
Abstract
Pseudohypoparathyroidism is a rare disorder caused by mutations and/or epigenetic changes at the complex GNAS locus on chromosome 20q13.3. This locus is capable of undergoing parent-specific methylation changes at several sites. GNAS encodes the alpha subunit of the stimulatory G protein (Gsα) and its several splice variants. Pseudohypoparathyroidism type 1a is caused by heterozygous inactivating mutations involving the maternal GNAS exons 1–13. Because paternal Gsα is not significantly expressed in the proximal renal tubule, thyroid, or pituitary, there is little or no normal Gsα protein in the presence of the maternal GNAS mutations, resulting in resistance to parathyroid hormone and consequent hyperparathyroidism, hypocalcemia, and hyperphosphatemia. Patients with pseudohypoparathyroidism type 1a have Albright’s hereditary osteodystrophy (AHO), with short stature, obesity, and shortened fourth and fifth metacarpal and metatarsal bones. When the same heterozygous inactivating mutations are present on the paternal GNAS allele, patients develop pseudopseudohypoparathyroidism, with features of AHO but without associated hyperparathyroidism, hypocalcemia, and hyperphosphatemia. Autosomal dominant pseudohypoparathyroidism type 1b is caused by heterozygous maternal deletion within GNAS or STX16, which are associated with loss of methylation at exon A/B alone or at all maternally methylated GNAS exons. Loss of methylation at exon A/B and the resulting biallelic expression of A/B transcripts reduce Gsα expression, leading to hormone resistance. Pseudohypoparathyroidism type 2 is thought to result from normal PTH/PTH-rp receptor-Gsα-adenylyl cyclase complex function but reduced action of the generated cAMP on downstream intracellular targets, such as sodium-phosphate cotransporters that mediate renal tubular phosphate reabsorption.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pseudohypoparathyroidism
- Hypocalcemia
- Hyperphosphatemia
- Hyperparathyroidism
- Albright’s hereditary osteodystrophy
Case Description
A 38-year-old healthy female was referred for evaluation and management of hypocalcemia discovered incidentally during medical evaluation. She complained of intermittent mild tingling paresthesias and occasional muscle cramps in her legs. She had never had tetany, bronchospasm, laryngospasm, cardiac rhythm disturbances, seizures, or loss of consciousness. She had been advised by her primary care physician to take supplemental calcium carbonate with 500 mg elemental calcium once a day and calcitriol 0.25 mcg twice a day, but she did not take these supplements on a regular basis.
Her physical evaluation showed normal stature of 68 in, weight 140 lb, and BMI 21.3 kg/m2. Her blood pressure was normal at 130/80 mm Hg, and her pulse was normal at 72 beats/min. Her physical exam showed a normal skeletal phenotype, without shortened fourth or fifth metacarpals or metatarsals or a dimple sign when she clenched her fists. She had no café au lait areas of macular hyperpigmentation, or subcutaneous calcifications, to suggest McCune-Albright syndrome.
Her initial serum calcium during evaluation was 7.5 mg/dL (normal, 8.9–10.1), with serum phosphorus upper normal at 4.5 mg/dL (normal, 2.5–4.5). Her serum creatinine was normal at 0.6 mg/dL (normal, 0.6–1.1). Her parathyroid hormone was increased at 540 pg/mL (normal, 15–65). Her serum 25-hydroxyvitamin D was optimal at 35 ng/mL (optimal, 20–50). Her serum magnesium was normal at 1.8 mg/dL (normal, 1.7–2.3).
Assessment for signs of hormone resistance other than to parathyroid hormone showed a normal sensitive TSH at 1.2 mIU/L (normal, 0.3–4.2). Her serum FSH was normal at 8.1 IU/L (normal, 1.8–22.5), with serum LH normal at 2.2 IU/L (normal, 1.2–100).
Her laboratory findings were consistent with pseudohypoparathyroidism. Because of her normal stature, lack of an apparent skeletal phenotype, or features of McCune-Albright syndrome, she was felt to most likely have pseudohypoparathyroidism type 1b and treated presumptively with calcium and calcitriol to try to lower her PTH level toward the normal range.
Introduction
Pseudohypoparathyroidism is a group of disorders characterized principally by proximal renal tubular resistance to parathyroid hormone (PTH) action [1]. These patients have impaired signaling by a number of hormones, particularly PTH, that activate cAMP-dependent pathways via Gsα proteins. Affected patients typically are characterized by hypocalcemia, hyperphosphatemia, and increased parathyroid hormone levels, with decreased serum 1,25-dihydroxyvitamin D levels. Patients with pseudohypoparathyroidism who are given exogenous biologically active PTH do not respond with an appropriate increase in urinary phosphate or cyclic adenosine monophosphate (cAMP). These patients do not demonstrate PTH resistance in other PTH target tissues, including the skeleton or thick ascending limb of the renal tubule [1].
Patients with pseudohypoparathyroidism usually develop neuromuscular irritability, manifesting as tingling paresthesias, muscle cramps, or seizures, unless treated with oral calcium supplementation and 1,25-dihydroxyvitamin D (calcitriol) [2]. Occasional patients are asymptomatic and have normal serum calcium and phosphate levels but maintain increased PTH levels. If these patients are not treated with calcium and/or calcitriol to lower their increased PTH secretion, they may develop significant bone disease over many years of chronic skeletal stimulation.
Pseudohypoparathyroidism was first described by Fuller Albright in 1942 as a disorder with target organ resistance to actions of PTH [3]. Since then a number of different variants have been described [4]. Patients with pseudohypoparathyroidism type 1 are characterized by unchanged serum cAMP and unchanged urinary phosphate and urinary cAMP after exogenous PTH injection. Patients with pseudohypoparathyroidism type 2 have increased plasma and urinary cAMP but unchanged urinary phosphate, after exogenous PTH injection.
This chapter will provide a broad overview of pseudohypoparathyroidism and discuss the clinical presentation, differential diagnosis, molecular pathophysiology, and treatment of the spectrum of disorders characterized by PTH resistance.
Epidemiology
Because of the rarity of pseudohypoparathyroidism, not much is known about the epidemiology of this disorder. Underbjerg et al. [5] identified all patients in Denmark with billing code diagnoses for pseudohypoparathyroidism through the Danish National Patient Registry and a prescription database. Billing code diagnoses were subsequently validated by records review. For each case, three age- (±2 years) and sex-matched controls were randomly selected from the general background population. A total of 60 cases of pseudohypoparathyroidism were identified, giving an estimated prevalence of 1.1 per 100,000 inhabitants. The average age at diagnosis in the cohort was 13 years (range, 1–62 years), and 42 of the patients were women. Only 14 patients had an identified mutation in their GNAS1 gene . Compared with controls, patients with pseudohypoparathyroidism had an increased risk of neuropsychiatric disorders (P < 0.01), infections (P < 0.01), seizures (P < 0.01), and cataracts (P < 0.01), whereas their risk of renal, cardiovascular, malignant disorders and fractures was comparable to the general background population. The same risks were found in a subgroup analysis in the 14 cases with genetically verified pseudohypoparathyroidism. The study concluded that patients with pseudohypoparathyroidism have an increased risk of neuropsychiatric disorders, infections, cataracts, and seizures, whereas mortality is comparable to that of the background population.
Clinical Presentation
Pseudohypoparathyroidism is typically defined by hypocalcemia, hyperphosphatemia, and increased PTH levels, associated with low-normal or decreased serum 1,25-dihydroxyvitamin D levels [1]. These biochemical abnormalities result from proximal renal tubular resistance to action of PTH. Physical symptoms have historically been attributed primarily to decreased ionized extracellular calcium, because studies have not demonstrated symptoms that correlated better with increased PTH levels than with low serum calcium.
Extracellular fluid hypocalcemia results in neuromuscular irritability, which may cause tingling paresthesias, muscle cramps, or tetany [6]. Patients may experience tingling sensations of their fingers, toes, lips, or tongue or nose tip and occasionally more diffuse tingling paresthesias over other facial areas. Symptoms can vary over time in the same patient, although many patients describe stereotypic symptoms that they learn to recognize as being due to low serum calcium. The level of serum calcium at which symptoms begin is quite variable between patients, but most patients describe symptoms with serum calcium below 7.5 mg/dL [7]. Some patients experience classical symptoms with serum calcium levels higher than this, and some seem to have few symptoms with serum calcium levels below 7.5 mg/dL.
Severe hypocalcemia usually presents in a more dramatic fashion, with seizures, bronchospasm, laryngospasm, cardiac rhythm disturbances, congestive heart failure, loss of consciousness, or, in extreme circumstances, sudden death.
Chvostek’s sign is a characteristic of latent neuromuscular irritability, in which tapping the facial nerve in front of the ear causes ipsilateral twitching of the upper lip [8]. This sign is not pathognomonic of hypocalcemia, however, as about 10–15% of the general healthy population demonstrates this also [9]. Trousseau’s sign is also characteristic of latent neuromuscular irritability, in which increasing pressure in a blood pressure cuff over the upper arm by 5 mm Hg above the systolic pressure will cause painful tetany in the arm below the blood pressure cuff within 3 min [10]. Trousseau’s sign is also not pathognomonic of hypocalcemia, as about 1–2% of the general healthy population may have this sign [11].
Patients with chronic hypocalcemia may develop features such as pseudo-papilledema, increased intracranial pressure, and dry rough skin [12]. Long-standing hypocalcemia and hyperphosphatemia associated with an increase in the calcium x phosphate product may cause cataracts or intracranial calcifications of the basal ganglia and other intracerebral structures [13]. Patients with extensive basal ganglia calcification may occasionally experience extrapyramidal dysfunction, but this is uncommon [14]. Spondyloarthropathy may occur rarely, causing significant joint pain and swelling and destruction [15]. Hypocalcemia may prolong the QT corrected interval on an electrocardiogram [16]. Congestive heart failure may occasionally develop due to prolonged severe hypocalcemia.
Patients with chronic mild to moderate hypocalcemia may adapt to their hypocalcemia fairly well and remain asymptomatic until low serum calcium is detected on routine blood testing.
Differential Diagnosis
Once other causes of hypocalcemia are ruled out, pseudohypoparathyroidism is usually easily diagnosed because of the classical biochemical changes. Patients with postsurgical or other forms of hypoparathyroidism have decreased serum calcium, upper-normal or increased serum phosphate, and lower PTH levels than expected for the simultaneously drawn serum calcium [17]. Patients with hypoparathyroidism demonstrate a significant increase in urinary phosphate and plasma and urinary cAMP after exogenous PTH administration [18]. In contrast, patients with pseudohypoparathyroidism type 1 have blunted increases in urinary phosphate and plasma and urinary cAMP after exogenous PTH, and patients with pseudohypoparathyroidism type 2 show increased plasma and urinary cAMP but blunted increases in urinary phosphate [19, 20] (Table 6.1).
Pseudohypoparathyroidism type 1 is caused by tissue deficiency of the alpha subunit of Gs (Gsα). Gsα is the signaling protein that couples stimulation of the PTH/PTH-rp receptor to stimulation of adenylyl cyclase [21]. Three forms of pseudohypoparathyroidism type 1 have been reported.
Pseudohypoparathyroidism type 1a (OMIM 103580) is the result of generalized deficiency of Gsα due to mutations within GNAS exons 1–13 [1]. Pseudohypoparathyroidism type 1b (OMIM 603233) is caused by more restricted deficiency of Gsα due to mutations affecting GNAS imprinting [22]. Patients with pseudohypoparathyroidism type 1a typically have resistance to multiple hormones and certain somatic features not seen in pseudohypoparathyroidism type 1b. Pseudohypoparathyroidism type 1c is thought to be a variant of type 1a in which resistance to multiple hormones is present without a defect in Gsα [23].
Pseudohypoparathyroidism type 1a is the most frequent type of pseudohypoparathyroidism and usually more easily identified than other types because of associated physical features. Patients with pseudohypoparathyroidism type 1a have Albright’s hereditary osteodystrophy (AHO), characterized by variable short stature, round facies, dental abnormalities, shortened fourth and fifth metacarpals and metatarsals, mild to moderate mental retardation, and subcutaneous calcifications [24]. Many of these patients have early-onset obesity, and some have sensory neuropathy, and they appear to have GNAS mutations causing abnormal Gsα signaling in the hypothalamus and central nervous system [25]. The obesity appears to be due to decreased expression of Gαs in imprinted regions of the hypothalamus [26], thereby leading to reduced energy expenditure rather than increased caloric intake.
Pseudohypoparathyroidism type 1a is caused by heterozygous mutations of the maternal allele of the imprinted GNAS gene on chromosome 20q13.2-q13.3, causing decreased expression or function of the Gsα protein. The requirement of normal expression of Gsα protein for signal transduction by many hormones and neurotransmitters leads to hormone resistance in patients with pseudohypoparathyroidism type 1a to not just PTH but also thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), calcitonin, and growth hormone-releasing hormone (GHRH) [27, 28], in tissues that express the maternal GNAS allele. Hormone resistance is not present in patients with pseudohypoparathyroidism type 1a in tissues where GNAS is not imprinted and both parental alleles are expressed. Because of this, response to adrenocorticotropic hormone (ACTH) and vasopressin is normal in these patients.
Patients with paternally inherited GNAS mutations have physical features of AHO but no evidence of resistance to PTH or other hormones. This disorder was first described in 1952 [29] and is known as pseudopseudohypoparathyroidism [30]. Kindreds with pseudopseudohypoparathyroidism without hormone resistance often have family members with pseudohypoparathyroidism with hormone resistance, with the variation in expression depending on the parental origin of the GNAS mutation.
Molecular Causes of Pseudohypoparathyroidism
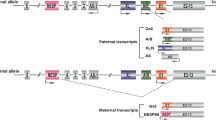
The GNAS gene maintains flexible expression in different tissues by using alternative first exons upstream of exon 1, alternative splicing of downstream exons, antisense mRNA transcripts, or reciprocal imprinting (Fig. 6.1). Gsα is encoded by exons 1–13 of the GNAS gene, with inclusion or exclusion of exon 3 leading to expression of either 52 kDa or 45 kDa proteins. These two Gsα isoforms both appear to function normally in signal transduction.

General organization and imprinting of GNAS. The maternal and paternal alleles of GNAS are shown. Methylated regions are indicated in gray, and active promoters are indicated by arrows in the direction of transcription. The alternative first exons and common exon 2 are shown as rectangles with exon 1 being the first coding exon for Gsα. The dashed line at the paternal Gsα promoter indicates tissue-specific imprinting of the gene. For clarity only the first exon for Nespas is shown, and the localization of DMR (differentially methylated regions) has been omitted in the figure. The figure also shows representation of the maternally derived deletions (double-headed arrows) found to cause familial PHP-Ib. The diagram is not drawn to scale. (Used with permission from Mantovani [1])
Different Gsα mRNA transcripts are produced by three alternative first exons upstream of exon 1, each splicing to exons 2–13. The first alternative exon XL is expressed only by the paternal allele and generates an mRNA transcript with overlapping open reading frames that encode XLsα [31] and ALEX. Both proteins are able to interact with each other and are expressed specifically in neuroendocrine cells. XLsα measures about 78 kDa and therefore is much larger than Gsα, which is either 52 or 45 kDa. XLsα interacts with PTH/PTH-rp receptors and other receptors in cell systems, but it is not yet clear whether this protein can interact with these receptors in the whole organism.
The second alternative first exon is encoded only by the maternal allele and generates a secretory protein called neuroendocrine secretory peptide 55 (NESP55) [32]. This protein has no sequence homology with Gsα.
The third alternative first exon 1A or A/B (associated first exon) is encoded only by the paternal allele. Transcripts from this alternative first exon may be translated from an initiator codon in the second exon and produce an N-terminal truncated protein that competitively inhibits Gsα [33].
These three alternative first exons for Gsα are associated with differentially methylated promotor regions. Methylation results in silencing of the affected allele. Unlike the three alternative first exons, the promotor for exon 1 is within a CpG island and remains unmethylated on both alleles in all tissues. Cis-acting elements that control tissue-specific paternal imprinting of Gsα are thought to be located within the primary imprint region in exon 1A [34].
Multiple kindreds with pseudohypoparathyroidism type 1a have four-base deletions in exon 7, and because the missense mutation A366S has been found in exon 13 in two unrelated boys, it is possible that exons 7 and 13 may represent sites of frequent GNAS mutations. About 80% of patients with AHO have been identified as having small deletions or point mutations in GNAS, whereas other cases have been reported to have larger rearrangements or uniparental disomy, where both GNAS alleles are inherited from the mother.
Patients with pseudohypoparathyroidism type 1b may have postzygotic somatic mutations in GNAS that increase the expression of the Gsα protein, with constitutive activation of adenylyl cyclase, resulting in proliferation and autonomous increased function of hormonally responsive tissues [35]. Activating mutations of the GNAS maternal allele expressed in imprinted tissues may lead to clinically significant effects. Variable Gsα activity in different tissues may help determine hormone action in the different tissues.
Patients with pseudohypoparathyroidism type 1b may have shortened fourth or fifth metacarpal or metatarsal bones but do not express most of the other features of AHO. Gsα activity is normal when measured in various tissues of these patients. Pseudohypoparathyroidism type 1b patients have PTH resistance and, in some cases, TSH resistance but lack resistance to other hormones. Changes in bone density over time in these patients correlate with serum PTH levels [36]. Patients with sporadic or inherited pseudohypoparathyroidism type 1b have switched their pattern of maternal methylation of the Gsα allele to a pattern of paternal methylation [37]. Mutations causing a switch in the maternal to paternal methylation pattern are found in most patients with pseudohypoparathyroidism type 1b. These mutations include two microdeletions in the STX16 gene located 220 kB centromeric to the GNAS exon 1A [38] and deletions of the differentially methylated region of the NESP55 exon and exons 3 and 4 of the antisense mRNA transcript. Inheritance of the mutation from the mother, or a spontaneous mutation of a maternally derived allele, negates the maternal GNAS methylation pattern. Patients with pseudohypoparathyroidism type 1b have not been reported to have small mutations, but uniparental disomy has been reported, where both GNAS alleles were inherited from the father. It is believed that conversion of the maternal GNAS allele epigenotype to the paternal GNAS allele epigenotype, or inheritance of two paternal GNAS alleles, results in transcriptional silencing of the Gsα promoter in imprinted tissues by both alleles, with resultant limited or absent expression of Gsα in these tissues.
Patients with pseudohypoparathyroidism type 1c have AHO features with resistance to multiple hormones, without evidence of Gsα or Giα signaling transduction abnormalities. These patients may have GNAS mutations resulting in functional defects in Gsα [39].
The heterotopic ossifications of AHO are a unique feature of this disorder [40]. These ossifications are not mature calcifications and not related to serum calcium or phosphorus levels or the calcium x phosphate product. These ossifications are areas of intramembranous bone that seem to develop without prior inflammation or trauma. It is thought that Gsα deficiency leads to expression of ectopic Cbfa1/Runx2 in mesenchymal stem cells in the skin, causing these cells to differentiate into osteoblasts that cause new bone to form in the skin.
Osteoma cutis and progressive osseous heteroplasia (POH) are two forms of AHO in which abnormal bone formation is the only feature present [41]. In osteoma cutis, abnormal bone formation occurs only in the skin, whereas in POH, abnormal bone formation occurs in the skin, subcutaneous tissues, muscles, tendons, and ligaments. Progressive osseous heteroplasia may result in limitation of joint or limb movement due to widespread calcification of the skin during childhood, followed by gradual conversion of skeletal muscles and deeper connective tissues into the bone. Bony nodules and strands of heterotopic bone may extend from the skin and superficial connective tissues into the subcutaneous fat and deeper connective tissues.
Patients with osteoma cutis and POH may have paternally inherited inactivating heterozygous GNAS mutations. These patients lack other features of AHO or pseudohypoparathyroidism.
Pseudohypoparathyroidism type 2 is thought to result from normal PTH/PTH-rp receptor-Gsα-adenylyl cyclase complex function but reduced action of the generated cAMP on downstream intracellular targets, such as sodium-phosphate cotransporters that mediate renal tubular phosphate reabsorption. Pseudohypoparathyroidism type 2 currently lacks a defined genetic or familial inheritance, with the clinical and biochemical presentation similar to severe vitamin D deficiency or vitamin D resistance. It is possible that cases of pseudohypoparathyroidism type 2 may result from unsuspected vitamin D deficiency [42].
Acrodysostosis is a form of pseudohypoparathyroidism type 2 characterized by increased basal and PTH-stimulated urinary cAMP excretion but lack of PTH-induced phosphate excretion [43]. This disorder has several forms, with type 1 due to mutations in protein kinase A regulatory subunit 1A and type 2 due to mutations in phosphodiesterase 4D.
Transient pseudohypoparathyroidism of the newborn may occur in infants within 5–7 days of birth [44]. Most infants developing hypocalcemia at this age have decreased PTH levels causing hypoparathyroidism, but as many as 25% may have increased PTH levels. These infants may have delayed development of the post-cAMP signaling pathway limited to the proximal renal tubule. Affected infants seem to recover from this delay in post-receptor signaling maturation within 6 months of birth.
Patients with pseudohypoparathyroidism type 1 commonly have an apparent dissociation between circulating bioactive PTH and serum immunoreactive PTH. Plasma from these patients may have reduced biological activity in in vitro cytochemical bioassays, implying inhibition of PTH action. A potential explanation for this inhibition may be accumulated fragments of PTH 7–84 and other fragments that inhibit the hypercalcemic and hypophosphatemic effects of PTH 1–84. PTH 7–84 fragments are often increased in patients with pseudohypoparathyroidism types 1a and 1b, with the proportion of these fragments increased compared to biologically active PTH 1–84. These and other fragments could accumulate in patients with pseudohypoparathyroidism type 1 due to the duration of their long-standing hyperparathyroidism and may not play a significant role in the pathogenesis of the disorder.
Hypomagnesemia and severe vitamin D deficiency may both cause biochemical findings of PTH resistance, so it is important to measure serum magnesium and 25-hydroxyvitamin D before confirming a suspected diagnosis of pseudohypoparathyroidism.
The AHO features seen in pseudohypoparathyroidism type 1a may be also seen in other genetic disorders such as Prader-Willi syndrome or Ullrich-Turner syndrome. Patients with small terminal deletions of chromosome 2q37 may appear to have AHO, but they have normal hormone function and normal Gsα activity.
The classical test for pseudohypoparathyroidism, the Ellsworth-Howard test, was later modified by Chase, Melson, and Aurbach [45, 46]. This test required intravenous infusion of 200–300 USP units of purified bovine PTH or parathyroid extract. Since purified bovine PTH is no longer available, this protocol has been modified to use teriparatide (human recombinant parathyroid hormone 1–34) either by intravenous infusion or subcutaneous injection. One version of this modified protocol [47] recommends having patients drink 250 mL of water each hour from 6 a.m. to 12 p.m. Two 30-min control urine collections are obtained before 9 a.m. Teriparatide is given at 9 a.m. at 0.625 mcg/kg body weight to a maximum of 25 mcg by intravenous infusion over 15 min or to a maximum of 40 mcg by subcutaneous injection. Thirty-minute urine collections are started at 9 a.m. and 9:30 a.m., and 1-h urine collections are started at 10 a.m. and 11 a.m. Serum phosphate and creatinine are drawn at 9 a.m. and 11 a.m. The urine samples from the different time points are measured for cAMP, phosphorus, and creatinine. Cyclic AMP results are expressed as nmol cAMP per 100 mL glomerular filtrate and renal tubular phosphate reabsorption as TmP/GFR. Normal healthy subjects typically demonstrate a 10–20-fold increase in urinary cAMP excretion and 20–30% decrease in TmP/GFR. Patients with pseudohypoparathyroidism types 1a and 1b show markedly blunted responses no matter how low their serum calcium.
Genetic testing may help in the diagnosis of pseudohypoparathyroidism type 1a but is not helpful in making a diagnosis of pseudohypoparathyroidism type 1b or other types of pseudohypoparathyroidism.
A new classification schema has recently been established by the EuroPHP network to cover all disorders of the PTH receptor and its signaling pathway [48, 49]. Inactivating PTH/PTH-related protein signaling disorder (iPPSD) is the new name proposed for these disorders. These disorders are divided into subtypes iPPSD1 through iPPSD6. PTH receptor inactivation mutations leading to Eiken and Blomstrand dysplasia are classified as iPPSD1. Inactivating Gsα mutations causing pseudohypoparathyroidism type 1a, pseudohypoparathyroidism type 1c, and pseudopseudohypoparathyroidism are classified as iPPSD2. Mutations leading to loss of methylation of GNAS disease-modifying regions leading to pseudohypoparathyroidism type 1b are classified as iPPSD3. Mutations of protein kinase A regulatory subunit 1A (PRKAR1A) leading to acrodysostosis type 1 are classified as iPPSD4. Mutations of phosphodiesterase 4D (PDE4D) leading to acrodysostosis type 2 are classified as iPPSD5. Mutations of phosphodiesterase 3A (PDE3A) causing autosomal dominant hypertension with brachydactyly are classified as iPPSD6. iPPSDx is the designation given for unknown molecular defects, and iPPSDn+1 is the term used for new molecular defects not yet described.
Treatment
Treatment of pseudohypoparathyroidism is focused on correcting the biochemical abnormalities of low serum calcium, high serum phosphorus, and associated hyperparathyroidism [50]. These goals include improving serum calcium to as close to the normal range as possible, thereby helping suppress PTH secretion as much as possible toward normal. The target PTH level is as close to the upper limit of normal as possible to minimize increased bone resorption leading to osteitis fibrosa cystica.
Because the distal renal tubular effects of PTH in patients with pseudohypoparathyroidism remain intact, calcium reabsorption from the glomerular filtrate occurs normally. This minimizes the amount of calcium supplement required to keep serum calcium normal and to suppress PTH secretion.
Because proximal renal tubular production of 1,25-dihydroxyvitamin D from 25-hydroxyvitamin D via 1α-hydroxylase is limited in pseudohypoparathyroidism, calcitriol supplementation is typically required [51]. Combination therapy with calcium and calcitriol usually does not increase urinary calcium because the distal renal tubular mechanisms for reabsorption of calcium from glomerular filtrate remain intact. Urinary calcium excretion may increase before serum calcium normalizes. The best way to prevent significant hypercalciuria in this situation is to target serum calcium in the low-normal range.
Low-phosphate diets or phosphate binders are sometimes necessary to reduce increased serum phosphate levels.
Calcium supplementation with 1–2 g elemental calcium each day is usually sufficient to increase serum calcium levels into the low-normal range, as well as to block intestinal phosphate absorption to limit hyperphosphatemia. Calcium supplements should be taken with meals to reduce intestinal phosphate absorption.
Calcitriol has a short half-life of 2–3 h and is usually given at starting doses of 0.25–0.50 mcg at least twice daily. Alfacalcidol is not approved for use in the USA but is available in Europe and much of the rest of the world. Alfacalcidol has a longer half-life and is usually given at starting doses of 0.50–1.00 mcg once daily. Vitamin D2 or D3 may be given in age-appropriate doses for skeletal health as per the Institute of Medicine 2011 dietary reference intakes [52] but should not be given in very large doses as in the past, as these very high doses may lead to markedly increased serum 25-hydroxyvitamin D levels that may become toxic and cause significant hypercalcemia and kidney dysfunction. Vitamin D2 or D3 toxicity may take weeks to months to resolve after vitamin D2 or D3 are stopped, during which renal dysfunction may worsen or become permanent.
Thiazide-type diuretics limit urinary calcium loss, and these may be helpful in stabilizing or improving serum calcium levels in pseudohypoparathyroidism. Because thiazide-type diuretics cause urinary potassium loss, potassium supplementation may be necessary to prevent hypokalemia with long-term thiazide-type diuretic use.
Cinacalcet has been used as adjunctive therapy to reduce very high levels of PTH secretion in at least one pediatric patient and one young man with pseudohypoparathyroidism type 1b when calcium and activated vitamin D supplementation in combination were not sufficient to control the hyperparathyroidism [53, 54].
Recombinant human growth hormone has been used to treat short stature in eight prepubertal children with pseudohypoparathyroidism type 1a for 3–8 years, with reported results similar to children with idiopathic growth hormone deficiency [55].
PTH treatment is not recommended for pseudohypoparathyroidism because serum PTH levels are significantly increased already. Tissue resistance to endogenous PTH secretion makes it difficult for exogenous PTH administration to have a significant beneficial effect.
Summary and Conclusions
Pseudohypoparathyroidism is a rare disorder that results from proximal renal tubular resistance to parathyroid hormone. This tissue resistance results in hyperparathyroidism associated with decreased serum calcium, increased serum phosphate, and decreased 1,25-dihydroxyvitamin D. A variety of forms of pseudohypoparathyroidism have been described. Pseudohypoparathyroidism type 1 results in inability of exogenous PTH to stimulate urinary cAMP and phosphate excretion. Pseudohypoparathyroidism type 2 is associated with increased urinary cAMP excretion but normal urinary phosphate excretion after administration of exogenous PTH. PTH resistance occurs in the proximal renal tubule but not in other PTH target tissues. Most patients develop symptoms of hypocalcemia, including tingling paresthesias, muscle cramps, tetany, or seizures without adequate supplementation. Calcium and calcitriol supplementation is recommended for all patients with pseudohypoparathyroidism to prevent sustained hyperparathyroidism and resultant bone disease.
References
Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab. 2011;96:3020–30.
Singla M, Garg G, Gupta A. Pseudohypoparathyroidism type 1a. QJM. 2018;111:331–3.
Albright F, Burnett CH, Smith PH, Parson W. Pseudohypoparathyroidism – an example of “Seabright-Bantam syndrome”. Endocrinology. 1942;39:922–32.
Cianferotti L, Brandi ML. Pseudohypoparathyroidism. Minerva Endocrinol. 2018;43:156–67.
Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. Pseudohypoparathyroidism – epidemiology, mortality and risk of complications. Clin Endocrinol. 2016;84:904–11.
Tafaj O, Jüppner H. Pseudohypoparathyroidism: one gene, several syndromes. J Endocrinol Invest. 2017;40:347–56.
Mantovani G, Spada A, Elli FM. Pseudohypoparathyroidism and Gsα-cAMP-linked disorders: current view and open issues. Nat Rev Endocrinol. 2016;12:347–56.
Athappan G, Ariyamuthu VK. Images in clinical medicine. Chvostek’s sign and carpopedal spasm. N Engl J Med. 2009;360:e24.
Kotowicz J. Occurrence of Chvostek’s sign in healthy people and its significance. Pol Med J. 1969;8:440–3.
Meininger ME, Kendler JS. Images in clinical medicine. Trousseau’s sign. N Engl J Med. 2000;343:1855.
van Bussel BC, Koopmans RP. Trousseau’s sign at the emergency department. BMJ Case Rep. 2016;2016.
Kuzel AR, Lodhi MU, Rahim M. Classic and non-classic features in pseudohypoparathyroidism: case study and brief literature review. Cureus. 2017;9:e1878.
Kobayashi Y, Tsuyuzaki J, Koizumi Y. Pseudohypoparathyroidism causing multiple brain calcifications. Intern Med. 2018;57:153–4.
Song CY, Zhao ZX, Li W, Sun CC, Liu YM. Pseudohypoparathyroidism with basal ganglia calcification: a case report of rare cause of reversible parkinsonism. Medicine (Baltimore). 2017;96:e6312.
Sharma M, Nolkha N, Sharma A, Bhadu D, Dhakad U, Singh R, Das SK. Spondyloarthropathy-like findings and diffuse osteosclerosis as the presenting feature of pseudohypoparathyroidism. J Clin Rheumatol. 2016;22:102–4.
Heemskerk CPM, Pereboom M, van Stralen K, Berger FA, van den Bemt PMLA, Kuijper AFM, van der Hoeven RTM, Mantel-Teeuwisse AK, Becker ML. Risk factors for QTc interval prolongation. Eur J Clin Pharmacol. 2018;74:183–91.
Bilezikian JP, Khan AA, Potts JT Jr, Brandi ML, Clarke BL, Shoback D, Jüppner H, D’Amour P, Fox J, Rejnmark L, Mosekilde L, Rubin MR, Dempster D, Gafni R, Collins MT, Sliney J, Sanders J. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res. 2011;26:2317–37.
Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med. 2008;359:391–403.
Chase LR, Melson GL, Aurbach. Pseudohypoparathyroidism: defective excretion of 3′,5′-AMP in response to parathyroid hormone. J Clin Invest. 1969;48:1832–44.
Drezner M, Neelon FA, Lebovitz HE. Pseudohypoparathyroidism type II: a possible defect in the reception of the cyclic AMP signal. N Engl J Med. 1973;289:1056–60.
Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory g protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705.
Mantovani G, Elli FM. Pseudohypoparathyroidism type Ib in 2015. Ann Endocrinol (Paris). 2015;76:101–4.
Brix B, Werner R, Staedt P, Struve D, Hiort O, Thiele S. Different pattern of epigenetic changes of the GNAS gene locus in patients with pseudohypoparathyroidism type Ic confirm the heterogeneity of underlying pathomechanisms in this subgroup of pseudohypoparathyroidism and the demand for a new classification of GNAS-related disorders. J Clin Endocrinol Metab. 2014;99:E1564–70.
Salemi P, Skalamera Olson JM, Dickson LE, Germain-Lee EL. Ossifications in Albright hereditary osteodystrophy: role of genotype, inheritance, sex, age, hormonal status, and BMI. J Clin Endocrinol Metab. 2018;103:158–68.
Grüters-Kieslich A, Reyes M, Sharma A, Demirci C, DeClue TJ, Lankes E, Tiosano D, Schnabel D, Jüppner H. Early-onset obesity: unrecognized first evidence for GNAS mutations and methylation changes. J Clin Endocrinol Metab. 2017;102:2670–7.
Chen M, Shrestha YB, Podyma B, Cui Z, Naglieri B, Sun H, Ho T, Wilson EA, Li YQ, Gavrilova O, Weinstein LS. Gsα deficiency in the dorsomedial hypothalamus underlies obesity associated with Gsα mutations. J Clin Invest. 2017;127:500–10.
Mantovani G, Maghnie M, Weber G, De Menis E, Brunelli V, Cappa M, Loli P, Beck-Peccoz P, Spada A. Growth hormone releasing hormone resistance in pseudohypoparathyroidism type ia: new evidence for imprinting of the Gs alpha gene. J Clin Endocrinol Metab. 2003;88:4070–4.
Germain-Lee EL, Groman J, Crane JL, Jan de Beur SM, Levine MA. Growth hormone deficiency in pseudohypoparathyroidism type 1a: another manifestation of multihormone resistance. J Clin Endocrinol Metab. 2003;88:4059–69.
Albright F, Forbes AP, Henneman PH. Pseudopseudohypoparathyroidism. Trans Assoc Am Phys. 1952;65:337–50.
Thiele S, Werner R, Grötzinger J, Brix B, Staedt P, Struve D, Reiz B, Farida J, Hiort O. A positive genotype-phenotype correlation in a large cohort of patients with Pseudohypoparathyroidism type Ia and pseudo-pseudohypoparathyroidism and 33 newly identified mutations in the GNAS gene. Mol Genet Genom Med. 2015;3:111–20.
Kehlenbach RH, Matthey J, Huttner WB. XLαs is a new type of G protein. Nature. 1995;372:804–9.
Ischia R, Lovisetti-Scamihorn P, Hogue-Angeletti R, Wolkersdorfer M, Winkler H, Fischer-Colbrie R. Molecular cloning and characterization of NESP55, a novel chromogranin-like precursor of a peptide with 5HT1B receptor agonist activity. J Biol Chem. 1997;272:11657–62.
Basteppe M, Pincus JE, Sugimoto T, Tojo K, Kanatami M, Azuma Y, Kruse K, Rosenbloom AL, Koshiyama H. Positional dissociation between the genetic mutation responsible for pseudohypoparathyroidism type Ib and the associated methylation defect at exon A/B: evidence for a long-range regulatory element within the imprinted GNAS1 locus. Hum Mol Genet. 2001;10:1231–41.
Williamson CM, Ball ST, Nottingham WE, Skinner A, Plagge A, Turner MD, Powles N, Hough T, Papworth D, Fraser WD, et al. A cis-acting control region is required exclusively for the tissue imprinting of Gnas. Nat Genet. 2004;36:894–9.
Elli FM, Bordogna P, Arosio M, Spada A, Mantovani G. Mosaicism for GNAS methylation defects associated with pseudohypoparathyroidism type 1B arose in early post-zygotic phases. Clin Epigenetics. 2018;10:16.
Chu X, Zhu Y, Wang O, Nie M, Quan T, Xue Y, Wang W, Jiang Y, Li M, Xia W, Xing X. Bone mineral density and its serial changes are associated with PTH levels in pseudohypoparathyroidism type 1b patients. J Bone Miner Res. 2018;33:743–52.
Bastepe M, Frolich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, Korkko J, Nakamoto JM, Rosenbloom AL, Slyper AH, et al. Autosomal dominant pseudohypoparathyroidism type 1b is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–63.
Linglart A, Gensure RC, Olney RC, Juppner H, Bastepe M. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type 1b redefines the boundaries of a cis-acting imprinting control element of GNAS. Am J Hum Genet. 2005;76:804–14.
Thiele S, de Sanctis L, Werner R, Grotzinger J, Aydin C, Juppner H, Bastepe M, Hiort O. Functional characterization of GNAS mutations found in patients with pseudohypoparathyroidism type 1c defines a new subgroup of pseudohypoparathyroidism affecting selectively Gsalpha-receptor interaction. Hum Mutat. 2011;32:653–60.
Bastepe M. GNAS mutations and heterotopic ossification. Bone. 2018;109:80–5.
Kaplan FS, Shore EM. Progressive osseous heteroplasia. J Bone Miner Res. 2000;15:2084–94.
Shriraam M, Bhansali A, Velayutham P. Vitamin D deficiency masquerading as pseudohypoparathyroidism type 2. J Assoc Physicians India. 2003;51:619–20.
Silve C. Acrodysostosis: a new form of pseudohypoparathyroidism? Ann Endocrinol (Paris). 2015;76:110–2.
Lee CT, Tsai WY, Tung YC, Tsau YK. Transient pseudohypoparathyroidism as a cause of late-onset hypocalcemia in neonates and infants. J Formos Med Assoc. 2008;107:806–10.
Chase LR, Melson GL. Aurbach GD Pseudohypoparathyroidism: defective excretion of 3′,5′-AMP in response to parathyroid hormone. J Clin Invest. 1969;48:1832–44.
Sohn HE, Furukawa Y, Yumita S, Miura R, Unakami H, Yoshinaga K. Effect of synthetic 1-34 fragment of human parathyroid hormone on plasma adenosine 3′,5′-monophosphate (cAMP) concentrations and the diagnostic criteria based on the plasma cAMP response in Ellsworth-Howard test. Endocrinol Jpn. 1984;31:33–40.
Rubin MR, Levine MA. Hypoparathyroidism and pseudohypoparathyroidism. In: Rosen CJ editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. 8th ed. Wiley; 2013. p. 579–89.
Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L, Elli FM, Freson K, Garin I, Grybek V, Hanna P, Izzi B, Hiort O, Lecumberri B, Pereda A, Saraff V, Silve C, Turan S, Usardi A, Werner R, de Nanclares GP, Linglart A. From pseudohypoparathyroidism to inactivating PTH/PTHrP signaling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. 2016;175:P1–P17.
Turan S. Current nomenclature of pseudohypoparathyroidism: inactivating parathyroid hormone/parathyroid hormone-related protein signaling disorder. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):58–68.
Lopes MP, Kliemann BS, Bini IB, Kulchetscki R, Borsani V, Savi L, Borba VZ, Moreira CA. Hypoparathyroidism and pseudohypoparathyroidism: etiology, laboratory features and complications. Arch Endocrinol Metab. 2016;60:532–6.
Ishii A, Imanishi Y, Kurajoh M, Nagata Y, Kobayashi K, Miki T, Inaba M, Nishizawa Y. The administration of an active vitamin D(3) analogue reduced the serum concentrations of 1-84 and truncated parathyroid hormone in pseudohypoparathyroidism type Ib patients. Endocr J. 2010;57:609–14.
IOM (Institute of Medicine). Dietary reference intakes for calcium and vitamin D. Washington, DC: The National Academies Press; 2011.
Srivastava T, Krudys J, Mardis NJ, Sebestyen-VanSickle J, Alon US. Cinacalcet as adjunctive therapy in pseudohypoparathyroidism type 1b. Pediatr Nephrol. 2016;31:795–800.
Koch M, Kohnle M. Successful off-label use of cinacalcet HCl after standard therapy failure in a young man with pseudohypoparathyroidism type 1b and vitamin D intoxication sequelae. Clin Nephrol. 2008;70:439–44.
Mantovani G, Ferrante E, Giavoli C, Linglart A, Cappa M, Cisternino M, Maghnie M, Ghizzoni L, de Sanctis L, Lania AG, Beck-Peccoz P, Spada A. Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type Ia: first study on the effect on growth. J Clin Endocrinol Metab. 2010;95:5011–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Clarke, B.L. (2019). Pseudohypoparathyroidism. In: Camacho, P. (eds) Metabolic Bone Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-03694-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-03694-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03693-5
Online ISBN: 978-3-030-03694-2
eBook Packages: MedicineMedicine (R0)