
Abstract
The translation and appropriate folding of proteins is critical for the maintenance of cellular function. This process is tightly controlled, and it can create a significant energy demand, particularly in secretory cells. Inadequate folding of proteins, as may occur with an insufficient energy supply, can cause unfolded, misfolded, or damaged proteins to accumulate in the endoplasmic reticulum. The consequent endoplasmic reticulum stress leads to activation of the unfolded protein response (UPR). In stressed mitochondria, a similar process is activated (mitochondrial unfolded protein response). The unfolded protein response is an ancient stress response that coordinates multiple functions in an attempt to restore metabolic homeostasis. In higher organisms, three signaling arms, driven respectively by PERK, ATF6, and IRE1α, may be activated. Together, the signaling from these arms coordinates specific cellular functions including autophagy, cell metabolism, and apoptosis. From a cell fate perspective, the outcome of activating the unfolded protein response can be either the restoration of homeostasis and normal cell function, or the failure to do so leading to aberrant cellular function (including neoplastic transformation) and/or the eventual initiation of an irreversible programmed cell death. Hence, activation of the unfolded proteins response can be either prosurvival or prodeath. This book covers many aspects of the unfolded protein response, from its roles in normal cell development and some aspects of immunity, through to those associated with neoplastic transformation and drug resistance in cancer. Also included is a chapter on the role of UPR-activated autophagy in specific neurodegenerative disorders. The primary focus of these chapters is the unfolded protein response as activated by an endoplasmic reticulum stress. While each chapter may be read independently, the reader will gain a much broader perspective of the critical roles of the unfolded protein response when the chapters are read collectively.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Autophagy
- ATF4
- ATF6
- Endoplasmic reticulum stress
- Cancer
- Canonical signaling
- Diabetes
- eIF2α
- GRP78
- Hematopoietic
- IRE1α
- Metabolism
- Neurodegenerative
- PERK
- Protein synthesis
- Ribosome
- Translation
- Transcription
Introduction
Living cells must synthesize the proteins that perform the functions that sustain their life. These proteins often require folding into a specific conformation(s) that enables them to act. Cells expend a significant amount of their available energy in executing the process of protein folding. Unfolded, misfolded, or damaged proteins must be eliminated because the accumulation of these proteins within the endoplasmic reticulum (EnR) causes the EnR to swell, become stressed (EnR stress), and fail to function optimally. A similar process and response can occur within mitochondria [1]. To address the challenges that this proteotoxic stress creates for cell function, EnR stress can activate a process that identifies and targets unfolded proteins for degradation. The process that is activated is called the unfolded protein response (UPR); in mitochondria, this response is called the mitochondrial UPR (UPRmt) [2].
Since the requirements to fold proteins properly and to eliminate unfolded or damaged proteins are fundamental to life, features of the UPR likely arose relatively early during evolution. For example, components of UPR action are readily detected in species as diverse as yeast and human. The UPR network has three signaling arms regulated, respectively, by protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring enzyme 1 alpha (IRE1α). Each arm of the three UPR may have evolved over different timeframes. These is some controversy over which is the oldest arm, with the IRE1α arm often being considered the older. However, orthologs of IRE1α are rarely seen in protozoans, where activities similar to PERK signaling are more often detected [3].
In higher eukaryotes, the UPR can help the cell respond to stresses that include the limitations in cellular energy and oxygen that are of particular relevance to cancer cells growing in poorly vascularized solid tumor microenvironments. For example, the instruction to degrade unfolded or damaged proteins releases their amino acids and other components, including sugars and fatty acids, for reuse within the cells. This recycling can save energy by supporting intermediate metabolism within the cell [4]. Moreover, activation of the UPR, for example as induced following EnR stress, integrates the signaling that controls multiple cellular functions with the goal of enabling cell survival or executing a programmed cell death.
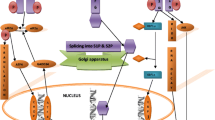
In its canonical signaling representations (Fig. 1.1), the three signaling arms of the UPR network are each regulated by the common upstream activator glucose-regulated protein-78 (GRP78), also known as binding immunoglobulin protein (BiP) or heat shock protein A5 (HSPA5; Human Genome Organization symbol for this gene). GRP78, a generally short-lived, EnR membrane-resident protein [5] is one of a series of molecular chaperones that bind to unfolded or misfolded proteins accumulating in the lumen of stressed EnR, and then targets these proteins for refolding or degradation. Unfolded, misfolded, or damaged proteins generally have features exposed on their surface that are recognized by chaperone proteins as being inappropriate. These features include cysteine residues that would normally be paired, hydrophobic regions that are often internal rather than on a protein’s surface, and/or immature glycans. Chaperone binding prevents unfolded proteins from aggregating and assists in their refolding or retrotranslocation back out of the EnR or mitochondria to the cytosol for degradation. When acting as a chaperone, GRP78 binding to misfolded proteins favors degradation in part by recognizing existing ubiquitin moieties and catalyzing additional ubiquitination [6]. Degradation of marked proteins involves the ubiquitin-proteasome system [7, 8]. An EnR-activated autophagy (ERAA) pathway also can be activated as a means to degrade unfolded proteins [9].

Simple representation of the structure of the canonical unfolded protein response (UPR) network. Upon activation, GRP78 (HSPA5, BiP) is released from each of the three sensors (PERK, ATF6, IRE1α) to act as a chaperone for unfolded, misfolded, or damaged proteins. Each sensor is activated upon release from GRP78 to initiate signaling in its respective UPR arm. Activated PERK controls eIF2α phosphorylation that can then inhibit translation and activate ATF4. ATF6 (90 kDa) is cleaved by site 1 and site 2 proteases to release a transcriptionally active cleaved ATF6 (50 kDa). Activated IRE1α removes a 26 bp fragment from unspliced XBP1 (XBP1U) creating a frame shift that encodes for a spliced XBP1 (XBP1S) that can now act as a nuclear transcription factor
GRP78 is normally bound to three EnR membrane-resident proteins (PERK, ATF6, and IRE1α), which are consequently maintained in an inactive state. When released from GRP78, which leaves to fulfill its molecular chaperone role in response to an EnR stress, PERK, ATF6, and IRE1α can regulate their respective signaling cascades. Unresolved UPR activation could lead to prolonged repression of transcription and translation, and to excessive autophagic degradation of critical cellular components, that can induce an irreversible cell death cascade [10]. Hence, feedback inhibition within the UPR can normally return signaling to its resting state once the stress has been resolved. The complexity of these and other control mechanisms within the UPR is still being defined. Perhaps the feedback control that has been most widely studied is that controlling the regulation of eukaryotic initiation factor 2-alpha (eIF2α) phosphorylation in the PERK arm of the UPR. eIF2α can control the rate of protein translation. When dephosphorylated by PP1, as regulated by activating transcription factor 4 (ATF4) and growth arrest and DNA damage-inducible protein-34 (GADD34 ; PPP1R15A), the eIF2α-driven inhibition of translation is relieved and the rate of translation can return to normal.
The importance of the UPR in managing protein load has been studied in both normal and diseased tissues, with key fundamental work evident in the pancreas with respect to insulin production in the normal and diabetic states [11, 12] and in hematopoietic cells [13, 14]. The importance of regulating unfolded proteins in prion diseases has recently begun to attract notable attention. Inappropriate protein aggregation is particularly problematic in the brain and contributes to several major neurological disorders [15]. The need to ensure appropriate control of protein production and folding is likely to be different in tissues that secrete large amounts of proteins when compared with those that are non-secretory and/or not proliferating.
In several cancers and other diseases, UPR components can be regulated differently from normal tissues. The control of cell proliferation and survival as determined by estrogen receptor alpha (ER; ESR1) activation in many breast cancers has major implications for cell fate determination [16]. The importance of endocrine regulation of the UPR, and loss of this regulation when cells become resistant to drugs that target ER activity, has been established [4, 10, 16,17,18]. In this example, aspects of canonical UPR signaling are evident in cancer cells. However, the control of individual features of canonical signaling can be modified by hormones, growth factors, or other changes in cells including those affected by other signals received from the tumor microenvironment. Together, these intrinsic and extrinsic activities contribute significantly to cell context-specific modifications to UPR regulation and execution within an individual cancer cell.
The timing of activation of each UPR arm, and the relative importance of each arm once activated, remains unclear and likely differs by cell context and the nature and/or potency of the stressor(s). UPR components activated differentially in duration or level of activity likely also contribute to whether the ultimate cell fate outcome is death versus survival and/or proliferation. Some cells can activate an anticipatory UPR, including estrogen responsive breast cancer cells [18]. Translational inhibitory activities often occur first and within minutes of UPR activation [19]. Blocking the translation of existing mRNA templates would have an immediate effect on the number of protein molecules requiring folding and produce a rapid decline in the energy needs within the cell. The speed of activation implies that the executors of this inhibition are already present and do not require transcription or translation. Inhibition of transcription occurs later, often concurrent with chromatin remodeling [19]. This sequencing allows for the general inhibition of mRNA and protein production and the preferential synthesis of any new proteins required (including molecular chaperones) and (ideally) only in the amounts the cell can manage. Concurrent chromatin remodeling would allow the cells to retain this pattern of expression for prolonged periods and possibly dampen the potential for the transcriptional regulation from UPR activation to cycle on and off inappropriately. For any cell already committed to completing a turn of the cell cycle, its progeny also would arise programmed to manage their protein production in a stressed environment, likely also reflecting any cell context-specific modifications.
A key consequence of UPR activation is its direction to degrade unfolded proteins through an EnR-associated degradation (ERAD) [20, 21]. For example, misfolded glycoproteins are managed by genes in the EDEM family (EnR degradation-enhancing α-mannosidase-like protein). This process targets the misfolded proteins for eventual degradation by the ubiquitin-proteasome [22]. Proteins bound to chaperones are also removed through chaperone-mediated autophagy; other proteins targeted for degradation may be collected from the cytosol through microautophagy [23, 24]. Macroautophagy can also be induced by UPR signaling [4]. It is through the degradation of unfolded or damaged proteins, or damaged or excess organelles, that the products so released can be used as substrates for intermediate metabolism to help restore metabolic homeostasis. Hence, signaling through the UPR network to control autophagy and cellular metabolism represents an example of the functional integration required to optimize the ability of a cell to respond to stress.
Within the UPR network, the signaling arms that are perhaps the most widely studied for their impact on cell fate are those for PERK and IRE1α. While the control of transcriptional responses to stress are complex [19], activation of the PERK signaling arm can essentially modify the rates of transcription, translation, and protein transport into the EnR to help align the resources available with the protein folding load and so restore homeostasis. This arm can also initiate cell death signaling through activation of C/EBP homologous protein (CHOP ; DDIT3). Conversely, activation of IRE1α leads to the unconventional splicing of X-box binding protein-1 (XBP1), which can drive prosurvival signaling through XBP1 regulation of select members of the BCL2 family and the coordinated regulation of autophagy and apoptosis [4, 10]. The balance of prodeath and prosurvival signaling is a major determinant of cell fate in normal and neoplastic cells. In this regard, the altered regulation of UPR signaling appears to be central to the process of neoplastic transformation and cancer cell survival [25], in addition to representing a fundamental component of cancer cells’ adaptive responses to the cellular stresses induced by many systemic anticancer therapies. Thus, the UPR likely also contributes to de novo and/or acquired resistance to some anticancer drugs.
A Brief Overview of This Book
The various activities of the UPR noted above are described and discussed in more detail throughout this book. Starting with the role of UPR in development and ending with a description of its regulation of autophagy and the consequences thereof for neuronal cell fate in neurodegenerative diseases, UPR network signaling and function are presented from multiple different perspectives. Each chapter in this book is written so that it can be read and understood independently, without necessarily referring to other chapters. While this leaves some ground covered more than once, this approach achieves two important goals. Firstly, it presents the UPR in different contexts while underscoring the central nature of this ancient response in cancer biology. Secondly, because each chapter can stand alone, the reader can choose the chapters of greatest relevance or interest without the need to read the book in its entirety. While it is not possible to cover all aspects of the UPR in normal development and in disease, the chapters in this book provide readers with insight from several perspectives into the importance of this stress response network. Moreover, components of the UPR that can affect other functions independent of the need to ameliorate multiple different forms of cellular stress are also considered.
The chapter by Dominicus et al. [26] addresses the role of EnR stress and the UPR in the context of development. Each of the major control components of the UPR is introduced (PERK, IRE1α, ATF6), providing an excellent overview of the canonical processes attributed to UPR signaling. In the context of development, the authors explore the role of the IRE1-XBP1 arm in embryonic liver and placenta, pancreas and stomach, B-cells and plasma cells, dendritic cells, osteogenesis, and in an epithelial-to-mesenchymal transition (EMT). The PERK-eIF2α-ATF4 arm is considered in the development of the pancreas, and the processes of osteogenesis and adipogenesis. While the ATF6 arm is perhaps the least well studied of the three UPR arms, its key role in the development of myoblasts, osteogenesis, and photoreceptor biology is presented. A short section also discusses the role of ATF6 paralogues in cellular development. Overall, it is now evident that the role of the UPR extends well beyond a canonical EnR stress response network for cells [27]. While it might be expected that these activities would be most evident in secretory cells, the UPR is also a key player in the development of non-secretory cells. This latter series of activities in non-secretory cells may be a broader reflection of the general need of all cells to maintain control over the use of their available resources to synthesize and fold the new proteins essential for the repair and replacement of damaged or aged organelles and to maintain basic cellular functions over time.
The roles of the UPR in the development of neoplasia and anticancer drug responsiveness are introduced in the chapter by Morreall et al. [28]. Following on from the observations from the development of normal cells in the prior chapter, these authors address the role of UPR signaling in enabling the maintenance of cell proliferation and neoplastic transformation, and of an EMT that has been widely implicated in the acquisition of a more aggressive, metastatic cancer phenotype. This chapter places the co-opting of the UPR at the center of these critical biological processes as they occur along the spectrum of changes that arise with the development of cancer, on through to enabling cancer cells to adapt to the stressors imposed by the systemic interventions applied during cancer treatment. As noted above, regulation of key components of the UPR can be altered in cancer cells. Examples include regulation of PERK activity by the oncogene MYC [29] and the requirement of MYC-transformed cells for XBP1 signaling to maintain growth [30]. MYC is frequently dysregulated in breast cancer, where MYC can regulate the UPR and both glucose and glutamine uptake [31]. Here, Morreall et al. provide critical insights into how the UPR can regulate key aspects of cellular metabolism and cell fate, with their text organized respectively by the UPR arms of PERK, ATF6, and then IRE1α. Reflecting the complexity of signaling within the UPR, coordination among PERK, ATF6, and IRE1α is introduced briefly with clarity and specific examples. Following a similar organization, the chapter continues by discussing pharmacological interventions for each UPR arm, concluding with valuable insights into what the future may hold for anticancer treatments that could more effectively target the prosurvival signaling of the UPR. The hypoxia present in tumor microenvironments is also introduced here, and discussed in depth with a focus on drug resistance in the following chapter by Singleton and Harris.
Areas of hypoxia are common in solid tumors, reflecting their often poor and heterogeneous vascularization [32]. Since low oxygen levels can affect cellular metabolism and the oxidative environment of the EnR, reduced capacity for protein folding occurs leading to the accumulation of unfolded proteins and UPR activation [33]. Focusing on the role of ATF4, Singleton and Harris [34] provide a clear and detailed review of the role of hypoxia and UPR activation in conferring resistance to chemotherapy and radiotherapy. The authors begin with an overview of hypoxia and the molecular responses to a low oxygen environment, most notably the hypoxia inducible gene family (HIF) and HIF-related signaling. Activation of ATF4 also introduces one of the key integration points between the UPR and the cellular process of autophagy. Further integration between autophagy and UPR is described in the chapters by Clarke (on ER+ breast cancer) and Moussa (on neurodegenerative diseases). Here, Singleton and Harris describe how ATF4 signaling contributes to redox metabolism, amino acid homeostasis, angiogenesis, and cell invasion and metastasis. The role ascribed to ATF4 in this latter process reflects its role in regulating an EMT, a relationship introduced by Dominicus et al. earlier in this book. How ATF4 can alter responsiveness to both chemotherapy and radiotherapy is then presented, followed by some emerging approaches to targeting eIF2α, PERK, and ATF4. While in the relatively early phases of discovery, these approaches to targeting key features of UPR signaling could offer important new therapeutic interventions that could be of significant value to some cancer patients.
UPR signaling initiated by activation of PERK leads to phosphorylation of the eIF2α complex. The consequences of acute and chronic activation of eIF2α are compared in the chapter by Sengupta et al. [35]. The initial goal of eIF2α regulation is to inhibit global protein translation, thereby reducing the load of newly synthesized proteins for folding within the EnR. When considered in the context of the concurrent removal, degradation, and recycling of damaged or unfolded proteins, this effect can enable the cell to better align its available resources with the number of protein molecules that need appropriate folding. If homeostasis is restored, the cell will not induce a programmed cell death. In marked contrast, chronic activation of eIF2α-related signaling can lead to initiation and execution of apoptosis [36]. Appropriate regulation of this pathway is one of the critical control features of the UPR and can explain, in part, why activation of the UPR can result in either cell survival or cell death. This chapter describes in detail how the PERK arm of the UPR regulates the protein translational machinery that plays a critical role in deciding a cell’s fate following an EnR stress.
The next two chapters address the roles of XBP1 in breast cancer. The first of these chapters describes the roles of unspliced XBP1 (XBP1-U) and spliced (XBP1-S). The second of these chapters looks more closely at the role of XBP1 in triple negative breast cancers (TNBC), those that lack detectable expression of ER, progesterone receptor (PR), and the oncogene HER2. A later chapter by Clarke includes XBP1 activities within the broader role of the UPR as induced by endocrine therapies in ER+ breast cancer cells and their responsiveness to these agents.
As described by Hu and Clarke [37], unlike almost all other mRNAs that are spliced, XBP1 splicing occurs outside spliceosome assemblies. Rather, the endoribonuclease activity of IRE1α removes a 26 bp intron from within the unspliced XBP1 transcript. Splicing produces a frame shift that creates a template for translating an XBP1 protein now capable of acting as a transcription factor. XBP1 proteins arising from both the spliced (29 kDa) and unspliced transcripts (56 kDa) can be detected in human cancer cells. While the XBP1-S protein is more stable and has a longer half-life, XBP1-U can act as an endogenous dominant negative of XBP1-S. This chapter details the relative importance of both XBP1-U and XBP1-S, including a discussion of XBP1-S activities that occur outside the canonical UPR signaling. Indeed, several chapters include discussions of activities that can regulate key components of the UPR in a manner outside what is currently thought of as being canonical UPR signaling.
The second chapter on XBP1 focuses on another important breast cancer molecular subtype. Representing approximately 15% of all newly diagnosed breast cancers, TNBCs are often highly aggressive. Moreover, unlike the remaining two molecular subgroups (ER+ and/or PR+; HER2+) there is no standard molecular targeted therapy for TNBCs [38, 39]. In the chapter by Zhao et al. [40], the authors provide powerful new insights into the role of XBP1 as a central driver of the TNBC phenotype. An overview of the role of the UPR in normal mammary gland development provides critical background for the subsequent discussion of its role in TNBCs. Importantly, the authors link the UPR to hypoxia, nutrient deprivation, and MYC activity, extending the discussion to other important cancer-associated genes that are often overexpressed or mutated in breast and other cancers (RAS, PI3K, TP53). Some of these subjects were introduced briefly in earlier chapters, further reinforcing the central role of the UPR in cancer biology. This chapter further details the role of the UPR in cell communication within the tumor microenvironment and presents some exciting new therapies that could be developed by targeting features of the UPR often upregulated in cancer cells.
The most commonly diagnosed breast cancer subtype is comprised of tumors that express ER and/or PR. These tumors often respond initially to an endocrine therapy that either targets the synthesis of 17β-estradiol (aromatase inhibitors such as Letrozole or Anastrozole) or inhibits ER activity (selective estrogen receptor modulators, SERMs, like Tamoxifen or selective estrogen receptor downregulators, SERDs, like Fulvestrant) [41]. The chapter by Clarke [42] describes a stress response to these endocrine therapies that is centered on activation of the UPR and its regulation of autophagy and apoptosis. There are multiple ways the UPR can be activated by these drugs including through altered glucose or glutamine metabolism and/or reduced ATP production, each of which may lead to the release of GRP78 and UPR activation. Other sensors of altered energy production including AMPK can also affect the activation of UPR and its control of autophagy and apoptosis. Key features of this integrated stress response may be affected by genes commonly mutated in ER+ breast cancers such as PI3K and AKT. However, unlike HER2 as a driver of HER2+ breast cancer biology, these mutations alone may not be particularly powerful drivers of endocrine responsiveness in many ER+ breast tumors because of complex feedback control signaling that may dampen their signaling. Thus, this chapter describes components of an integrated network that can explain the role of known gene mutations but that does not require these to explain the role of the UPR as an integrator of endocrine responsiveness, and how UPR signaling may be affected by unique features of the ER+ cellular context.
While canonical signaling within the UPR has been well documented, as noted above cellular context can alter how the signals flow through the entire UPR network. Adding to the complexity of UPR-related signaling, several of its key components are regulated by activities that fall outside canonical representations. The insightful chapter by Cook [43] addresses these issues from the perspective of GRP78, the common regulator of each of the three canonical arms of the UPR. Here, the role of plasma membrane bound GRP78, a feature of many cancers [44], is explored in detail. Similarly, GRP78 location on the mitochondria and secreted GRP78 are discussed in the context of their respective contributions to cancer cell biology. These observations have clear implications for how protein subcellular localization may affect the ability to interpret data correctly. Our understanding of the role of immunity in response to cancer and its treatment has begun to advance rapidly in recent years. What is now evident in cancer immunology research is the relative importance of how the UPR may affect multiple aspects of innate and adaptive immunity with clear implications for tumor biology [45]. Hence, the final sections of this chapter begin to address an important feature of the UPR that is less fully covered in the preceding chapters, that is the role of the UPR in immunity.
Several of the earlier chapters in this book have emphasized the critical relationship between the UPR and autophagy. While most chapters have addressed different aspects of the role of the UPR in cancer biology, the inappropriate accumulation of proteins is a notable feature of some neurodegenerative diseases [46, 47]. For example, PERK activation is closely associated with the progression of several neurodegenerative diseases, offering a potential target for therapeutic interventions [48]. The chapter by Moussa [49] provides readers with a perspective beyond cancer by addressing the role of the UPR in the biology of neurodegenerative diseases, with a primary focus on the consequences of its regulation of autophagy. The author carefully introduces the role of an inappropriate accumulation of proteins in neurons. Targeting this accumulation could lead to potentially transformative treatments for diseases that, for the moment, remain intractable therapeutically where the intent is to achieve a cure. Subsequent sections describe the process of autophagy in detail. These sections will be particularly useful to readers unfamiliar with the cellular process of autophagy, independent of the neurodegenerative disease setting in which it is presented here.
Clearly, the UPR plays a critical role in maintaining the balance between the energy needed to fold proteins and, particularly in secretory cells, the need to manage a high load of proteins without compromising cell function and survival. For cancers that arise in secretory tissues, such as breast, prostate, pancreas, salivary gland, and some immune cells, the ability to use the UPR in an attempt to manage the stresses applied from therapeutic interventions is already hard-wired as part of their respective biology [4]. Thus, targeting the UPR, perhaps specifically those features that are uniquely regulated in a cell context-specific manner, may offer the opportunity to develop more effective and more personalized treatments for some of the most common and most lethal cancers.
When taken together, the work presented in this book will provide readers with a detailed but accessible introduction to the UPR and its potential as a target for the development of new anticancer strategies. The section below provides some insight into some future research directions in areas where current knowledge may be limited.
Looking Forward
As is evident from each of the chapters in this book, signal flow through the UPR network is complex. Moreover, the extent to which a canonical representation is useful to understand this signaling, and particularly how this is affected by cell context (including stress-specific responses), is not always clear. Crosstalk among the three UPR signaling arms may also be commonplace and cell context specific, both in nature (e.g., which nodes use which connections) and in time (e.g., when are specific interactions or signaling paths used and when not). As a general guide and as a place to start, static canonical models clearly have their place [16]. However, in dynamic signaling networks with complex feedback and feedforward control and crosstalk among signaling pathways, signaling is often difficult (or impossible) to interpret or predict heuristically. For example, the effects of some signals can be non-linear, where small changes in one component produce a much larger change in another [50]. Mathematical models can be very useful in this setting, particularly where it is important to predict the likely consequences of a measured change in the expression or activation state of key genes. Given the known complexity of signaling and the often sparsity of data, there are relatively few such models of the UPR and these tend to be relatively high level [16, 51]. Moreover, the extent to which canonical signaling is modified by cellular context is often unclear, as is the degree and nature of coordination and integration of signaling crosstalk among the three UPR arms. For complex disease states, including those evident in heterogeneous tumor cell microenvironments, there is likely much yet to be discovered about how the UPR is differentially regulated. Predicting the consequences of UPR activation for the survival and proliferation of cancer cells, and their adaptive responses to systemic treatment interventions, is an area that likely also requires considerable additional research. This line of research may be greatly facilitated by appropriate computational and mathematical modeling [50].
While activation of the UPR can modify rates of transcription and translation, the proteins a cell needs to function normally, particularly in response to different stressors, likely varies significantly with time and with exposure to cell intrinsic and extrinsic stress. How the UPR is directed to ensure that adequate amounts of the most critical proteins are made in a cell context-specific manner, is another area where there are likely gaps in current knowledge. Indeed, it remains to be seen what level of specificity is required and achieved in this regard beyond those events regulated specifically within the UPR network, at least as this network is understood canonically. Some of these decisions may be made outside the canonical UPR (or the cell) yet affect UPR network signaling. For example, the expression of some genes may be controlled by transcription factors that respond to signals from the cell’s microenvironment. In cancer cells, intrinsic events may alter signal flow through the UPR network. Signal flow through a locality of the network may be altered when a driver mutation is acquired during neoplastic transformation or tumor progression. Even if the UPR network connections retain features present in current canonical signaling representations, the weight or importance of each connection may then differ. Changes in the use of different individual nodes is often seen in complex signaling networks that can exhibit small world properties where each node (e.g., mRNA, protein, metabolite) can be reached by connecting through only a small number of other nodes [52]. Thus, there may be cell context-specific preferred signaling routes for UPR activation and execution. Here again, mathematical models may be useful in understanding critical nuances in UPR network signal flow. These issues become more important when looking for drug combinations to block prosurvival signaling through the UPR, as would usually be the case in developing new cancer treatment modalities. Several chapters in this book discuss potential drugs that may be useful in developing new intervention strategies for cancer patients. Developing appropriate drug combinations and schedules may require a greater understanding of UPR signaling, including the dynamical features of signal flow (most signaling is inherently directed and dynamic), and features of redundancy and degeneracy that may confer apparently emergent properties on UPR signaling as are often evident in other signaling networks [50].
Timing of UPR induction is clearly critical if a cell is to be proactive rather than only reactive. Since it is evident that the UPR is not merely a stress response network, it is not surprising that some signals may induce the UPR in advance of the build-up of proteins for folding in the EnR. For example, in secretory cells that receive an external signal to initiate the production of proteins for secretion, waiting for the cell to become stressed from an increased protein load before activating homeostatic regulatory functions could put the cell’s ability to survive at risk. Prewiring of the UPR is evident in response to heat shock [19] and is generally called an anticipatory UPR response [53]. Thus, the UPR is activated in advance of increased protein translation [54]. The anticipatory prewiring of key UPR stress response features is a relatively new and exciting area of research [18]. Moreover, the anticipatory aspects of the UPR are likely to have broad implications beyond the cell models in which they were first reported.
While much of the research described in this book tends to treat the UPR in the context of its action within the EnR, it is evident that the UPR can also be activated in mitochondria [1]. As the primary energy source for a cell, preservation of mitochondrial function is critical for cell survival. Hence, mitochondrial UPR (UPRmt) is induced in response to the accumulation of unfolded or damaged proteins within mitochondria. To maintain integrity and functionality of the mitochondrial proteome, UPRmt activates retrograde signaling that coordinates actions within both the mitochondrial and nuclear genomes [1]. Importantly, the UPRmt ensures continued oxidative phosphorylation through signaling that involves accumulation of ATFS-1 [55]. Perhaps reflecting its role in several aspects of the immune response, UPRmt can initiate a protective innate immune response to eliminate pathogens that attack mitochondrial function [56]. A greater research focus on the UPRmt is anticipated in the near future.
The UPR is a highly coordinated network that controls and/or integrates multiple cellular functions that can support cell development and restore key cellular functions to homeostasis during stress. Whether initiated and executed within the EnR or mitochondria of cancer cells, key components of its signaling offer targets for novel therapeutic intervention. Research into the UPR and its role in cancer biology continues to receive increasing attention. Thus, it is hoped that this volume will provide a useful introduction and reference for its readers.
References
Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response-synchronizing genomes. Curr Opin Cell Biol. 2015;33:74–81.
Arnould T, Michel S, Renard P. Mitochondria retrograde signaling and the UPR mt: where are we in mammals? Int J Mol Sci. 2015;16:18224–51.
Hollien J. Evolution of the unfolded protein response. Biochim Biophys Acta. 2013;1833:2458–63.
Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, Warri A, Shajahan AN. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321–31.
Shim SM, Choi HR, Sung KW, Lee YJ, Kim ST, Kim D, Mun SR, Hwang J, Cha-Molstad H, Ciechanover A, Kim BY, Kwon YT. The endoplasmic reticulum-residing chaperone BiP is short-lived and metabolized through N-terminal arginylation. Sci Signal. 2018;11. pii: eaan0630.
Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata K. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell. 2008;19:1328–36.
Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–52.
Shiber A, Ravid T. Chaperoning proteins for destruction: diverse roles of Hsp70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome. Biomol Ther. 2014;4:704–24.
Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–50.
Clarke R, Shajahan AN, Wang Y, Tyson JJ, Riggins R, Weiner LM, Baumann WT, Xuan J, Zhang B, Facey C, Aiyer H, Cook K, Hickman FE, Tavassoly I, Verdugo A, Chen C, Zwart A, Wärri A, Hilakivi-Clarke LA. Endoplasmic reticulum stress, the unfolded protein response, and gene network modeling in antiestrogen resistant breast cancer. Horm Mol Biol Clin Invest. 2011;5:35–44.
Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–93.
Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–33.
Mohrin M, Widjaja A, Liu Y, Luo H, Chen D. The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell. 2018;17:e12756.
Sigurdsson V, Miharada K. Regulation of unfolded protein response in hematopoietic stem cells. Int J Hematol. 2018;107:627.
Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nat Rev Genet. 2018;19:405–18.
Tyson JJ, Baumann WT, Chen C, Verdugo A, Tavassoly I, Wang Y, Weiner LM, Clarke R. Dynamic modeling of oestrogen signalling and cell fate in breast cancer cells. Nat Rev Cancer. 2011;11:523–32.
Rajapaksa G, Thomas C, Gustafsson JA. Estrogen signaling and unfolded protein response in breast cancer. J Steroid Biochem Mol Biol. 2016;163:45–50.
Livezey M, Kim JE, Shapiro DJ. A new role for estrogen receptor alpha in cell proliferation and cancer: activating the anticipatory unfolded protein response. Front Endocrinol. 2018;9:325.
Vihervaara A, Duarte FM, Lis JT. Molecular mechanisms driving transcriptional stress responses. Nat Rev Genet. 2018;19:385–97.
Kondratyev M, Avezov E, Shenkman M, Groisman B, Lederkremer GZ. PERK-dependent compartmentalization of ERAD and unfolded protein response machineries during ER stress. Exp Cell Res. 2007;313:3395–407.
Tsai YC, Weissman AM. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer. 2010;1:764–78.
Kleiger G, Mayor T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014;24:352–9.
Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365–81.
Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet. 2007;16:618–29.
Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V, Hetz C, Chevet E. Endoplasmic reticulum proteostasis in glioblastoma-from molecular mechanisms to therapeutic perspectives. Sci Signal. 2017;10. pii: eaal2323.
Dominicus CS, Patel V, Chambers JE, Malzer E, Marciniak SJ. Endoplasmic reticulum stress signaling during development. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Arensdorf AM, Diedrichs D, Rutkowski DT. Regulation of the transcriptome by ER stress: non-canonical mechanisms and physiological consequences. Front Genet. 2013;4:256.
Morreal J, Hong F, Li Z. Regulation of the unfolded protein response and its roles in tumorigenesis and cancer therapy. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Bu Y, Diehl JA. PERK integrates oncogenic signaling and cell survival during cancer development. J Cell Physiol. 2016;231:2088–96.
Xie H, Tang CH, Song JH, Mancuso A, Del Valle JR, Cao J, Xiang Y, Dang CV, Lan R, Sanchez DJ, Keith B, Hu CC, Simon MC. IRE1alpha RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J Clin Invest. 2018;128:1300–16.
Shajahan-Haq AN, Cook KL, Schwartz-Roberts JL, Eltayeb AE, Demas DM, Warri AM, Facey CO, Hilakivi-Clarke LA, Clarke R. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol Cancer. 2014;13:239.
Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92.
Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5:723–8.
Singleton DC, Harris AL. ATF4, hypoxia and treatment resistance in cancer. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Sengupta S, Jordan VC, Clarke R. Role of protein translation in the unfolded protein response. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Aarti I, Rajesh K, Ramaiah KV. Phosphorylation of eIF2 alpha in Sf9 cells: a stress, survival and suicidal signal. Apoptosis. 2010;15:679–92.
Hu R, Clarke R. Roles of spliced and unspliced XBP1 in breast cancer. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–90.
Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M, Sinha N. Therapeutic targets of triple-negative breast cancer: a review. Br J Pharmacol. 2015;172:4228–37.
Zhao N, Peng F, Chen X. The unfolded protein response in triple-negative breast cancer. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer – an overview and update. Mol Cell Endocrinol. 2015;418:220–34.
Clarke R. The unfolded protein response as an integrator of response to endocrine therapy in estrogen receptor positive breast cancer. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Cook KL. Outside the endoplasmic reticulum: non-canonical GRP78 signaling. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Schwarze S, Rangnekar VM. Targeting plasma membrane GRP78 for cancer growth inhibition. Cancer Biol Ther. 2010;9:153–5.
Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, Auffray C, Albrengues J, Egeblad M, Iacobuzio-Donahue CA, Lyons SK, Fearon DT. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. 2018;360. pii: eaao4908.
Scheper W, Hoozemans JJ. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 2015;130:315–31.
Scheper W, Hoozemans JJ. A new PERKspective on neurodegeneration. Sci Transl Med. 2013;5:206fs37.
Hughes D, Mallucci GR. The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J. 2018.
Moussa C. Autophagy and the unfolded protein response in neurodegenerative diseases. In: Clarke R, editor. Introduction to the unfolded protein response. New York: Springer; 2018.
Clarke, R., Tyson, J. J., Tan, M., Baumann, W. T., Xuan, J., and Wang, Y. Systems biology: perspectives on multiscale modeling in research on endocrine-related cancers. Endocr Relat Cancer. 2018; in revision.
Parmar JH, Cook KL, Shajahan-Haq AN, Clarke PA, Tavassoly I, Clarke R, Tyson JJ, Baumann WT. Modelling the effect of GRP78 on anti-oestrogen sensitivity and resistance in breast cancer. Interface Focus. 2013;3:20130012.
Ma’ayan A. Insights into the organization of biochemical regulatory networks using graph theory analyses. J Biol Chem. 2009;284:5451–5.
Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6.
Shapiro DJ, Livezey M, Yu L, Zheng X, Andruska N. Anticipatory UPR activation: a protective pathway and target in Cancer. Trends Endocrinol Metab. 2016;27:731–41.
Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. 2015;58:123–33.
Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014;516:414–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Clarke, R. (2019). Introduction: The Unfolded Protein Response. In: Clarke, R. (eds) The Unfolded Protein Response in Cancer. Cancer Drug Discovery and Development. Humana Press, Cham. https://doi.org/10.1007/978-3-030-05067-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-05067-2_1
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-030-05065-8
Online ISBN: 978-3-030-05067-2
eBook Packages: MedicineMedicine (R0)