
Abstract
Estrogens play an important role not only in the reproductive system but in the central nervous system as well. Major events of ontogenesis that occur earlier in pregnancy are connected to the formation of estrogen receptors and expression of estrogens leading to the normal physiological development of the central nervous system, though development of the brain by itself is a complex process and lasts during the whole pregnancy. Estetrol (E4) is a recently described natural estrogen with four hydroxyl groups that is synthesized exclusively during pregnancy by the human fetal liver. Its role in the central nervous system is not fully understood. Our studies showed for the first time and proved impressive antioxidative effects of E4 in vitro and proved its tremendous neuroprotective, promyelinating, neurogenic, and cerebro-angiogenic properties in vivo. E4 decreases brain damage markers (S100B and GFAP) in blood assuming that E4 attenuates neonatal hypoxic-ischemic encephalopathy in vivo. We have also shown that the combined use of E4 with other steroids does not have any priority over the single use of E4. E4’s antioxidative actions mostly depend on ERα and ERβ, whereas neurogenesis and possibly promyelinating activities might be realized through ERβ. Taken together our studies suggest importance of E4 treatment possibly not only in neonates but in adults with different neurological diseases like that opening new directions for the use of E4 in clinical practice in neurological diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Estetrol
- Estrogen receptors
- Neonatal hypoxic-ischemic encephalopathy
- Neurogenesis
- Cerebro-angiogenesis
- Myelination
- Hippocampus
- Cortex
- S100B
- GFAP
4.1 Development of the Nervous System
Development of the nervous system in humans during pregnancy is passing through critical and complex periods of morphological and functional differentiation. During ontogenesis newly developed structures of the nervous system, which differ by function and localization, are unified in one complete functional system.
Main steps in human organogenesis are taking place before the eighth week of fertilization. Development of the brain by itself includes several major stages and lasts during the whole pregnancy. Major events in human brain development include primary neurulation, prosencephalic development, neuronal proliferation and migration, organization, and myelination [1]. The neuronal tube formation is already finished by the 20th day of fertilization [2], and the formation of the cortical plate takes place between 7 and 16 weeks of gestation though the cortex increases in thickness until neurogenesis is completed after midgestation. The total number of neurons in the central nervous system (CNS) reaches a maximum in the first 20–24 weeks of the antenatal period and remains relatively constant up to adulthood, only slightly decreasing in early postnatal period.
By the 9th week of gestation, ERα is detected in the proliferating zones and the cortical plate [2, 3]. In contrast to the expression patterns of ERα, ERβ is detected at 15 weeks of gestation in proliferating zones and at 16–17 weeks of gestation in the cortical plate. From the same period of time, both receptors are expressed in different subregions of the hippocampus [3]. In the rat pups, ERs are present in the developing as well as adult hippocampus [4, 5] with a peak in binding at postnatal day 4 declining to adult levels already by postnatal day 15 [6, 7]. Thus, ERα plays a role in early developmental processes, whereas ERβ might be more important for later events of corticogenesis [8]. Two pairs of internal carotid and vertebral arteries, connected by the circle of Willis, supply the brain with blood [2]. The internal carotid arteries develop quite early, by the 4th week of gestation, whereas by the 5th week of gestation, most of arteries are developed by forming a specific pattern [2]. At 16 weeks of gestation, the anterior, middle, and posterior cerebral arteries are already well established. In premature newborns between 22 and 30 weeks of gestation, the blood vessels of the germinal and periventricular zone and the perforating ventriculopetal vessels are particularly vulnerable to perinatal asphyxia [2], whereas between 30 and 34 weeks of gestation, the fetal white matter is vulnerable to hypoxic ischemic injury, and the injury leads to the formation of focal hemorrhagic lesions and periventricular leukomalacia (PVL) [often resulting in infarction (necrosis) and cavitation], respectively [2].
Myelination in the CNS is performed by oligodendrocytes and is a slow process which is a significative mark of the maturity of the CNS. Notably, in the brain stem, myelination starts at the 8th week of gestation though not completed until after birth, and the rate of myelin deposition is greatest during the first 2 postnatal years [9].
4.2 Estrogens, Estrogen Receptors (ERs), and the Brain
4.2.1 Estrogens
Successful maintenance of pregnancy requires the coordinated secretion of hormones. Indeed, placenta, feto-placental unit, and fetus become the main sources of estrogens: estrone (E1), estradiol (E2), estriol (E3), and estetrol (E4). Appearance of each estrogen in maternal plasma after 9 weeks of gestation coincides with main events of the brain development, pointing out the importance of estrogens in formation of brain morphology and its functionality. For example, we can easily follow the manifestation of unconjugated estradiol (E2) in maternal plasma by the 9th week of gestation and the detection of ERα in the proliferating zones and the cortical plate [2, 3], like that showing the importance of E2 in early corticogenesis. Plasma concentrations of E1, E2, E3, and E4 increase as human pregnancy progresses [10, 11] implicating importance of estrogens in fetal development during pregnancy in general as well as in parturition.
Some recent studies already have shown role of estrogens in (1) fetal neurogenesis, (2) prevention of neuronal cell death, (3) axonal sprouting, and (4) synaptic transmission [12, 13]. Others implicated estrogens as major players for fetal cerebral angiogenesis and cerebral blood flow maintenance due to (1) neurovascular sharing of major signaling pathways and the development of blood-brain barrier, (2) interaction between neurons and vessels mediated by Ca2+ ions released from astrocytes, (3) direct effects of estrogens on cerebral vessels [14,15,16,17,18], (4) decrease in water permeability of the blood-brain barrier (BBB) [19], and (5) upregulation of vascular endothelial growth factor (VEGF) expression from neuronal cells [20].
VEGF is an angiogenic protein with neurotrophic and neuroprotective effects which stimulate neurogenesis in vitro and in vivo in the subventricular zone (SVZ) and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) [21] and promotes proliferation of cortical neuron precursors by regulating E2F expression (the family of transcription factors, a key regulator of the cell cycle machinery) [22]. Usually neuronal VEGF expression correlates with angiogenesis in postnatal developing rat brain and might be upregulated by hypoxia [23], but it has importance in the developing brain as well. As it was already evidenced, loss of VEGF expression by CNS neurons impairs vascularization, curbs neuronal expansion, and results in neuronal apoptosis in the developing brain, pointing out that VEGF-induced blood vessel growth is essential for nervous tissue growth during embryonic development [24, 25].
According to different studies, general impact of estrogens on the CNS includes (1) neuromodulatory effect by affecting neuron excitability, synaptic plasticity, and neurotransmitter system; (2) neurotrophic effect by influencing glial morphology and functions, neurite outgrowth and sprouting, and cell viability; and (3) neuroprotective effect by exerting proneurogenic, antiexcitatory, antioxidative, antiapoptotic, and anti-inflammatory profile [26, 27].
Estrogen administration in animal models and clinical studies of Parkinson’s and Alzheimer’s diseases, ischemic stroke, spinal cord injury, and multiple sclerosis has already demonstrated neuroprotective effect [12, 28,29,30,31,32], suggesting the early protective effect of estrogens administered particularly in younger patients (50–62 years) [33] or in women soon after menopause [34]. Even short-term estrogen treatment increases dopamine transporters in the caudate putamen [35] and amyloid beta-protein (Aβ) uptake by microglia. It also prevents Aβ peptide formation by neurons [36, 37] and protects against loss of DA neurons [38, 39]. According to the “healthy cell bias of estrogen action hypothesis,” if estrogens are administered too late in the disease, they are not protective [40, 41].
At cellular and molecular levels, estrogens might have different important actions: (1) increase of astrocyte ability to uptake glutamate and like that preventing neuronal loss due to glutamate toxicity [42, 43], (2) direct neuroprotective effect on mitochondria [44,45,46,47,48], (3) induction of expression of genes regulating cytoskeleton of neuron cells (e.g., neurofilaments, microtubule-associated proteins), and (4) increase of aerobic glycolysis, respiratory efficiency, ATP generation, and Ca2+ load tolerance leading to antioxidant defense [49].
4.2.2 Estrogen Receptors (ERs)
Estrogen actions are realized through specific estrogen receptors (ERs).
GPER is a membrane-bound receptor, though some research groups found it in the Golgi complex [50] or at the endoplasmic reticulum [51] or even intracellularly in some transfection experiments [52]. Estrogens may affect serotonin signaling in the hypothalamus [53] and dopamine efflux in PC-12 cells [54] or mechanical hyperalgesia in nociceptive neurons of rat dorsal root ganglia [55] and control the energy homeostasis in the hypothalamus [56] by employing GPER.
ERs have specific regions, activation function 1 and 2 (AF1 and AF2), which are responsible for formation of initial transcriptional complexes. AF1 and AF2 are situated in the amino-terminal and carboxyl-terminal domains of the receptors, respectively [57]. These functions characterize most of nuclear receptors and correspond to two active domains which are responsible for recruitment of specific co-regulator proteins, and these proteins in turn might modulate transcriptional activity of ERα. The level of AF1 activity does not depend on the presence of ligand, whereas the activity of AF2 is ligand-dependent [57]. Inactive receptors are linked to the heat shock proteins, and binding of ligand to the ERs leads to dissociation of heat shock proteins from ERs followed by dimerization of ERs. ERs are forming homo- and heterodimers [58]. Zinc (Zn2+) fingers of ligand-ERs complex bind to DNA at specific ERs elements (EREs) which are located in the regulatory regions of target genes and where they act as a hub for a large transcriptional complex including coactivators and corepressors resulting in gene transcription. EREs can alter transcription indirectly by interacting with other transcription factors (AP1, C/EBPβ, and SP1) [57, 58].
According to different studies, estrogen receptors (ERs ), ERα, ERβ, and G protein-coupled estrogen receptor 1 (GPER , also known as G protein-coupled receptor 30 (GPR30)), may coexist in many brain areas, although their expression levels and distribution patterns are different and sometimes gender specific [59,60,61,62,63]. Like that in adult human brain, ERα, ERβ, and GPER coexist in the basal forebrain, hypothalamus, and hippocampus; ERα and ERβ are expressed in the prefrontal cortex, amygdala, locus coeruleus, and raphe nucleus; ERβ and GPER coexist in the thalamus, whereas only ERβ is expressed in the posterior cingulate [63]. During development ERα and ERβ display distinct chronology and distribution patterns that undergo dynamic changes in the course of corticogenesis as already discussed above.
In terms of subcellular localization, there is a difference between ER localization during development and in adults. During development ERα and ERβ are located in the cell nuclei, whereas in the adult human brain, the ERα staining is localized in both cell compartments (cytoplasm and nucleus), and ERβ has exclusively cytoplasmic localization [64, 65]. The ERα staining is clearly cytoplasmic in the pyramidal cells of Ammon’s horn (CA) and in layer II of the entorhinal cortex, whereas it is more nuclear in the dentate gyrus (DG) and in layer V of the entorhinal cortex and temporal cortex [59, 64, 65]. Some recent investigations already have shown localization patterns of ERα, ERβ, and GPER in different neuronal cells pointing out involvement of these receptors in neurogenesis and myelination. In astrocytes and microglia, expression of ERα and ERβ is observed in the nuclei, whereas ERβ is expressed in the cytoplasm; nuclei of the axonal bodies express ERα and ERβ, whereas ERα, ERβ, and GPER are manifested in the cytoplasm; basal dendrites, axonal (initial) segments, and myelin sheaths are rich with ERβ [63]. Another question that became the main direction of research is subcellular localization of ERs in the dentate gyrus (DG) region of the hippocampus which is important for neurogenesis and synaptic remodeling as well as neuroprotection and realization of cognitive function. In DG, a subset of GABAergic interneurons contains nuclear ERα, whereas granule cells, newly born cells, and some GABAergic interneurons contain cytosolic and plasma membrane-associated ERβ [64]. Dendritic spines, mostly originating from granule cells, contain ERα and ERβ. A few dendritic spines in the hilus of DG, originating from mossy cells, contain ERα and ERβ. Interestingly, some ERα-containing axon terminals are cholinergic, whereas some ERβ-containing terminals are monoaminergic. Astrocytes, mostly in the molecular layer, also contain ERα and ERβ [64].
In general, expression of ERα mRNA in the neonatal cortex, olfactory bulb and cerebellum suggests its role in the regulation of early postnatal differentiation and development of these brain areas by estrogens, since ERα is supposed closely related to cellular differentiation and sexual differentiation of developing brain [6].
Different studies already have demonstrated the presence of ERs in rat pial arteries and intracerebral blood vessels [65] and also proved expression of ERα in nuclei, membranes, and mitochondria of endothelial and vascular smooth muscles of cerebral arteries [19, 65]. Although ERβ was detected in immunoblots of cerebral artery lysates, the definitive role of ERβ is not fully understood [65], though ERα and ERβ may modulate each other’s activity [66].
According to some studies, ER activation might offer neuroprotection, in part, through transcriptional mechanisms affecting the apoptotic cascade including BCl2, caspases, and Apaf-1 like that limiting cell death [67,68,69,70]. ERs can also directly activate signal transduction pathways involving MAP kinase resulting in neuroprotection that is receptor-mediated [71]. As it was shown in Parkinson’s disease animal model of 6-hydroxydopamine (6-OHDA), estradiol can also act indirectly by activating the insulin-like growth factor-1 (IGF-1) receptor to protect against 6-OHDA-induced neuronal loss [72].
If the abovementioned effects of estrogens and ERs in the CNS were prominent mainly for E2, information on how E4 affects the CNS became available only few years ago based on studies performed by our research group.
4.2.3 Estetrol
Estetrol (E4) is a steroid hormone, discovered in 1965 by Egon Diczfalusy and co-workers [73]. Structurally, estetrol is an estrogenic steroid with four hydroxyl groups, explaining the acronym E4. Estetrol is produced in nature by the human fetal liver, since its synthesis requires two hydroxylases (15α- and 16α-hydroxylase) only expressed by the fetal liver during pregnancy. Substrates for E4 are estradiol (E2), requiring both 15- and 16-hydroxylation, and estriol (E3), requiring 15-hydroxylation only. Estetrol is an end product of steroid metabolism . There is no metabolism backward to E3, E2, or E1, and there are no active metabolites [74]. The chemical name of estetrol is estra-1,3,5(10)-trien-3,15a,16a,17b-tetrol, and it is known under CAS No. 15183-37-6. The molecular formula of E4 is C18H24O4, and it has a molecular weight of 304.38. Its physical appearance is that of a white to off-white solid. Estetrol has a melting point in the range of 240–245 °C [73]. Experience so far indicates that E4 is very stable, even under nonoptimal storage conditions. E4 might be slightly hygroscopic. Storage conditions of E4 should therefore be optimized to prevent moisture and water uptake. Both in pure water and in phosphate buffers, E4 is highly soluble. In water, the solubility amounted to 1.0 mg/ml. The octanol-water partition coefficient (Pow) is a measure of the lipophilic or hydrophilic properties of a compound and is expressed as the logarithm of Pow. The lipophilic and hydrophilic properties largely determine the passive gastrointestinal absorption, the distribution through the body, and the passive passage of the blood-brain barrier. In two sets of experiments, using different methods to determine the partition coefficient of E4, the observed log Pow values were 1.470 and 1.695 [73, 74]. This means that concentrations in the octanol phase were about 30–50 times higher compared to those in the water phase. A log Pow of about 2.0 is considered optimal to allow passage through the blood-brain barrier [74].
E4 is found in maternal urine as early as 9 weeks of gestation, increasing substantially as pregnancy progresses [74, 75]. Estetrol produces a number of biological changes in the rodent uterus, such as weight increase, progesterone receptor stimulation, enzyme induction, and histological and ultrastructural changes. From a teleological viewpoint, it seems likely that an estrogenic steroid produced in such significant quantities by the male and female human fetal liver during pregnancy is safe and has physiological significance. As it was concluded, genomic clinical effects of E4 will most likely occur through the estrogen receptors. E4 has a moderate affinity for human ERα and ERβ, with Ki values of 4.9 + 0.567 nmol/l and 19 + 1 nmol/l, respectively, demonstrating a four- to fivefold preference for the ERα (lower Ki value) [76]. Estetrol has high selectivity for the estrogen receptors. Binding to the glucocorticoid, progesterone, and testosterone receptors was only 11–15% at a concentration of 10 mmol/l, and further profiling of E4 in a set of 124 receptors and enzymes demonstrated inactivity toward 123 molecular targets. The single target showing interaction with E4 was the adrenergic α1β receptor (weak binding) [76]. It is concluded that genomic clinical effects of E4 will most likely occur through the estrogen receptors. The high selectivity of E4 suggests a low risk of unexpected side effects [77, 78]. E4 could be a safe and efficacious candidate for the treatment of early brain damage in newborn. The use of E2 unlike the use of E4 might have diverse effects on inflammation and immune responses [79], cardiovascular complications, venous thrombosis, and stroke [80,81,82], even the initiation/progression of several endocrine-related cancers (e.g., breast, prostate, ovarian, and endometrial cancer) [83].
Main properties of E4 are as follows: (1) slow metabolism and the long half-life; (2) strong antioxidant properties; (3) no binding to sex hormone-binding globulins (SHBG), suggesting that E4 may not influence the plasma levels of SHBG; and (4) log Pow index about 1.470–1.695 which is enough for any compound to pass the blood-brain barrier [75,76,77]. These properties of E4 might be important to assume the possible neuroprotective actions of E4.
4.3 Neonatal Hypoxic-Ischemic Encephalopathy
Neonatal encephalopathy (NE) is a clinically defined syndrome of disturbed neurologic function in the earliest days of life in an infant born at or beyond 35 weeks of gestation, manifested by a subnormal level of consciousness or seizures and often accompanied by difficulty with initiating and maintaining respiration and depression of tone and reflexes [84]. Neonatal hypoxic-ischemic encephalopathy is associated with increased lethality and long-term morbidity. Mortality and the neurodevelopmental outcomes in infants with moderate and severe HIE are as follows: 23–27% of infants die prior to discharge from the neonatal IC unit (NICU), whereas 37–38% die at follow-up 18–22 months later. The neurodevelopmental outcome at 18 months includes mental and psychomotor development retardation, cerebral palsy, epilepsy, blindness, and hearing impairment [85, 86]. Usually neonates with HIE have different complications manifested with the different degree of severity as follows: mental development index (MDI) <70 (39%), psychomotor development index (PDI) <70 (35–41%), disabling cerebral palsy (30%), epilepsy (16%), blindness (14–17%), and severe hearing impairment (6%) [85, 86], and HIE is the fifth largest cause of death of children before age 5 [87,88,89].
Perinatal asphyxia or birth asphyxia , more appropriately known as neonatal hypoxic-ischemic encephalopathy (HIE) or hypoxic and ischemic brain injury in the newborn, is characterized by clinical and laboratory evidence of acute or subacute brain injury (encephalopathy) due to intrapartum or late antepartum brain hypoxia and ischemia [90, 91]. A common but crucial problem is the inability to time the onset, duration, magnitude, and single or repetitive nature of the exact insult that causes brain injury resulting in neonatal encephalopathy. The uncertain timing and etiology of brain injury in most cases of neonatal encephalopathy also fuel birth injury malpractice litigation. It is usually unknown whether the ultimate brain injury is caused by the events only around delivery or by cumulative insults throughout pregnancy [84].
Health factors that influence the risk of neonatal encephalopathy include maternal diseases, multiple pregnancy, gestational age at delivery, malformations within or outside the nervous system, intrauterine growth restriction, congenital infections, intrapartum hypoxic-ischemic events, metabolic problems, and stroke [84,85,86]. Type and timing of contributing factors that are consistent with an acute peripartum or intrapartum events include sentinel hypoxic or ischemic event occurring immediately before or during labor and delivery (a ruptured uterus, severe abruption placentae, umbilical cord prolapsed, amniotic fluid embolus with coincident severe and prolonged maternal hypotension and hypoxemia, maternal cardiovascular collapse, fetal exsanguinations from either vasa previa or massive feto-maternal hemorrhage) [84]. More precisely, there are several risk factors associated with the development of perinatal HIE: preconceptual (e.g., diabetes mellitus type 1, thyroid disease, fertility treatment, nulliparity, advanced maternal age), antepartum (severe preeclampsia, placental abruption, multiple pregnancy, antepartum hemorrhage, fetal growth restriction), and intrapartum (e.g., breech and malpresentation, cord prolapse, caesarean section, maternal pyrexia, induction) [84, 91, 92]. Given the history of the understanding of NE and HIE, it’s not surprising that HIE has been most commonly studied in vitro and in vivo. Consequently we will focus on the understanding of the cellular mechanisms of HIE because this is the pathway to NE that has been best studied [84].
The principal pathogenetic mechanism underlying most of the neuropathological conditions leading to hypoxia-ischemia is a failure of compensatory mechanisms and impaired cerebral blood flow (CBF). At the cellular level, hypoxia-ischemia initially causes energy failure, reperfusion and oxidative and nitrosative stress (immediate phase), and then the loss of mitochondrial function and caspase activation (delayed phase) [88, 93]. Step by step, the primary energy failure is accompanied by glutamate-mediated excitotoxicity. Excitotoxic cellular injury occurs via excess activation of glutamate receptors, which leads to necrotic cell death within 6 h after insult and is more prominent within 1.5 h after insult. There are four receptor types for glutamate, but the N-methyl-d-aspartate (NMDA) receptors are the most avid and physiologically active. The channels activated by NMDA receptors are voltage-dependent and calcium-permeable. Their activation causes neuron depolarization. Repeated depolarization of a neuron by unregulated glutamate release results in accumulation of intracellular calcium. Consequently, an increase of intracellular calcium sets off additional pathologic cascades [88, 93] including oxidative stress and interaction with nitric oxide pathway to produce reactive nitrogen species—peroxynitrites leading to peroxynitrite-induced neurotoxicity, lipid peroxidation, mitochondrial damage and remodeling, depletion of antioxidant reserve, and DNA damage [94]. Between 6 and 72 h after insult, development of mitochondrial dysfunction leads to caspase activation and to the apoptotic cell death which is more prominent during the first 6–8 h after insult [88]. This is the point of “no return.” Inflammatory mediators (cytokines and chemokines) have been implicated in the pathogenesis of hypoxic-ischemic encephalopathy and may represent a final common pathway of brain injury. As it was shown, NF-kB activation in neurons could provide survival, whereas activation in glial cells enhances neuronal cell death [95].
Assuming that hypoxic-ischemic encephalopathy represents the clinical condition affecting mostly the brain, brain lesions have been reported in many studies. Pial arteriolar vasodilatation is a constant finding in the brain of asphyxiated newborns. It is simply evidenced at panoramic view, and it is mainly related to the loss of microvascular reactivity in cerebral vessels [95, 96]. Endothelial damage represents probably the most important change in the brain of asphyxiated newborns. All the endothelial lesions previously reported may be encountered at the histological examination of brain samples [97]. Endothelial swelling represents a peculiar feature in the small intracerebral vessels. Given the narrow lumen of the intravascular capillaries, endothelial swelling may lead to the occlusion of the vascular lumen, leading to the block of the intracerebral circulation, aggravating brain hypoxia. The endothelial damage is followed by the dysfunction of the neurovascular unit that contributes to subsequent neuronal cell death [97, 98]. Neuronal cell death represents a major pathological finding in the interpretation of the severity of the hypoxic encephalopathy. Apoptosis is the most frequent type of cell death occurring in the brain of asphyxiated newborns. At histology, affected neurons show shrinkage, increased eosinophilia of the cytoplasm, nuclear pyknosis, and karyorrhexis, ending with the formation of roundish eosinophilic globules that appear intermingled with preserved neurons [97]. Neuronal apoptosis may be encountered, in the clinical setting of asphyxia, in all the cerebral regions. In our experience, neurons of the brain stem, basal nuclei, and cerebellum appear as the most frequently affected by apoptosis, often in association with apoptosis of the cerebral cortical neurons. Recently, the increased expression of pro-apoptotic proteins—including BAX, cytoplasmic cytochrome C and caspase-3—has been reported in the cortex and thalamus of the brain of mice affected by birth hypoxia [97, 99], suggesting the use of these antibodies in cases in which histology could not clearly evidence the typical features of neuronal cell death. The hippocampus should be always sampled for histological studies, given the frequent functional compromise of this brain region in newborns affected by asphyxia, particularly in female infants [96, 100]. In a recent study, all 16 full-term asphyxiated infants displayed neuronal cell damage and glial reactivity in the hippocampus [96, 101]. If we are talking about the patterns of neonatal HIE at term, there are five patterns of brain injury identified by imaging studies in neonates with this pathological condition: pattern I, basal ganglia and thalami lesions associated with severe white matter damage; pattern II, basal ganglia and thalami lesions with mild or moderate white matter changes; pattern III, isolated thalamic injury; pattern IV, moderate white matter damage only; and pattern V, mild white matter changes or normal findings. Usually infants with patterns III and IV had developmental delay and diplegic cerebral palsy, respectively, and pattern V is associated with normal outcomes [102]. Thus, the basal ganglia and thalami lesions are the imaging signature in term neonates exposed to hypoxic-ischemic sentinel events, and in general, patterns of central gray matter and secondary white matter injury are associated with higher risks of severe morbidity and death [102].
Clinical manifestation of neonatal encephalopathy varies depending on encephalopathy severity.
Neonates with suspected encephalopathy are classified according to the Sarnat staging system, which evaluates the level of consciousness, muscle tone, tendon reflexes, complex reflexes, and autonomic function and classifies HIE into the following three categories: stage I (mild), stage II (moderate), and stage III (severe) [103, 104].
Stage I: Mild Encephalopathy Muscle tone may be slightly increased, and deep tendon reflexes may be brisk during the first few days. Transient behavioral abnormalities, such as poor feeding, irritability, or excessive crying or sleepiness (typically in an alternating pattern), may be observed [88, 103, 104].
Stage II: Moderate Encephalopathy The infant is lethargic, with significant hypotonia and diminished deep tendon reflexes. The grasping, Moro, and sucking reflexes may be sluggish or absent. The infant may experience occasional periods of apnea. Seizures typically occur early within the first 24 h after birth [103]. Full recovery within 1–2 weeks is possible and is associated with a better long-term outcome. An initial period of well-being of mild encephalopathy may be followed by sudden deterioration, suggesting ongoing brain cell dysfunction, injury, and death; during this period, seizure intensity might increase [88, 103, 104].
Stage III: Severe Encephalopathy Stupor or coma is typical. The infant may not respond to any physical stimulus. Breathing may be irregular, and the infant often requires ventilatory support. Generalized hypotonia and depressed deep tendon reflexes are common. Neonatal reflexes (e.g., sucking, swallowing, grasping, Moro) are absent [88, 103, 104]. Disturbances of ocular motion, such as a skewed deviation of the eyes, nystagmus, bobbing, and loss of “doll’s eye” (i.e., conjugate) movements may be revealed by cranial nerve examination [88]. Pupils may be dilated, fixed, or poorly reactive to light. Seizures are delayed, can be severe, and may be initially resistant to conventional treatments. The seizures are usually generalized, and their frequency may increase during the 24–48 h after onset, correlating with the phase of reperfusion injury. As the injury progresses, seizures subside, and the EEG becomes isoelectric or shows a burst suppression pattern. At that time, wakefulness may deteriorate further, and the fontanelle may bulge, suggesting increasing cerebral edema [88].
Irregularities of heart rate and blood pressure (BP) are common during the period of reperfusion injury, as is death from cardiorespiratory failure. Multiple organ dysfunction also presents [88]. Multi-organ systems involvement is a hallmark of NE associated with perinatal asphyxia [88]. Organs involved following a hypoxic-ischemic events include the heart (43–78%) with reduced myocardial contractility, severe hypotension, passive cardiac dilatation, and tricuspid regurgitation; lungs (71–86%) with severe pulmonary hypertension requiring assisted ventilation; kidneys (46–72%) with renal failure presenting as oliguria and, during recovery, as high-output tubular failure, leading to significant water and electrolyte imbalances; liver (80–85%) with elevated liver function test results, hyperammonemia, and coagulopathy; and gastrointestinal system with poor peristalsis and delayed gastric emptying, and necrotizing enterocolitis is rare, and intestinal injuries may not be apparent in the first few days of life or until feeds are initiated; hematologic disturbances (32–54%) include increased nucleated red blood cells (RBCs), neutropenia or neutrophilia, thrombocytopenia, and coagulopathy [88]. Severely depressed respiratory and cardiac functions and signs of brain stem compression suggest a life-threatening rupture of the vein of Galen (i.e., great cerebral vein) with a hematoma in the posterior cranial fossa.
NE is often reported to be the most frequent cause of neonatal seizures [88]. Large, unilateral infarcts occur with neonatal seizures in as many as 80% of patients. Infants with multiple or diffuse lesions and cerebral venous infarcts often have multifocal or migratory seizures observed even during physical examination [88].
A single valuable test for the diagnosis of HIE does not exist. It is important to assess the neonate at birth for detection of signs consistent with an acute peripartum or intrapartum event and the designation of perinatal asphyxia severe enough to result in acute neurologic injury [84]. The following parameters should be taken into consideration: (1) Apgar score of less than 5 at 5 and 10 min after birth; (2) umbilical artery pH less than 7.0, or base deficit greater than or equal to 12 mmol/l, or both; (3) neonatal neurologic sequelae (e.g., seizures, coma, hypotonia); and (4) multiple organ involvement (e.g., renal injury, hepatic injury, hematologic abnormalities, cardiac dysfunction, metabolic derangements, and gastrointestinal injury, or a combination of them) [84, 88]. Although the presence of organ dysfunction increases the risk of HIE in the setting of neonatal encephalopathy, the severity of brain injury seen on neuroimaging does not always correlate with the degree of injury to other organ systems [84]. Nowadays, magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS), as the most sensitive neuroimaging modalities, are extensively used for the monitoring and evaluation of neonates with NE [84]. Distinct patterns of neuroimaging abnormalities, including deep nuclear gray matter or watershed cortical injury, are recognized in hypoxic-ischemic cerebral injury and have prognostic value for predicting later neurodevelopmental impairments. Early MRI obtained between 24 and 96 h of life may be more sensitive for the delineation of the timing of perinatal cerebral injury, whereas an MRI undertaken optimally at 10 days of life (with an acceptable window between 7 and 21 days of life) will best delineate the full extent of cerebral injury [84]. Use of electroencephalography (EEG) might have some limitations due to hypothermia: it may depress the amplitude-integrated EEG (aEEG) and thus limit the early predictive ability of aEEG. Improvement in aEEG tracings may be delayed until the patient undergoes rewarming and is no longer sedated [105]. Some recent investigations showed that S100B protein is a good indicator of brain damage [106]. Furthermore, the serum GFAP levels during the first week of life were increased in neonates with HIE and were predictive of brain injury on MRI. Biomarkers such as S100B and glial fibrillary acidic protein (GFAP) could help triage neonates with HIE to treatment, measure treatment efficacy, and provide prognostic information [106, 107].
According to the contemporary treatment strategy for HIE, initial resuscitation and stabilization are followed by the following steps: (1) neuroprotective strategy, (2) support of adequate ventilation and perfusion, (3) careful fluid management, (4) avoidance of hypo- and hyperglycemia, (5) avoidance of hypotension [a mean blood pressure (BP) above 35–40 mmHg is necessary to avoid decreased cerebral perfusion], and (6) treatment of seizures [108, 109].
At present, hypothermia therapy (HT) is considered as the best neuroprotective strategy for mild to moderate HIE. Mild hypothermia in the range of 33.5–35.0 °C is used. The two types of treatment are used: whole-body hypothermia and selective head cooling. Some basic studies showed far greater histological and electrophysiological protection if hypothermia was initiated within 1.5 h than if it was started 5.5 h after the cerebral insult [110]. According to recent trials, neonates undergoing earlier cooling therapy (within 180 min of birth) had better outcomes compared with those who underwent the therapy later (180–360 min after birth) [111]. The rate of death or severe disability in infants with HIE is decreased from 60% to 46% after cooling [112].
Although cooling is safe, it results in some adverse effects which include a slightly lower baseline heart rate, a marginally significant increase in the need for blood pressure support, and a platelet count below 150 × 109/l [113]. In general, lowering the core temperature can impact hemodynamic status, respiratory physiology, fluid and electrolyte balance, and hematologic factors. In addition, pharmacokinetics and pharmacodynamics of a number of drugs commonly used in asphyxiated neonates are affected by hypothermia. Careful attention to physiologic parameters, laboratory tests, and drug dosing is essential to assure optimum outcomes for neonates undergoing hypothermia therapy [114]. There are some risks associated with return from hypothermia to normothermia as follows: (1) apnea, (2) the risk for seizures (increases due to rewarming leading to peripheral vasodilatation and the intravascular blood volume increase) [115], (3) hypotension (may occur if the vascular bed is underfilled), and (4) alteration in the cardiac function (as a result of the initial hypoxic event may play a contributing role) [110].
Importance of searching for new safe neuroprotective strategy alone or in combination with hypothermia therapy became crucial. We paid our attention to already described specific properties of E4 which were important to come up with the hypothesis that E4 might have neuroprotective effect.
4.4 Estetrol as a New Drug to Treat Neonatal HIE
4.4.1 Estetrol Attenuates Neonatal HIE
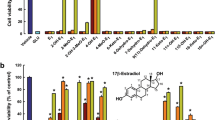
Our primary goal was to define effects of E4 in primary hippocampal cell cultures. In vitro experiments with primary hippocampal neuronal cell cultures showed impressive antioxidative and cell survival effects of different doses of E4 (notably, 650 μl, 3.25 mM, and 6.5 mM) used before or after induction of oxidative stress (Fig. 4.1a, b, c, and d, respectively) [116]. More precisely, E4 at a dose of 3.25 and 6.5 mM significantly downregulated the LDH activity in both sets of experiments (Fig. 4.2a, c), showing the dose-dependent differences at higher doses of E4 (3.25 and 6.5 mM) after induction of oxidative stress (Fig. 4.2c), whereas the same doses of E4 demonstrated significant upregulation of cell proliferation (Fig. 4.2b, d) [116].


Effect of E4 on LDH activity and the cell viability in primary hippocampal neuronal cultures subjected to the H2O2-induced oxidative stress. (a, b) E4 pretreatment. Primary hippocampal cell cultures were treated either with vehicle or with 650 μM, 3.25 mM, and 6.5 mM of E4 (1 h) before induction of oxidative stress by 100 μM of H2O2 (30 min). (a) LDH activity in untreated, H2O2-treated, and E4-pretreated cell cultures; (b) cell viability in untreated, H2O2-treated, and E4-pretreated groups; (c, d) E4 treatment. Primary hippocampal cell cultures were treated with 650 μM, 3.25 mM, and 6.5 mM of E4 (1 h) after induction of oxidative stress by 100 μM of H2O2 (30 min). (c) LDH activity in untreated, treated by H2O2, and E4-treated cell cultures; (d) cell viability in untreated cells as well as cells treated by H2O2 and different concentrations of E4. All measurements are expressed as mean ± SEM. *p < 0.05. Reproduced from Experimental Neurology (Tskitishvili et al., 2014, 261:298–307)

Hematoxylin-eosin staining of coronal brain sections in the rat pups pretreated or treated with E4. Brains of the rat pups were removed upon sacrifice at P14; paraformaldehyde-fixed and paraffin-embedded samples were processed for sectioning at the hippocampus region and hematoxylin-eosin staining. Brain coronal sections of the rat pups (scale bar, 2 mm) with hippocampus region (scale bar, 500 μm) and cortex (scale bar, 100 μm) are shown. E4 pretreatment: sham, vehicle, 1 mg E4/kg/day, 5 mg E4/kg/day, 10 mg E4/kg/day, and 50 mg E4/kg/day. E4 treatment: sham, vehicle, 1 mg E4/kg/day, 5 mg E4/kg/day, 10 mg E4/kg/day, and 50 mg E4/kg/day groups. Reproduced from Experimental Neurology (Tskitishvili et al., 2014, 261:298–307)
Also our aim was to study the effect of E4 on brain damage in vivo . In vivo studies of hypoxic-ischemic brain injury were performed according to two protocols in newborn rat pups. A preventive model was tested with different doses of E4 injected to the rat pups before ischemic injury. E4 was administered intraperitoneally from postnatal day 4 (P4) to postnatal day 7 (P7). Subsequently, the left common carotid artery was ligated and cut. After recovery, the pups were exposed to low oxygen tension (8%) for 30 minutes, in a closed chamber. The therapeutic model consisted, first in the ligation at P7, of the common carotid artery and exposure to 8% oxygen for 30 minutes, to induce an hypoxic-ischemic insult (HI). The various doses of E4 were injected upon retrieval of the rat pups from the hypoxia chamber. All manipulations were performed at 37 °C. The sham group was also operated, but the carotid artery was neither ligated nor cut and the pups were not exposed to hypoxia. Rat pups recovered with their dams and reared normally until being sacrificed at P14 [116,117,118].
E4 did affect neither body weight nor brain weight or body temperature of the rat pups in both sets of experiments when E4 was applied before or after induction of HI events [116]. Histochemical studies of brain coronal sections revealed massive damage of the hippocampus and the cortex at the left (damaged) side of the brain (Fig. 4.2a2, b2) along with dilation of the central and the lateral ventricles in the vehicle-pretreated/treated groups [116].
Usually a good predictor of the neuroprotective activity of the compound is the counting of intact neuronal cells per visual field. Obviously, among E4-pretreated/treated groups, the hippocampus and the cortex were more preserved in sham, 5, 10, and 50 mg/kg/day E4 groups compared to the vehicle group in each region (Fig. 4.2a(1, 4–6), b(1, 4–6)). Intact cell counting was significantly different between the groups pretreated by E4 before induction of experimental hypoxic-ischemic brain injury, in the hippocampal and the cortical regions: vehicle and 5 mg/kg/day E4 groups (DG); vehicle and 5 and 10 mg/kg/day groups or sham and 1 and 50 mg/kg/day E4 groups (SGZ); sham and 1 and 10 mg/kg/day E4 groups (CA1); vehicle and sham, 50 mg/kg/day, or sham and 1 mg/kg/day E4 groups; whereas in the cortex, the intact cell counting was significantly higher in 50 mg/kg/day E4-pretreated group (Table 4.1) like that proving impressive neuroprotective effect of E4 in the brain [116].
E4 treatment after induction of hypoxic-ischemic brain injury in newborn rat pups resulted in significant difference of intact cell counting in the hippocampal and the cortical regions between the groups as follows: vehicle and 1 mg/kg/day, sham and 5, 10, and 50 mg/kg/day E4 groups (CA1); vehicle and 10 mg/kg/day or sham and 1, 5, 10 mg/kg/day, and 50 mg/kg/day E4 groups (CA2/CA3); whereas in the cortex, the intact cell counting was significantly different between the vehicle and 1 mg/kg/day or sham and 5, 10, and 50 mg/kg/day E4 groups (Table 4.1) like that proving the importance of therapeutic effect of E4 [116].
Contemporary basic and translational research studies in the field of neurology frequently employ different technics and methodologies from proof of concept to fully operational status. Among different issues studies of the gray and white matter and neuro- and cerebro-angiogenesis by using specific markers have paramount importance.
As it was already demonstrated earlier, microtubule-associated protein 2 (MAP-2), as a cytoskeleton protein, has its value in the growth, differentiation, and plasticity of neurons, playing key roles in neuronal responses to growth factors, neurotransmitters, synaptic activity, and neurotoxins [119]. It is frequently used as a marker of early gray matter loss in immunohistochemistry studies.
As shown in Fig. 4.3A, B in both models of in vivo hypoxic-ischemic brain injury in the vehicle groups, MAP-2 staining was negative in the hippocampus of the left hemisphere extended to the cortex (Fig. 4.3A(b), B(b)). Further calculations demonstrated that the ratio of the MAP-2-positive area was significantly higher not only in the sham-operated animals (Fig. 4.3A(a), B(a)) but in animals pretreated/treated by different concentrations of E4 (Fig. 4.3A(c)–(f), B(c)–(f)) compared to the vehicle groups in both models (Fig. 4.4a(g), b(g)). Thus, E4 can preserve the early gray matter loss when administered before or after induction of experimental hypoxic-ischemic brain injury [116].

MAP-2 staining of brain coronal sections in the rat pups pretreated or treated with E4 The sections were processed for immunohistochemical detection of neuronal cytoskeletal disruption. (A) E4 pretreatment: sham group (a), vehicle-treated group (b), 1 mg/kg/day E4 (c), 5 mg/kg/day E4 (d), 10 mg/kg/day E4 (e), and 50 mg/kg/day E4 (f) groups (g); (B) E4 treatment: sham group (a), vehicle-treated group (b), 1 mg/kg/day E4 (c), 5 mg/kg/day E4 (d), 10 mg/kg/day E4 (e), and 50 mg/kg/day E4 (f) groups. (g) The ratio of the MAP-2 positive areas was calculated as the MAP-2 positive area of the ipsilateral hemisphere divided by the MAP-2 positive area of the contralateral hemisphere. Ten samples from each group were analyzed. The ratio of the MAP-2 positive area in sham-operated animal group was considered by default as 1.0. All measurements are expressed as mean ± SEM. *p < 0.05. Reproduced from Experimental Neurology (Tskitishvili et al., 2014, 261:298–307)

Myelin basic protein (MBP) staining of brain coronal sections in rat pups pretreated with estetrol. (a) MBP staining of brain coronal sections (scale bar, 2 mm) is shown. (b) MBP staining of cingulum of the left hemisphere is shown (scale bar, 2 mm). (c) The ratio of the MBP-positive areas OD ratio was calculated as the MBP-positive area OD of the ipsilateral hemisphere divided by the MBP-positive area OD of the contralateral hemisphere. Ten samples from each study group were analyzed. The ratio of the MBP-positive area OD in the sham group was considered by default as 1.0. The MBP-positive area OD ratio was significantly higher in sham-operated animals and the 5 and 50 mg/kg/day E4-pretreated groups compared to the vehicle group. All measurements are expressed as mean ± SEM. *p < 0.05. Reproduced from J Endocrinol (Tskitishvili et al., 2017, 232(1):85–95)
Definitely, for us, it was important to show the possible promyelinating activity of E4. Myelin basic protein (MBP), as a marker of white matter damage/demyelination, is frequently used in studies connected to brain damage [120].
There was a loss of MBP staining at the damaged left side (Figs. 4.4a and 4.5a) which was more prominent in main white matter regions [the subcortical region and the cingulum (Figs. 4.4b and 4.5b)]. Promyelinating effect of E4 was significantly upregulated in groups treated by 5 and 50 mg/kg/day E4 before induction of hypoxia-ischemia (Fig. 4.4c), whereas all the groups treated by different doses of E4 after induction of experimental brain injury had significantly higher MBP-positive area OD ratio along with the sham group (Fig. 4.5c) [118]. Significant positive correlation was observed between the myelination and the brain weights (r = 0.707, p = 0.0198) in the vehicle group of the neuroprotective in vivo model [118].

Myelin basic protein (MBP) staining of brain coronal sections in rat pups treated with estetrol. (a) MBP staining of brain coronal sections (scale bar, 2 mm) is shown. (b) MBP staining of cingulum of the left hemisphere is shown (scale bar, 2 mm). (c) The ratio of the MBP-positive areas OD ratio was calculated as the MBP-positive area OD of the ipsilateral hemisphere divided by the MBP-positive area OD of the contralateral hemisphere. Ten samples from each study group were analyzed. The ratio of the MBP-positive area OD in the sham group was considered by default as 1.0. The MBP-positive area OD ratio was significantly higher in sham-operated animals and the 1, 5, 10, and 50 mg/kg/day E4-treated groups compared to the vehicle group. All measurements are expressed as mean ± SEM. *p < 0.05. Reproduced from J Endocrinol (Tskitishvili et al., 2017, 232(1):85–95)
Next step in studies of E4 and its neurological effects in brain was connected to the possible neurogenic- and cerebro-angiogenic effects of E4. Doublecortin (DCX), as a marker of neurogenesis, and VEGF as a marker for angiogenesis were used (Fig. 4.6). Obviously, pretreatment/treatment with different doses of E4 upregulated expression of the abovementioned markers in different regions of the brain and in different manner in all the study groups, showing co-localization of both markers in the cortical region of the 10 mg/kg/day E4-treated group (Fig. 4.4) [116]. In general, E4 pretreatment caused a significant upregulation of neurogenesis and cerebro-angiogenesis in the DG region, with 10 mg/kg for doublecortin and with all doses of E4 for VEGF (Table 4.2); in the CA1 region, in 5 mg/kg/day, 10 mg/kg/day, and 10 mg/kg/day E4 groups, respectively (Table 4.2); in the CA2/CA3 region, in 5 and 10 mg/kg/day E4 groups, respectively (Table 4.2); and in the cortex, in 10 mg/kg/day E4 (Fig. 4.4). Treatment by E4 after hypoxic-ischemic insult (Table 4.2) showed significant upregulation of neuro- and cerebro-angiogenesis in the CA1 region—in 10 and 50 mg/kg/day and in 5, 10, and 50 mg/kg/day E4 groups, respectively; in the CA2/CA3 region, neurogenesis was significantly upregulated in the 10 mg/kg/day E4 group alone, whereas in the cortex, upregulation of neuro- and cerebro-angiogenesis were more prominent in 5, 10, and 50 mg/kg/day and 10 mg/kg/day E4 groups, respectively (Table 4.2) [116] (Fig. 4.6).

Representative views of double-labeled immunofluorescence in the hippocampus and cortex of the rat pups pretreated or treated with E4. Double immunofluorescent staining was performed to determine the localization and expression of doublecortin (DCX) and vascular endothelial growth factor (VEGF) in different regions of hippocampus (dentate gyrus (DG), cornu ammonis 1 (CA1), cornu ammonis 2/3 (CA2/CA3), and cortex. Arrows denote DCX positively stained cells (red). Arrowheads denote VEGF positively stained cells (green). Asterisks indicate co-localization of DCX and VEGF positively stained cells. Scale bar: 200 μm. Reproduced from Experimental Neurology (Tskitishvili et al., 2014, 261:298–307)
Another issue was connected to the question whether pretreatment/treatment by E4 can affect the expression of brain damage markers in blood. Pretreatment by E4 in neuroprotective model resulted in significant downregulation of brain damage markers (S100B and GFAP) at a concentration of 50 mg/kg/day E4, whereas treatment by E4 after induction of experimental hypoxic-ischemic insult led to significant decrease of S100B and GFAP expression in all E4-treated groups (Table 4.3) [116].
4.4.2 Can We Use Estetrol in Combination with Other Steroids for Attenuation of HIE?
Dynamic changes in neurological field proposed new treatment strategies employing different compounds/steroids for attenuation of some neurological diseases. Like that several investigations already have demonstrated the neuroprotective efficacy of estradiol (E2) and progesterone (pregn-4-ene-3,20-dione) (P4) alone or in combination in different experimental models and clinical studies of neurological diseases: Parkinson’s and Alzheimer’s diseases, ischemic stroke, spinal cord injury, traumatic brain injury (TBI), and multiple sclerosis [12, 28, 29, 31, 32, 34, 37, 121,122,123,124,125]. P4-dependent neuroprotection is partly realized through attenuation of oxidative stress resulting from glutamate and glucose deprivation-induced toxicity [126,127,128,129]. Besides, in vitro, in primary hippocampal cell cultures, P4 might have protective effect against FeSO4 and amyloid β-peptide-induced toxicity [130, 131]. Clinical studies in extremely preterm infants demonstrated reduction of the risk for cerebral palsy, spasticity, and ametropia at 5 years neurodevelopmental follow-up due to postnatal E2 and P4 combined replacement therapy [132]. Results of some studies employing combination of E2 and P4 are still controversial: though some studies have suggested that P4 does not affect the positive effects of E2 [128, 133, 134], others still argue that P4 might antagonize the positive effects of E2 [135,136,137,138,139,140].
As we know, the rat forebrain expresses high levels of progesterone receptors (PR) as early as E17–E18 in regions with important cognitive, motor, and visual functions; thus, the hippocampus has importance in establishment of early cortical circuitry. P4 neuroprotective effects are based on activation of inflammatory and oxidative mechanisms and the repair processes that usually follow the injury. As it was shown recently, the upregulation of nitric oxide synthase 2 (NOS-2), involved in production of nitric oxide free radicals, and pro-inflammatory IL-1β after ischemic events caused by MCAO is inhibited by progesterone treatment [141]. In adults after traumatic brain injury, P4 has an ability to reduce the proliferation of reactive astrocytes and inflammatory prostaglandin synthesis further leading to the reduction of edema and the blood-brain barrier leakage [122, 142]. P4 may induce a neuroprotective effect by upregulating expression of brain-derived neurotrophic factor (BDNF) or promoting increase of myelin basic protein expression (MBP) [143], upregulating the inhibitory transmitter GABAa, and reducing the apoptosis by downregulation of NFκB [144,145,146]. Taken together, P4 along with E2 plays a critical role in neuronal developmental processes not only in prenatal period but in adulthood as well [147, 148].
Before starting a new stage of our studies with E4 and other steroids, it was important to define the working concentrations of P4 and E2. First, primary hippocampal neuronal cell cultures were treated after induction of the oxidative stress by P4 and E2 solely at doses starting from 1 nM until up to 1 mM. A significant downregulation of the LDH activity was observed at P4 concentration of 1 mM and E2 concentration of 100 nM (data not shown). Next we have performed treatment of primary hippocampal cell cultures after induction of oxidative stress with previously defined successful concentrations of E4 [116] which showed tremendous antioxidative and cell proliferative effects (650 μM, 3.25 mM, and 6.5 mM) alone or in combination with 1 mM PROG and/or E2 100 nM [117]. LDH activity as a marker of oxidative stress was significantly downregulated in all cultures exposed to steroids, especially in cultures exposed to different concentrations of E4 with E2 and P4 (Fig. 4.7a, b). Similar pattern of LDH activity was observed in cultures treated either by 6.5 mM E4 with E2 (Fig. 4.7a) or by different concentrations of E4 with P4 (Fig. 4.7b) compared to cultures treated by E4 alone [117]. The cell survival rate was significantly increased in cultures treated either by 6.5 mM E4 with/without E2 (Fig. 4.7c) or by 6.5 mM E4 with/without P4 (Fig. 4.7d) or by high doses of E4 with E2 and P4 (Fig. 4.7c, d). Furthermore, cells exposed to 6.5 mM E4 with/without E2 had significantly higher cell survival rate than the cultures treated by 650 μM E4 with/without E2 (Fig. 4.7c), though the dose-dependent pattern was more prominent when different concentrations of E4 were used with/without P4 (Fig. 4.7d). Cells treated by 6.5 mM E4 and P4 or treated by 6.5 mM E4 with E2 and P4 had significantly higher survival rate than the cells treated only by E4 (Fig. 4.7d) or those cells treated by 6.5 mM E4 with/without E2 (Fig. 4.7c) and by the lower doses of E4 combined with E2 and P4, respectively (Fig. 4.7c, d) [117].


Effect of E4 alone or in combination with P4 and/or E2 on LDH activity and cell viability in primary hippocampal cell cultures subjected to the H2O2-induced oxidative stress. (a–d) Primary hippocampal neuronal cells were treated with 650 μM, 3.25 mM, and 6.5 mM of estetrol alone or in combination with 100 nM E2 and/or 1 mM P4 for 1 h after induction of oxidative stress by 100 μM of H2O2 for 30 min. (a, b). LDH activity was significantly downregulated in all the study groups compared to the H2O2-treated group. The LDH activity level was significantly lower in cultures treated either with 6.5 mM E4 or 100 nM E2 (a) or in cultures treated by any dose of E4 along with 1 mM P4 than in cultures treated by E4 alone (b) as well as in cultures combinedly treated by any dose of E4 along with 1 mM P4 and 100 nM E2 than in cultures treated by E4 alone (a, b). (c) Cell survival was significantly upregulated in cultures treated by 6.5 mM E4 with/without 1 mM P4 or by 3.25 mM and 6.5 mM E4 with 100 nM E2 and 1 mM P4 in comparison with H2O2-treated cultures. Cells exposed to 6.5 mM E4 with/without 100 nM E2 had significantly higher cell survival rate than the cultures treated by 650 μM E4 with/without 100 nM E2. Cells combinedly treated by 6.5 mM E4 with 100 nM E2 and 1 mM P4 had significantly higher survival level than the cells treated by 6.5 mM E4 with/without 100 nM E2. (d) Cell survival was significantly upregulated in cultures treated by 6.5 mM E4 with/without 1 mM P4 or by 3.25 mM and 6.5 mM E4 with 100 nM E2 and 1 mM P4 than in H2O2-treated cultures. The dose-dependent pattern was observed when 650 μM, 3.25 mM, and 6.5 mM E4 were used with/without 1 mM P4. (c, d) Cell cultures combinedly treated by 6.5 mM E4 with 100 nM E2 and 1 mM P4 had significantly higher survival level than the cells treated by 650 μM and 3.25 mM E4 in combination with 100 nM E2 and 1 mM P4. All measurements are expressed as mean ± SEM. *p ≤ 0.05. Reproduced from Oncotarget (Tskitishvili et al., 2016, 7(23):33722–43)
Interesting observations were monitored in vivo. In neuroprotective model, when steroids were used before induction of brain injury, rectal temperature immediately after hypoxic-ischemic (HI) insult (at 0 h time point) was significantly increased only in animals from the vehicle group, whereas 2 h later the rectal temperature was significantly decreased in groups pretreated by combination of 5 mg/kg/day E4 and 1.6 mg/kg/day P4 with/without 136 ng/kg/day E2 (Fig. 4.8a) [117]. In therapeutic model, 2 h after HI insult, groups treated by combination of any dose of E4 with 16 mg/kg/day P4 plus 136 ng/kg/day E2 had significantly decreased rectal temperature than the vehicle group or the groups treated by the same doses of E4 with 1.6 mg/kg/day P4 and E2. Combination of 10 mg/kg/day E4 with 16 mg/kg/day P4 with or without 136 ng/kg/day E2 also 10 mg/kg/day E4 alone or combined with 1.6 mg/kg/day P4 significantly downregulated the rectal temperature (Fig. 4.8b) [117]. At 4 h after HI insult, animals treated by E4 and E2 with/without 1.6 mg/kg/day/16 mg/kg/day P4 had significantly decreased rectal temperature compared to animals treated by single doses of E4 (Fig. 4.8b). Treatment by 5 mg/kg/day E4 with 16 mg/kg/day P4 and E2 significantly decreased the rectal temperature than the treatment by the same combination of compounds with 1.6 mg/kg/day P4 (Fig. 4.8b) [117].


Postoperative rectal temperature and body weight of rat pups. (a). In neuroprotective model, immediately after hypoxic-ischemic (HI) insult (at 0 h), the rectal temperature was significantly increased only in the vehicle group than in the sham group, whereas 2 h later the rectal temperature was significantly decreased in pretreated by 5 mg/kg/day E4 and 1.6 mg/kg/day P4 with/without 136 ng/kg/day E2 groups compared to the sham group. Four hours later no significant difference was observed among the study groups. (b) In therapeutic model, between the study groups immediately after HI insult, no significant differences were detected, whereas 2 h later groups treated by combination of 5 mg/kg/day or 10 mg/kg/day E4 with 16 mg/kg/day P4 plus 136 ng/kg/day E2 had significantly decreased rectal temperature than the vehicle group or the groups treated by the same doses of E4 with 1.6 mg/kg/day P4 and E2. Moreover, combination of 10 mg/kg/day E4 with 16 mg/kg/day P4 and 136 ng/kg/day E2 significantly downregulated the rectal temperature compared to the sham group or the group treated by 10 mg/kg/day E4 and 16 mg/kg/day P4 (Fig. 4.2b). Also, the groups treated by 10 mg/kg/day E4 alone or combined with 1.6 mg/kg/day P4 had significantly decreased rectal temperature compared to the group treated by 10 mg/kg/day E4 with 1.6 mg/kg/day P4 and 136 ng/kg/day E2. At 4 h after HI event, animals treated by 5 mg/kg/day or 10 mg/kg/day E4 and 136 ng/kg/day E2 with/without 16 mg/kg/day P4 had significantly decreased rectal temperature along with the sham group compared to animals treated by single doses of E4 (Fig. 4.2b). The same pattern was observed between groups treated by 10 mg/kg/day E4 with 1.6 mg/kg/day P4 and 136 ng/kg/day E2 and the group treated by E4 alone. Treatment by 5 mg/kg/day E4 with 16 mg/kg/day P4 and 136 ng/kg/day E2 significantly decreased the rectal temperature than the treatment by the same combination of compounds with 1.6 mg/kg/day P4 (Fig. 4.2b). All measurements are expressed as mean ± SEM. *p ≤ 0.05. Reproduced from Oncotarget (Tskitishvili et al., 2016, 7(23):33722–43)
As it is shown in Table 4.4, animals pretreated by a combination of 5 mg/kg/day E4 and 16 mg/kg/day P4 before experimental brain injury had significantly higher body weight than animals from the vehicle, sham, and combinedly pretreated by 5 mg/kg/day E4 and E2 groups, and the brain-body weight ratio was significantly higher in groups pretreated by 5 mg/kg/day E4 and 136 ng/kg/day E2 in combination either with 1.6 or 16 mg/kg/day P4. In groups treated by steroids after experimental brain damage, only animals from 10 mg/kg/day E4 group had significantly higher brain weight compared to the vehicles (Table 4.4) without affecting the brain-body weight ratio [117].
Histochemical studies of coronal sections from rat pups’ brains pretreated/treated by the vehicle showed obvious injury of the hippocampus at the left carotid artery occlusion (damaged) side which was extended to the cortex at the same side (Figs. 4.9 and 4.10A(b), B(b)). It was also interesting to observe the damage of the cortex of the left hemisphere in animals that were pretreated/treated by combination of E4 and E2 (Fig. 4.9) [117].

Representative views of hematoxylin-eosin-stained brain coronal sections from rat pups pretreated/treated by E4 alone or in combination with P4 and/or E2. Paraffin-embedded brain samples were sliced into 5-μm-thick coronal sections at the hippocampus level. Sections were deparaffinized and rehydrated, and hematoxylin and eosin staining was performed. Brain coronal sections (scale bar, 2 mm) with hippocampus region (scale bar, 500 μm) and the cortex (scale bar, 100 μm) from pretreated (a) and treated (b) study groups are presented. Reproduced from Oncotarget (Tskitishvili et al., 2016, 7(23):33722–43)

Hematoxylin-eosin staining of the brain coronal sections from rat pups pretreated/treated by E4 alone or in combination with P4 and/or E2. Brain coronal sections (scale bar, 2 mm) with hippocampus region (scale bar, 500 μm) and cortex (scale bar, 100 μm) from pretreated A and treated B study groups are shown: sham (a), vehicle (b), 5 mg/kg/dayE4 (c), 5 mg/kg/day E4 + 16 mg/kg/day P4 + 136 ng/kg/day E2 (d), 5 mg/kg/day E4 + 136 ng/kg/day E2 (e), 10 mg/kg/day E4 (f), 10 mg/kg/day E4 + 16 mg/kg/day P4 (g), 10 mg/kg/day E4 + 136 ng/kg/day E2 (h). Reproduced from Oncotarget (Tskitishvili et al., Use of estetrol with other steroids for attenuation of neonatal hypoxic-ischemic brain injury: to combine or not to combine? Oncotarget. 2016, 7(23):33722–43)
Cell counting per visual field in pretreated groups showed (Table 4.5) that in the DG region there were significant differences between the study groups as follows: sham and the vehicle groups or groups pretreated by 5 mg/kg/day E4 and E2 with/without 16 mg/kg/day P4 (Fig. 4.9a and 4.10A(c), (d), respectively) or by 10 mg/kg/day E4 with E2 and 16 mg/kg/day P4 (Fig. 4.9a). In the same region of the hippocampus, significantly higher number of intact cells was observed in animals pretreated by 5 mg/kg/day E4 (Fig. 4.10A(c)) and 10 mg/kg/day E4 (Fig. 4.10A(f)) alone or in combination with 1.6 mg/kg/day P4 and/or E2, also in animals pretreated with 10 mg/kg/day E4 and 16 mg/kg/day P4 (Table 4.5), as well as between animals pretreated by 10 mg/kg/day E4 alone or in combination with E2 and/or any concentration of P4, and the sham group (Table 4.5) [117]. In the SGZ the sham group had significantly higher intact cell counting than animals pretreated by 5 mg/kg/day E4 with E2 (Figs. 4.9a and 4.10A(h)), whereas the number of intact cells was significantly upregulated in the groups pretreated by different doses of E4 alone or combined with 16 mg/kg/day P4. The same pattern of significant difference was observed in animals pretreated by 5 mg/kg/day E4 in combination with 16 mg/kg/day P4 plus E2 and the vehicle group (Table 4.5) [117].
In treated groups (Figs. 4.9b and 4.10B), in the DG region, the number of intact cells was significantly different between the groups: vehicle and sham, also groups treated by combination of different doses of E4 either with any dose of P4 or E2. Intact cell number was significantly downregulated in animals combinedly treated by 10 mg/kg/day E4 and E2 (Fig. 4.10B(h)) compared to the sham group (Table 4.5). In the SGZ region, significant differences were observed between the vehicle group and the animals treated by different doses of E4 with 1.6 mg/kg/day P4 or E2, also the animals treated by 10 mg/kg/day E4 with E2 and different doses of P4 (Fig. 4.9b and Table 4.5). In the CA2/CA3 region, significant differences were observed only among animals from sham and the vehicle groups, whereas in the cortex, significantly higher number of intact cells was detected except for the sham group in groups treated either by different doses of E4 in combination with 16 mg/kg/day P4 or 10 mg/kg/day E4 alone (Fig. 4.10B) and the vehicle group (Table 4.5) [117].
By using the MAP staining, we have evaluated the gray matter loss in study groups. MAP-2 negatively stained areas corresponded to the damaged areas in the left hemisphere (the hippocampus and the cortex) (Fig. 4.9a, b) [117]. MAP-2 positively stained area ratio was significantly upregulated in animals pretreated by 10 mg/kg/day E4 alone along with animals from sham group (Figs. 4.11a and 4.12a), whereas after treatment with different combinations of steroids, MAP-2 positive area ratio was significantly higher in groups treated by E4 alone or in combination with 16 mg/kg/day P4 compared to the vehicle group (Figs. 4.11b and 4.12b). The similar pattern showed animals combinedly treated by 5 mg/kg/day E4 with 1.6 mg/kg/day P4 and E2. Treatment with 5 mg/kg/day E4 alone or either with 1.6 mg/kg/P4 and E2 or 16 mg/kg/day P4 restored the MAP-2 positive area ratio almost to the sham level (Figs. 4.11b and 4.12b) [117].

Representative views of MAP-2-stained coronal brain sections from groups pretreated or treated with E4 alone or in combination with P4 and/or E2. From left to right are presented MAP-2-stained sections from pretreated (a) and treated (b) groups. In sections from the vehicle-pretreated/treated animals was observed an existence of MAP-2 negatively stained areas in the hippocampus and the cortex at the left, damaged side. Scale bar: 2 mm. Reproduced from Oncotarget (Tskitishvili et al., 2016, 7(23):33722–43)


MAP-2 staining of brain coronal sections from rat pups pretreated/treated by E4 alone or in combination with P4 and/or E2. For evaluation of gray matter loss, MAP-2 staining was performed. (a) Among pretreated groups the MAP-2 positively stained area ratio was significantly upregulated in animals pretreated by 10 mg/kg/day E4 alone than in the vehicles as well as in animals from sham group. (b) After treatment with different combinations of steroids, MAP-2-positive area ratio was significantly higher along with the sham group in groups treated by 5 or 10 mg/kg/day E4 alone or in combination with 16 mg/kg/day P4 compared to the vehicle group. The similar pattern was observed in animals combinedly treated by 5 mg/kg/day E4 with 1.6 mg/kg/day P4 and 136 ng/kg/day E2. Ten samples from each group were analyzed. The ratio of the MAP-2-positive area in sham-operated animals was considered as 1.0 by default. All measurements are expressed as mean ± SEM. *p ≤ 0.05. Reproduced from Oncotarget (Tskitishvili et al., 2016, 7(23):33722–43)
We have also studied the possible effect of combined use of E4 with other steroids on neuro- and cerebro-angiogenesis by using specific markers DCX and VEGF, respectively, as previously (Fig. 4.13) [117]. In groups pretreated by different combinations of steroids before experimental HI insult in the DG region, neurogenesis and angiogenesis were significantly upregulated in 5 mg/kg/day E4 and 10 mg/kg/day E4 along with 1.6 mg/kg/day P4 and E2, respectively (Table 4.6); in the CA1 region, significant differences in DCX expression were observed between the sham and the vehicle groups, though VEGF expression was significantly increased in animals combinedly pretreated by 5 mg/kg/day E4 and 16 mg/kg/day P4 (Table 4.6); in the CA2/CA3 region, expressions of DCX and VEGF were significantly different only between the sham and the vehicle groups, whereas in the cortex between the sham and 5 mg/kg/day E4 plus 16 mg/kg/day P4 groups (Table 4.6) [117]. Notably, pretreatment by combination of E4 and E2 did not show any positive result for neuro- and angiogenesis (Fig. 4.13) [117].

Representative views of double-labeled immunofluorescent sections from different regions of the hippocampus and the cortex from groups pretreated/treated with E4 alone or in combination with P4 and/or E2. To determine the localization and expression of DCX and VEGF in different regions of the hippocampus and the cortex, the double immunofluorescent staining was performed. Red cells denote the DCX positively stained cells, whereas green cells denote the VEGF positively stained cells. Asterisks indicate co-localization of DCX and VEGF positively stained cells. Scale bar: 200 μm. Reproduced from Oncotarget (Tskitishvili et al., 2016, 7(23):33722–43)
Treatment of animals after HI insult with different combinations of steroids resulted in significant upregulation of neurogenesis in the hippocampus in animals treated by 5 mg/kg/day E4 alone or with 16 mg/kg/day P4 (Fig. 4.13). Also, sham group showed significantly higher number of DCX positively stained cells than the groups combinedly treated either by 5 mg/kg/day with 1.6 mg/kg/day P4 or 10 mg/kg/day E4 with 16 mg/kg/day P4 and E2 as well as groups treated by E4 in combination with 1.6 mg/kg/day P4 and E2 (Table 4.6) [117]. In the same region, angiogenesis was significantly upregulated in the sham-operated animals and in groups treated by 5 mg/kg/day E4 alone or in combination with 16 mg/kg/day P4; in the CA1 region, neurogenesis was significantly upregulated along with the sham group in animals treated by 5 mg/kg/day E4 along with E2 than in the vehicles, whereas angiogenesis was significantly upregulated in sham group and in animals treated by 10 mg/kg/day E4 with 1.6 mg/kg/day P4 (Table 4.6) [117]; In the CA2/CA3 region expressions of DCX and VEGF were significantly different between sham and the vehicle groups. In the CA2/CA3 region significant differences in DCX expression was detected between sham group, the animals treated by 10mg/kg/d E4 with 1.6mg/kg/d P4 and the vehicles as well as between sham group, the animals treated by 5mg/kg/d E4 with 16mg/kg/d P4 and the group treated by 5mg/kg/d E4 alone (Table 4.6); in the same region, VEGF was significantly more expressed in sham group than in the vehicles; in the cortex neuro- and angiogenesis were significantly more upregulated only in sham group compared to the vehicle group (Table 4.6); in general, combination of E4 with E2 resulted in low DCX and VEGF expression levels in the cortex (Fig. 4.13) [117].
The next step was evaluation of brain damage marker (GFAP) as it was discussed previously [117]. Combined pretreatment by 5 mg/kg/day E4 with P4 and E2 resulted in significant downregulation of GFAP expression compared to the vehicle group (Table 4.7). Different patterns of GFAP expression were observed in different groups. Significant downregulation of GFAP concentration was also observed in animals pretreated either by 5 mg/kg/day E4 alone or in combination with different doses of P4 and E2 or combined with 16 mg/kg/day P4 and in sham group than in animals pretreated by 5 mg/kg/day E4 and E2 (Table 4.7), also between groups pretreated by 10 mg/kg/day E4 alone or in combination with different doses of P4 and E2 or with 16 mg/kg/day P4 than in animals pretreated by 10 mg/kg/day E4 and E2 (Table 4.7) [117]. Treatment by E4 with P4 and/or E2 resulted in significant decrease of GFAP protein concentration in 10 mg/kg/day E4 along with the sham groups compared to animals treated by combination of 10 mg/kg/day E4 and E2 (Table 4.7). In vivo, in pretreated/treated groups, the combination of E4 and E2 showed significantly higher levels of GFAP, suggesting a negative cooperativity of these steroids upon cell survival (Table 4.7) [118].
After taking into account all the observations and experimental results, we have defined that combined use of E4 with other steroids has no benefit over the single use of E4 [117].
4.4.3 How E4 Is Realizing Its Neuroprotective Effects ?
Recent studies in different cells and tissues showed that E4 acts as a selective estrogen receptor modulator (SERM) by activating the nuclear ERα, inhibiting its membrane form and blocking the membrane initiated steroid signaling by E2 [149]. E4 may have a synergistic role with E2 (through activation of nuclear ERα) or an antiestrogenic effect by blocking membrane ERα and its activation by E2 depending on the respective role of nuclear and membrane forms of ERα in target organs. Thus, E4 has biological activities distinct from E2, depending on the tissues and cells and the selective binding to the nuclear/membrane form of ERα [149]. In general, palmitoylation regulates 17β-estradiol-induced ERα degradation and transcriptional activity [150] and may explain the ability of ERα to associate to plasma membrane making possible E2-dependent rapid functions [151], and the same might be plausible for E4-dependent rapid functions. Recent studies also have shown that ERβ expression in oligodendrocytes is important for the attenuation of clinical disease by an ERβ ligand, like that pointing an importance of this receptor in myelination [152].
As far we were going in our research as meticulously, we were trying to identify the exact mechanism of E4-dependent neuroprotective actions. Recent studies showed that, in general, the neuroprotective actions of estrogens among other factors also depend on their strong antioxidant properties. All estrogens have a phenolic moiety in their structure, the free phenolic OH group, which has been considered the quintessential feature in conferring protection against oxidative stress [153]. E4 has the highest number (four) of free phenolic hydroxyl groups in its structure, thus pointing out the possibility to have stronger antioxidant properties than other estrogens. Thus, one more explanation for E4 neuroprotective effect might be attributed to its strong antioxidant effect as well, which is demonstrated by our previous studies [116, 117], but it is not enough to explain the full spectrum of impressive results of action of E4 in the CNS.
For in vitro studies, we have used one of the most successful concentrations of E4 (3.25 mM) already defined from our previous research [116] alone or in combination with different estrogen receptor inhibitors and/or palmitoylation inhibitor after induction of oxidative stress in primary hippocampal neuronal cell cultures. The antioxidative activity of E4 and the expression of LDH were completely blocked only by concomitant treatment of cells with E4, MPP (inhibitor of ERα), and PHTTP (inhibitor of ERβ) (Fig. 4.14a, b) [118]. Inhibition of palmitoylation alone with 2-BR or in combination with MPP significantly decreased LDH activity, suggesting that the combined blockage of ERα and palmitoylation is not sufficient to inhibit the E4-dependent effects (Fig. 4.14c), whereas combination of E4 with 2-BR, MPP, and PHTTP completely blocked the antioxidative effects of E4 once again suggesting the role of both receptors, ERα and ERβ (Fig. 4.14c). Inhibition of GPR30 receptor did not block the E4 actions (Fig. 4.14d) [118].


Effect of E4 in combination with different receptor inhibitors on LDH activity in primary hippocampal neuronal cultures subjected to the H2O2-induced oxidative stress. Primary hippocampal cell cultures were exposed to 3.25 mM E4 alone or in combination with MPP, PHTTP, G15, and/or 2-BR after induction of oxidative stress. (a) LDH activity was significantly decreased by treatment with E4 alone or in combination with ERα inhibitor MPP compared to the H2O2-treated cell cultures or cultures combinedly treated by E4 + MPP + PHTTP. Combined use of MPP and PHTTP significantly increased the LDH activity compared to the cells treated by E4 alone or in combination with MPP. (b) LDH activity was significantly decreased by treatment with E4 alone or in combination with ERβ inhibitor PHTTP compared to the H2O2-treated cell cultures or cultures combinedly treated by E4 + MPP + PHTTP. Combined use of MPP and PHTTP significantly increased the LDH compared to the cell cultures treated by E4 alone or in combination with PHTTP. (c) Inhibition of palmitoylation alone or in combination with MPP significantly downregulated LDH activity compared to the H2O2-treated cells or to those treated by E4 alone. Combination of E4 with 2-BR, MPP, and PHTTP significantly upregulated LDH activity compared to the cell cultures treated by E4 or 2-BR alone or in combination with MPP. (d) Cell cultures treated by E4 alone or in combination with GPR30 inhibitor G15 had significantly lower LDH activity compared to the cultures treated by H2O2 alone. No significant difference was observed between the cells treated by E4 alone or in combination with G15. Reproduced from J Endocrinol (Tskitishvili et al., 2017, 232(1):85–95)
Cell survival rate was significantly downregulated only by inhibition of ERβ alone (Fig. 4.15b). All cells treated either by E4 alone or in combination with different combinations of 2-BR, MPP, and PHTTP had significantly higher cell survival rate, and inhibition of palmitoylation along with inhibition of ERα activity resulted in a significantly higher cell survival rate suggesting that ERα (probably membrane form of the receptor) does not affect the E4-dependent cell survival/proliferation actions (Fig. 4.15c). Inhibition of GPER did not affect the cell survival rate (Fig. 4.15d) [118].


Effect of E4 in combination with different receptor inhibitors on cell survival in primary hippocampal neuronal cultures subjected to the H2O2-induced oxidative stress. Primary hippocampal cell cultures were exposed to 3.25 mM E4 alone or in combination with MPP, PHTTP, G15, and/or 2-BR after induction of oxidative stress. (a) Cell survival rate was significantly upregulated in cells treated by E4 alone or in combination either with MPP or MPP + PHTTP compared to cells solely treated by H2O2. (b) Cultures treated either by E4 alone or with PHTTP with/without MPP had significantly upregulated cell survival rate compared to cells treated by H2O2 alone. Cells combinedly treated by E4 with PHTTP had significantly lower cell survival rate than the cell cultures treated by E4 alone. (c) Cells treated either by E4 alone or in combination with 2-BR, MPP, and/or PHTTP had significantly higher cell survival rate compared to the cells solely treated by H2O2. Treatment of cultures by E4 and 2-BR along with MPP resulted in significant upregulation of cell survival compared to the cultures treated by 2-BR alone or in combination with MPP and PHTTP. No significant difference was observed between the cells treated by E4 alone or those treated by different combinations of E4, 2-BR, MPP, and/or PHTTP. (d) Treatment of cell cultures by E4 alone or in combination with G15 significantly upregulated the cell survival rate compared to cell cultures treated by H2O2. No significant difference was observed between cells treated by E4 alone or in combination with G15. Reproduced from J Endocrinol (Tskitishvili et al., 2017, 232(1):85–95)
As we have already discussed earlier, the expression of ERα and ERβ displays different spatial-temporal patterns during human cortical and hippocampal development, and knowledge of the region-specific expression of each ER subtype is critical to better understand the actions of estrogens on the human brain [3]. Even though, the genomic effects of ERs are mostly studied, we have to pay attention to the rapid cellular signaling (non-genomic) effects that are thought to be mediated primarily by membrane-associated forms of these receptors [154]. These non-genomic signaling events are dependent to the estrogen-binding sites on intracellular membranes, whereas some reports suggest that palmitoylation or phosphorylation [150] may target classical ERs to the cytoplasmic side of the plasma membrane. In general, palmitoylation is necessary for ERα transcriptional activity, and inhibition of ERα palmitoylation constitutively addresses ERα to the nuclear matrix resulting in the basal degradation of the neo-synthesized ERα [155], though we did not observe any significant effect of palmitoylation inhibition on cell survival/proliferation [118].
It was already defined that the potential role of ERβ expression in cells of oligodendrocyte (OL) lineage in ERβ ligand-mediated neuroprotection is important, and it results in the upregulation of myelination [152]. Also, neuroprotection might be mediated through ERα in astrocytes exclusively [156]. Our studies prove that the E4-mediated activities in the CNS are realized through ERα and ERβ, like that enlightening the important role of E4 as a selective estrogen receptor modulator (SERM) with neurosteroid actions.
4.5 Conclusion
Summarizing our findings we can admit that for the first time we proved impressive antioxidative, neuroprotective, promyelinating, and neuro-, and angiogenesis effects of estetrol in the CNS by employing in vitro and in vivo studies. We believe that our investigation will open new horizons for the development of new perinatal treatment strategies not only for HIE but for periventricular leukomalacia (PVL) as well. Our research could also contribute to a better understanding of the pathophysiology and treatment of other neurological diseases such as Alzheimer’s and Parkinson’s diseases, traumatic brain injury, and multiple sclerosis.
References
Charles H. Rodeck, Martin J. Whittle, Fetal medicine: basic science and clinical practice. 2nd edition, 2009, Elsevier Health Sciences, London.
Shiota K. Prenatal development of the human central nervous system, normal and abnormal. Donald School J Ultrasound Obstet Gynecol. 2015;9(1):61–6.
Gonzalez M, Cabrera-Socorro A, Perez-Garcia CG, Fraser JD, Lopez FJ, Alonso R, et al. Distribution patterns of estrogen receptor alpha and beta in the human cortex and hippocampus during development and adulthood. J Comp Neurol. 2007;503(6):790–802. https://doi.org/10.1002/cne.21419.
Hart SA, Patton JD, Woolley CS. Quantitative analysis of ER alpha and GAD colocalization in the hippocampus of the adult female rat. J Comp Neurol. 2001;440:144–55. https://doi.org/10.1002/cne.1376.
Shughrue PJ, Merchenthaler I. Evidence for novel estrogen binding sites in the rat hippocampus. Neuroscience. 2000;99:605–12. https://doi.org/10.1210/endo.139.12.6525.
O’Keefe JA, Li Y, Burgess LH, Handa RJ. Estrogen receptor mRNA alterations in the developing rat hippocampus. Brain Res Mol Brain Res. 1995;30:115–24. https://doi.org/10.1016/0169-328X(94)00284-L.
Solum DT, Handa RJ. Localization of estrogen receptor alpha (ER-alpha) in pyramidal neurons of the developing rat hippocampus. Brain Res Dev Brain Res. 2001;28:165–75. https://doi.org/10.1016/S0165-3806(01)00171-7.
Nomura M, Korach KS, Pfaff DW, Ogawa S. Estrogen receptor b (ERb) protein levels in neurons depend on estrogen receptor a (ERa) gene expression and on its ligand in a brain region-specific manner. Mol Brain Res. 2003;110(2003):7–14. https://doi.org/10.1016/S0169-328X(02)00544-2.
Van der Knaap MS, Valk J. Magnetic resonance of myelin, myelination and myelin disorders. 2nd ed. Berlin: Springer; 1995.
Oakey RE. The progressive increase in oestrogen production in human pregnancy: an appraisal of the factors responsible. Vitam Horm. 1970;28:1.
Levitz M, Young BK. Estrogens in pregnancy. Vitam Horm. 1977;35:109.
Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007;72:381–405. https://doi.org/10.1016/j.steroids.2007.02.003.
McCarty MM. Estradiol and the developing brain. Physiol Rev. 2008;88(1):91–124.
Lee HS, Han J, Baim HJ, Kim KW. Brain angiogenesis in developmental and pathological processes: regulation, molecular and cellular communication at the neurovascular interface. FEBS J. 2009;276(17):4622–35. https://doi.org/10.1111/j.1742-4658.2009.07174.x.
Park JA, Choi KS, Kim SY, Kim KW. Coordinated interaction of the vascular and nervous systems: from molecule- to cell-based approaches. Biochem Biophys Res Commun. 2003;311:247–53. https://doi.org/10.1016/j.bbrc.2003.09.129.
Gordon GR, Mulligan SJ, MacVicar BA. Astrocyte control of the cerebrovasculature. Glia. 2007;55:1214–21. https://doi.org/10.1002/glia.20543.
Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–9. https://doi.org/10.1038/nature02827.
Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. https://doi.org/10.1038/nn980.
Krause DN, Duckles SP, Pelligrino DA. Influence of sex steroid hormones on cerebrovascular function. J Appl Physiol. 2006;101(4):1252–61. https://doi.org/10.1152/japplphysiol.01095.2005.
Barouk S, Hintz T, Li P, Duffy AM, MacLusky NJ, Scharfman HE. 17β-estradiol increases astrocytic vascular endothelial growth factor (VEGF) in adult female rat hippocampus. Endocrinology. 2011;152(5):1745–51. https://doi.org/10.1210/en.2010-1290.
Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A. 2002;99(18):11946–50. https://doi.org/10.1073/pnas.182296499.
Zhu Y, Jin K, Mao XO, Greenberg DA. Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. FASEB J. 2003;17(2):186–93. https://doi.org/10.1096/fj.02-0515com.
Ogunshola OO, Stewart WB, Mihalcik V, Solli T, Madri JA, Ment LR. Neuronal VEGF expression correlates with angiogenesis in postnatal developing rat brain. Brain Res Dev Brain Res. 2000;119(1):139–53. https://doi.org/10.1016/S0165-3806(99)00125-X.
Haigh JJ, Morelli PI, Gerhardt H, Haigh K, Tsien J, Damert A, et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGFA paracrine signaling. Dev Biol. 2003;262:225–41. https://doi.org/10.1016/S0012-1606(03)00356-7.
Raab S, Beck H, Gaumann A, Yüce A, Gerber HP, Plate K, et al. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91:595–605. https://doi.org/10.1160/TH03-09-0582.
Wise PM. Estrogens and neuroprotection. Trends Endocrinol Metab. 2002;6:229–30.
Dubal DB, Wisel PM. Estrogen and neuroprotection: from clinical observations to molecular mechanisms. Dialogues Clin Neurosci. 2002;4(2):149–61.
Cho JJ, Iannucci FA, Fraile M, Franco J, Alesius TN, Stefano GB. The role of the estrogen in neuroprotection: implications for neurodegenerative diseases. Neuro Endocrinol Lett. 2003;24:141–7.
Garcia-Segura LM, Azcoitia I, DonCarlos LL. Neuroprotection by estradiol. Prog Neurobiol. 2001;63:29–60. https://doi.org/10.1016/S0301-0082(00)00025-3.
Gold SM, Voskuhl RR. Estrogen treatment in multiple sclerosis. J Neurol Sci. 2009;286(1–2):99–103. https://doi.org/10.1016/j.jns.2009.05.028.
Samantaray S, Matzelle DD, Ray SK, Banik NL. Physiological low dose of estrogen-protected neurons in experimental spinal cord injury. Ann N Y Acad Sci. 2010;1199:86–9. https://doi.org/10.1111/j.1749-6632.2009.05360.x.
Suzuki S, Brown CM, Wise PM. Neuroprotective effects of estrogens following ischemic stroke. Front Neuroendocrinol. 2009;30:201–11. https://doi.org/10.1016/j.yfrne.2009.04.007.
Henderson VW, Benke KS, Green RC, Cupples LA, Farrer LA, MIRAGE study Group. Postmenopausal hormone therapy and Alzheimer’s disease risk: interaction with age. J Neurol Neurosurg Psychiatry. 2005;76:103–5. https://doi.org/10.1136/jnnp.2003.024927.
Currie LJ, Harrison MB, Trugman JM, Bennett JP, Wooten JF. Postmenopausal estrogen use affects risk for Parkinson disease. Arch Neurol. 2004;61:886–8. https://doi.org/10.1001/archneur.61.6.886.
Gardiner SA, Morrison MF, Mozley PD, Mozley LH, Brensinger C, Bilker W. Pilot study on the effect of estrogen replacement therapy on brain dopamine transporter availability in healthy, postmenopausal women. Am J Geriatr Psychiatry. 2004;12:621–30. https://doi.org/10.1176/appi.ajgp.12.6.621.
Li R, Shen Y, Yang LB, Lue LF, Finch C, Rogers J. Estrogen enhances uptake of amyloid beta-protein by microglia derived from the human cortex. J Neurochem. 2000;75:1447–54. https://doi.org/10.1046/j.1471-4159.2000.0751447.x.
Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, et al. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat Med. 1998;4:447–51.
Kenchappa RS, Diwakar L, Annepu J, Ravindranath V. Estrogen and neuroprotection: higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. FASEB J. 2004;18:1102–4. https://doi.org/10.1096/fj.03-1075fje.
Ramirez AD, Liu X, Menniti FS. Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology. 2003;77:223–31. https://doi.org/10.1159/000070277.
Nilsen J. Estradiol and neurodegenerative oxidative stress. Front Neuroendocrinol. 2008;9(4):463–75. https://doi.org/10.1016/j.yfrne.2007.12.005.
Zhang QG, Wang RM, Scott E, Han D, Dong Y, Tu JY, et al. C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proc Natl Acad Sci U S A. 2011;108:E617–E24. https://doi.org/10.1093/brain/awt046.
Arevalo MA, Santos-Galindo M, Bellini M, Azcoitia I, Garcia-Segura LM. Actions of estrogens on glial cells: implications for neuroprotection. Biochim Biophys Acta. 2010;1800:1106–12. https://doi.org/10.1016/j.bbagen.2009.10.002.
Dhandapani KM, Brann DW. Role of astrocytes in estrogen-mediated neuroprotection. Exp Gerontol. 2007;42(1–2):70–5. https://doi.org/10.1016/j.exger.2006.06.032.
Brinton RD. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008;31(10):529–37. https://doi.org/10.1016/j.tins.2008.07.003.
Irwin RW, Yao J, Hamilton RT, Cadenas E, Brinton RD, Nilsen J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology. 2008;149(6):3167–75. https://doi.org/10.1210/en.2007-1227.
Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2004;3(4):297–13.
Nilsen J, Chen S, Irwin RW, Iwamoto S, Brinton RD. Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci. 2006;7:74. https://doi.org/10.1186/1471-2202-7-74.
Nilsen J, Irwin RW, Gallaher TK, Brinton RD. Estradiol in vivo regulation of brain mitochondrial proteome. J Neurosci. 2007;27:14069–77. https://doi.org/10.1523/JNEUROSCI.4391-07.2007.
Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer’s disease. Adv Drug Deliv Rev. 2008;60(13–14):1504–11. https://doi.org/10.1016/j.addr.2008.06.003.
Sakamoto H, Matsuda K, Hosokawa K, Nishi M, Morris JF, Prossnitz ER, et al. Expression of G protein-coupled receptor-30, a G protein-coupled membrane estrogen receptor, in oxytocin neurons of the rat paraventricular and supraoptic nuclei. Endocrinology. 2007;148(12):5842–50. https://doi.org/10.1210/en.2007-0436.
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–30. https://doi.org/10.1126/science.1106943.
Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;46(3):904–10. https://doi.org/10.1016/j.bbrc.2006.05.191.
Xu H, Qin S, Carrasco GA, Dai Y, Filardo EJ, Prossnitz ER, et al. Extra-nuclear estrogen receptor GPR30 regulates serotonin function in rat hypothalamus. Neuroscience. 2009;158(4):1599–607. https://doi.org/10.1016/j.neuroscience.2008.11.028.
Alyea RA, Laurence SE, Kim SH, Katzenellenbogen BS, Katzenellenbogen JA, Watson CS. The roles of membrane estrogen receptor subtypes in modulating dopamine transporters in PC-12 cells. J Neurochem. 2008;106(4):1525–33. https://doi.org/10.1111/j.1471-4159.2008.05491.x.
Kuhn J, Dina OA, Goswami C, Suckow V, Levine JD, Hucho T. GPR30 estrogen receptor agonists induce mechanical hyperalgesia in the rat. Eur J Neurosci. 2008;27(7):1700–9. https://doi.org/10.1111/j.1460-9568.2008.06131.x.
Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, et al. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci. 2006;26(21):5649–55. https://doi.org/10.1523/JNEUROSCI.0327-06.2006.
Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Functional differences between the amino-terminal domains of estrogen receptors alpha and beta. Mol Pharmacol. 2000;58(3):584–90. https://doi.org/10.1124/mol.58.3.584.
He S, Nelson ER. 27-Hydroxycholesterol, an endogenous selective estrogen receptor modulator. Maturitas. 2017;104:29–35. https://doi.org/10.1016/j.maturitas.2017.07.014.
Li X, Schwartz PE, Rissman EF. Distribution of estrogen receptor β-like immunoreactivity in rat forebrain. Neuroendocrinology. 1997;66:63–7.
Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–25. https://doi.org/10.1002/(SICI)1096-9861(19971201)388:4<507::AID-CNE1>3.0.CO;2-6.
Zhang JQ, Cai WQ, Zhou de S, Su BY. Distribution and differences of estrogen receptor beta immunoreactivity in the brain of adult male and female rats. Brain Res. 2002;935(1–2):73–80. https://doi.org/10.1016/S0006-8993(02)02460-5.
Mitra SW, Hoskin E, Yudkovitz J, Pear L, Wilkinson HA, Hayashi S, et al. Immunolocalization of estrogen receptor-α in the mouse brain: comparison with estrogen receptor β. Endocrinology. 2003;144(5):2055–67. https://doi.org/10.1210/en.2002-221069.
Brinton RD. Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol Sci. 2009;30(4):212–22. https://doi.org/10.1016/j.tips.2008.12.006.
Hajszan T, Milner TA, Leranth C. Sex steroids and the dentate gyrus. Prog Brain Res. 2007;163:399–416. https://doi.org/10.1016/S0079-6123(07)63023-4.
Dan P, Cheung JC, Scriven DR, Moore ED. Epitope-dependent localization of estrogen receptor-alpha, but not -beta, in en face arterial endothelium. Am J Physiol Heart Circ Physiol. 2003;284:H1295–306. https://doi.org/10.1152/ajpheart.00781.2002.
Mazzucco CA, Lieblich SE, Bingham BI, Williamson MA, Viau V, Galea LAM. Both estrogen receptor alpha and estrogen receptor beta agonists enhance cell proliferation in the dentate gyrus of adult female rats. Neuroscience. 2006;141(4):1793–800. https://doi.org/10.1016/j.neuroscience.2006.05.032.
Alkayed NJ, Murphy SJ, Traystman RJ, Hurn PD, Miller VM. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke. 2000;31(1):161–8. https://doi.org/10.1161/01.STR.31.1.161.
Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23(7):291–7. https://doi.org/10.1016/S0166-2236(00)01581-2.
Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine. 2001;14(3):407–15. https://doi.org/10.1385/ENDO:14:3:407.
Zaidi AU, D’Sa-Eipper C, Brenner J, Kuida K, Zheng TS, Flavell RA, et al. Bcl-X(L)-caspase-9 interactions in the developing nervous system: evidence for multiple death pathways. J Neurosci. 2001;21(1):169–75.
Wade CB, Dorsa DM. Estrogen activation of cyclic adenosine 5′-monophosphate response element-mediated transcription requires the extracellularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology. 2003;144:832–8. https://doi.org/10.1210/en.2002-220899.
Quesada A, Micevych PE. Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxydopamine lesions. J Neurosci Res. 2004;75(1):107–16. https://doi.org/10.1002/jnr.10833.
Hagen AA, Barr M, Diczfalusy E. Metabolism of 17β-oestradiol-4-14C in early infancy. Acta Endocrinol. 1965;49:207–20. https://doi.org/10.1530/acta.0.0490207.
Warmerdam EG, Visser M, Coeling Bennink HJ, Groen M. A new route of synthesis of estetrol. Climacteric. 2008;11(Suppl 1):59–63. https://doi.org/10.1080/13697130802054078.
Holinka CF, Diczfalusy E, Coeling Bennink HJTC. Estetrol: a unique steroid in human pregnancy. J Steroid Biochem Mol Biol. 2008;110(1–2):138–43. https://doi.org/10.1016/j.jsbmb.2008.03.027.
Visser M, Foidart J-M, Coelingh Bennink HJT. In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism. Climacteric. 2008;11(Suppl 1):64–8. https://doi.org/10.1080/13697130802050340.
Coeling Bennink HJTC, Skouby S, Bouchard P, Holinka CF. Ovulation inhibition by estetrol in an in vivo model. Contraception. 2008;77(3):186–90. https://doi.org/10.1016/j.contraception.2007.11.014.
Coelingh Bennink HJTC, Holinka CF, Diczfalusy E. Estetrol review: profile and potential clinical applications. Climacteric. 2008;11(Suppl 1):47–58. https://doi.org/10.1080/13697130802040077.
Hirano S, Furutama D, Hanafusa T. Physiologically high concentrations of 17beta-estradiol enhance NF-kappaB activity in human T cells. Am J Physiol Regul Integr Comp Physiol. 2007;292(4):R1465–71. https://doi.org/10.1152/ajpregu.00778.2006.
Koh KK, Yoon BK. Controversies regarding hormone therapy: insights from inflammation and hemostasis. Cardiovasc Res. 2006;70(1):22–30. https://doi.org/10.1016/j.cardiores.2005.12.004.
Herrington DM, Klein KP. Invited review: Pharmacogenetics of estrogen replacement therapy. J Appl Physiol (1985). 2001;91(6):2776–84. https://doi.org/10.1152/jappl.2001.91.6.2776.
Arnal JF, Valéra MC, Payrastre B, Lenfant F, Gourdy P. Structure-function relationship of estrogen receptors in cardiovascular pathophysiological models. Thromb Res. 2012;130(Suppl 1):S7–11. https://doi.org/10.1016/j.thromres.2012.08.261.
Deroo BJ, Korach KS. Estrogen receptors and human disease: an update. Arch Toxicol. 2012;86(10):1491–504. https://doi.org/10.1007/s00204-012-0868-5.
ACOG. Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123(4):896–901.
Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicenter randomised trial. Lancet. 2005;65(9460):663–70. https://doi.org/10.1016/S0140-6736(05)17946-X.
Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353(15):1574–84. https://doi.org/10.1056/NEJMcps050929.
Robertson CM, Perlman M. Follow-up of the term infant after hypoxic-ischemic encephalopathy. Paediatr Child Health. 2006;11(5):278–82.
Zanelli SA. http://emedicine.medscape.com/article/973501-overview?src=refgatesrc1#a5. Accessed 16 Jan 2015.
Bryce J, Boschi-Pinto C, Shibuya K, Black RE, WHO Child Health Epidemiology Reference Group. WHO estimates of the causes of death in children. Lancet. 2005;365(9465):1147–52.
Badawi N, Kurinczuk JJ, Keogh JM, Alessandri LM, O’Sullivan F, Burton PR, et al. Antepartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ. 1998;317(7172):1549–53. https://doi.org/10.1136/bmj.317.7172.1549.
Badawi N, Kurinczuk JJ, Keogh JM, Alessandri LM, O’Sullivan F, Burton PR, et al. Intrapartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ. 1998;317(7172):1554–8. https://doi.org/10.1136/bmj.317.7172.1554.
Graham EM, Ruis KA, Hartman AL, Northington FJ, Fox HE. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am J Obstet Gynecol. 2008;199(6):587–95. https://doi.org/10.1016/j.ajog.2008.06.094.
Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15(4):961–73. https://doi.org/10.1016/0896-6273(95)90186-8.
Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. https://doi.org/10.1152/physrev.00029.2006.
Chang YC, Huang CC. Perinatal brain injury and regulation of transcription. Curr Opin Neurol. 2006;19(2):141–7. https://doi.org/10.1097/01.wco.0000218229.73678.a8.
Domoki F, Kis B, Nagy K, Farkas E, Busija DW, Bari F. Diazoxide preserves hypercapnia-induced arteriolar vasodilation after global cerebral ischemia in piglets. Am J Physiol Heart Circ Physiol. 2005;289:H368–73. https://doi.org/10.1152/ajpheart.00887.2004.
Gerosa C, Fanni D, Puddu M, Locci G, Obinu E, Fanos V, et al. Histological markers of neonatal asphyxia: the relevant role of vascular changes. J Pediatr Neonat Individual Med. 2014;3(2):e030275.
Olah O, Toth-Szuki V, Temesvari P, Bari F, Domoki F. Delayed neurovascular dysfunction is alleviated by hydrogen in asphyxiated newborn pigs. Neonatology. 2013;104:79–86. https://doi.org/10.1159/000348445.
Ireland Z, Castillo-Melendez M, Dickinson H, Snow R, Walker DW. A maternal diet supplemented with creatine from mid-pregnancy protects the newborn spiny mouse brain from birth hypoxia. Neuroscience. 2011;194:372–9. https://doi.org/10.1016/j.neuroscience.2011.05.012.
Fleiss B, Coleman HA, Castillo-Melendez M, Ireland Z, Walker DW, Parkington HC. Effects of birth asphyxia on neonatal hippocampal structure and function in the spiny mouse. Int J Dev Neurosci. 2011;29:757–66. https://doi.org/10.1016/j.ijdevneu.2011.05.006.
Schiering IA, de Haan TR, Niermeijer JM, Koelman JH, Majoie CB, Reneman L, et al. Correlation between clinical and histologic findings in the human neonatal hippocampus after perinatal asphyxia. J Neuropathol Exp Neurol. 2014;73:324–34. https://doi.org/10.1097/NEN.0000000000000056.
Okereafor A, Allsop J, Counsell SJ, Fitzpatrick J, Azzopardi D, Rutherford MA, et al. Patterns of brain injury in neonates exposed to perinatal sentinel events. Pediatrics. 2008;121(5):906–14. https://doi.org/10.1542/peds.2007-0770.
Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress: a clinical and electroencephalographic study. Arch Neurol. 1976;33(10):696–705. https://doi.org/10.1001/archneur.1976.00500100030012.
Douglas-Escobar M, Weiss MD. Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA Pediatr. 2015;169(4):397–403. https://doi.org/10.1001/jamapediatrics.2014.3269.
Thoresen M, Hellstrom-Westas L, Liu X, de Vries LS. Effect of hypothermia on amplitude-integrated electroencephalogram in infants with asphyxia. Pediatrics. 2010;126(1):e131–9. https://doi.org/10.1542/peds.2009-2938.
Rothermundt M, Peters M, Prehn JH, Arolt V. S100B in brain damage and neurodegeneration. Microsc Res Tech. 2003;60(6):614–32. https://doi.org/10.1002/jemt.10303.
Ennen CS, Huisman TA, Savage WJ, Northington FJ, Jennings JM, Everett AD, et al. Glial fibrillary acidic protein as a biomarker for neonatal hypoxic-ischemic encephalopathy treated with whole-body cooling. Am J Obstet Gynecol. 2005;205(3):251.e1–7. https://doi.org/10.1016/j.ajog.2011.06.025.
Shankaran S. The postnatal management of the asphyxiated term infant. Clin Perinatol. 2002;29(4):675–92.
Stola A, Perlman J. Post-resuscitation strategies to avoid ongoing injury following intrapartum hypoxia-ischemia. Semin Fetal Neonatal Med. 2008;13(6):424–31. https://doi.org/10.1016/j.siny.2008.04.011.
Hoehn T, Hansmann G, Bührer C, Simbruner G, Gunn AJ, Yager J, et al. Therapeutic hypothermia in neonates. Review of current clinical data, ILCOR recommendations and suggestions for implementation in neonatal intensive care units. Resuscitation. 2008;78:7–12. https://doi.org/10.1016/j.resuscitation.2008.04.027.
Thoresen M, Tooley J, Liu X, Jary S, Fleming P, Luyt K, et al. Time is brain: starting therapeutic hypothermia within three hours after birth improves motor outcome in asphyxiated newborns. Neonatology. 2013;104(3):228–33. https://doi.org/10.1159/000353948.
Edwards DA, Azzopardi DV, Gunn AJ. Neonatal neural rescue: a clinical guide. Cambridge, UK: Cambridge University Press; 2013.
Ballot DE. Cooling for newborns with hypoxic ischaemic encephalopathy: RHL commentary (last revised: 1 October 2010). The WHO Reproductive Health Library. Geneva: World Health Organization (WHO).
Zanelli S, Buck M, Fairchild K. Physiologic and pharmacologic considerations for hypothermia therapy in neonates. J Perinatol. 2011;31(6):377–86. https://doi.org/10.1038/jp.2010.146.
Thoresen M, Whitelaw A. Therapeutic hypothermia for hypoxic-ischaemic encephalopathy in the newborn infant. Curr Opin Neurol. 2005;18(2):111–6. https://doi.org/10.1097/01.wco.0000162850.44897.c6.
Tskitishvili E, Nisolle M, Munaut C, Pequeux C, Gerard C, Noel A, et al. Neonatal estetrol attenuates neonatal hypoxic-ischemic brain injury. Exp Neurol. 2014;261:298–307. https://doi.org/10.1016/j.expneurol.2014.07.015.
Tskitishvili E, Pequeux C, Munaut C, Viellevoye R, Nisolle M, Noël A, et al. Use of estetrol with other steroids for attenuation of neonatal hypoxic-ischemic brain injury: to combine or not to combine? Oncotarget. 2016;7(23):33722–43. https://doi.org/10.18632/oncotarget.9591.
Tskitishvili E, Pequeux C, Munaut C, Viellevoye R, Nisolle M, Noël A, et al. Estrogen receptors and estetrol-dependent neuroprotective actions: a pilot study. J Endocrinol. 2017;232(1):85–95. https://doi.org/10.1530/JOE-16-0434.
Johnson GV, Jope RS. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J Neurosci Res. 1992;33(4):505–12. https://doi.org/10.1002/jnr.490330402.
Bartosik-Psujek H, Stelmasiak Z. Biochemical markers of damage of the central nervous system in multiple sclerosis. Ann Univ Mariae Curie Sklodowska Med. 2001;56:389–92.
Roof RL, Duvdevani R, Braswell L, Stein DG. Progesterone facilitates cognitive recovery and reduces secondary neuronal loss caused by cortical contusion injury in male rats. Exp Neurol. 1994;129:64–9. https://doi.org/10.1006/exnr.1994.1147.
Roof RL, Hoffman SW, Stein DG. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol Chem Neuropathol. 1997;31(1):1–11.
Roof RL, Hall E. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17(5):367–88. https://doi.org/10.1089/neu.2000.17.367.
Stein D. Brain damage, sex hormones and recovery: a new role for progesterone and estrogen? Trends Neurosci. 2001;24(7):386–91. https://doi.org/10.1016/S0166-2236(00)01821-X.
Gold SM, Voskuhl RR. Estrogen and testosterone therapies in multiple sclerosis. Prog Brain Res. 2009;175:239–51. https://doi.org/10.1016/S0079-6123(09)17516-7.
Kaur P, Jodhka PK, Underwood WA, Bowles CA, de Fiebre NC, de Fiebre CM, et al. Progesterone increases brain-derived neurotrophic factor expression and protects against glutamate toxicity in a mitogen-activated protein kinase- and phosphoinositide-3 kinase-dependent manner in cerebral cortical explants. J Neurosci Res. 2007;85(11):2441–9. https://doi.org/10.1002/jnr.21370.
Nilsen J, Brinton RD. Impact of progestins on estradiol potentiation of the glutamate calcium response. Neuroreport. 2002;13(6):825–30.
Nilsen J, Brinton RD. Impact of progestins on estrogen induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology. 2002;143(1):205–12. https://doi.org/10.1210/endo.143.1.8582.
Nilsen J, Brinton RD. Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen activated protein kinase signaling. Proc Natl Acad Sci U S A. 2003;100(18):10506–11. https://doi.org/10.1073/pnas.1334098100.
Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–44. https://doi.org/10.1046/j.1471-4159.1996.66051836.x.
Singh M, Su C. Progesterone and neuroprotection. Horm Behav. 2013;63(2):284–90. https://doi.org/10.1016/j.yhbeh.2012.06.003.
Trotter A, Steinmacher J, Kron M, Pohlandt F. Neurodevelopmental follow-up at five years corrected age of extremely low birth weight infants after postnatal replacement of 17-estradiol and progesterone. J Clin Endocrinol Metab. 2012;97(3):1041–7. https://doi.org/10.1210/jc.2011-2612.
Lorenz L, Dang J, Misiak M, Tameh Abolfazl A, Beyer C, Kipp M. Combined 17beta-oestradiol and progesterone treatment prevents neuronal cell injury in cortical but not midbrain neurones or neuroblastoma cells. J Neuroendocrinol. 2009;21(10):841–9. https://doi.org/10.1111/j.1365-2826.2009.01903.x.
Mannella P, Sanchez AM, Giretti MS, Genazzani AR, Simoncini T. Oestrogen and progestins differently prevent glutamate toxicity in cortical neurons depending on prior hormonal exposure via the induction of neural nitric oxide synthase. Steroids. 2009;74(8):650–6. https://doi.org/10.1210/me.2008-0408.
Aguirre CC, Baudry M. Progesterone reverses 17betaestradiol-mediated neuroprotection and BDNF induction in cultured hippocampal slices. Eur J Neurosci. 2009;29(3):447–54. https://doi.org/10.1111/j.1460-9568.2008.06591.x.
Aguirre C, Jayaraman A, Pike C, Baudry M. Progesterone inhibits estrogen-mediated neuroprotection against excitotoxicity by down-regulating estrogen receptor-beta. J Neurochem. 2010;115(5):1277–87. https://doi.org/10.1111/j.1471-4159.2010.07038.x.
Carroll JC, Rosario ER, Pike CJ. Progesterone blocks estrogen neuroprotection from kainate in middle-aged female rats. Neurosci Lett. 2008;445(3):229–32. https://doi.org/10.1016/j.neulet.2008.09.010.
Jayaraman A, Pike CJ. Progesterone attenuates oestrogen neuroprotection via downregulation of oestrogen receptor expression in cultured neurones. J Neuroendocrinol. 2009;21(1):77–81. https://doi.org/10.1111/j.1365-2826.2008.01801.x.
Rosario ER, Ramsden M, Pike CJ. Progestins inhibit the neuroprotective effects of estrogen in rat hippocampus. Brain Res. 2006;1099(1):206–10. https://doi.org/10.1016/j.brainres.2006.03.127.
Yao J, Chen S, Cadenas E, Brinton RD. Estrogen protection against mitochondrial toxin-induced cell death in hippocampal neurons: antagonism by progesterone. Brain Res. 2011;1379:2–10. https://doi.org/10.1016/j.brainres.2010.11.090.
Gibson C, Constantin D, Prior M, Bath P, Murphy S. Progesterone suppresses the inflammatory response and nitric oxide synthase-2 expression following cerebral ischemia. Exp Neurol. 2005;193(2):522–30. https://doi.org/10.1016/j.expneurol.2005.01.009.
Grossman K, Goss C, Stein D. Effects of progesterone on the inflammatory response to brain injury in the rat. Brain Res. 2004;1008(1):29–39. https://doi.org/10.1016/j.brainres.2004.02.022.
Labombarda F, Gonzalez S, Gonzalez Deniselle MC, Garay L, Guennoun R, Schumacher M, et al. Progesterone increases the expression of myelin basic protein and the number of cells showing NG2 immunostaining in the lesioned spinal cord. J Neurotrauma. 2006;23(2):181–92. https://doi.org/10.1089/neu.2006.23.181.
Pierson RC, Lyons AM, Greenfield LJ Jr. Gonadal steroids regulate GABAA receptor subunit mRNA expression in NT2-Nneurons. Brain Res Mol Brain Res. 2005;138(2):105–15. https://doi.org/10.1016/j.molbrainres.2004.10.047.
Mani SK. Signaling mechanisms in progesterone neurotransmitter interactions. Neuroscience. 2006;138(3):773–81. https://doi.org/10.1016/j.neuroscience.2005.07.034.
Pettus EH, Wright DW, Stein DG, Hoffman SW. Progesterone treatment inhibits the inflammatory agents that accompany traumatic brain injury. Brain Res. 2005;1049(1):112–9. https://doi.org/10.1016/j.brainres.2005.05.004.
Quadros PS, Pfau JL, Wagner CK. Distribution of progesterone receptor immunoreactivity in the fetal and neonatal rat forebrain. J Comp Neurol. 2007;504(1):42–56.
Jahagirdar V, Wagner CK. Ontogeny of progesterone receptor expression in the subplate of fetal and neonatal rat cortex. Cereb Cortex. 2010;20(5):1046–52. https://doi.org/10.1002/cne.21427.
Abot A, Fontaine C, Buscato M, Solinhac R, Flouriot G, Fabre A, et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor modulation, uncoupling nuclear and membrane activation. EMBO Mol Med. 2014;6(10):1328–46. https://doi.org/10.15252/emmm.201404112.
La Rosa P, Pesiri V, Leclerq G, Marino M, Acconcia F. Palmitoylation regulates 17β-estradiol-induced estrogen receptor-α degradation and transcriptional activity. Mol Endocrinol. 2012;26(5):762–74. https://doi.org/10.1210/me.2011-1208.
Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun. 2004;316:878–83. https://doi.org/10.1016/j.bbrc.2004.02.129.
Khalaj AJ, Yoon J, Nakai J, Winchester Z, Moore SM, Yoo T, et al. Estrogen receptor (ER) β expression in oligodendrocytes is required for attenuation of clinical disease by an ERβ ligand. Proc Natl Acad Sci U S A. 2013;110(47):19125–30. https://doi.org/10.1073/pnas.1311763110.
Prokai L, Prokai-Tatrai K, Perjesi P, Simpkins JW. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev Res. 2005;66(2):118–25. https://doi.org/10.1002/ddr.20050.
Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726–41. https://doi.org/10.1210/er.2007-0022.
Suzuki S, Gerhold LM, Bottner M, Rau SW, Dela Cruz C, Yang E, et al. Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors a and b. J Comp Neurol. 2007;500:1064–75. https://doi.org/10.1002/cne.21240.
Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, et al. Neuroprotection mediated through estrogen receptor-α in astrocytes. Proc Natl Acad Sci U S A. 2011;108:8867–72. https://doi.org/10.1073/pnas.1103833108.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 International Society of Gynecological Endocrinology
About this chapter

Cite this chapter
Tskitishvili, E., Foidart, J.M. (2019). Estetrol and Its Effects on the Damaged Brain. In: Brinton, R., Genazzani, A., Simoncini, T., Stevenson, J. (eds) Sex Steroids' Effects on Brain, Heart and Vessels. ISGE Series. Springer, Cham. https://doi.org/10.1007/978-3-030-11355-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-11355-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11354-4
Online ISBN: 978-3-030-11355-1
eBook Packages: MedicineMedicine (R0)