
Abstract
A novel dual-phase gradient material of a high-entropy alloy (HEA) FeCoCrNiMo0.15 was fabricated by spark plasma sintering (SPS) processing. After SPS, the HEA specimens consisted of a single face-centred cubic (FCC) phase in the centre but dual FCC and body-centred cubic (BCC) structures near the surface. Surprisingly, the sintering pressure was sufficient to influence the proportion of phases, and thus, the properties of HEA samples. The hardness of the specimen sintered under a pressure of 30 MPa increased gradually from 167 HV in the centre to 634 HV near the surface due to the gradual increase in the fraction of the transformed BCC phase. This phase transformation , which could be influenced by pressure indicated a major possibility for designing gradient HEA materials.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
Introduction
Due to the rapid development of modern engineering and manufacturing industries, the high-performance alloys need to be urgently developed. High-entropy alloys (HEAs) constitute a unique class of alloys exhibiting high strength and hardness , decent wear and corrosion resistance , and other attractive mechanical properties for both scientific research and practical applications [1,2,3,4]. For further studies on HEAs, phase transformations could be crucial for controlling their microstructures to obtain superior properties [5,6,7]. A significant amount of research on HEAs has been performed to study phase transformation using vacuum arc melting [8], and this procedure was usually restricted to laboratory settings. Using spark plasma sintering (SPS) to consolidate mechanically alloyed HEA powders is a promising method to obtain high-performance bulk HEAs [9, 10]. The sintering pressure is one of the most important parameters in SPS method [11]. Moreover, pressure can influence the equilibrium between gaseous and liquid phases. The influence of pressure is typically neglected for the equilibrium of two or more solid phases. Standard phase diagrams involve only composition and temperature as the relevant variables [12]. This approach is based on phase transformations involving only solid phases not being associated with significant volume changes unlike changes from liquid to gas. According to this theory, if the pressure could be controlled and further influence the phase transformation , it would represent a breakthrough in the field of controlling structure and performance of HEAs.
In the present study, a FeCoCrNiMo0.15 HEA was successfully prepared using the SPS method under different pressures. The gradient distribution of microstructures and mechanical properties in the SPS samples was investigated. The face-centred cubic to body-centred cubic (BCC) phase transformation was also studied. This phase transformation of FCC to BCC structure which can be influenced by SPS pressure indicates a major possibility to design a novel gradient materials of HEAs.
Experimental
The investigated alloy with a nominal composition FeCoCrNiMo0.15 (in at.%) was prepared using powder metallurgy . Powders consisting of particles under 200 mesh in size were prepared using gas atomization and were mechanically milled using conventional planetary milling equipment. Details on the milling process can be found in our recent study [13]. The milled powders were then added into a graphite die 40 in mm diameter and consolidated by using an HPD 25/3 SPS equipment under reduced pressure (10−3 Pa). The sintering temperature was 1150 °C and the pressures were 30, 35 and 40 MPa. After a holding time of 480 s, the sintered billets were cooled down to room temperature in the furnace . The bending strength of the samples (size: 12 × 2 × 30 mm3) was determined by Instron 3369 mechanical testing facility using the three-point method. Finally, the samples were annealed at 1473 K for 5 h in a sealed silica tube filled with Ar atmosphere. After annealing , the samples were quickly cooled down. Thanks to this last processing step, it is assumed that the studied alloys represent the high temperature stable state. Samples were prepared by mechanical grinding using 1200 to 4000 grit SiC papers followed by a final polishing step. An FEI Quanta FEG 250 scanning electron microscope (SEM) equipped with an energy dispersive X-ray (EDX) analyser was used to investigate the microstructure and chemical compositions of the sintered specimens. The phase constitution of the specimens was characterized using a Rigaku D/MAX-2550 VB + 18 Kw X-ray diffractometer (XRD) utilising Cu Kα radiation. The hardness of the alloy was determined using Buehler 5104 hardness tester under a 200 g load for 15 s and was averaged from three measurements.
Results and Discussion
Bending Test
Figure 1a–c shows the cross section of the HEAs sintered at 1150 °C under different pressures after bending tests. Judging from the contrast images, the microstructure did not consist of a single phase as those obtained after lower temperature sintering in our previous study [13]. The microstructure gradually changed along the radial direction, towards the centre of the samples. After sintering at different pressures, the samples exhibited different cracks after bending test without breaking.

Cross section of the FeCoCrNiMo0.15 HEA after bending tests sintered at 1150 °C under different pressures: a 30, b 35, c 40 MPa and d SEM image of the FeCoCrNiMo0.15 HEA after sintering at 1150 ℃ and 30 MPa. P1–P5 represent the points of the microhardness tests
The phase transformation occurred on the upper and lower surfaces of the three samples during SPS. The transformed region was the thickest in the immediate vicinity of the crack, and the thickness decreased gradually along the longitudinal direction of the cylinder towards both ends.
Hardness
Under the sintering pressure of 30 MPa, the microstructure exhibited a distinct gradient distribution as illustrated in Fig. 1d. No micro-cracks and micro-holes were observed. This indicates that the raw powders completely reacted with each other, and multiple phase structures were successfully synthesized during SPS. Moreover, the sample exhibited unique mechanical properties : its microhardness gradually changed.
The centre of the specimen consisted of a single-phase structure with an average microhardness of approximately 167 HV. As an increasing amount of the new structure precipitated, the hardness gradually increased as the distance from the surface decreased, and the hardness increased to 634 HV at 150 μm from the surface. The results of the microhardness tests are listed in Table 1.
Phase Identification
To understand the improved bending stress and increase in hardness due to the phase transformation , the phase constitution of the specimens was investigated using a combination of electron backscattered diffraction (EBSD ), EDS and XRD techniques.
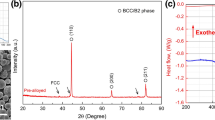
As shown in Fig. 2a, elemental Fe, Co, Cr, Ni and Mo could be obviously identified in the samples. The precipitated phase was a Cr-rich phase, containing relatively low amounts of elemental Co and Ni. Because no elemental Al was added, no intermetallic compound was formed; therefore, the precipitate was a hard, Cr-rich BCC phase. The distribution of the BCC structure in the specimen can be identified using XRD analysis as shown in Fig. 2b. The centre of the specimen that underwent SPS under 30 MPa of pressure was a single FCC structure. While gradually transitioning towards the surface of the specimen, the (111) FCC diffraction peak became less obvious than that of the centre. However, the peaks corresponding to the (110) and (200) planes could be identified, indicating the formation of the BCC structure. The high hardness of 633 HV of this structure after SPS (Table 1) could be mainly attributed to the effect of the hard BCC phases. In addition, EDX analysis (Table 2) showed that the FCC matrix was rich in Mo and Ni while the precipitated BCC structure was rich in Cr, indicating that the matrix was represented by the FCC phase and the precipitate was the disordered BCC phase. These results were in agreement with the XRD analysis results. Previous studies on the phase transformation of HEAs caused by SPS or the vacuum arc melting method reported that the BCC phases were composed of a spinodally modulated matrix, and precipitates exhibiting near-equiaxed shape were distributed uniformly throughout the HEA. The FCC phases exhibiting net-like structure were located at the boundaries of the BCC phases [14, 15]. However, the distribution of the BCC and FCC structures we observed was significantly different than those described in previous studies. Interestingly, the microstructure presented a significant gradient distribution as the volume fraction of the BCC structure increased. The mixed structure did not present a net-shaped, nor a dendritic form. This suggested that the FCC phase primarily formed during sintering, while the BCC phase precipitated. The BCC phase is a solid solution precipitate which dispersed and distributed in the FCC matrix.

Identifying the phase constitution of HEA using a combination of EBSD , EDX and XRD analysis. a SEM image, EBSD phase map (blue-BCC structure and red-FCC structure), and elemental distribution map of Fe, Co, Cr, Ni and Mo and b XRD analysis of the selected area
From Table 2, we learned that the Mo content of the BCC phase was smaller than 9%, while that of the FCC phase was approximately 25%. Thus, the FCC phase was enriched in Mo. This was due to the good inter-solubility of Cr, Fe, Co, and Ni, while the solubility of Mo in the other elements was poor. Therefore, during the dissolution process of the solid solution, Mo, as a solute element, was repelled towards the FCC phase and redistributed along with Cr, while Cr entered the solid solution. The precipitated BCC phase became rich in Cr and poor in Mo.
According to the above analysis, the precipitates were identified as the BCC structures that were transformed from the initial FCC structure during SPS. Therefore, it can be inferred that the BCC structure was much harder than the FCC structure. As can be seen in Fig. 1, the thickness of the BCC precipitates decreased gradually as the SPS pressure increased from 30 to 40 MPa. In addition, the results of the bending tests demonstrated that the SPS sample, which contained a larger volume fraction of BCC structure could resist greater bending stress. This observation was in agreement with Tsai’s work, who found that the hardness of the Al0.3CrFe1.5MnNi0.5 HEA nearly tripled after the sigma phase formed [16] since sigma phase is a very hard phase [17].
Heat Treatment (HT)
After HT the SPS specimens exhibited notable differences both in microstructures and phase compositions. Figure 3a shows the homogenization of the microstructure of the SPS specimen (P5 in Fig. 1d). The absence of the massive BCC phase was recorded after HT and was most likely due to its dissolution into the FCC phase during heating. The hardness of the SPS FeCoCrNiMo0.15 HEA specimens after HT no longer presented gradient-like distribution. However, the specimens maintained a moderate hardness of approximately 250 HV. This occurred because the samples were mainly single-phase after HT. However, the Cr-rich BCC particles (marked as B), which were approximately 5 μm in size, were not completely eliminated and were uniformly distributed at the grain boundaries, as shown in Fig. 3b. The dissolved BCC phase enhanced the Cr content of the FCC phase of the matrix (marked as A in Fig. 3b). Compared to the information in Table 2, due to the increase in Cr content in the matrix from 11.88 to 25.5% illustrated in Fig. 3b, the hardness of the FCC phase increased correspondingly.

a Microstructure of specimens collected from the centre of the SPS FeCoCrNiMo0.15 HEA after HT at 1200°C for 5 h; (b) Magnified microstructure in (a)
It is worth noting that the BCC phase dissolved into the FCC matrix again at the temperature of 1200 °C but it was still not completely transformed. So, it can be inferred that the temperature of FCC to BCC phase transformation was beyond 1200 °C. In fact, the phase transition temperature of FeCoCrNiMo0.15 HEA was about 1260 °C which was measured by DSC . So, the original FCC structure broke after pressure treatment in sintering and exhibited microstructure refinement which inevitably accompanied with dislocations. This made the increase in density of grain boundaries and dislocations, and therefore provided more locations and diffusion channels for nucleation of new nuclei in the phase transformation of FCC to BCC. The phase transformation became easier, which reduced the transformation temperature and shortened the time of transformation. Compared with Fig. 1(a), under the sintering pressure of 30 MPa, the FCC structure occurred about 50% volume fraction of phase transformation at 1150 °C, so the sintering pressure significantly reduced the phase transformation temperature. In addition, the density of BCC structure was less than FCC structure, however, the pressure treatment could increase the density of metallic materials and would lead to greater resistance of FCC to BCC phase transformation . This explained when sintering pressure rose to 35 and 40 MPa, the volume fraction of the transformed BCC structure decreased. Accordingly, the gradient distribution of microstructure is caused by the sintering pressure and by adjusting the sintering pressure , the volume fraction of phase transformation can be changed to obtain the required gradient materials.
Conclusion
The mechanical properties of the FeCoCrNiMo0.15 HEA exhibited a significantly gradual change because of gradient distribution of the mixed FCC and BCC structure. This gradient distribution was mainly caused by the sintering pressure . The hard, Cr-rich BCC phase was homogeneously distributed throughout the FCC matrix, and the volume fraction of the BCC structure increased from the centre to the surface of the SPS sample like it would for gradient materials. The volume fraction of transformed BCC structure can be easily adjusted by sintering pressure . The implication for control of properties via changing the phase balance in HEAs will provide a strong technical base for the potential applications of a novel gradient material.
References
Yeh JW, Chen SK, Lin SJ, Gao MC, Dahmen KA, Liaw PK, Lu ZP (2004) Advanced Engineering Material 6(5):299
Zhang Y, Zuo TT, Tang Z, Gao MC, Dahmen KA, Liaw PK, Lu ZP (2014) Prog Mater Sci 61:1–93
Tsai MH, Yeh JW (2014) Mater Res Lett 2(3):107
Chuang MH, Tsai MH, Wang WR, Lin SJ, Yeh JW (2011) Acta Mater 59:6308–6317
He JY, Wang H, Huang HL, Xu XD, Chen MW, Wu Y, Liu XJ, Nieh TG, An K, Lu ZP (2016) Acta Mater 102:187–196
Shun TT, Du YC (2009) J. Alloys Compd. 479:157–160
Li Z, Pradeep KG, Deng Y, Raabe D, Tasan CC (2016) Nature 534:227–230
Roy U, Roy H, Daoud H, Glatzel U, Ray KK (2014) Mater Lett 132:186–189
Praveen BS, Murty RS (2012) Mater Sci Eng A 534:83–89
Fu ZQ, Chen WP, Wen HM, Zhang DL, Chen Z, Zheng BL, Zhou YZ, Lavernia EJ (2016) Acta Mater 107:59–71
Mamedov V (2002) Powder Metall 45(4):322–328
Huang A, Hu D, Loretto MH, Mei J, Wu XH (2007) Scripta Mater 54(4):253–256
Zhang M, Zhang W, Liu Y, Liu B, Wang J (2018) Powder Metall 61(2):1–8
Wang J, Niu SZ, Guo T, Kou HC, Li JS (2017) J. Alloys Compd. 710:144–150
Zhang A, Han J, Meng J, Su B, Li P (2016) Mater Lett 181:82–85
Tsai MH, Chang KC, Li JH, Tsai RC, Cheng AH (2016) Mater Res Lett 4(2):1–6
Tsai MH, Yuan H, Cheng GM (2013) Intermetallics 33(2):81–86
Acknowledgements
This work was supported by the XUCHANG Fellowship Program [grant number: XW2017-40].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 The Minerals, Metals & Materials Society
About this paper

Cite this paper
Zhang, W., Zhang, M., Liu, F., Peng, Y., Hu, S., Liu, Y. (2019). A Novel Dual-Phase Gradient Material of High-Entropy Alloy Prepared by Spark Plasma Sintering. In: TMS 2019 148th Annual Meeting & Exhibition Supplemental Proceedings. The Minerals, Metals & Materials Series. Springer, Cham. https://doi.org/10.1007/978-3-030-05861-6_120
Download citation
DOI: https://doi.org/10.1007/978-3-030-05861-6_120
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-05860-9
Online ISBN: 978-3-030-05861-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)