
Abstract
Lipid-based and polymeric nanotechnologies are poised to dramatically alter the landscape of treatment options for cancer and may hold unique potential for easily accessible, oral chemotherapy. A growing consensus points to nanoscale drug delivery systems as a promising therapeutic modality with enhanced efficacy and diminished side effects and with increasing evidence that these platforms can be engineered to facilitate transport of poorly bioavailable drug compounds and target neoplastic tissue with precision. Significant design and process challenges remain however. The emergence of oral chemotherapeutics in cancer treatment and the role lipid and polymer nanotechnologies play in its development are discussed in this chapter. Several recent research results provide rules of thumb for design and optimization of nanoparticles (i.e., physicochemical and surface properties) to achieve the goals of enhancing intestinal permeability, decreasing immunogenicity and extending circulation half-life, tumor targeting, and minimizing aggregation. Finally, characterization methods to assess drug release and pharmacokinetics will be examined, including dialysis systems, in vitro intestinal co-culture models, microfluidic artificial organs, and in vivo preclinical models.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Aggregation
- Bioavailability
- Chemical permeation enhancer
- Dialysis systems
- Excipients
- Flash nanoprecipitation
- Freezing
- Drying
- Immunoliposome
- In vitro intestinal co-culture model
- Liposome
- Lipid nanocapsule
- Microfluidic artificial organs
- Mucoadhesion
- Mucopenetration
- Nanoprecipitation
- Nanostructured lipid carriers
- Nanotherapeutics
- Oral absorption
- Ostwald ripening
- Secondary crystallization
- Solid lipid nanocapsules
- Stealth liposome
- Surface design
- Transmucosal permeability
- Two-phase sink condition
1 Oral Chemotherapeutic Drug Formulations: Overview of Current Challenges, Issues, and Opportunities for Oral Absorption
The guiding principle for formulation scientists , pharmaceutical directors, and clinicians alike is to prioritize pills and tablets over intravenous infusions. Certainly, these pronouncements are wholly justified because orally administered medicines have robust data to confirm their clinical efficacy: their dosing regimens are uncomplicated, not requiring a visit to the hospital or specially trained clinicians for administration. They are also minimally invasive and do not cause the patient excessive pain. Taken together, oral administration is the most facile and least invasive strategy for drug therapy [1, 2]. However, certain newly discovered antineoplastic agents represent a particular formulation challenge because of their physicochemical sensitivity, poor absorption, and significant side effects, virtually precluding the application of current design strategies employed in the pharmaceutical industry including stalwart processes like hot-melt extrusion [3].
Biologic therapies, pharmaceuticals that are manufactured in bioreactors using recombinant protein engineering, are poised to supersede conventional small molecule medicines as the first line of therapy for a number of conditions [4]. These medications present unique formulation challenges and must be given intravenously or subcutaneously to be properly absorbed [5].
Notwithstanding, significant capital investment in cancer research and development has enabled several oral chemotherapeutic medicines to be approved and marketed in the United States, Europe, and elsewhere for several years [6]. For example, imatinib (Gleevec®, Novartis) is an effective therapy against chronic myelogenous leukemia (CML) and countless other cancers; it nevertheless manifests a series of severe side effects traditionally encountered with other oncolytic medicines [7].
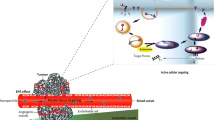
One of the complications in the design of an efficacious oral cancer therapy is the mode in which the gastrointestinal tract functions simultaneously as both a chemical and a physical barrier to drug absorption compared to parenteral administration routes (Fig. 9.1). The stomach churns with juices of high acidity that rapidly degrade pH-sensitive compounds [8]. A plethora of enzymes hydrolyze proteins and peptides, lipids, polysaccharides, and esters. More importantly, the epithelium acts as a semipermeable membrane, allowing the transport of only valuable nutrients, but preventing the absorption of large hydrophilic molecules and some hydrophobic compounds as well [9]. Moreover, it also secretes a layer of several micron-thick viscoelastic fluid filled with a cross-linked network of proteins that readily expels foreign particles out of the body [10].

Graphical description of the structural components of the mucosal barrier and drug transport mechanism across intestinal cells. The mucus layer acts simultaneously as both a physical and a chemical barrier, through the action of secretory immunoglobulins (secretory IgAs) and antimicrobial peptides. There are several possible mechanisms of drug transport through the intestinal mucosal barrier. The first pathway is the passive transcellular route. Through this pathway, drug permeates the cell passively by partitioning to cell membranes at both apical and basolateral sides. The second pathway is the paracellular route, where the drug transports through the tight intercellular junctions mediated by protein-protein interaction at different regions. The third pathway is the active transport route, where the drug is recognized by transporters and shuttled from the apical to the basolateral side. The fourth pathway is the mechanism of inhibiting drug permeation by efflux pumps. The efflux pumps expel the drug from cell membranes during the cell membrane partition process
As such, the first-pass metabolism of active pharmaceutical ingredients (APIs) given orally is frequently much more significant than after parenteral administration, which therefore requires oral dosages to be many folds higher to achieve the equivalent serum concentrations of the drug. For example, docetaxel’s oral bioavailability is low because it is extensively degraded by gastric and hepatic cytochromes and prevented from permeating the intestinal epithelium by a glycoprotein transporter. Docetaxel is therefore intravenously formulated to mitigate this extensive first-pass metabolism. However, when dosed orally concomitantly with the antibiotic cyclosporine, which competes for cytochrome binding and inhibits the epithelial protein, serum docetaxel concentrations dramatically improve [11].
Additionally, simply reformulating parenteral chemotherapies for oral administration at higher dosages in order to attain commensurate blood levels may be impractical and raises significant toxicological issues. Upon transit of the gastric epithelium , all orally ingested drugs enter the circulation only to be transported directly to the liver [12]. Overloading the intestinal mucus barrier with a chemotherapeutic in order to trigger drug permeation may thus lead to potentially toxic hepatic accumulation, e.g., with oral tamoxifen citrate [13]. Mindful of such scenarios, researchers have primarily focused on oral chemotherapies for colon cancer (e.g., oral capecitabine) [14], where transmucosal permeability is not crucial for treatment efficacy. In addition, oral bioavailability of these drugs can be further improved by using small molecule receptor tyrosine kinase (RTK) inhibitors [15].
Despite the formidable obstacles facing oral administration of novel chemotherapeutic medicines, a number of approaches are currently being investigated to overcome these formulation challenges beyond the current conventional clinical practice. It was demonstrated in small and big animals that polymeric nanoparticles with controlled physicochemical properties can dramatically improve hydrophobic drug oral availability [16,17,18,19,20,21]. O. M. Farokhzad et al. at MIT and Harvard Medical School have achieved a breakthrough in the oral delivery of biologic medicines by focusing on protein transporters distributed on the intestinal epithelium. By conjugating an antibody to a biodegradable polymeric nanoparticle containing the biologic medicine, the drug delivery system allows for the therapy to enter the bloodstream [22]. Chemical permeation enhancers (CPEs) such as surfactants and lipid compounds are another delivery strategy used to modify the morphology of the gastrointestinal epithelium to enhance drug bioabsorption by orders of magnitude [23]. Perhaps even more promising for the pharmaceutical industry, nanotechnologies have enabled the invention of intelligent drug delivery vehicles employing natural lipid molecules and biocompatible synthetic polymers (e.g., platforms like phospholipid vesicles and PLGA nanoparticles, respectively) especially suited to transport drug cargoes to the targeting sites, promising enhanced efficacy and significantly reduced side effects [24]. Lipid- and polymer-based technology platforms for oral delivery of cancer chemotherapeutics will be discussed in the section below, with special emphasis placed on tailoring nanoparticle structure to modulate therapeutic function and in vitro assessments as a predictor of (and as a means to construct) in vivo pharmacological models.
2 The Big Picture: How Do Nanotherapeutic Systems Enable Oral Absorption?
Nanoscale drug delivery systems (DDSs) (with at least one dimension in the order of hundreds of nanometers or smaller) provide many advantages to current industry design strategies with regard to the formulation of orally administered cancer chemotherapies. Chemotherapeutic nanoparticles may actively or passively accumulate at the tumor site [25]. The APIs can be protected by immune assault and premature metabolism [26], which enhances the bioavailability of the APIs [27]. With these functional design elements, the lipid-based and polymeric nanotherapeutics explored throughout this chapter promise to outperform conventional oncolytic medicines . However, significant challenges remain in the development of easily accessible oral formulations.
Nanoscopic assemblies of lipids and polymers with hydrophobic constituents can be used to encapsulate poorly water-soluble drugs in their hydrophobic cores in order to minimize free energy. Rational design of these drug delivery systems can then ensure that the particles will be well dispersed in aqueous media and the drugs are absorbable either in the gastric mucosa or in the circulatory system, using structures with hydrophilic shells or other materials with amphipathic or surface active moieties for kinetic stability [28].
Free from the bulk solvent environment in the particle core, drug compounds are shielded from biochemical attack in the form of enzymatic hydrolysis or degradation, mitigating first-pass metabolism, for example, after oral ingestion. Chemically safeguarded in the particle interior, the compound’s pharmacokinetics and pharmacodynamics are drastically altered, allowing for prolonged or enhanced therapeutic effect [29].
Decoration of the particle shell with polymers or biomolecules that favorably interact and entangle in the cross-linked mucin hydrogel comprising the intestinal mucous layer will promote drug release adjacent to the epithelial tight junctions to enhance absorption of poorly bioavailable compounds [30]. In instances where the partitioning of the drug is so unfavorable, e.g., in the case of a very hydrophilic therapeutic peptide, then it is in practice and impossible for the compound to be transported across the epithelium even with the aid of a mucoadhesive carrier. In such scenarios the nanoscale drug delivery system (DDS) can be designed to penetrate completely through the GIT into the circulatory system [31]. Functionalization with polyethylene glycol (PEG) chains of certain molecular weights and at proper surface densities enables the DDS to penetrate certain vulnerable regions of the intestinal endothelium without becoming entangled in the mucus’s proteinaceous web [32].
Equipped with an array of design strategies and precise control over the physical, chemical, and biological function of these nanoscale therapies, researchers and clinicians can successfully innovate efficacious lipid- and polymer-based DDSs for oral cancer chemotherapy. A review of the current state of the art and recognition of the challenges still remaining are discussed below.
3 Lipid Formulations and Technology Platforms
The application of lipid-based nanoformulations (Fig. 9.1) as drug delivery platforms overcomes several of the contemporary challenges encountered in the oral administration of oncolytic medicines: serving as a depot for hydrophobic and hydrophilic drugs alike, acting as a barrier to proteolytic and chemical degradation, and, perhaps most significantly, as a vehicle across tissues to target cells. Simultaneously, these nanotherapies offer a relatively benign and biocompatible alternative to other approaches employing synthetic polymers and chemicals as part of their formulary. In the following sections, nanoformulations incorporating both phospholipids and other related components are delineated. The most commonly studied type of lipid formulation is liposomes encapsulating hydrophobic or hydrophilic drug compounds.
Liposomes are spherical vesicles—hollow spheres consisting of one or more phospholipid bilayer shells ranging from tens of nanometers to microns in diameter. Resembling cells voided of their organelles, these vesicles naturally emerged as a primary drug delivery vehicle platform. With a facile synthetic route, they spontaneously self-assemble in aqueous conditions above their critical micelle concentrations (CMC). Due to their biomimetic structure—an aqueous core and selectively permeable bilayer membrane—liposomes could potentially encapsulate a diversity of drug compounds due to the vesicle’s architecture.
Translation of liposomal formulations to clinical applications has achieved the most material progress to date in cancer chemotherapy [33]. Y Barenholz introduced Doxil® (nanoformulated doxorubicin), the first liposomal medication to gain regulatory approval by the Food and Drug Administration (FDA) in 1995 for Kaposi’s sarcoma, a cancer manifested most frequently with AIDS patients [34]. The liposome shell consists of a very high melting temperature (Tm) of phosphocholine (HSPC) and a small amount of PEGylated phosphoethanolamine (DSPE-PEG2000) to mitigate premature leakage of the drug and to confer it with “stealth” properties [26]. Despite the preconceived benefits of the liposomal formulation over the free compound, Doxil® has not been more universally adopted for treatment because of dose-dependent hypersensitivity reactions (HSRs) and other unforeseen side effects since its introduction more than two decades ago [35].
A myriad of other liposomal nanomedicines are in clinical development or have gained regulatory approval in North America and Europe (Table 9.1) [40, 45]. Amphotericin B, a potent antifungal agent which is poorly bioavailable and displays a severe number of side effects when administered intravenously, has been formulated into AmBisome® by Gilead, where the anti-infective compound is intercalated into the bilayer [36]. Epaxal is a liposomal vaccine, or “virosome,” for hepatitis A. Inflexal V is a virosomal medication for influenza vaccination [40]. Myocet®, developed by Elan Pharmaceuticals and marketed by Cephalon in the European Union and Canada, is a non-pegylated formulation of doxorubicin for breast cancer treatment [43]. In addition, several other chemotherapeutic compounds have been successfully encapsulated as vesicle medicines. LipoCurc is a liposomal formulation of the poorly bioavailable anticancer compound curcumin and is currently in phase II trials for glioblastoma [40]. Daunorubicin (DaunoXome®, Galen Pharma) was approved for intravenous administration for Kaposi’s sarcoma like its counterpart Doxil® [37]. The FDA approved a liposomal nanoformulation of vincristine (Marqibo®, CASI Pharmaceuticals) in 2012 for blood cancers [42]. Liposomal cytarabine (DepoCyt®, Pacira Pharmaceuticals) recently gained approval in 2010 for meningitis related to breast metastases [38]. PEGylated (“stealth”) nanoformulations gaining entry to the market include nanoformulated cisplatin (Lipoplatin®, Regulon) for lung cancers [41], and a formulation of the topoisomerase I inhibitor Belotecan (S-CKD 602®, Chong Kun Dang) is in late-stage clinical trials [44]. However, none of the current liposome formulations is available for oral administration.
Beyond the inclusion of lipopolymers such as PEGylated lipids for prolonging circulation half-life, efforts to develop third-generation liposomal nanoformulations are focusing on platforms to develop therapies with additional or ancillary biochemical functionalities to home in on target histologies. Rather than relying on conventional passive methods such as enhanced permeation and retention (EPR) effects [25, 46], these next-generation smart medicines aim to exploit nonlinear, stimuli-driven, or threshold-switchable biochemical networks by responding to unique biochemical or environmental signals at their target organs. “Immunoliposomes” are phospholipid vesicles that have been chemically functionalized or physically decorated with immunoglobulins [47]. Chemically modified phospholipid head groups can easily be covalently linked to generic polypeptides via maleimide coupling chemistry under aqueous conditions if sulfhydryl groups are present [48]. Once decorated with antibodies, immunoliposomes can then home in on host cells that overexpress their target antigens [49]. However, developing oral available liposome-based therapy has to overcome the problem that liposomes are not stable under large fold dilution [51].
4 Polymeric Nanoparticle Formulations and Technology Platforms
Exceptional advances in the manipulation, design, and synthesis of biocompatible and biodegradable soft matter materials to nanometer length scales including functional block copolymers such as polyethylene glycol (PEG), poly(lactic-co-glycolic acid) (PLGA), polylactic acid (PLA), and polycaprolactone (PCL) have heralded a sea change in pharmaceutical research and development efforts, with significant financial and intellectual capital directed toward the invention and implementation of drug delivery platforms employing these materials [50]. Although >700 articles have been published on “polymeric nanoparticle cancer chemotherapy” according to the Web of Science to date, no platform has entered the US or European market (even though several are in early-stage human clinical trials) due to lingering questions about their possible presentation in humans [45]. Polymeric nanoparticles (Fig. 9.1) are advantageous for oral administration because they are structurally resilient [51] and kinetically stable (even under high dilution) [52], due to the super low CMC of the polymers. These nanoplatforms have the potential to be mucopenetrative [31] or actively transported [22] and can be made to be anti-immunogenic, biodegradable, and biocompatible [50].
5 Surface Design: Mucoadhesion and Mucopenetration
Although the encapsulation of therapeutic molecules in nanoscale lipid-based and polymeric drug delivery vehicles resolves many of the challenges encountered in the oral administration of chemotherapeutic drugs, including limited solubility, chemical degradation or proteolysis, pH sensitivity, etc., significant obstacles remain in transiting through the gastrointestinal barrier, so that the drug can be incorporated into the circulation and reach its target tissue. The viscous, gel-like secretions from the endothelium in the gastrointestinal tract consist of complex dispersions of biomolecules including lipids and glycoproteins (proteins posttranslationally modified with carbohydrates) which guard the human body against intrusion by malicious foreign actors like bacteria and virions while simultaneously allowing for the transit of valuable nutrients from food [9].
Mucin is the most abundant glycosylated protein produced by mucus-secreting cells and is primarily responsible for mucus’s ability to modulate gastrointestinal transit of ingested matter, conferring mucus with rheological properties akin to a sticky and flexible spider’s web. The mucin proteins act as monomers to assemble a cross-linked network of glycoproteins. Other constituents in the secreted hydrogel such as phospholipids, electrolytes, and other carbohydrates and proteins are relied upon to act in concert with the mucin web to adhere a wide diversity of molecular entities by exerting van der Waals, electrostatic, hydrophobic, steric, hydrodynamic, and mechanical forces [10]. Polymeric nanoparticles, for instance, which typically have long polymer chains extending from their corona, can easily become entangled in the mucin mesh [53].
Once entrapped in the polymeric network, peristaltic forces act to expeditiously expel adhered foreign bodies [54]. Even if a nanoscale particle can release its drug payload once it is entangled, a sufficiently large burst release must occur to reach therapeutic concentrations in the blood stream [55]. However, this requires that the pharmaceutical compound possesses high permeability. Peptides and other small molecules which bear the biopharmaceutics classification class of II or IV will never reach the circulation at therapeutic concentrations after a robust burst release from mucoadherent nanoparticles [56]. Indeed, a large proportion of newly identified leads with oncolytic therapeutic potential are proteins such as immunoglobulins, hydrophilic peptides, and small molecules with extremely low bioavailability and permeability. In order to deliver these drugs as an oral formulation, mucopenetrating nanotechnologies are currently being sought and under development in the early preclinical stages [57].
Learning from nature’s successes in circumventing this barrier will lead to a rationally designed drug delivery platform with mucus-penetrating properties. PEG is a hydrophilic polymer frequently utilized in parenteral drug delivery for its anti-immunogenic and biocompatible attributes. However, there was a lack of consensus in the scientific community regarding the interaction between mucus membranes and PEGylated micro- and nanoparticles. Justin Hanes and colleagues at Johns Hopkins University systematically investigated the effects of PEG on particle transit through the GIT using PEG molecular weight and surface density as variables. Using robust and sophisticated tracking instrumentation and analysis to calculate the effective diffusivity of the particles, it was concluded that low molecular weight (2000 Da) polymers decorated on particle surfaces at maximum numbers possess the ability to transit the mucus layer quite effectively. However, longer PEG chains (~10,000 Da) may become entrapped in the mucin network, whereas nanoparticles of lower PEG surface coverage interact electrostatically with the mucus layer, promoting elimination [32, 58].
6 In Vitro and In Vivo Assessments of the Orally Administrated Nanotherapeutics
6.1 Assessment of Release of Therapeutics and Transport of Nanocarriers Using In Vitro Models
Development of experimental platforms that predict quantitatively the in vivo drug release and response behaviors of nanoparticle drug delivery systems from in vitro measurements is a formidable task, but it is paramount in implementing and characterizing nanotherapies that will be efficacious in the clinical setting [59]. Numerous techniques have already been devised to quantify release rates including dialysis, two-phase systems with artificial membranes, and more exotic analytical tools such as artificial organs employing microfluidic architectures. These in vitro methods for characterizing nanoparticle drug delivery systems for oral administration will be discussed more in detail below after an introduction to two important processes involved in these assessments—transport and transmucosal permeability .
Mass transport of therapeutic compounds from the particle interior into the circulation involves manifold steps (Fig. 9.2). For a nanoparticle that is not mucous penetrating (Fig. 9.2a), the drug first must diffuse out from the nanoparticle. The magnitude of this transport can vary wildly as well depending on the hydrophilicity or ionizability of its functional groups. Having arrived in the bulk medium, the therapy must now cross the intestinal epithelium to reach the blood circulation and ultimately its target [60]. In this case, the main goal is to maintain the sustained release of the drug from the nanoparticle and drug delivery across the intestinal epithelium without a reduction in the drug concentration originally encapsulated in the nanoparticle.

(a) Mucoadhesive polymeric nanoparticles are entangled in the mucin mesh near the intestinal epithelium. The active pharmaceutical ingredient (API) is released in a burst and transits the gastrointestinal barrier into the circulation. (b) Mucus-penetrating nanoparticles are able to gain entry into the circulation and may provide efficacious vectors for peptides such as insulin and biologics for oral administration
Frequently, the most significant bottleneck in drug compound transport materializes when the molecule is transiting the mucus layers lining the epithelium. In instances of oral administration of nanomedicines , drug compounds must be delivered through the intestinal epithelium via the thick mucus lining to gain entry to the vasculature. Transmucosal permeability describes the propensity of a chemical compound to transit through the protective mucus layers surrounding epithelial tissue. The fraction of therapeutic medicine able to permeate the mucus lining can vary by several orders of magnitude depending on the drug’s physicochemical properties and biological metabolism [61]. Both transport and transmucosal permeability are therefore aspects of the intestinal epithelium that are modeled to gain a greater understanding of how these processes can be manipulated to improve oral administration and delivery of nanotherapeutics.
6.1.1 In Vitro Intestinal Co-culture Models
Anne des Rieux and colleagues devised a paradigm to study nanoparticle release and transport through specialized areas of the intestinal epithelium into regions particularly vulnerable to transit of foreign bodies such as Peyer’s patches. The epithelium protecting these tissues is more permeable to nanoparticulate entry and is called the follicle-associated epithelium (FAE). Co-culturing of two different cell lines, Caco-2 and Raji, precipitated a transformation of some Caco-2 into M cells, a histology resembling the FAE, as confirmed by differential expression of β1-integrin measured via immunofluorescence. Flow cytometry was then employed to interrogate the rate of transport of nanoparticles decorated with various moieties. It was determined that negatively charged particles (such as carboxylated particles) more facilely crossed the epithelial barrier compared to positively charged particles with amine functionalizations [62]. Sizeable improvements can be realized after upturning the Caco-2 culture before introducing Raji cells. This single modification significantly increases the conversion of Caco-2 into M cells and leads to more robust and reproducible NP transport results [63]. Other researchers have attempted to construct more advanced analogues of the intestinal mucous layer by including other cell types. Filipa Antunes and colleagues developed a co-culture with Caco-2, Raji, and mucous-secreting cells and investigated the transport of peptide drug delivery systems [64].
6.1.2 Semipermeable Artificial Membranes
Measuring in vitro drug release kinetics using semipermeable membranes is a well-established and robust technique for nanoparticulate drug delivery platforms, which can be either a diffusion-controlled one-phase system (dialysis ) or a two-phase setup with sink conditions. In dialysis, a colloidal nanotherapy suspension is separated by a semipermeable membrane from another chamber containing only solvent. A concentration gradient is utilized to transport the drug compound from the particle core to the bulk solvent and across the semipermeable membrane into the second compartment. Drug release kinetics are characterized by measuring the drug concentration in the second compartment as a function of time [65]. Dialysis methods have been employed extensively for nanoparticle chemotherapies in particular. P.K. Gupta et al . in 1987 employed dynamic dialysis to measure the rates of doxorubicin release from protein microparticles synthesized from bovine serum albumin (BSA) [66]. Eliana Leo and coworkers investigated doxorubicin release from gelatin nanoparticles utilizing the same technique in the presence of an intestinal protease trypsin which helps to hydrolyze proteins in the human digestive system [67]. However, release kinetics measurement by using these dialysis methods is often limited by drug solubility.
Quantification of drug release kinetics from nanoparticles encapsulating hydrophobic compounds can be interrogated by exploiting both diffusive transport and the molecule’s partition coefficient simultaneously. In two-phase sink conditions, the nanocarrier is dispersed in an aqueous suspension which is segregated by a semipermeable, artificial membrane from an organic phase miscible with the drug . The compound’s low water solubility acts as a driving force for it to partition into the sink compartment containing the organic phase [21]. Drug release kinetics are subsequently determined in a manner equivalent to conventional single-phase dialysis techniques [68].
6.1.3 Microfluidic Artificial Organs
Dialysis and two-phase sink systems crudely simulate the permeation of pharmaceutical ingredients through the gastrointestinal mucosa and their release kinetics in the body. However, even the most complex intestinal co-culture models fail to closely approximate the in vivo pharmacokinetics and pharmacodynamics because the cellular surroundings are so foreign to its physiological environment. In these experimental systems, epithelial and immune cell histologies such as Caco-2 and Raji cells are grown in culture dishes as single layers lacking the three-dimensional architecture and morphological complexity of the intestinal villi from which they are derived.
In the past decade, scientists and researchers have initiated an effort to construct environments which more closely mimic human physiology using novel fabrication and machining techniques such as microfluidic technology to construct miniature artificial organs to study the feasibility of newly invented drug delivery platforms before they enter the marketplace or begin human clinical trials (Fig. 9.3) [69]. A successful example has been realized in the HuMiX platform, a cellular co-culture system exploiting a modular microfluidic technology (Fig. 9.3) [69]. Some research groups have even fabricated devices with multiple major organ mimetics such as the liver, spleen, brain, and lungs in an effort to create a micro-human in silico [70].

The HuMiX platform, a modular microfluidics-based co-culture device [69]. (a) Schematic illustration of the key features of the HuMiX platform; (b) expanded view of the HuMiX device; (c) image of the assembled HuMiX device with the scale bar equivalent to 1 cm; (d) diagram of the experimental setup
6.2 In Vivo Assessments of Efficacy
Once lipid-based and polymeric nanotherapeutic platforms have been systematically evaluated using robust and predictive in vitro drug release and permeability models like dialysis and the intestinal co-culture model, research may progress into the succeeding preclinical stages with animal models of cancer. Below, an outline of the current endeavors and the accompanying analytical methods to assess oral bioavailability, pharmacokinetics/pharmacokinetics (PK/PD), and drug toxicity is discussed.
Typical approaches to evaluating in vivo efficacy entail grafting tumors on model animals such as nude mice, orally administering a nanoparticle suspension by gavage and sacrificing the animals at various time intervals. Overall efficacy is evaluated by the reduction in tumor volume versus a suitable control [75,76,77]. The blood and major organs (e.g., liver, lungs, spleen) are harvested, and the drug and its key metabolites are measured to determine crucial pharmacokinetic and pharmacodynamic parameters, including those to verify that the drug is within the therapeutic concentration, and determine where the nanoparticles/drugs accumulate (Fig. 9.4). Chromatographic methods, such as high-performance liquid chromatography (HPLC) and mass spectrum protocols, are often relied upon heavily for these PK/PD and toxicological assessments. Although a majority of the market-available anticancer drugs are delivered via intravenous administration, several nanotechnologies have shown promising results from oral administration. The Liu group at the University of Illinois at Chicago used a scalable process to generate high-drug-loaded PLGA nanoparticles to orally deliver SR13668, a cancer-preventive compound. Compared with Labrasol®, the nanoformulations helped to achieve two orders of higher oral bioavailability of SR13668 in mice and one order of higher bioavailability in beagle dogs [20, 21](Fig. 9.5). S. Bisht et al . demonstrated satisfactory efficacy of rapamycin-loaded polymeric nanoparticles using a mouse model of pancreatic cancer. In vivo pharmacokinetic studies demonstrate that their nanorapamycin formulation had superior or equivalent serum rapamycin levels over a 24-h period compared with conventional oral rapamycin [74]. Golla et al. encapsulated doxorubicin into lactoferrin (polypeptide) nanoparticles and evaluated the preclinical outcomes in rats grafted with a hepatic tumor. The measured outcomes were greatly improved in comparison to conventional doxorubicin therapy, as the cancer was specifically targeted by the nanoparticles [71]. O.M. Farokhzad of MIT and Harvard Medical School is a leader in oral delivery of nanoparticle-encapsulated biologic medicines, employing active transmucosal transport. Nanoparticles encapsulating insulin transported across the intestinal epithelium by the FcRn receptor had permeations in an order of magnitude greater than passively loaded nanoparticles [22]. Vong and colleagues disregard transmucosal permeability and target colon cancer with their redox nanoparticles. Of course, colon cancer is a less formidable target for oral chemotherapy than other malignant neoplasms, as the drug does not need to exit the gastrointestinal tract to reach the tumor location. However, the drug must still retain its integrity in the low pH conditions of the stomach and sustained released in the colon. The RNPOs effectively neutralized oxygen radicals in neoplastic tissue in the large intestine, leading to positive outcomes in mice. Endoscopy was used to evaluate experimental outcomes [73]. Recently, the clever design of nanocapsules by Benita and colleagues, that were administered orally and used the lymphatic system to improve systemic circulation, led to a significant increase in docetaxel oral bioavailability in rats and an increase lung cancer treatment efficacy [78]. A similar strategy is to activate the immune response by using bacteria [79]. Several notable oral nanotechnologies in preclinical development for cancer and other indications are listed in Table 9.2.

In vivo evaluation of multicomponent microemulsions administered orally for tumor targeting and anti-multidrug-resistant (anti-MDR) breast cancer treatment [75]. The potent chemotherapeutic etoposide was co-formulated in the microemulsions with coix seed oil and ginsenoside Rh2 to promote synergic antineoplastic activity against aggressive, drug-resistant breast cancer. The emulsion system successfully targeted anti-MDR breast cancer in a nude mouse model

SR13668 mean levels in beagle dog (a) plasma and (b) blood. Dogs were orally dosed at drug level of 2.8 mg/kg. Comparisons were between PLGA nanoformulation, Labrasol®, and drug in 0.5% methylcellulose (as the unformulated neat compound) [20]
7 Decision Crossroads and Best Practices
Exponential advances in information technology and automation have engendered a new paradigm in drug discovery efforts, enabling high-throughput analysis of hundreds of thousands of compounds per day to identify leads [80]. This increase in productivity in the initial screening stages however does not entirely translate into an increased rate of efficacious drug products entering the marketplace [81]. The subsequent phase requires modification of the lead compound’s physicochemical properties in an effort to balance its solubility, bioavailability, and toxicity, without comprising its pharmacological activity, and often remains a formidable challenge to medicinal chemists, especially in the case of highly hydrophobic drugs. Further formulation of these poorly soluble molecules into oral dosage forms is thus prohibitive with current industrial processing technologies [82]. A number of currently available antineoplastic agents, as well as many of the next cohorts of leads identified in screening efforts, fall into this category and thus are not economical to formulate for oral administration with current industry practices [83]. Moreover, safety concerns relating to hepatotoxicity of orally administered chemotherapeutics have yet to be addressed with these conventional oral dosage formulations.
Lipid-based and polymeric drug delivery systems are arguably the next tools in the oral cancer chemotherapy arsenal, with the capability to circumvent the current obstacles in drug formulation and development [24]. These nanotechnologies facilitate oral absorption by functioning as a depot for highly hydrophobic drugs, effectively enhancing their solubility, delaying their metabolism, and altering their release kinetics. Oral nanoformulations would therefore mitigate toxicological issues, as smaller dosages would be required to achieve equivalent serum concentrations and minimize impact on the liver. Passive and active targeting to the tumor location mitigates cytotoxicity without initiating undesired responses in other organs throughout the body. Finally, enhanced efficacy is gained through controlled release characteristics accomplished by intelligently tailored particle design.
Lipid nanocarriers such as liposomes, liquid crystalline nanoparticles, and solid lipid nanoparticles (SLNs) are at the forefront in cancer chemotherapy, predating other next-generation delivery systems with entrance to the market [84]. Lipid-based delivery platforms offer several advantages over other systems requiring synthetic materials with their inherent biocompatibility, facile synthetic schemes, and potential stealth properties [33]. Nonetheless, lipid-based nanocarriers often lack structural integrity and stability after high dilution in the bloodstream to deliver therapeutic concentrations of oncolytic medicines to their targets effectively [51]. Further, the majority of lipid-based nanotherapeutics approved by the FDA are approved for parenteral administration (Table 9.1) due to their hydrophobicity, with only solid lipid nanoparticles (SLNs) having the potential to be orally formulated [85].
Polymer-based nanovehicles have garnered considerable attention as a feasible alternative to lipid-based systems, enabled by the design of polymers and copolymers that have received generally recognized as safe (GRAS) status from major regulatory bodies [86] and the development of sustainable processes that do not require the use of toxic organic solvents. Polymer drug delivery platforms such as micelles, polymersomes, and nanoparticles are kinetically and structurally stable, even under high shear or strain and extreme concentration dilution below their critical micelle concentration, therefore being more naturally suited for oral administration [52]. However, considerable uncertainty about the clinical manifestations that may present in human patients is hindering the approval of these platforms, with no such treatment approved to date. Promisingly, however, several polymeric nanoparticulate therapies are undergoing human clinical trials at the time of this writing [45].
Lipid-based and polymeric nanoparticle delivery platforms may appear transformative in their ability to treat human disease, yet questions still remain about whether these newly engineered technology platforms could feasibly supersede conventional oral chemotherapies in the future. Human clinical trials may only be successful for parentally administered nanoparticle formulations, as gastrointestinal transit may prove an insurmountable challenge. Further, the unique biology of cancer compared to other chronic diseases states may limit the usefulness of nanoparticulate drug delivery platforms as simply alternative prophylactic regimens. Moreover, reproducibility and scalability of nanoparticle synthesis are essential to ensure the translation of fundamental research to clinical applications. However, the discussion in this chapter provides substantial evidence to the contrary that researchers have reached a tipping point in the development of a portfolio of oral nanoparticle-based medicines. The tools currently exist to effectively transit the gastrointestinal tract, identify and target neoplastic tissue, and characterize its pharmacokinetics and pharmacodynamics. The conditions that must be fulfilled to realize adoption of oral nanotherapeutics for widespread clinical use are becoming more apparent every day: (1) scale-up and manufacturing of platforms with appropriate economies of scale and quality expectations are coming more into view; and (2) additional evaluations relating to the safety of nanotherapeutics and the ramifications of the use of nanotechnologies, more broadly speaking, must still be addressed by appropriate regulatory bodies.
With the virtues of an abundance of nanotechnologies currently being reported in the literature, the path to choose the most suitable platform for specific oral cancer chemotherapies may seem challenging at first pass. Nonetheless, lipid-based and polymeric drug delivery technology platforms have evolved past the proof-of-principle stage into clinical practice in recent years, with complementary attributes available to the formulation of scientist and clinician alike to ensure both proper medication design and effective clinical outcomes for a diverse array of antineoplastic compounds with varying physicochemical properties and pharmacological activity. Antiangiogenic peptides could be encapsulated in the hydrophobic core of a mucous-penetrating block copolymer nanoparticle to directly transit into the blood stream. Amphipathic small molecules could be incorporated into the transmembrane compartment of a solid lipid nanoparticle or the phospholipid bilayer of a stealth liposome . These laboratory techniques function together as the next toolkit in fighting cancer.
References
Findlay, M., von Minckwitz, G., Wardley, A.: Effective oral chemotherapy for breast cancer: pillars of strength. Ann. Oncol. 19(2), 212–222 (2008). https://doi.org/10.1093/annonc/mdm285
Aisner, J.: Overview of the changing paradigm in cancer treatment: Oral chemotherapy. Am. J. Health Syst. Pharm. 64, S4–S7 (2007). https://doi.org/10.2146/ajhp070035
Lu, Y., Park, K.: Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int. J. Pharm. 453(1), 198–214 (2013). https://doi.org/10.1016/j.ijpharm.2012.08.042
Kinch, M.S.: An overview of FDA-approved biologics medicines. Drug Discov. Today. 20(4), 393–398 (2015). https://doi.org/10.1016/j.drudis.2014.09.003
Truong-Le, V., Lovalenti, P.M., Abdul-Fattah, A.M.: Stabilization challenges and formulation strategies associated with oral biologic drug delivery systems. Adv. Drug Deliv. Rev. 93, 95–108 (2015). https://doi.org/10.1016/j.addr.2015.08.001
Thanki, K., Gangwal, R.P., Sangamwar, A.T., Jain, S.: Oral delivery of anticancer drugs: Challenges and opportunities. J. Control. Release. 170(1), 15–40 (2013). https://doi.org/10.1016/j.jconrel.2013.04.020
Wu, P., Nielsen, T.E., Clausen, M.H.: Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov. Today. 21(1), 5–10 (2016). https://doi.org/10.1016/j.drudis.2015.07.008
Yun, Y., Cho, Y.W., Park, K.: Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv. Drug Deliv. Rev. 65(6), 822–832 (2013). https://doi.org/10.1016/j.addr.2012.10.007
Cone, R.A.: Barrier properties of mucus. Adv. Drug Deliv. Rev. 61(2), 75–85 (2009). https://doi.org/10.1016/j.addr.2008.09.008
Bansil, R., Turner, B.S.: Mucin structure, aggregation, physiological functions and biomedical applications. Curr. Opin. Colloid Interface Sci. 11(2–3), 164–170 (2006). https://doi.org/10.1016/j.cocis.2005.11.001
Malingre, M.M., Richel, D.J., Beijnen, J.H., Rosing, H., Koopman, F.J., Huinink, W.W.T.B., Schot, M.E., Schellens, J.H.M.: Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J. Clin. Oncol. 19(4), 1160–1166 (2001)
Varma, M.V., Obach, R.S., Rotter, C., Miller, H.R., Chang, G., Steyn, S.J., El-Kattan, A., Troutman, M.D.: Physicochemical space for optimum oral bioavailability: contribution of human intestinal absorption and first-pass elimination. J. Med. Chem. 53(3), 1098–1108 (2010). https://doi.org/10.1021/jm901371v
Jain, A.K., Swarnakar, N.K., Godugu, C., Singh, R.P., Jain, S.: The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials. 32(2), 503–515 (2011). https://doi.org/10.1016/j.biomaterials.2010.09.037
Van Cutsem, E., Twelves, C., Cassidy, J., Allman, D., Bajetta, E., Boyer, M., Bugat, R., Findlay, M., Frings, S., Jahn, M., McKendrick, J., Osterwalder, B., Perez-Manga, G., Rosso, R., Rougier, P., Schmiegel, W.H., Seitz, J.F., Thompson, P., Vieitez, J.M., Weitzel, C., Harper, P., Grp, X.C.C.S.: Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: Results of a large phase III study. J. Clin. Oncol. 19(21), 4097–4106 (2001)
Herbrink, M., Nuijen, B., Schellens, J.H.M., Beijnen, J.H.: Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat. Rev. 41(5), 412–422 (2015). https://doi.org/10.1016/j.ctrv.2015.03.005
Hu, X.Y., Huang, F., Szymusiak, M., Liu, Y., Wang, Z.J.: Curcumin attenuates opioid tolerance and dependence by inhibiting Ca2+/Calmodulin-Dependent Protein Kinase II alpha activity. J. Pharmacol. Exp. Ther. 352(3), 420–428 (2015). https://doi.org/10.1124/jpet.114.219303
Hu, X.Y., Huang, F., Szymusiak, M., Tian, X.B., Liu, Y., Wang, Z.J.: PLGA-Curcumin Attenuates Opioid-Induced Hyperalgesia and Inhibits Spinal CaMKII alpha. Plos One. 11(1), e0146393 (2016). https://doi.org/10.1371/journal.pone.0146393
Shen, H., Hu, X.Y., Szymusiak, M., Wang, Z.J., Liu, Y.: Orally administered nanocurcumin to attenuate morphine tolerance: comparison between negatively charged PLGA and partially and fully PEGylated nanoparticles. Mol. Pharm. 10(12), 4546–4551 (2013). https://doi.org/10.1021/mp400358z
Szymusiak, M., Hu, X.Y., Plata, P.A.L., Ciupinski, P., Wang, Z.J., Liu, Y.: Bioavailability of curcumin and curcumin glucuronide in the central nervous system of mice after oral delivery of nano-curcumin. Int. J. Pharm. 511(1), 415–423 (2016). https://doi.org/10.1016/j.ijpharm.2016.07.027
Banerjee, A.A., Shen, H., Hautman, M., Anwer, J., Hong, S., Kapetanovic, I.M., Liu, Y., Lyubimov, A.V.: Enhanced oral bioavailability of the hydrophobic chemopreventive agent (Sr13668) in Beagle Dogs. Curr. Pharm. Biotechnol. 14(4), 464–469 (2013)
Shen, H., Banerjee, A.A., Mlynarska, P., Hautman, M., Hong, S., Kapetanovic, I.M., Lyubimov, A.V., Liu, Y.: Enhanced oral bioavailability of a cancer preventive agent (SR13668) by employing polymeric nanoparticles with high drug loading. J. Pharm. Sci. 101(10), 3877–3885 (2012). https://doi.org/10.1002/jps.23269
Pridgen, E.M., Alexis, F., Kuo, T.T., Levy-Nissenbaum, E., Karnik, R., Blumberg, R.S., Langer, R., Farokhzad, O.C.: Transepithelial transport of Fc-Targeted nanoparticles by the Neonatal Fc receptor for oral delivery. Sci. Transl. Med. 5(213), 213ra167 (2013). https://doi.org/10.1126/scitranslmed.3007049
Williams, A.C., Barry, B.W.: Penetration enhancers. Adv. Drug Deliv. Rev. 64, 128–137 (2012). https://doi.org/10.1016/j.addr.2012.09.032
Davis, M.E., Chen, Z., Shin, D.M.: Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug Discov. 7(9), 771–782 (2008). https://doi.org/10.1038/nrd2614
Torchilin, V.: Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 63(3), 131–135 (2011). https://doi.org/10.1016/j.addr.2010.03.011
Mamidi, R.N.V.S., Weng, S., Stellar, S., Wang, C., Yu, N., Huang, T., Tonelli, A.P., Kelley, M.F., Angiuoli, A., Fung, M.C.: Pharmacokinetics, efficacy and toxicity of different pegylated liposomal doxorubicin formulations in preclinical models: is a conventional bioequivalence approach sufficient to ensure therapeutic equivalence of pegylated liposomal doxorubicin products? Cancer Chemother. Pharmacol. 66(6), 1173–1184 (2010). https://doi.org/10.1007/s00280-010-1406-x
Shen, H., Hong, S.Y., Prud'homme, R.K., Liu, Y.: Self-assembling process of flash nanoprecipitation in a multi-inlet vortex mixer to produce drug-loaded polymeric nanoparticles. J. Nanopart. Res. 13(9), 4109–4120 (2011). https://doi.org/10.1007/s11051-011-0354-7
Sun, T.M., Zhang, Y.S., Pang, B., Hyun, D.C., Yang, M.X., Xia, Y.N.: Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. Engl. 53(46), 12320–12364 (2014). https://doi.org/10.1002/anie.201403036
Parveen, S., Misra, R., Sahoo, S.K.: Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 8(2), 147–166 (2012). https://doi.org/10.1016/j.nano.2011.05.016
Ponchel, G., Montisci, M.J., Dembri, A., Durrer, C., Duchene, D.: Mucoadhesion of colloidal particulate systems in the gastro-intestinal tract. Eur. J. Pharm. Biopharm. 44(1), 25–31 (1997). https://doi.org/10.1016/S0939-6411(97)00098-2
Tang, B.C., Dawson, M., Lai, S.K., Wang, Y.Y., Suk, J.S., Yang, M., Zeitlin, P., Boyle, M.P., Fu, J., Hanes, J.: Biodegradable polymer nanoparticles that rapidly penetrate the human mucus barrier. Proc. Natl. Acad. Sci. U. S. A. 106(46), 19268–19273 (2009). https://doi.org/10.1073/pnas.0905998106
Wang, Y.Y., Lai, S.K., Suk, J.S., Pace, A., Cone, R., Hanes, J.: Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “Slip” through the human mucus barrier. Angew Chem Int Ed Engl. 47(50), 9726–9729 (2008). https://doi.org/10.1002/anie.200803526
Allen, T.M., Cullis, P.R.: Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 65(1), 36–48 (2013). https://doi.org/10.1016/j.addr.2012.09.037
Barenholz, Y.: Doxil (R) - The first FDA-approved nano-drug: lessons learned. J. Control. Release. 160(2), 117–134 (2012). https://doi.org/10.1016/j.jconrel.2012.03.020
Szebeni, J., Baranyi, L., Savay, S., Milosevits, J., Bunger, R., Laverman, P., Metselaar, J.M., Storm, G., Chanan-Khan, A., Liebes, L., Muggia, F.M., Cohen, R., Barenholz, Y., Alving, C.R.: Role of complement activation in hypersensitivity reactions to doxil and hynic PEG liposomes: experimental and clinical studies. J. Liposome Res. 12(1–2), 165–172 (2002). https://doi.org/10.1081/LPR-120004790
Adlermoore, J.: Ambisome targeting to fungal-infections. Bone Marrow Transplant. 14, S3–S7 (1994)
Rosenthal, E., Poizot-Martin, I., Saint-Marc, T., Spano, J.P., Cacoub, P., Grp, D.S.: Phase IV study of liposomal daunorubicin (DaunoXome) in AIDS-related Kaposi sarcoma. Am. J. Clin. Oncol. 25(1), 57–59 (2002). https://doi.org/10.1097/00000421-200202000-00012
Mayer, A.M.S., Glaser, K.B., Cuevas, C., Jacobs, R.S., Kem, W., Little, R.D., McIntosh, J.M., Newman, D.J., Potts, B.C., Shuster, D.E.: The odyssey of marine pharmaceuticals: a current pipeline perspective. Trends Pharmacol. Sci. 31(6), 255–265 (2010). https://doi.org/10.1016/j.tips.2010.02.005
Gabizon, A., Shmeeda, H., Barenholz, Y.: Pharmacokinetics of pegylated liposomal doxorubicin - Review of animal and human studies. Clin. Pharmacokinet. 42(5), 419–436 (2003). https://doi.org/10.2165/00003088-200342050-00002
Grimaldi, N., Andrade, F., Segovia, N., Ferrer-Tasies, L., Sala, S., Veciana, J., Ventosa, N.: Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 45, 6520 (2016)
Boulikas, T.: Clinical overview on Lipoplatin (TM): a successful liposomal formulation of cisplatin. Expert Opin. Investig. Drugs. 18(8), 1197–1218 (2009). https://doi.org/10.1517/13543780903114168
Silverman, J.A., Deitcher, S.R.: Marqibo (R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 71(3), 555–564 (2013). https://doi.org/10.1007/s00280-012-2042-4
Leonard, R.C.F., Williams, S., Tulpule, A., Levine, A.M., Oliveros, S.: Improving the therapeutic index of anthracycline chemotherapy: Focus on liposomal doxorubicin (Myocet (TM)). Breast. 18(4), 218–224 (2009). https://doi.org/10.1016/j.breast.2009.05.004
Schell, R.F., Sidone, B.J., Caron, W.P., Walsha, M.D., White, T.F., Zamboni, B.A., Ramanathan, R.K., Zamboni, W.C.: Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomedicine. 10(1), 109–117 (2014). https://doi.org/10.1016/j.nano.2013.07.005
Wang, A.Z., Langer, R., Farokhzad, O.C.: Nanoparticle delivery of cancer drugs. Annu. Rev. Med. 63(63), 185–198 (2012). https://doi.org/10.1146/annurev-med-040210-162544
Maruyama, K.: Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 63(3), 161–169 (2011). https://doi.org/10.1016/j.addr.2010.09.003
Torchilin, V.P.: Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 4(2), 145–160 (2005). https://doi.org/10.1038/nrd1632
Kirpotin, D., Park, J.W., Hong, K., Zalipsky, S., Li, W.L., Carter, P., Benz, C.C., Papahadjopoulos, D.: Sterically stabilized Anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry. 36(1), 66–75 (1997). https://doi.org/10.1021/bi962148u
Maruyama, K., Ishida, O., Takizawa, T., Moribe, K.: Possibility of active targeting to tumor tissues with liposomes. Adv. Drug Deliv. Rev. 40(1–2), 89–102 (1999). https://doi.org/10.1016/S0169-409x(99)00042-3
Kumari, A., Yadav, S.K., Yadav, S.C.: Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces. 75(1), 1–18 (2010). https://doi.org/10.1016/j.colsurfb.2009.09.001
Bermudez, H., Brannan, A.K., Hammer, D.A., Bates, F.S., Discher, D.E.: Molecular weight dependence of polymersome membrane structure, elasticity, and stability. Macromolecules. 35(21), 8203–8208 (2002). https://doi.org/10.1021/ma020669l
Discher, D.E., Ahmed, F.: Polymersomes. Annu. Rev. Biomed. Eng. 8, 323–341 (2006). https://doi.org/10.1146/annurev.bioeng.8.061505.095838
Ensign, L.M., Cone, R., Hanes, J.: Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 64(6), 557–570 (2012). https://doi.org/10.1016/j.addr.2011.12.009
Muller, C., Perera, G., Konig, V., Bernkop-Schnurch, A.: Development and in vivo evaluation of papain-functionalized nanoparticles. Eur. J. Pharm. Biopharm. 87(1), 125–131 (2014). https://doi.org/10.1016/j.ejpb.2013.12.012
Tao, Y.Y., Lu, Y.F., Sun, Y.J., Gu, B., Lu, W.Y., Pan, J.: Development of mucoadhesive microspheres of acyclovir with enhanced bioavailability. Int. J. Pharm. 378(1–2), 30–36 (2009). https://doi.org/10.1016/j.ijpharm.2009.05.025
Pauletti, G.M., Gangwar, S., Knipp, G.T., Nerurkar, M.M., Okumu, F.W., Tamura, K., Siahaan, T.J., Borchardt, R.T.: Structural requirements for intestinal absorption of peptide drugs. J. Control. Release. 41(1–2), 3–17 (1996). https://doi.org/10.1016/0168-3659(96)01352-1
Bakhru, S.H., Furtado, S., Morello, A.P., Mathiowitz, E.: Oral delivery of proteins by biodegradable nanoparticles. Adv. Drug Deliv. Rev. 65(6), 811–821 (2013). https://doi.org/10.1016/j.addr.2013.04.006
Lai, S.K., O'Hanlon, D.E., Harrold, S., Man, S.T., Wang, Y.Y., Cone, R., Hanes, J.: Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl. Acad. Sci. U. S. A. 104(5), 1482–1487 (2007). https://doi.org/10.1073/pnas.0608611104
Obach, R.S., Baxter, J.G., Liston, T.E., Silber, B.M., Jones, B.C., MacIntyre, F., Rance, D.J., Wastall, P.: The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 283(1), 46–58 (1997)
Amidon, G.L., Lennernas, H., Shah, V.P., Crison, J.R.: A theoretical basis for a biopharmaceutic drug classification - the correlation of in-Vitro drug product dissolution and in-Vivo bioavailability. Pharm. Res. 12(3), 413–420 (1995). https://doi.org/10.1023/A:1016212804288
Martinez, M.N., G L, A.: A mechanistic approach to understandingthe factors affecting drug absorption:a review of fundamentals. J. Clin. Pharmacol. 42, 620–643 (2002)
des Rieux, A., Ragnarsson, E.G., Gullberg, E., Préat, V., Schneider, Y.-J., Artursson, P.: Transport of nanoparticles across an in vitro model of the human intestinal follicle associated epithelium. Eur. J. Pharm. Sci. 25(4), 455–465 (2005)
des Rieux, A., Fievez, V., Théate, I., Mast, J., Préat, V., Schneider, Y.-J.: An improved in vitro model of human intestinal follicle-associated epithelium to study nanoparticle transport by M cells. Eur. J. Pharm. Sci. 30(5), 380–391 (2007)
Antunes, F., Andrade, F., Araújo, F., Ferreira, D., Sarmento, B.: Establishment of a triple co-culture in vitro cell models to study intestinal absorption of peptide drugs. Eur. J. Pharm. Biopharm. 83(3), 427–435 (2013)
Modi, S., Anderson, B.D.: Determination of drug release kinetics from nanoparticles: overcoming pitfalls of the dynamic dialysis method. Mol. Pharm. 10(8), 3076–3089 (2013). https://doi.org/10.1021/mp400154a
Gupta, P.K., Hung, C.T., Perrier, D.G.: Quantitation of the Release of Doxorubicin from Colloidal Dosage Forms Using Dynamic Dialysis. J. Pharm. Sci. 76(2), 141–145 (1987). https://doi.org/10.1002/jps.2600760211
Leo, E., Cameroni, R., Forni, F.: Dynamic dialysis for the drug release evaluation from doxorubicin-gelatin nanoparticle conjugates. Int. J. Pharm. 180(1), 23–30 (1999)
Mogollon, C.: In Vitro Release of Curcumin from Polymeric Nanoparticles Using Two-Phase System. University of Illinois at Chicago (2016)
Shah, P., Fritz, J.V., Glaab, E., Desai, M.S., Greenhalgh, K., Frachet, A., Niegowska, M., Estes, M., Jager, C., Seguin-Devaux, C., Zenhausern, F., Wilmes, P.: A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat. Commun. 7, 11535 (2016). https://doi.org/10.1038/ncomms11535
Eisenstein, M.: ARTIFICIAL ORGANS Honey, I shrunk the lungs. Nature. 519(7544), S16–S18 (2015)
Golla, K., Bhaskar, C., Ahmed, F., Kondapi, A.K.: A Target-Specific Oral Formulation of Doxorubicin-Protein Nanoparticles: Efficacy and Safety in Hepatocellular Cancer. J. Cancer. 4(8), 644–652 (2013). https://doi.org/10.7150/jca.7093
Jain, S., Kumar, D., Swarnakar, N.K., Thanki, K.: Polyelectrolyte stabilized multilayered liposomes for oral delivery of paclitaxel. Biomaterials. 33(28), 6758–6768 (2012). https://doi.org/10.1016/j.biomaterials.2012.05.026
Vong, L.B., Yoshitomi, T., Matsui, H., Nagasaki, Y.: Development of an oral nanotherapeutics using redox nanoparticles for treatment of colitis-associated colon cancer. Biomaterials. 55, 54–63 (2015). https://doi.org/10.1016/j.biomaterials.2015.03.037
Bisht, S., Feldmann, G., Koorstra, J.B.M., Mullendore, M., Alvarez, H., Karikari, C., Rudek, M.A., Lee, C.K., Maitra, A., Maitra, A.: In vivo characterization of a polymeric nanoparticle platform with potential oral drug delivery capabilities. Mol. Cancer Ther. 7(12), 3878–3888 (2008). https://doi.org/10.1158/1535-7163.Mct-08-0476
Qu, D., Wang, L.X., Liu, M., Shen, S.Y., Li, T., Liu, Y.P., Huang, M.M., Liu, C.Y., Chen, Y., Mo, R.: Oral Nanomedicine Based on Multicomponent Microemulsions for Drug-Resistant Breast Cancer Treatment. Biomacromolecules. 18(4), 1268–1280 (2017). https://doi.org/10.1021/acs.biomac.7b00011
Groo, A.C., Bossiere, M., Trichard, L., Legras, P., Benoit, J.P., Lagarce, F.: In vivo evaluation of paclitaxel-loaded lipid nanocapsules after intravenous and oral administration on resistant tumor. Nanomedicine. 10(4), 589–601 (2015). https://doi.org/10.2217/nnm.14.124
Wang, Y.C., Zhang, D.R., Liu, Z.P., Liu, G.P., Duan, C.X., Jia, L.J., Feng, F.F., Zhang, X.Y., Shi, Y.Q., Zhang, Q.: In vitro and in vivo evaluation of silybin nanosuspensions for oral and intravenous delivery. Nanotechnology. 21(15), 155104 (2010). https://doi.org/10.1088/0957-4484/21/15/155104
Attili-Qadri, S., Karra, N., Nemirovski, A., Schwob, O., Talmon, Y., Nassar, T., Benita, S.: Oral delivery system prolongs blood circulation of docetaxel nanocapsules via lymphatic absorption. Proc. Natl. Acad. Sci. U. S. A. 110(43), 17498–17503 (2013). https://doi.org/10.1073/pnas.1313839110
Hu, Q.L., Wu, M., Fang, C., Cheng, C.Y., Zhao, M.M., Fang, W.H., Chu, P.K., Ping, Y., Tang, G.P.: Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett. 15(4), 2732–2739 (2015). https://doi.org/10.1021/acs.nanolett.5b00570
Claus, B.L., Underwood, D.J.: Discovery informatics: its evolving role in drug discovery. Drug Discov. Today. 7(18), 957–966. doi: Pii S1359-6446(02)02433-9 (2002). https://doi.org/10.1016/S1359-6446(02)02433-9
Kaitin, K.I., DiMasi, J.A.: Pharmaceutical innovation in the 21st Century: new drug approvals in the First Decade, 2000-2009. Clin. Pharmacol. Ther. 89(2), 183–188 (2011). https://doi.org/10.1038/clpt.2010.286
Chaubal, M.V.: Application of formulation technologies in lead candidate selection and optimization. Drug Discov. Today. 9(14), 603–609. doi: Pii S1359-6446(04)03171-X (2004). https://doi.org/10.1016/S1359-6446(04)03171-X
Torchilin, V.P., Lukyanov, A.N.: Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov. Today. 8(6), 259–266. doi: Pii S1359-6446(03)02623-0 (2003). https://doi.org/10.1016/S1359-6446(03)02623-0
Allen, T.M., Cullis, P.R.: Drug delivery systems: Entering the mainstream. Science. 303(5665), 1818–1822 (2004). https://doi.org/10.1126/science.1095833
Sarmento, B., Martins, S., Ferreira, D., Souto, E.B.: Oral insulin delivery by means of solid lipid nanoparticles. Int. J. Nanomedicine. 2(4), 743–749 (2007)
Knop, K., Hoogenboom, R., Fischer, D., Schubert, U.S.: Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 49(36), 6288–6308 (2010). https://doi.org/10.1002/anie.200902672
Acknowledgment
The research of nanoparticle design and production of Ying Liu is supported by NSF CMMI Nanomanufacturing Program (NSF CAREER 1350731).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply
About this chapter

Cite this chapter
Donovan, A.J., Liu, Y. (2019). Oral Nanotherapeutics for Cancer with Innovations in Lipid and Polymeric Nanoformulations. In: Rai, P., Morris, S.A. (eds) Nanotheranostics for Cancer Applications. Bioanalysis, vol 5. Springer, Cham. https://doi.org/10.1007/978-3-030-01775-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-01775-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-01773-6
Online ISBN: 978-3-030-01775-0
eBook Packages: EngineeringEngineering (R0)