
Abstract
Behçet’s disease (BD) is a complex disease characterized by recurrent episodes of oral and genital ulcers and inflammatory lesions in a variable number of vessels throughout the body which lead to significant organ involvement. This condition, given its low prevalence, is classified as a rare systemic vasculitis. Although its etiology remains unclear, it is known that it is a multifactorial and immune-mediated disease in which imbalances of the immune response, triggered by environmental factors in genetically predisposed individuals, may be the underlying mechanisms of the disease. Throughout this chapter, we will review the current knowledge of the genetic component identified in this disorder. Firstly, we will focus on the HLA region which harbors the strongest known susceptibility factors for this disease. Additionally, we will review the available data in non-HLA regions, highlighting the confirmed risk loci and summarizing those that are only suggested. Finally, we will provide an overview of the main molecular pathways involved in the development of this pathology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Introduction
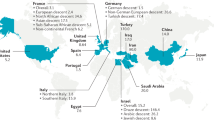
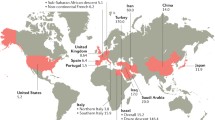
Behçet’s disease (BD) is a complex systemic syndrome characterized by inflammatory lesions of blood vessels throughout the body, being small vessels the most frequently involved. This pathology is a rare and debilitating vasculitis, which presents a wide range of clinical phenotypes. The main clinical features are genital ulceration, ocular involvement (mainly uveitis), and skin lesions, but patients also can suffer gastrointestinal involvement, arthritis, and neurological disorders, among other symptoms, which lead to significant morbidity and mortality [1]. The lack of a pathognomonic sign and the absence of specific biomarkers of the disease make difficult the diagnosis of BD which is based on criteria and classification systems being the most widely used those proposed by the International BD Study Group (ICBD) in 1990 [2]. BD is found worldwide; however, its prevalence varies along the different geographical regions. The highest prevalence is found in Turkey, followed by Japan and Iran, and it is very low in North America and in Western countries; in fact, BD is also known as “Silk Road disease” given its particular geographical distribution overlap with the ancient trading route stretching from China to the Mediterranean area [3]. Regarding the gender distribution and onset, both genders are affected equally, although with geographical differences, and the disease typically arises in the third or fourth decade of life, being uncommon in children or patients above 50s [4].
With respect to the immunological data, multiple alterations have been described in the homeostasis of the T cells in BD patients. Accordingly, activation of γ/δ T lymphocytes in both peripheral blood and mucous lesions has been described [5, 6]. Besides, imbalances in T helper (Th) cell populations have been extensively studied in BD, and Th1 infiltrates have been observed in oral and genital ulcers and cutaneous and gastrointestinal lesions. Consistently, an increase in Th1 cytokines has also been found in blood [7,8,9]. In addition, high levels of IL23 and IL17 have been described in peripheral blood mononuclear cells of BD patients [10]. IL23 induces the production of IL17 by T lymphocytes, and this cytokine promotes a neutrophil-mediated inflammatory response. Therefore, high levels of IL23 are consistent with the hyperactivation of neutrophils observed in the early phases of the lesion infiltration [11], which leads to the increase in the levels of reactive oxygen species, endothelial adhesion, chemotaxis, and phagocytosis [12,13,14,15]. All these data suggest that the Th17/Th1 balance plays an important role in the regulation of the inflammation in BD [16].
Despite high efforts, the etiology of BD remains unclear. However, cumulative evidences suggest that certain infectious agents and environmental factors may trigger the disease in genetically predisposed individuals. It has been proposed that different virus and bacteria could play a role in the BD development. Of special note are Herpes simplex virus I, which DNA has been isolated from genital ulcers and saliva samples of patients [17], and Streptococcus sanguinis that has been related with the formation of recurrent aphthous lesions [18]. Nevertheless, the present chapter focuses on the genetic component, in which great advances have been made in recent years.
3.2 Genetic Component of Behçet’s Disease
The substantial genetic contribution to the pathogenesis of this disease is strongly supported by several facts. In addition to the aforementioned geographical variation in prevalence [3], familial aggregation has been extensively reported. The results of these studies revealed a higher frequency of the cases among the relatives of the patients than in the general population [19,20,21,22] with the highest sibling recurrence risk ratio, in the Turkish population (between 11.4 and 52.5) [20]. Besides, although there are few studies in BD, the disease concordance rate was higher in monozygotic twins compared with dizygotic ones [23]. Finally, specific associations of different genes with BD susceptibility have been robustly described [24]. From a genetic point of view, BD is considered as a complex disease, in which multiple genes are involved, each of them with a modest effect in the disease, being able to be related with the onset as well as with its severity and progression.
We will examine throughout this chapter the current knowledge of the BD genetic background on the basis of the recent advances in this subject. Firstly, the HLA region (which represents the main genetic contributor to the disease) will be thoroughly reviewed together with the main confirmed associated loci outside the HLA region. Besides, we summarized the results of other studies that propose new susceptibility genetic factors, and, finally, we will highlight the most important molecular pathways implicated in the disease.
3.2.1 HLA Region
The major histocompatibility complex (MHC) region includes the largest number of genetic associations for a wide range of pathological conditions, including most of the immune-mediated diseases. The earliest association of BD with the human MHC (HLA) was reported in the 1970s [25]. These initial studies, using serological typing method, revealed that HLA-B51 had a relevant relationship with the disease, so that it was detected with a relative small sample size. Decades later, with the availability of DNA-based methods and larger study cohorts, the specific association of HLA-B*51 with the disease was well established and repeatedly contrasted in different ethnic groups [24]. Multiple studies exploring other additional susceptibility loci in the HLA region have suggested the association of diverse HLA molecules with the disease. In this sense, in addition to other HLA-B molecules [26, 27], some studies have reported other classic class I HLA molecules (HLA-A and HLA-C) as associated to the disease, although in general these results were less consistent [28,29,30]. As we stated above, the association between HLA-B*51 and BD is well established worldwide as the strongest genetic risk factor for this condition, but the functional basis of this association in the pathogenesis of BD has not been elucidated yet. For this reason, other loci located within the HLA region have been proposed as major contributors. Specifically, a study published in 1999 in the Japanese population proposed for the first time that the causative gene of the HLA region was MICA , a gene located very close to HLA-B with strong linkage disequilibrium (LD) [31,32,33]. Although this idea was embraced by the scientific community at the beginning, later studies performed in different populations were inconsistent [34,35,36,37,38]. The problems encountered to clarify which gene is true responsible of the association are related to the complexity of the HLA region, which is particularly dense in genes related with the immune system and shows a strong LD, making difficult to clarify whether the identified risk signals are independent each other or whether they are reflections of the primary association. This problem is aggravated in the case of rare diseases, such as BD, because the sample size, relatively low compared with other pathologies with more prevalence, limits the statistical power of the individual studies.
Given the advance of the new typing approaches during the last years, new data have been raised from fine-mapping studies, thus allowing the typing of the entire region. Four independent studies performed in different populations, and following a comprehensive approach that combines high-throughput genomics with the novel algorithms of HLA imputation [39, 40], have been published [41,42,43,44]. All of these studies concluded that the HLA-B*51 haplotype represents the strongest association factor with the disease but also discarded an association of any HLA class II molecule, something that was previously proposed using older methodologies [41,42,43,44]. However, these studies showed discrepancies regarding the independent association of other HLA class I factors. In the context of the HLA-B locus, HLA-B*15 and HLA-B*27 were identified as risk factors and HLA-B*49 as a protective factor with independent effects of HLA-B*51 in one study in the Turkish population [42]. Additionally, the allelic group HLA-B*57 was shown to confer susceptibility to BD in two studies conducted in the Spanish and Turkish populations [42, 43]. With regard to the HLA-A locus, variation within the gene was also associated with disease predisposition in different studies. Specifically, three of these studies found that the HLA-A*03 group is an independent protective factor for BD [42,43,44]. In addition, one of these studies reported suggested evidence that HLA-A*26 could be also an independent risk factor for BD [42]. Only one study described an independent association of the HLA-C locus, specifically HLA-C*1602 [41], which had been previously suggested by two smaller studies from Southern European [45, 46]. However, it should be noted that this independent association of the HLA-C haplotype was not consistent with the rest of the studies that evaluated the contribution of this locus [26, 42,43,44]. On the other hand, although the results of one of these studies returned the idea of the MICA gene as the causal susceptibility marker for BD [41], the results of the other large-scale genetic studies did not support an independent association of the MICA gene with BD [34,35,36,37,38, 42, 43].
Many of the HLA class I molecules have a dual function, and they present peptides to the CD8 T cells, but they also control the activity of the natural killer cells because they are ligands of some of their receptors (KIR). The relevant amino acid positions in one or the other function are located in different parts of the molecule. Therefore, deciphering the amino acid positions involved in BD susceptibility may definitively help to better understand the functional implications of the HLA system in the disease pathophysiology. In an effort to identify the motifs that may explain the variety of protective and risk effects conferred by HLA class I molecules in BD, the possible association of polymorphic amino acid residues has been analyzed in some studies [41, 43]. One of them proposed a model comprising five amino acids located in three positions of HLA-B, 67 (glutamic acid, Glu, or phenylalanine, Phe), 97 (threonine, Thr), and 116 (leucine, Leu), as well as one position in HLA-A, 161 (Glu) [41]. According to one of those studies in which an omnibus test was performed, the most relevant amino acid position for disease risk was HLA-B 97. Six possible residues can be harbored at this position, with two of them conferring risk (Thr and valine, Val), another two conferring protection (arginine, Arg, and serine, Ser), and the remaining two being neutral (asparagine, Asn, and tryptophan, Trp). In addition, according to this model, the position 66 of HLA-A represented an independent association (with lysine, Lys conferring risk and Asn protection) [43]. Interestingly, the four more relevant positions (HLA-B 67, 97, 116 and HLA-A 66) are located in the binding groove of their corresponding molecules. These data brought additional evidences supporting the importance of the peptide binding by the class I HLA molecules in BD. Nevertheless, the study of the most relevant amino acid positions has to be interpreted with caution, and it is evident that each HLA-B molecule has a specific set of amino acids in its polymorphic positions, with many of them in LD with each other, thus increasing the difficulty to evaluate dependency at the amino acid level. Therefore, it should be noted that dependency does not exclude biological influence. Therefore, only by complementing the knowledge gained by this type of approaches with those provided by functional studies, it would be possible to elucidate the precise etiopathogenic role of these molecules in disease, which would be essential for a personalized medicine [43].
3.2.2 Non-HLA Region
The contribution of the HLA region to the genetic component of BD has been estimated to be approximately 20% [47], which indicates that other genes outside the region may have to be involved in this pathology. For many years, a large number of candidate gene studies tried to unravel the complex genetic architecture of BD outside of the HLA region. However, those studies yielded contradictory results that were not usually replicated in independent populations. The identification of the genetic factors involved in the susceptibility to complex diseases represents an enormous challenge, given that the effect of each gene in the development of this type of diseases is relatively low independently. To detect genes with low or medium effects, very large sample sizes are needed, and, given the condition of BD as a rare disease, most of candidate gene studies performed did not have enough statistical power. Additionally, the lack of replication among studies could be also related to specific population associations due to particular genetic architectures. Due to the above, few consistent associations with BD have been identified to date (Table 3.1).
3.2.2.1 Confirmed Risk Loci
Interleukin 23 receptor ( IL23-R ). This gene represents the most consistently associated non-HLA locus with BD, as it has been repeatedly identified as a risk factor for this disease in different populations and multiple independent studies [26, 43, 44, 48,49,50,51,52,53,54,55,56]. It is worth mentioning that this gene is a known risk factor for a multitude of immune-related diseases [57,58,59]. IL23R encodes a subunit of the IL-23 receptor which is expressed on the surface of Th17 cells and macrophages and binds the subunit p19 of IL-23, a pro-inflammatory cytokine composed by p19 and p40 (which is common with IL-12 and binds to IL12RB1). The IL-23/IL-23R complex promotes the polarization of the T cells to Th17 and increases the levels of inflammatory cytokines such as IL-1, IL-6, IL-17, and TNFα [60]. Although there are strong evidences supporting that the variants of IL23R influence BD susceptibility through their effects on IL-23R itself, an additional role of these variants as markers of other nearby genes, such as IL12RB2, cannot be discarded [49]. IL12RB2 gene encodes the IL-12 receptor beta 2, and the complex IL-12/IL12RB2 has a crucial role in Th1 cell differentiation. Thus, although the causal mechanism of the association remains unclear, all these evidences support an important role of the IL-23/IL-17 pathway in the pathophysiology of immune-mediated diseases, including BD.
Interleukin 10 ( IL10 ). This gene was identified together with IL23R as susceptibility factor for BD, and this association has been subsequently replicated in different populations [26, 44, 48,49,50,51, 55, 61, 62]. IL-10 is a potent anti-inflammatory molecule that inhibits the activation of macrophages and the synthesis of pro-inflammatory cytokines (including IL-1, IL-6, and TNFα); therefore, it suppresses Th1 cell activation [63]. Imbalances in the regulation of Th1 activation could cause deviations toward a Th1 profile, which could predispose to the disease. Interestingly, several genetic variants within this gene have been associated with the levels of expression. Specifically, the SNP reported by the Remmers group is associated with a decrease of the IL10 expression levels in monocytes [49, 64, 65]. Besides, it has been demonstrated that a low expression of IL10 in mouse leads to inflammation processes [66].
Interleukin 12A ( IL12A ). This gene encodes a subunit of IL-12, a cytokine that plays an important role in the polarization of the immune response toward Th1 and also in the production of IFNγ by both the T lymphocytes and the NK cells, so it is related to the production of pro-inflammatory cytokines [67]. Several studies reported association of this gene with BD [43, 44, 55, 68, 69], although further investigation is needed to clarify the causal variant.
The signal transducer and activator of transcription-4 ( STAT4 ). This gene represents a shared genetic susceptibility factor for several autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and Sjögren’s syndrome, among others [70, 71]. Regarding BD, the association with this gene has been extensively reported in different populations [53, 55, 69, 72]. STAT4 is a transcription factor that is activated by cytokines such as IL-12 and IL-23, which, as stated above, are involved in the differentiation of lymphocytes into Th1 and Th17 [73]. Two of the identified risk variants of this gene have been implicated in changes of STAT4 mRNA expression [69, 74], although further experiments are needed to better understand the way in which this genetic variation affect the pathogenesis of BD.
The endoplasmic reticulum aminopeptidase 1 ( ERAP1 ). The association of a missense variant of ERAP1, p.Arg725Gln, was described for the first time by Kirino and colleagues, whose data suggested that the associated variant contributes to disease susceptibility through a strong interaction or epistasis with HLA-B51 [69]. After that, additional studies have replicated these first results [55, 75, 76]. It is noteworthy that the association of this gene with other HLA class I-related diseases such as ankylosing spondylitis (AS) and psoriasis has been thoroughly investigated in the last years, and the implication of ERAP1 with these diseases has been always reported through an epistatic interaction with the corresponding associated HLA allele in each case [57, 77,78,79]. This gene encodes an aminopeptidase with an ubiquitous distribution, which plays an important role trimming the N-terminal end of the peptides in the endoplasmic reticulum, a critical step of the processing of the peptides to optimize their length for HLA I molecule binding [80].
Fucosyltransferase 2 ( FUT2 ). This association was firstly reported by Xavier and collaborators [81] but also in a recently published large-scale study [44]. This gene encodes the alpha [1, 2] fucosyltransferase, a molecule that produces in fluids and intestinal mucosa the secreted H antigen, which is the precursor of the ABO histo-blood group antigens [82]. It has been described that homozygosity in two missense variants (p.Trp143Ter and p.Ile129Phe) leads to an ABO nonsecretor phenotype [82]. Of note, these variants are also linked with other immune-mediated disorders such as Crohn’s disease and type 1 diabetes [82,83,85]. The nonsecretor phenotype has also been associated with resistance to several infectious agents [86, 87] and the gut microbiome composition [88, 89]. These evidences support the hypothesis that relationship between infectious agents and the genetic component is crucial for the development of BD.
Killer cell lectin-like receptor C4 ( KLRC4 ). This gene has been found as a susceptibility locus in two large-scale genetic studies. Two non-synonymous variants in high LD (p.Ile29Ser and p.Asn104Ser) seem to be part of the susceptibility haplotype for BD [55, 69]. The KLRC4 gene, also known as NKG2F, encodes a c-type lectin receptor expressed on NK cells. Although the specific function of this molecule is unknown, the haplotype related with the disease has reported to be associated with a high natural cytotoxic activity on peripheral blood cells [90].
CCR1-CCR3 . This locus harbors a cluster of chemokine receptor genes with a high LD among them [69, 91]. Through binding to its ligands, these receptors act as a key regulator in leukocyte trafficking and in the homeostasis of the immune system [92]. The risk allele reported by Hou et al. has been associated with a reduced expression of both, CCR1 and CCR3, in peripheral blood mononuclear cells (PBMCs) [91] and another variant located in the same region was also related with a lower expression of CCR1 in human primary monocytes [69].
3.2.2.2 Suggested Risk Loci
The number of genes identified in large-scale genetic studies is higher than that exposed in the previous section, and includes genes such as KIAA1529, CPVL, LOC100129342, UBASH3A, UBAC2 [93], GIMAP [94], JRKL-CNTN5 [43], IL1A-IL1B, IRF8, CEBPB-PTPN1 [44]. However, the association of these loci with BD remains unconfirmed. In some cases, specific replication studies have been performed in other populations but the results obtained are contradictories [26, 95]. In other cases the association has recently been described in only one population [44].
New approaches such as next-generation sequencing (NGS) have being recently implemented for the investigation of the rare polymorphisms. In a recent study, 21 candidate genes were evaluated for BD association through deep exonic resequencing with the aim of identifying low-frequency non-synonymous variants [56]. The association of rare variants in four genes (IL23R, NOD2, TLR4, and MEFV) with BD is supported by the results obtained in this work. In a later study, seven genes related with immune-mediated diseases were analyzed using NGS. The findings of this second study suggested the influence of rare variants of, at least, NOD2, PSTPIP1, and MVK in the pathogenesis of BD [96]. More independent studies performed in other populations and/or with other approaches are necessary to confirm or discard these suggested associations.
3.3 Molecular Pathways
In the last years, large achievements have been accomplished to understand the genetic basis of BD. Although multiple studies will still be necessary for a fully comprehension of the pathophysiology underlying this disorder, given the last advances, we can outline a model that integrates the main molecular pathways involved in the development of this disease.
As it is described before, several functional studies have established that the T lymphocytes are the most important cell population involved in the immunopathogenesis of BD. These data are in concordance with the results yielded by genetic studies, because several of the BD susceptibility genes (e.g., STAT4, IL12, IL23R) are involved in the differentiation of naïve CD4+ T cells into mature Th1 effector cell or in the maintenance of Th17 cells [97] and others in the balance of Th1 cells (IL10). In addition, the association of the IL-23/IL-17 pathway with BD is also supported by genetic data and provides evidences of the essential role that this pathway has in the pathophysiology of multiple immune-mediated diseases, especially BD.
The genetic association of ERAP1, FUT2, and KLRC4 supports the hypothesis that the disease would be triggered by environmental agents in which microorganisms would play a key role. On this sense, the association of FUT2 could be related with the immune response to invasive microorganisms and the microbiota composition.
3.4 Conclusions and Future Perspectives
Despite the impressive increase in our knowledge of the genetic basis of BD during the last years, the list of the confirmed risk loci for this type of vasculitis remains significantly lower than other immune-mediated diseases [98, 99]. One of the main limitations in the genetic study of this disease is the lack of statistical power, which is conditioned by the low prevalence of this disorder and that does not permit to identify susceptibility signals with modest effects for which large sample size is required. Therefore, additional strategies are necessary to unravel the genetic component underlying BD. In this sense, one new approach which is been successfully applied is the combination of the genetic data from different diseases with similar features considering them as a single phenotype (cross-phenotype meta-analysis), and numerous shared genetic components have been described in the last years using this methodology [99,100,102].
On the other hand, the way in which the information of genetics variants is translated into pathogenetic mechanisms remains unclear for most of the variants associated with BD, which are located mostly in noncoding region, as occurs in many immune-mediated diseases. This fact suggests that these variants could affect different regulatory elements in the genome. Thus, further studies should be focused on the effects that the associated variants produce. In this sense, the role of epigenetics in the pathogenesis of immune-mediated diseases seems now undeniable, and the contribution of epigenetic dysregulation in vasculitis is increasingly recognized. The genetic-epigenetic relationships are taking on great importance in a field in which functional data are emerging [103]. Expanding our knowledge of how these epigenetic mechanisms interact with the polymorphisms will help to better understand the pathogenesis of this disease.
References
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Criteria for diagnosis of Behçet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078–80.
Sakane T, Takeno M, Suzuki N, Inaba G. Behçet’s disease. N Engl J Med. 1999;341(17):1284–91.
Yurdakul S, Yazici H. Behçet’s syndrome. Best Pract Res Clin Rheumatol. 2008;22(5):793–809.
Hamzaoui K, Hamzaoui A, Hentati F, Kahan A, Ayed K, Chabbou A, et al. Phenotype and functional profile of T cells expressing gamma delta receptor from patients with active Behçet’s disease. J Rheumatol. 1994;21(12):2301–6.
Freysdottir J, Hussain L, Farmer I, Lau SH, Fortune F. Diversity of gammadelta T cells in patients with Behcet’s disease is indicative of polyclonal activation. Oral Dis. 2006;12(3):271–7.
Ben Ahmed M, Houman H, Miled M, Dellagi K, Louzir H. Involvement of chemokines and Th1 cytokines in the pathogenesis of mucocutaneous lesions of Behçet’s disease. Arthritis Rheum. 2004;50(7):2291–5.
Dalghous AM, Freysdottir J, Fortune F. Expression of cytokines, chemokines, and chemokine receptors in oral ulcers of patients with Behcet’s disease (BD) and recurrent aphthous stomatitis is Th1-associated, although Th2-association is also observed in patients with BD. Scand J Rheumatol. 2006;35(6):472–5.
Ferrante A, Ciccia F, Principato A, Giardina AR, Impastato R, Peralta S, et al. A Th1 but not a Th17 response is present in the gastrointestinal involvement of Behçet’s disease. Clin Exp Rheumatol. 2010;28(4 Suppl 60):S27–30.
Chi W, Zhou S, Yang P, Chen L. CD4+ T cells from Behcet patients produce high levels of IL-17. Eye Sci. 2011;26(2):65–9.
Eksioglu-Demiralp E, Direskeneli H, Kibaroglu A, Yavuz S, Ergun T, Akoglu T. Neutrophil activation in Behçet’s disease. Clin Exp Rheumatol. 2001;19(5 Suppl 24):S19–24.
Kobayashi M, Ito M, Nakagawa A, Matsushita M, Nishikimi N, Sakurai T, et al. Neutrophil and endothelial cell activation in the vasa vasorum in vasculo-Behçet disease. Histopathology. 2000;36(4):362–71.
Carletto A, Pacor ML, Biasi D, Caramaschi P, Zeminian S, Bellavite P, et al. Changes of neutrophil migration without modification of in vitro metabolism and adhesion in Behçet’s disease. J Rheumatol. 1997;24(7):1332–6.
Efthimiou J, Addison IE, Johnson BV. In vivo leucocyte migration in Behçet’s syndrome. Ann Rheum Dis. 1989;48(3):206–10.
Neves FS, Carrasco S, Goldenstein-Schainberg C, Gonçalves CR, de Mello SB. Neutrophil hyperchemotaxis in Behçet’s disease: a possible role for monocytes orchestrating bacterial-induced innate immune responses. Clin Rheumatol. 2009;28(12):1403–10.
Kim J, Park JA, Lee EY, Lee YJ, Song YW, Lee EB. Imbalance of Th17 to Th1 cells in Behçet’s disease. Clin Exp Rheumatol. 2010;28(4 Suppl 60):S16–9.
Studd M, McCance DJ, Lehner T. Detection of HSV-1 DNA in patients with Behçet’s syndrome and in patients with recurrent oral ulcers by the polymerase chain reaction. J Med Microbiol. 1991;34(1):39–43.
Cho SB, Zheng Z, Ahn KJ, Choi MJ, Cho S, Kim DY, et al. Serum IgA reactivity against GroEL of Streptococcus sanguinis and human heterogeneous nuclear ribonucleoprotein A2/B1 in patients with Behçet disease. Br J Dermatol. 2013;168(5):977–83.
Koné-Paut I, Geisler I, Wechsler B, Ozen S, Ozdogan H, Rozenbaum M, et al. Familial aggregation in Behçet’s disease: high frequency in siblings and parents of pediatric probands. J Pediatr. 1999;135(1):89–93.
Gül A, Inanç M, Ocal L, Aral O, Koniçe M. Familial aggregation of Behçet’s disease in Turkey. Ann Rheum Dis. 2000;59(8):622–5.
Fietta P. Behçet’s disease: familial clustering and immunogenetics. Clin Exp Rheumatol. 2005;23(4 Suppl 38):S96–105.
Yilmaz S, Cimen KA. Familial Behçet’s disease. Rheumatol Int. 2010;30(8):1107–9.
Masatlioglu S, Seyahi E, Tahir Turanli E, Fresko I, Gogus F, Senates E, et al. A twin study in Behçet’s syndrome. Clin Exp Rheumatol. 2010;28(4 Suppl 60):S62–6.
de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behçet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009;61(10):1287–96.
Ohno S, Aoki K, Sugiura S, Nakayama E, Itakura K, Aizawa M. Letter: HL-A5 and Behçet’s disease. Lancet. 1973;2(7842):1383–4.
Montes-Cano MA, Conde-Jaldón M, García-Lozano JR, Ortiz-Fernández L, Ortego-Centeno N, Castillo-Palma MJ, et al. HLA and non-HLA genes in Behçet’s disease: a multicentric study in the Spanish population. Arthritis Res Ther. 2013;15(5):R145.
Gül A, Uyar FA, Inanç M, Ocal L, Barrett JH, Aral O, et al. A weak association of HLA-B*2702 with Behçet’s disease. Genes Immun. 2002;3(6):368–72.
Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behçet’s disease: a comprehensive review. J Autoimmun. 2015;64:137–48.
Meguro A, Inoko H, Ota M, Katsuyama Y, Oka A, Okada E, et al. Genetics of Behçet disease inside and outside the MHC. Ann Rheum Dis. 2010;69(4):747–54.
Kang EH, Kim JY, Takeuchi F, Kim JW, Shin K, Lee EY, et al. Associations between the HLA-A polymorphism and the clinical manifestations of Behcet’s disease. Arthritis Res Ther. 2011;13(2):R49.
Yabuki K, Mizuki N, Ota M, Katsuyama Y, Palimeris G, Stavropoulos C, et al. Association of MICA gene and HLA-B*5101 with Behçet’s disease in Greece. Invest Ophthalmol Vis Sci. 1999;40(9):1921–6.
Mizuki N, Ota M, Katsuyama Y, Yabuki K, Ando H, Goto K, et al. Association analysis between the MIC-A and HLA-B alleles in Japanese patients with Behçet’s disease. Arthritis Rheum. 1999;42(9):1961–6.
Ota M, Mizuki N, Katsuyama Y, Tamiya G, Shiina T, Oka A, et al. The critical region for Behçet disease in the human major histocompatibility complex is reduced to a 46-kb segment centromeric of HLA-B, by association analysis using refined microsatellite mapping. Am J Hum Genet. 1999;64(5):1406–10.
González-Escribano MF, Rodríguez MR, Aguilar F, Alvarez A, Sanchez-Roman J, Núñez-Roldán A. Lack of association of MICA transmembrane region polymorphism and Behçet’s disease in Spain. Tissue Antigens. 1999;54(3):278–81.
Mizuki N, Ota M, Yabuki K, Katsuyama Y, Ando H, Palimeris GD, et al. Localization of the pathogenic gene of Behçet’s disease by microsatellite analysis of three different populations. Invest Ophthalmol Vis Sci. 2000;41(12):3702–8.
Salvarani C, Boiardi L, Mantovani V, Olivieri I, Ciancio G, Cantini F, et al. Association of MICA alleles and HLA-B51 in Italian patients with Behçet’s disease. J Rheumatol. 2001;28(8):1867–70.
Park SH, Park KS, Seo YI, Min DJ, Kim WU, Kim TG, et al. Association of MICA polymorphism with HLA-B51 and disease severity in Korean patients with Behcet’s disease. J Korean Med Sci. 2002;17(3):366–70.
Cohen R, Metzger S, Nahir M, Chajek-Shaul T. Association of the MIC-A gene and HLA-B51 with Behçet’s disease in Arabs and non-Ashkenazi Jews in Israel. Ann Rheum Dis. 2002;61(2):157–60.
Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–23.
Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One. 2013;8(6):e64683.
Hughes T, Coit P, Adler A, Yilmaz V, Aksu K, Düzgün N, et al. Identification of multiple independent susceptibility loci in the HLA region in Behçet’s disease. Nat Genet. 2013;45(3):319–24.
Ombrello MJ, Kirino Y, de Bakker PI, Gül A, Kastner DL, Remmers EF. Behçet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc Natl Acad Sci U S A. 2014;111(24):8867–72.
Ortiz-Fernández L, Carmona FD, Montes-Cano MA, García-Lozano JR, Conde-Jaldón M, Ortego-Centeno N, et al. Genetic analysis with the Immunochip Platform in Behçet disease. Identification of residues associated in the HLA class I region and new susceptibility loci. PLoS One. 2016;11(8):e0161305.
Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behçet’s disease susceptibility. Nat Genet. 2017;49(3):438–43.
Bettencourt A, Pereira C, Carvalho L, Carvalho C, Patto JV, Bastos M, et al. New insights of HLA class I association to Behçet’s disease in Portuguese patients. Tissue Antigens. 2008;72(4):379–82.
Sanz L, González-Escribano F, de Pablo R, Núñez-Roldán A, Kreisler M, Vilches C. HLA-Cw*1602: a new susceptibility marker of Behçet’s disease in southern Spain. Tissue Antigens. 1998;51(1):111–4.
Yazici H, Fresko I, Yurdakul S. Behçet’s syndrome: disease manifestations, management, and advances in treatment. Nat Clin Pract Rheumatol. 2007;3(3):148–55.
Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet’s disease susceptibility loci. Nat Genet. 2010;42(8):703–6.
Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat Genet. 2010;42(8):698–702.
Carapito R, Shahram F, Michel S, Le Gentil M, Radosavljevic M, Meguro A, et al. On the genetics of the Silk Route: association analysis of HLA, IL10, and IL23R-IL12RB2 regions with Behçet’s disease in an Iranian population. Immunogenetics. 2015;67(5–6):289–93.
Xavier JM, Shahram F, Davatchi F, Rosa A, Crespo J, Abdollahi BS, et al. Association study of IL10 and IL23R-IL12RB2 in Iranian patients with Behçet’s disease. Arthritis Rheum. 2012;64(8):2761–72.
Jiang Z, Yang P, Hou S, Du L, Xie L, Zhou H, et al. IL-23R gene confers susceptibility to Behcet’s disease in a Chinese Han population. Ann Rheum Dis. 2010;69(7):1325–8.
Kim ES, Kim SW, Moon CM, Park JJ, Kim TI, Kim WH, et al. Interactions between IL17A, IL23R, and STAT4 polymorphisms confer susceptibility to intestinal Behcet’s disease in Korean population. Life Sci. 2012;90(19–20):740–6.
Yalçin B, Atakan N, Dogan S. Association of interleukin-23 receptor gene polymorphism with Behçet disease. Clin Exp Dermatol. 2014;39(8):881–7.
Sousa I, Shahram F, Francisco D, Davatchi F, Abdollahi BS, Ghaderibarmi F, et al. Brief report: association of CCR1, KLRC4, IL12A-AS1, STAT4, and ERAP1 with Behçet’s disease in Iranians. Arthritis Rheumatol. 2015;67(10):2742–8.
Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behçet disease. Proc Natl Acad Sci U S A. 2013;110(20):8134–9.
Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80(2):273–90.
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–3.
Rueda B, Orozco G, Raya E, Fernandez-Sueiro JL, Mulero J, Blanco FJ, et al. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2008;67(10):1451–4.
Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116(5):1218–22.
Wu Z, Zheng W, Xu J, Sun F, Chen H, Li P, et al. IL10 polymorphisms associated with Behçet’s disease in Chinese Han. Hum Immunol. 2014;75(3):271–6.
Kang EH, Choi JY, Lee YJ, Lee EY, Lee EB, Song YW. Single nucleotide polymorphisms in IL-10-mediated signalling pathways in Korean patients with Behçet’s disease. Clin Exp Rheumatol. 2014;32(4 Suppl 84):S27–32.
Wallace GR, Kondeatis E, Vaughan RW, Verity DH, Chen Y, Fortune F, et al. IL-10 genotype analysis in patients with Behçet’s disease. Hum Immunol. 2007;68(2):122–7.
Temple SE, Lim E, Cheong KY, Almeida CA, Price P, Ardlie KG, et al. Alleles carried at positions -819 and -592 of the IL10 promoter affect transcription following stimulation of peripheral blood cells with Streptococcus pneumoniae. Immunogenetics. 2003;55(9):629–32.
Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet. 1997;24(1):1–8.
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765.
Chang JT, Shevach EM, Segal BM. Regulation of interleukin (IL)-12 receptor beta2 subunit expression by endogenous IL-12: a critical step in the differentiation of pathogenic autoreactive T cells. J Exp Med. 1999;189(6):969–78.
Kappen JH, Medina-Gomez C, van Hagen PM, Stolk L, Estrada K, Rivadeneira F, et al. Genome-wide association study in an admixed case series reveals IL12A as a new candidate in Behçet disease. PLoS One. 2015;10(3):e0119085.
Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013;45(2):202–7.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357(10):977–86.
Korman BD, Alba MI, Le JM, Alevizos I, Smith JA, Nikolov NP, et al. Variant form of STAT4 is associated with primary Sjögren’s syndrome. Genes Immun. 2008;9(3):267–70.
Hou S, Yang Z, Du L, Jiang Z, Shu Q, Chen Y, et al. Identification of a susceptibility locus in STAT4 for Behçet’s disease in Han Chinese in a genome-wide association study. Arthritis Rheum. 2012;64(12):4104–13.
Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C, et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci U S A. 2002;99(19):12281–6.
Abelson AK, Delgado-Vega AM, Kozyrev SV, Sánchez E, Velázquez-Cruz R, Eriksson N, et al. STAT4 associates with systemic lupus erythematosus through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk. Ann Rheum Dis. 2009;68(11):1746–53.
Conde-Jaldón M, Montes-Cano MA, García-Lozano JR, Ortiz-Fernández L, Ortego-Centeno N, González-León R, et al. Epistatic interaction of ERAP1 and HLA-B in Behçet disease: a replication study in the Spanish population. PLoS One. 2014;9(7):e102100.
Takeuchi M, Ombrello MJ, Kirino Y, Erer B, Tugal-Tutkun I, Seyahi E, et al. A single endoplasmic reticulum aminopeptidase-1 protein allotype is a strong risk factor for Behçet’s disease in HLA-B*51 carriers. Ann Rheum Dis. 2016;75(12):2208–11.
Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39(11):1329–37.
Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42(11):985–90.
Sun LD, Cheng H, Wang ZX, Zhang AP, Wang PG, Xu JH, et al. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat Genet. 2010;42(11):1005–9.
Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3(12):1169–76.
Xavier JM, Shahram F, Sousa I, Davatchi F, Matos M, Abdollahi BS, et al. FUT2: filling the gap between genes and environment in Behçet’s disease? Ann Rheum Dis. 2015;74(3):618–24.
Ferrer-Admetlla A, Sikora M, Laayouni H, Esteve A, Roubinet F, Blancher A, et al. A natural history of FUT2 polymorphism in humans. Mol Biol Evol. 2009;26(9):1993–2003.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–25.
Hu DY, Shao XX, Xu CL, Xia SL, Yu LQ, Jiang LJ, et al. Associations of FUT2 and FUT3 gene polymorphisms with Crohn’s disease in Chinese patients. J Gastroenterol Hepatol. 2014;29(10):1778–85.
Smyth DJ, Cooper JD, Howson JM, Clarke P, Downes K, Mistry T, et al. FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes. 2011;60(11):3081–4.
Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, Lindblad L, et al. Human susceptibility and resistance to Norwalk virus infection. Nat Med. 2003;9(5):548–53.
Ruiz-Palacios GM, Cervantes LE, Ramos P, Chavez-Munguia B, Newburg DS. Campylobacter jejuni binds intestinal H(O) antigen (Fuc alpha 1, 2Gal beta 1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J Biol Chem. 2003;278(16):14112–20.
Wacklin P, Mäkivuokko H, Alakulppi N, Nikkilä J, Tenkanen H, Räbinä J, et al. Secretor genotype (FUT2 gene) is strongly associated with the composition of bifidobacteria in the human intestine. PLoS One. 2011;6(5):e20113.
Rausch P, Rehman A, Künzel S, Häsler R, Ott SJ, Schreiber S, et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A. 2011;108(47):19030–5.
Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res. 2006;66(1):563–70.
Hou S, Xiao X, Li F, Jiang Z, Kijlstra A, Yang P. Two-stage association study in Chinese Han identifies two independent associations in CCR1/CCR3 locus as candidate for Behçet’s disease susceptibility. Hum Genet. 2012;131(12):1841–50.
Marzio PD, Sherry B, Thomas EK, Franchin G, Schmidtmayerova H, Bukrinsky M. beta-Chemokine production in CD40L-stimulated monocyte-derived macrophages requires activation of MAPK signaling pathways. Cytokine. 2003;23(3):53–63.
Fei Y, Webb R, Cobb BL, Direskeneli H, Saruhan-Direskeneli G, Sawalha AH. Identification of novel genetic susceptibility loci for Behçet’s disease using a genome-wide association study. Arthritis Res Ther. 2009;11(3):R66.
Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, et al. Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behcet’s disease. Ann Rheum Dis. 2013;72(9):1510–6.
Ortiz-Fernández L, Conde-Jaldón M, García-Lozano JR, Montes-Cano MA, Ortego-Centeno N, Castillo-Palma MJ, et al. GIMAP and Behçet disease: no association in the European population. Ann Rheum Dis. 2014;73(7):1433–4.
Burillo-Sanz S, Montes-Cano MA, García-Lozano JR, Ortiz-Fernández L, Ortego-Centeno N, García-Hernández FJ, et al. Mutational profile of rare variants in inflammasome-related genes in Behçet disease: a next generation sequencing approach. Sci Rep. 2017;7(1):8453.
Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–42.
Carmona FD, González-Gay MA, Martín J. Genetic component of giant cell arteritis. Rheumatology (Oxford). 2014;53(1):6–18.
Eyre S, Orozco G, Worthington J. The genetics revolution in rheumatology: large scale genomic arrays and genetic mapping. Nat Rev Rheumatol. 2017;13(7):421–32.
Ortiz-Fernández L, Carmona FD, López-Mejías R, González-Escribano MF, Lyons PA, Morgan AW, et al. Cross-phenotype analysis of Immunochip data identifies. Ann Rheum Dis. 2018;77(4):589–95.
Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. 2016;48(5):510–8.
Carmona FD, Coit P, Saruhan-Direskeneli G, Hernández-Rodríguez J, Cid MC, Solans R, et al. Analysis of the common genetic component of large-vessel vasculitides through a meta-Immunochip strategy. Sci Rep. 2017;7:43953.
Coit P, Direskeneli H, Sawalha AH. An update on the role of epigenetics in systemic vasculitis. Curr Opin Rheumatol. 2018;30(1):4–15.
Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419(6906):480–3.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ortiz-Fernández, L., González-Escribano, M.F. (2019). Behçet’s Disease. In: Martín, J., Carmona, F. (eds) Genetics of Rare Autoimmune Diseases. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-030-03934-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-03934-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03933-2
Online ISBN: 978-3-030-03934-9
eBook Packages: MedicineMedicine (R0)