner and Wagner has suggested the necessity for “…reconsidering steric effects” because in many cases sterically demanding hydrophobic groups, often leading to formation of untypical homopolar C–H•••H–C non-covalent interactions , are truly London dispersion donors which can easily overcompensate Pauli repulsion [6]. Furthermore, Liptrot and Power nicely reviewed the importance of London dispersion forces in organometallic and inorganic complexes [7]. One could also reference that bulky ligands might control directly catalytic activity [10].
Despite significant progress in the identification of untypical homopolar X–H•••H–X non-covalent interactions in various systems including transition metal complexes, one must admit that this subject is still the matter of some discussion in the literature particularly as far as intramolecular X–H•••H–X contacts are taken into account. Here, one can cite the following debates on the stability of biphenyl (planar versus bent), [11,12,13,14,15,16] 2-butene isomers [17,18,19] or the nature of inter-molecular homopolar B–H•••H–B (hydride-hydride) and other similar contacts in hydrogen storage systems [20,21,22,23,24,25,26,27]. Apart from the above non-covalent interactions and well established now polar dihydrogen X–Hδ+•••–δH–Y (X ≠ Y) bonds (named also as proton–hydride contacts) [28,29,30,31,32], there are other non-conventional and quite unintuitive weak interactions, which have been discovered in recent time, such as anion•••π interactions [33,34,35] or various types of σ/π-hole bonds [36,37,38,39,40]. All these types of weak interactions as well as typical hydrogen bonds, π•••π stacking and others are now crucial forces for various branches of chemistry including transition metal complexes [41,42,43,44,45].
This chapter covers selected contributions which allowed to determine how various non-covalent interactions including controversial homopolar dihydrogen contacts X–H•••H–X and other types of chemical bonds affect the stability and other properties of selected transition metal complexes. In order to shed light on the bonding situations, various methods/descriptors will be applied including the quantum theory of atoms in molecules (QTAIM) [46], interacting quantum atoms (IQA) energy decomposition scheme [47], non-covalent Interactions (NCI) method [48], and (ETS-NOCV) energy decomposition scheme [49].
2 Methods
2.1 ETS-NOCV Charge and Energy Decomposition Scheme
The ETS-NOCV consists of the ETS energy decomposition scheme and NOCV method which allows for partitioning orbital charge delocalization term into chemically meaningful constituents (σ, π, δ, etc.) [49].
In ETS scheme, the total bonding energy (ΔEtotal) is partitioned into the following contributions:
where ΔEelstat covers electrostatic interactions between fragments, ΔEPauli is responsible for the repulsion between electrons carrying the same spin, ΔEorb reflects stabilizing component due to electron density reorganization upon bond formation, whereas positive ΔEdist describes changes in fragments geometries due to the formation of a bond. Dispersion term ΔEdisp is accounted through the semi-empirical Grimme’s D3 correction [8, 9].
Natural orbitals for chemical valence (NOCV) denoted as Ψi are eigenvectors diagonalizing the deformation density matrix ΔP = P – P0 (P—molecule’s density matrix, P0—promolecular density matrix):
Ci is a vector of coefficients expanding NOCVs in the basis of fragment orbitals. Pairs (Ψ−i, Ψi). NOCVs decompose the differential density (Δρ) into chemically meaningful contributions (Δρk):
Combining ETS and NOCV, it is possible to determine the energy corresponding to each eigenvalue pair and deformation density channel:
where \( F_{i,i}^{TS} \) are Kohn–Sham matrix elements for the transition state, as formulated by ETS methodology [49].
2.2 Non-covalent Index (NCI)
Reduced density gradient (s) plots against electronic density (ρ):
have been shown to be a useful tool to identify the presence of both inter- and intramolecular non-covalent interactions. On said plots, a characteristic spike at the low values on s and ρ indicates the existence of interaction. Sign of the eigenvalues (λi) of the Hessian (∇2ρ = λ1 + λ2 + λ3), precisely the sign of λ2, indicates whether the interaction is bonding (λ2 < 0) or not (λ2 > 0). Also, plots of the contour of s colored by the sign of λ2 are very informative since they show electronic exchange channels [48].
2.3 Quantum Theory of Atoms in Molecules (QTAIM)
In the QTAIM theory, molecular electron density ρ(r) is divided into atomic basins based on the zero-flux surface criterion [46]. Interacting atoms, each possessing own basin and electronic density maximum in the position of nucleus, are connected by the atomic interaction line (AIL)—a line of local maximum density, called also as a bond path. Due to a diagonalization of a Hessian matrix, one can obtain critical points of ρ(r) [e.g., maximum of ρ(r)] including very often used a bond critical point (BCP)—the presence of BCP between atoms is attributed to the existence of bonding interactions. It allows further to create a molecular graph which shows which atoms are bonded to each other. Furthermore, values of electron density (and its laplacian) at BCPs are often discussed in terms of a bond strength.
2.4 Interacting Quantum Atoms (IQA) Energy Decomposition Scheme
IQA method [47] divides the total electronic energy E into atomic (\( E_{\text{self}}^{A} \)) and diatomic (\( E_{\text{int}}^{\text{AB}} \)) contributions:
Diatomic part covers all interactions between particles of different atoms A and B: nucleus–nucleus (\( V_{\text{nn}}^{\text{AB}} \)), nucleus–electron (\( V_{\text{ne}}^{\text{AB}} \)) electron–nucleus (\( V_{\text{en}}^{\text{AB}} \)), and electron–electron (\( V_{\text{ee}}^{\text{AB}} \)):
where \( V_{\text{eeC}}^{\text{AB}} \) and \( V_{\text{eeX}}^{\text{AB}} \) are Coulomb and exchange contributions of \( V_{\text{ee}}^{\text{AB}} \).
2.5 Computational Details
Zinc complexes with NTA and NTPA were optimized at the B3LYP/6-311++G(d,p) level of theory in the Gaussian03.D01 software, with the solvent effects (water environment) taken into account by means of the CPCM model with UAKS cavities [50]. Minima were confirmed by frequency calculations. Topological AIM calculations were performed in the AIMALL package at the same level of theory (B3LYP/6-311++G(d,p)) [50]. ETS-NOCV analyses were performed in the Amsterdam density functional (ADF) package with BP86 functional in DZP basis set for all atoms except metal, where TZP basis was utilized [50].
Other zinc complexes, with the bipyridyl as ligands, were optimized by means of X3LYP/ATZP as implemented in ADF2010 package [51]. COSMO model was used to model a solvent in calculations. Results were confronted with the B97-D/ATZP and MP2/6-311++G(d,p) calculations to ensure their validity. Wavefunctions for QTAIM, IQA (later performed in AIMALL), and NCI (NCIPLOT) analyses were obtained with Gaussian09.B with X3LYP/6-311++G(d,p) [51].
Complexes of nickel were calculated in the geometry taken from the crystal cif files’ data [52]. They were also reoptimized to confirm they are correct minima in B97-D3, B3LYP-D3, and M06. ETS-NOCV calculations were performed at BLYP-D3/TZP level of theory [52].
Main group metal complexes were also calculated in the geometry taken from the crystal structures [23]. They were also reoptimized to confirm they are correct minima in PBE-D3, BP86-D3, M06-2X, wB97XD, and MP2. ETS-NOCV calculations were performed at BLYP-D3/TZP level of theory [23].
3 Results and Discussion
It is known experimentally that Zn(II) ion prefers binding with five-member chelate rings (e.g., nitrotriacetic acid , NTA) rather than with six-member units (e.g., nitrotri-3-propanoic acid, NTPA)—the experimentally determined formation constants in water are logK = 5.3 for ZnNTPA complex and logK = 10.45 for ZnNTA, Fig. 1 [53, 54, 50]. Such difference in the stability is intuitively explained by the increased steric crowding (due to C–H•••H–C contacts between the adjacent C–H bonds) in the case of ZnNTPA [53, 54]. In order to shed some light on the origin of different stability between ZnNTPA and ZnNTA, we have performed an in-depth study of bonding situations in both complexes by the charge and energy decomposition method ETS-NOCV as well as by the QTAIM approach [50].

Reprinted with permission from [50]. Copyright (2011) American Chemical Society
Lowest energy conformers of ZnNTA and ZnNTPA complexes from B3LYP/6-311++G(d,p) in solvent (CPCM/UAKS).
At first stage, the lowest energy conformations have been found (Fig. 1) followed by the computational determination of the formation constants—the higher stability of ZnNTA versus ZnNTPA has been reproduced as indicated by the computed logK = 5.3 (ZnNTA) versus 3.83 (ZnNTPA) [50]. Then, the complexes have been subjected to in-depth bonding analyses.
According to our QTAIM-based results, non-covalent interactions are only found in the case of ZnNTPA, Table 1. Namely, the classical intramolecular hydrogen bonds are formed between the ligand’s C–H bonds and water species: CH35–O3H and CH24–O3H as indicated by the presence of the corresponding bond critical points, Table 1, Fig. 1. The most interestingly, the QTAIM revealed also bond critical points corresponding to non-classical homopolar dihydrogen interactions of the type CH31–H32C and CH27–H28C, Table 1, Fig. 1. It is very interesting result taking into account that lower stability of ZnNTPA versus ZnNTA is intuitively attributed to steric C–H•••H–C clashes [53, 54].
We have further confirmed due to the ETS-NOCV charge and energy decomposition-based study that there are indeed deformation density channels corresponding to homopolar C–H•••H–C interactions (H31•••H32, H27•••H28, H24•••H35) in ZnNTPA complex, Fig. 2. Clearly, an outflow of electron density from the occupied σ(C–H) bonds and the accumulation in the interatomic H•••H region is seen upon fragmentation of ZnNTPA into CH2CH2COO– arm and the rest of the molecule, black line in Fig. 2—such fragmentation allows to extract C–H•••H–C charge delocalizations between the adjacent carboxylic moieties within NTPA. They correspond to the overall stabilization by ca. ΔEorb = –4.13 kcal/mol, Fig. 2.

Reprinted with permission from [50]. Copyright (2011) American Chemical Society
Selected deformation density contributions together with the corresponding energies characterizing intramolecular interactions. Black circle lines in the structure of ZnNTPA indicate the fragmentation used in the bonding analysis. For clarity of the fragmentation, the carbon atoms of NTPA were labeled according to numbering in Fig. 1.
In order to extract ETS-NOCV-based information on typical dative bonds Zn–N and Zn–O, the following fragmentation patterns are applied L| Zn(H2O)2 and H2O| Zn(H2O)L (where L = NTA, NTPA), respectively. It is found that both the ETS-NOCV-based and QTAIM-based results demonstrate stronger binding of NTPA versus NTA, Table 2. Namely, the calculated interaction energies are ΔEint = –743.1 kcal/mol for ZnNTPA versus ΔEint = –732.8 kcal/mol for ZnNTA. The electron densities of Zn–N BCPs follow the same relation (0.058 a.u. vs. 0.061 a.u.) [50]. It nicely correlates with the calculated Zn–N bonds, lengths which are longer for ZnNTA, by ca. 0.03Å. Furthermore, the vertical water molecule (labeled in Fig. 1 as O4H5H6 in ZnNTA and O21H23H24 in ZnNTPA) is also less efficiently bonded to Zn(II) ion in the case of ZnNTA, by ΔEint = 2.04 kcal/mol, Fig. 3. Furthermore, both types of dative bonds Zn–N and Zn–O are clearly mostly ionic (the dominance of the electrostatic terms ΔEelstat) and the charge delocalization covalent-type channel is significantly less important, Fig. 3 and Table 2. All these results together with the already identified non-covalent interactions apparently would suggest higher stability of ZnNTPA than ZnNTA.

Reprinted with permission from [50]. Copyright (2011) American Chemical Society
ETS energy decomposition results describing the interaction of “vertical” (right) and “horizontal” (left) water molecules with the rest of the complex in ZnNTPA and ZnNTA complexes (panel a). In addition, the leading deformation densities (Δρ1) together with the corresponding energies (ΔEorb[1]) are presented based on ETS-NOCV method (panel b).
However, the situation changes dramatically when considering an energy penalty/distortion ΔEdist which is required to change the optimal geometries of NTA/NTPA (ΔEdist-NTA/NTPA) and Zn-fragments (\( \Delta E_{{{\text{dist-Zn}}({\text{H}}_{2} {\text{O}})_{2} }}\)) to those adopted in the complexes, Table 2. It could be added, that, although significant geometry reorganization of NTPA versus NTA is quite expected (81.7 kcal/mol vs. 38.8 kcal/mol, respectively), please note, that the energy cost required to change the geometry of Zn-fragments is also significant [38.7 kcal/mol (ZnNTPA) vs. 23.4 kcal/mol (ZnNTA)], Table 2. They both contribute to the summarized distortion term ΔEdist = 120.4 kcal/mol for ZnNTPA and 62.2 kcal/mol for ZnNTA, Table 2. It leads to the overall bonding energy ΔEtotal in favor of ZnNTA; ΔEtotal = –670.6 kcal/mol for ZnNTA versus ΔEtotal = –622.7 kcal/mol for ZnNTPA (gas phase), Table 2. An inclusion of solvent effects alleviates the energy differences obtained in the gas phase, but still the overall bonding energy is more negative by ca. 4.1 kcal/mol in favor of ZnNTA versus ZnNTPA. These results point at crucial role of the ligand’s strain energy as well as stronger binding of the horizontal water molecules in explaining larger stability of ZnNTA versus ZnNTPA, despite the formation of non-covalent interactions (C–H•••O and C–H•••H–C) in the latter case. The latter weak interactions are clearly unable to overcome the large destabilization from the distortion term and the Pauli repulsion contribution which are discovered in ZnNTPA.
2,2′-bipyridyl ligand (abbreviated as BPy) is known to form a number of chelate complexes with transition metals—in order to do so, these ligands can adopt cis-configuration in which very close CH•••HC contacts (~2 Å) between 3,3′-hydrogen atoms are enforced [53,54,55]. The existence of purportedly repulsive CH•••HC contacts in BPy is often applied to rationalize trends in stability constants [55]. The forthcoming paragraphs provides in-depth analyses of weak non-covalent interactions and dative bonds in the complexes [Zn(BPy)(H2O)4]2+, [Zn(BPy)2(H2O)2]2+ and [Zn(BPy)3]2+ (for simplicity, the abbreviations are applied ZnL, ZnL2, and ZnL3) [51], Fig. 4 (top). For the first time, four totally different bonding descriptors will be applied—[1] the quantum theory of atoms in molecules (QTAIM) [2, 46], the interacting quantum atoms (IQA) energy decomposition scheme [3, 47], the non-covalent Interactions (NCI) [48] method, and [4] (ETS-NOCV) [49] energy decomposition scheme.

Reprinted with permission from [51]. Copyright (2014) American Chemical Society
(Top) Ball and stick representations of ZnL, ZnL2 and ZnL3 complexes. (Bottom) ZnL2 complex together with the fragmentation patterns applied in ETS-NOCV analyses: a (7)-pyr, b (2)-bpy, and c (2)-OH2 (the numbers denote a number of fragments).
It is determined, consistently through QTAIM, IQA, NCI , and ETS-NOCV, that the strength of dative bonds Zn–N systematically decreases from ZnL through ZnL2 to ZnL3 [51]. The same trend is valid for Zn–O connections [51]. The selected ETS-NOCV-based data in Table 3 clearly demonstrates a drop in Zn–N stabilization from ΔEorb = –26.00 kcal/mol (for ZnL), through ΔEorb = –22.46 kcal/mol (for ZnL2) up to ΔEorb = –18.70 kcal/mol (for ZnL3). Zn–O connections appeared to be weaker than Zn–N and similarly, their strength decrease from ΔEorb = –11.33 kcal/mol (for ZnL), to ΔEorb = –8.57 kcal/mol (for ZnL2), Table 3. The same trend is valid when the overall Zn–N and Zn–O interaction energies are considered, what nicely correlates with the computed elongation of these distances when going from ZnL (e.g. Zn–N: 2.107Å) to ZnL3 (Zn–N:2.218Å) [51].
Apparently, taking solely dative bonds into consideration would suggest the smallest stability constant for the most crowded ZnL3 as compared with ZnL—the exactly opposite relation is valid experimentally where the stability decreases in the order ZnL3 > ZnL2 > ZnL [55, 56]. The most important and striking findings, discovered consistently from ETS-NOCV, IQA, NCI , and QTAIM methods, are increasing a number of typical CH•••O, CH•••N, and unintuitive CH•••HC non-covalent interactions when going from ZnL to ZnL3 [51]. Namely, the example ETS-NOCV results in Table 4 demonstrate the charge delocalization channels corresponding to efficient stabilization in ZnL3 from CH•••HC contacts (CH8•••12HC, CH26•••30HC, CH44•••48HC), by ca. ΔEorb = –7.36 kcal/mol, Table 4. Similar interactions are found in ZnL and ZnL2, and they amount to ΔEorb = –5.52 kcal/mol, ΔEorb = –4.27 kcal/mol, respectively, Table 4. Similar trend in valid for intramolecular CH•••O interactions is shown in Table 4.
It is crucial to highlight that the real space-based IQA energy decomposition scheme consistently revealed exactly the same trends and also identified various non-covalent interactions including the stabilizing CH•••HC, Table 5. Furthermore, the strength of a single CH•••HC bond appeared to increase from ΔEint = –2.5 kcal/mol (ZnL) up to –2.88 kcal/mol for ZnL3, Table 5. The same trend, but with more efficient overall stabilization which ranges from –13 kcal/mol up to –16 kcal/mol depending on the system), is true for CH•••O and CH•••N, Table 5. It has been further determined that the quantum mechanical exchange-correlation contribution (XC) makes the overall CH•••HC interactions negative (stabilizing), Table 5. This component often correlates well with the orbital interaction term from the ETS-NOCV analysis [14]. The latter allowed to observe that formation of CH•••HC contacts leads to the outflow of electrons from the σ(C–H) bonds engaged in CH•••HC and the accumulation in the interatomic H•••H region. It has been further confirmed by the calculated NMR spin–spin [1] J(C–H) coupling constants which decrease from 177.06 Hz (ZnL) to 173.87 Hz (ZnL3) in support of an increase in the local CH•••HC stabilization from ZnL to ZnL3 found from QTAIM, IQA, and ETS-NOCV [51]. These results shed novel light on factors which might determine the relative stability of ZnLn complexes in the context of orthodox steric-based interpretation of CH•••HC contacts—namely, it is possible that an increase in a number of non-covalent interactions CH•••O, CH•••N, and CH•••HC (and their strength) together with the same trend in π-bonding and electrostatic contributions might overcompensate a decrease in the strength of Zn–N/Zn–O dative bonds resulting accordingly in an enhanced stability of ZnL3 with respect to both ZnL2 and ZnL [51].
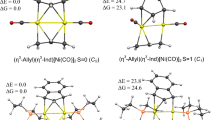
In the previous paragraphs, we have identified various intramolecular non-covalent interactions in the Zn(II)-based complexes. In the forthcoming sections, inter-molecular CH•••HC and other types of bonds will be discussed in the newly synthesized quasi-tetrahedral nickel complex abbreviated as cis-NiL2–hexane (L–thiourea -based ligand), Fig. 5 (top) [52]. It is formed by the reaction of N-thiophosphorylated thioureas containing iso-propyl (iPr) units with Ni(II) salt in basic condition crystallized from hexane solvent [52]. It has been also demonstrated that the crystals constituted from fully planar trans-NiL2 units (without hexane molecules) can be formed provided that other more polar solvents are applied during crystallization [52]. ETS-NOCV calculations allowed to determine the stabilizing character of CH•••HC formed between hexane and the iPr units of cis-NiL2—the overall CH•••HC interaction energies ΔEint appeared to vary between –2 kcal/mol and –10 kcal/mol (depending on XC functional containing the Grimme D3 correction) [52]. The main contributor (~73%) is the dispersion term followed by the similarly important (~13.5%) electrostatic and charge delocalization terms, Fig. 5 [52]. As far the latter contribution is concerned, the charge outflow from the σ(C–H) bonds engaged in CH•••HC is clearly visible (Fig. 5b) together with the accumulation in the interatomic H•••H region. It is necessary to point out that our conclusions herein on relative weighs of various contributions to inter-molecular CH•••HC are in accord with the recent topical literature findings [6, 7, 10, 25, 57,58,59,60,61,62,63,64]. For example, recent findings by the groups of Echeverría and Shaik [57, 59] allowed to highlight that, although London dispersion forces are crucial for CH•••HC interactions, other bonding components including charge delocalization term are also important.

The comparison between the dimeric model of [6]-graphanes (six CH2 units) bonded through CH•••HC with the larger one [65]-graphanes leads to the amplification of dispersion from ~5 kcal/mol up to ~90 kcal/mol, and at the same time, the two-way charge transfers σ(C–H) → σ*(C–H) cover ~15% of the overall stabilization which can reach the regime of typical covalently bonded species [59]. Echeverria has studied a series of homopolar dihydrogen interactions and also concluded on the importance of dispersion forces [24, 26, 27]. Furthermore, we have confirmed the stability of the Mcryst-Hex adduct bonded through CH•••HC by ab initio Born–Oppenheimer molecular dynamics simulations (DFT/BLYP-D3/TZP)—hexane was found to dynamically glide in the proximity of iPr ligands, and no spontaneous drifting apart was observed [52]. Furthermore, the calculations have shown that the sterically crowded cis-isomer exhibiting quasi-tetrahedral geometry is more stable than the corresponding planar trans-conformation at both energy and free energy levels, Fig. 6a. It is very striking result in the light of intuitively expectable steric repulsion between closely located iPr units in the case of cis-NiL2, Fig. 6a. Substitution of bulky iPr units by smaller Me groups leads not only to the planar conformation of cis-NiL2, but also the trans-isomer is now thermodynamically preferred [52], Fig. 6c. Therefore, the tetrahedral geometry of cis-NiL2 originates from the existence of numerous cooperative intramolecular non-covalent interactions: CH•••HC, CH•••S CH•••Ni, Fig. 6b. In turn, it is related to the bulky iPr units that are close to each other in the cis-NiL2. It is very beautiful example showing how two quite bulky groups being close to each other, classically considered as the source of steric repulsion, lead not only to overall stabilization, but also to the determination of the complex geometry. These data are perfectly in accord with recent topical findings on the importance of CH•••HC interactions and London dispersion forces in various branches of chemistry [6, 7, 10, 25, 52, 57,58,59,60,61,62,63,64]. It must be emphasized that dispersion contribution has been also recently recognized as a crucial factor (in addition to well-established charge transfer term) for agostic interactions (C–H•••metal) due to elegant and accurate energy decomposition DLPNO-CCSD(T) implemented in the Orca program [64].

Relative energies (and free energies) of cis-NiL2 versus trans-NiL2 together with dihedral angles which measure degree of planarity (part a). In part b, the QTAIM molecular graphs are depicted demonstrating formation of various non-covalent interactions. Part c depicts the relative energies (and free energies) of the models where the bulky OiPr units are replaced by OMe
Ammonia borane, named also as borazane, is considered nowadays as one of the most promising hydrogen storage materials predominantly due to significant hydrogen content (19.6%) as well as high melting point (104 °C). The latter property is attributed in the literature to the existence of polar (proton–hydride) dihydrogen bonds N–Hδ+•••−δH–B between AB monomers. These types of interactions are crucial for hydrogen storage materials [28, 29, 31]. Very recently, McGrady and coworkers have published a series of high-quality papers which demonstrate thepreparation of various hydrogen storage materials including LiN(CH3)2BH3 and KN(CH3)2BH3 in which untypical hydride–hydride interactions B–Hδ−•••−δH–B are observed from the QTAIM results [22, 66, 67]. These are very interesting suggestions since hydrogen atoms involved in such homopolar contacts B–Hδ−•••−δH–B carry negative partial charges, what intuitively shall lead to overall repulsion due to destabilizing electrostatic contribution. In order to shed some light on the role of B–Hδ−•••−δH–B and other types of chemical bonds in LiN(CH3)2BH3 and KN(CH3)2BH3, we have performed a comprehensive in-depth study of bonding situation based on ETS-NOCV, IQA, NCI methods, and molecular electrostatic potentials [23].
ETS-NOCV method allowed to determine that the major inter-molecular bonding in LiN(CH3)2BH3 stems from B–H•••Li contacts—it is dominated by the electrostatics which covers 55% (ΔEelstat = –35.56 kcal/mol) of the total stabilization, followed by the orbital interaction (ΔEorb = –18.17 kcal/mol) and dispersion (17%, ΔEdisp = –11.38 kcal/mol) terms, Fig. 7. Electrostatics-based bonding is in line with the molecular electrostatic potential of the monomer—the borane moiety is negatively charged, while lithium cation is electrophilic, Fig. 8a. The bottom of Fig. 8 clearly identifies the ancillary intra-molecular B–H•••Li charge transfer in the monomers constituting the crystal.

Adopted from [23]
Optimized tetrameric cluster model of LiN(CH3)2BH3 with fragmentation pattern and ETS energy decomposition results (part a) along with the two most important deformation density contributions describing B–H•••Li interactions.

Adopted from [23]
Monomer of LiN(CH3)2BH3 with its Molecular Electrostatic Potential in a.u. units (part a) and two most important deformation density channels depicting B–N bond and BH3•••Li interacion (part b).
The two NOCV channels which mostly (~85%) contribute to ΔEorb describe the outflow of electron density from σ(B–H) orbitals and inflow into the proximity of Li+ ions, what leads additionally to the charge accumulation into the BH•••HB bay region, Δρ2, Fig. 7. It is fully consistent with the QTAIM result of McGrady et al. [66] where bond critical points corresponding to BH•••HB are discovered. However, we have determined that removing the two monomers not engaged in B–H•••H–B interactions results in the formation of a system with the positive overall interaction energy ΔEtotal = +4.9 kcal/mol, Fig. 9. Although some stabilization from the orbital overlapping exists (ΔEorb = –1.86 kcal/mol), as well as from dispersion effects (ΔEdisp = –2.30 kcal/mol), in line with the QTAIM data [66], significant electrostatic and Pauli repulsion (ΔEelstat = +4.90 kcal/mol, ΔEPauli = +4.16 kcal/mol), overcompensate the stabilizing effect leading to positive (destabilizing) ΔEtotal = +4.9 kcal/mol, Fig. 9. The same conclusion is reached by us when considering the point charges (which mimic the Li ions) [23]. Moreover, no stabile minimum featuring solely B–H•••H–B interactions have been found upon geometry optimization. To this end, these ETS-NOCV-based data points at rather destabilizing nature of B–H•••H–B interactions in this system, contrary to the analogous C–H•••H–C contacts which are found to be significantly stabilizing in LiN(CH3)2BH3 and KN(CH3)2BH3, Figs. 10, 11 [23]. It is determined herein that the overall C–H•••H–C interaction energy in LiN(CH3)2BH3 is ΔEint = –4.34 kcal/mol and ΔEint = –17.45 kcal/mol for KN(CH3)2BH3 (Figs. 10, 11), which is quite comparable to typical hydrogen bonds [e.g., ΔEint for water dimer is ~ –5 kcal/mol, for adenine–thymine base pair is ~ –12 kcal/mol, and for FH•••HLi is ~ –13 kcal/mol] [44].

Adopted from [23]
Dimer of LiN(CH3)2BH3 (part b) from the optimized tetramer as indicated in part a. ETS energy decomposition is presented in part b.

Adopted from [23]. The contour value is 0.001a.u
Cluster model containing eight monomers of LiN(CH3)2BH3 along with energy decomposition results describing CH•••HC interactions between the two selected fragments (marked by black line), part a. In part b, the overall deformation density Δorb is depicted together with the corresponding stabilization ΔEorb.

Adopted from [23]. The contour value is 0.001a.u
Cluster model containing eight monomers of KN(CH3)2BH3 along with energy decomposition results describing CH•••HC interactions between the two selected fragments (marked by black line), part a. In part b, the overall deformation density Δorb is depicted together with the corresponding stabilization ΔEorb.
Further studies of the tetrameric model by means of IQA method were carried out in order to further characterize Li•••HB CH•••HC and BH•••HB interactions, Table 6. Expectedly, the strongest bond appeared to be Li•••HB with Eint(Li•••H–B) = –98.8 kcal/mol. In line with the ETS-NOCV results, homopolar CH•••HC is weakly stabilizing, whereas the similar B–H•••H–B interactions are indeed destabilizing as indicated by the large positive value of Eint(BH•••HB) =+49.4 kcal/mol, Table 6. It is due to electrostatic electron–electron repulsion term, Table 6, which is in line with the ETS-NOCV-based results (Fig. 9). These outcomes [23] pointing at destabilizing nature of B–H•••H–B in LiNMe2BH3 are in accord with the numerous experimental papers [20, 21, 68, 69, 65]. The excellent review by McGrady et al. [22] on possible role of stabilizing hydride–hydride interactions in hydrogen storage materials has been recently published. Therefore, definitely more works (from both theoretical and experimental laboratories) are needed to identify and fully unveil the nature of different types of X–H•••H–X contacts in various systems.
4 Conclusions
It has been demonstrated in this chapter that non-covalent interactions including untypical homopolar C–H•••H–C, despite being relatively weak (as compared to typical dative or covalent bonds), might play very important role in transition metal systems. Zn(II) complexes with various ligands NTA, NTPA, and BPy have been stabilized not only by typical electrostatically dominated dative–covalent bonds (e.g. Zn–N, Zn–O), but additionally through a number of typical hydrogen bonds CH•••O, CH•••N and predominantly unintuitive CH•••HC interactions—the latter have been shown to be stabilizing as opposed to traditional steric repulsion-based interpretation [23, 50, 51, 52]. We have determined that dispersion dominated CH•••HC can be as strong as typical hydrogen bonds [23, 50, 51, 52]. Although London dispersion forces are the prevailing factor, the charge delocalization contribution (outflow of electrons from the σ(C–H) bonds engaged in CH•••HC and the accumulation in the interatomic H•••H region) and electrostatic term are also non-negligible [23, 50, 51, 52]. Interestingly, similar to CH•••HC, hydride–hydride interactions BH•••HB in LiNMe2BH3 are found to be repulsive [23]. We have further proven that the two bulky alkyl groups being close to each other in Ni(II) complex, classically considered as the source of steric repulsion, lead not only to overall stabilization (due to the formation of multitude non-covalent interactions including CH•••HC), but also to determination of the complex geometry [52]. These results perfectly fit very recent topical findings which highlight the crucial role of non-covalent interactions including London dispersion forces in various branches of chemistry including transition metal complexes [6, 7]. Although significant progress has been made recently in terms of identification of non-covalent interactions in real materials [6, 7], there are still many known systems where the importance of London dispersion forces has not been yet recognized as nicely emphasized by Liptrot and Power [7].
References
(a) Dewar, MJS (1951) Bull Soc Chim 18:C71–C79 (b) Chatt J, Duncanson JA (1953) J Chem Soc 3:2939–2943 (c) Frenking G, Shaik S (eds) (2014) The chemical bond. Fundamental aspects of chemical bonding (part 1), chemical bonding across the periodic table (part 2), Wiley-VCH, Weinheim
Zhao L, von Hopffgarten M, Andrada DM, Frenking G (2018) WIREs Comput Mol Sci 8:e1345. https://doi.org/10.1002/wcms.1345
Brocławik E, Załucka J, Kozyra P, Mitoraj MP, Datka J (2011) Catal Today 169(1):45–51
Broclawik E, Załucka J, Kozyra P, Mitoraj MP, Datka J (2010) J Phys Chem C 114(21):9808–9816
Rejmak P, Mitoraj MP, Broclawik E (2010) Phys Chem Chem Phys 12(10):2321–2330
Wagner JP, Schreiner PR (2015) Angew Chem Int Ed 54:14
Liptrot DJ, Power PP (2017) Nat Rev Chem 1:0004
Grimme S, Antony J, Ehrlich S, Krieg H (2010) J Chem Phys 132:154104
Grimme S, Ehrlich S, Goerigk L (2011) J Comput Chem 32:1456
Wolters LP, Koekkoek R, Bickelhaupt FM (2015) ACS Catal 5:5766
Matta CF, Hernández-Trujillo J, Tang TH, Bader RFW (2003) Chem Eur J 9:1940
Poater J, Solà M, Bickelhaupt FM (2006) Chem Eur J 12:2889
Bader RFW (2006) Chem Eur J 12:2896
Pendás AM, Francisco E, Blanco MA, Gatti C (2007) Chem Eur J 13:9362
Eskandari K, Alsenoy CV (2014) J Comput Chem 35:1883
Cukrowski I (2015) Comput Theor Chem 1066:62
Weinhold F, Schleyer PR, McKee WC (2014) J Comput Chem 35:1499
Matta CF, Sadjadi SA, Braden DA, Frenking G (2016) J Comput Chem 37:143
Cukrowski I, Sagan F, Mitoraj MP (2016) J Comput Chem 37:2783
Ravindran P, Vajeeston P, Vidya R, Kjekshus A, Fjellvåg H (2002) Phys Rev Lett 89:106403
Schouwink P, Hagemann H, Embs JP, Anna VD, Černy R (2015) J Phys: Condens Matter 27:265403
Wolstenholme DJ, Dobson JL, McGrady GS (2015) Dalton Trans 44:9718 and references therein
Sagan F, Filas R, Mitoraj MP (2016) Crystals 6:28
Echeverría J, Aullón G, Alvarez S (2017) Dalton Trans 46:2844
Yourdkhani S, Jabłoński M, Echeverría J (2017) Phys Chem Chem Phys 19:28044
Echeverría J (2017) Cryst Growth Des 17:2097
Echeverría J, Aullón G, Alvarez S (2017) Int J Quantum Chem 117:e25432
Custelcean R, Jackson JE (2001) Chem Rev 101:1963
Mitoraj MP (2011) J Phys Chem A 115:14708
Grabowski SJ, Ruipérez F (2016) Phys Chem Chem Phys 18:12810
Sagan F, Piękoś Ł, Andrzejak M, Mitoraj MP (2015) Chem Eur J 21:15299
Belkova NV, Epstein LM, Filippov OA, Shubina ES (2016) Chem Rev 116:8545
Gamez P, Mooibroek TJ, Teat SJ, Reedijk J (2007) Acc Chem Res 40:435
Frontera A, Gamez P, Mascal M, Mooibroek TJ, Reedijk J (2011) Angew Chem Int Ed. 50:9564
Safin DA, Pialat A, Leitch AA, Tumanov NA, Korobkov I, Filinchuk Y, Brusso JL, Murugesu M (2015) Chem Commun 51:9547
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748
Scheiner S (2013) Acc Chem Res 46:280
Bauzá A, Mooibroek T, Frontera A (2016) Chem Rec 16:473
Grabowski SJ, Sokalski WA (2017) Phys Chem Chem Phys 18:1569
Gleiter R, Haberhauer G, Werz DB, Rominger F, Bleiholder C (2018) Chem Rev 118:2010
Grabowski SJ (2006) Hydrogen bonding—new insights. Springer, Dordrecht
Scheiner S (1997) Hydrogen bonding a theoretical perspective. Oxford University Press Inc., New York
Grabowski SJ (2016) Crystals 6:59
Grabowski SJ (2011) Chem Rev 111:2597 and references therein
Petrović P, Djukic JP, Hansen A, Bannwarth C, Grimme S (2016) Non‐covalent stabilization in transition metal coordination and organometallic complexes, Editors(s): Abel M. MaharramovKamran T. MahmudovMaximilian N. KopylovichArmando J. L. Pombeiro, Wiley, Print ISBN:9781119109891
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, Oxford
Blanco MA, Pendás AM, Francisco E (2005) J Chem Theory Comput 1:1096
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W (2010) J Am Chem Soc 132:6498
Mitoraj M, Michalak A, Ziegler T (2009) J Chem Theory Comput 5:962
Cukrowski I, Govender KK, Mitoraj MP, Srebro M (2011) J Phys Chem A 115:12746
Cukrowski I, de Lange JH, Mitoraj MP (2014) J Phys Chem A 118:623
Safin DA, Babashkina MG, Robeyns K, Mitoraj MP, Kubisiak P, Garcia Y (2015) Chem Eur J 21:16679
Hancock RD, de Sousa AS, Walton GB, Reibenspies JH (2007) Inorg Chem 46:4749
Hambley TW (1986) J Chem Soc Dalton Trans 565
Hancock RD, Martell AE (1989) Chem Rev 89:1875
Smith RM, Martell AE (eds) (2004) NIST standard reference database 46. NIST critically selected stability constants of metal complexes database; Version 8.0; US Department of Commerce, National Institute of Standards and Technology: Gaithersburg, MD
Echeverría J, Aullón G, Danovich D, Shaik S, Alvarez S (2011) Nat Chem 3:323
Krapp A, Frenking G, Uggerud E (2008) Chem Eur J 14:4028
Wang C, Mo Y, Wagner JP, Schreiner PR, Jemmis ED, Danovich D, Shaik S (2015) J Chem Theory Comput 11:1621
Jahiruddin S, Mandal N, Datta A (2018) Chem Phys Chem 19:67
Dutta B, Pratik SM, Jana S, Sinha C, Datta A, Hedayetullah Mir M (2018) ChemistrySelect 3:4289
Mandal N, Pratik SM, Datta A (2017) J Phys Chem B 121:825
de Almeida LR, Carvalho Jr PS, Napolitano HB, Oliveira SS, Camargo AJ, Figueredo AS, de Aquino GLB, Carvalho-Silva VH (2017) Cryst Growth Des 17(10):5145
(a) Rösel S, Quanz H, Logemann C, Becker J, Mossou E, Canadillas-Delgado L, Caldeweyher E, Grimme S, Schreiner PR (2017) J Am Chem Soc 139(22):7428 (b) Schneider WB, Bistoni G, Sparta M, Saitow M, Riplinger C, Auer AA, Neese FJ (2016) Chem Theory Comput 12:4778–4792 (c) Lu Q, Neese F, Bistoni G (2018) Angew Chem Int Ed 57:4760–4764
Černý R, Ravnsbæk DB, Schouwink P, Filinchuk Y, Penin N, Teyssier J, Smrčok L, Jensen TR (2012) J Phys Chem C 116:1563
Wolstenholme DJ, Flogeras J, Che FN, Decken A, McGrady GS (2013) J Am Chem Soc 135:2439
Wolstenholme DJ, Traboulsee KT, Hua Y, Calhoun LA, McGrady GS (2012) Chem Commun 48:2597
Černý R, Kim KC, Penin N, D’Anna V, Hagemann H, Sholl DS (2010) J Phys Chem C 114:19127
Ravnsbæk D, Filinchuk Y, Cerenius Y, Jakobsen HJ, Besenbacher F, Skibsted J, Jensen TR (2009) Angew Chem Int Ed 48:6659
Acknowledgements
DFT calculations were partially performed using the PL-Grid Infrastructure and resources provided by the ACC Cyfronet AGH (Cracow, Poland). M. P. M. acknowledges the financial support of the Polish National Science Center within the Sonata Bis Project 2017/26/E/ST4/00104.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sagan, F., Mitoraj, M.P. (2019). Non-covalent Interactions in Selected Transition Metal Complexes. In: Broclawik, E., Borowski, T., Radoń, M. (eds) Transition Metals in Coordination Environments. Challenges and Advances in Computational Chemistry and Physics, vol 29. Springer, Cham. https://doi.org/10.1007/978-3-030-11714-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-11714-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11713-9
Online ISBN: 978-3-030-11714-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)