
Abstract
Diffuse large B-cell lymphomas (DLBCLs) comprise a diverse group of aggressive lymphoid tumors characterized by distinct genetic, phenotypic, and clinical features, with over ten entities recognized in the updated WHO classification of hematopoietic and lymphoid tissues. Among them, DLBCL not otherwise specified (NOS) represents the most common diagnosis, accounting for 25–30% of all B cell lymphoma cases. In recent years, significant progress has been made in our understanding of the molecular pathogenesis of these diseases, thanks to the development of powerful genomic technologies that enabled the definition of multiple phenotypic and molecular subtypes of the disease, often associated with discrete clinical outcomes. These studies revealed a multitude of genetic alterations that contribute to the malignant transformation process by disrupting functional programs critical for the biology of normal germinal center B cells, i.e., the normal counterpart of most DLBCL types. These include epigenetic remodeling, blockade of B-cell differentiation, escape from immune surveillance, and the dysregulated expression of several transcription factors/signal transduction pathways. This wealth of new information is offering unique opportunities for the development of improved diagnostic and prognostic tools that could assist in the clinical management of DLBCL patients. Importantly, a number of the identified mutated genes are potentially actionable targets that are currently being explored for the development of novel therapeutic strategies. In this chapter, we summarize current knowledge on the pathology, biology, and genetic basis of these diseases, with emphasis on its most common types.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Diffuse large B-cell lymphoma
- Double-hit lymphoma
- Central nervous system
- Primary mediastinal B-cell lymphoma
- Germinal center
- Immunophenotype
- Genetic alterations
- BCL6
- NF-κB
- Immune escape
- Epigenetic modifications
1 Introduction
Diffuse large B-cell lymphoma (DLBCL) represents a heterogeneous group of aggressive lymphoid neoplasms that originate, in most cases, from the malignant transformation of B cells within the germinal center (GC) and feature distinct genetic, phenotypic, and clinical behavior. The 2017 update of the fourth edition of the WHO classification of lymphoid, histiocytic, and dendritic cell neoplasms reflects this heterogeneity by recognizing over ten large B-cell lymphoma entities, including, among others, the most common DLBCL not otherwise specified (NOS), primary mediastinal large B-cell lymphoma (PMBCL), intravascular large B-cell lymphoma, and a few new categories or provisional entities (Table 2.1) [1, 2]. Of importance, the revised classification requires the distinction of DLBCL-NOS into two “cell-of-origin” molecular subtypes, i.e., germinal center B-cell-like (GCB) and activated B-cell-like (ABC) DLBCL, incorporating the notion that these tumors are addicted to distinct oncogenic pathways and differ in clinical outcome. Moreover, a separate category has been introduced for DLBCL harboring chromosomal translocations of both MYC and BCL2 and/or BCL6 (the so-called double-hit or triple-hit lymphomas).
From a genetic standpoint, fundamental new insights have been gained over the past decade, particularly regarding the mutational landscape of DLBCL-NOS and PMBCL. These studies led to the identification of multiple genes and pathways that are recurrently disrupted by genetic lesions in these tumors and likely play significant roles in their pathogenesis. Of note, a number of the affected genes represent potentially actionable targets that could be exploited for precision medicine approaches.
This chapter will cover the pathology and genetics of DLBCL and related entities, according to the definition of the updated 2017 WHO classification, with special emphasis on the most common and molecularly better characterized DLBCL-NOS and PMBCL.
2 Diffuse Large B-Cell Lymphoma, NOS
DLBCL not otherwise specified (NOS) constitutes the most common type of B-cell lymphoma in adults, accounting for approximately 30% of all diagnoses, and includes cases that cannot be assigned to any other specific subtype or disease entity. Morphologically, these tumors consist of large lymphoid cells with basophilic cytoplasm and vesicular nuclei that are at least twice the size of normal lymphocytes and exhibit a diffuse growth pattern in the large majority of cases. DLBCLs are mainly nodal lymphomas, although roughly 40% of cases are considered primary extranodal in origin [3]. Among them, the most common extranodal site is the gastrointestinal tract; however, DLBCL can involve virtually every lymphoid and nonlymphoid organ, including the Waldeyer’s ring, spleen, bones, gonads, thyroid, liver, and kidneys. DLBCL cases localized to some of these sites are considered separate entities, as it is the case for primary DLBCL of the central nervous system (PCNSL) and primary mediastinal (thymic) large B-cell lymphoma (PMBCL). Leukemic spread is rare in DLBCL.
The etiology of DLBCL remains largely unknown; however, a few specific risk factors have been recognized, such as immunodeficiency and chronic inflammation. In particular, DLBCL NOS arising in the setting of immunodeficiency is associated with infection by the Epstein-Barr virus (EBV) in a large number of cases, a finding that is relatively uncommon in sporadic DLBCL. While most DLBCL cases arise de novo, they can also be seen in the setting of transformation from preexisting low-grade lymphoid tumors, including chronic lymphocytic leukemia (so-called Richter’s syndrome) and follicular lymphoma.
Patients with DLBCL can be asymptomatic or present with systemic symptoms such as fatigue, fever, and night sweats. Often, a rapidly growing mass is found at the site of involvement, and after appropriate clinical staging, only 25–30% of patients show localized (stage I or II) disease. In many cases, specific symptoms are related to the site of origin and/or the site of the predominantly manifesting tumor mass [4].
2.1 Pathology
In lymph nodes, tumor infiltrates may totally efface the underlying architecture of the parenchyma, while partial infiltration is found more rarely. The perinodal soft tissues are often affected as well. In extranodal localizations, the pattern of infiltration is generally related to the specific site involved, e.g., a diffuse infiltration of the lung parenchyma by PMBCL or the confinement of the infiltrate to the luminal parts of the bowel wall in primary intestinal lymphoma. Common morphological variants of DLBCL include the centroblastic, immunoblastic, and anaplastic variants (Fig. 2.1). Of these, the most common one is represented by centroblastic DLBCL, which consists of medium- to large-sized cells with scant to slightly basophilic cytoplasm, an open vesicular chromatin structure, and several small membrane-bound nucleoli (Fig. 2.1a). This classical tumor cell type (referred to as the monomorphic variant) may be admixed with immunoblasts (the so-called polymorphic variant of DLBCL-centroblastic), and nuclei with a conspicuously multilobated shape may be observed. Roughly 60–70% of centroblastic-type DLBCL cases show a germinal center B-cell-like gene expression profile (GEP) (see next paragraph). Immunoblastic DLBCLs are composed of large cells with broad basophilic cytoplasm and large vesicular nuclei showing a single central nucleolus (Fig. 2.1b). In this variant, partial plasmablastic differentiation may be observed, and most cases show an activated B-cell-like GEP [5]. The anaplastic DLBCL variant is characterized by large to very large cells with equally large and pleomorphic, sometimes bizarre nuclei resembling Hodgkin or Reed-Sternberg-like cells, and a cohesive growth pattern with occasional intrasinusoidal spread (Fig. 2.1c). CD30 expression is frequently observed. In many cases, immunohistochemistry is needed in order to differentiate this variant from anaplastic large cell lymphoma of T-/null-cell linage [6,7,8]. Often, however, DLBCL shows a mixed composition of these cell types, and no specific variant can be diagnosed. In addition, a varying number of accompanying interspersed T cells and histiocytes can be seen in the background, reflecting different interactions of the tumor cells with the surrounding microenvironment.

Morphological variants of DLBCL, NOS. (a) Centroblastic variant of DLBCL, characterized by medium-sized to large cells with a narrow, slightly basophilic cytoplasm and round to oval, or lobulated, nuclei with vesicular chromatin and several membrane-bound nucleoli. (b) Immunoblastic lymphoma, displaying a broader, basophilic cytoplasm and round nuclei with loose chromatin and a single, prominent nucleolus. (c) Anaplastic variant of DLBCL, characterized by large and at times very pleomorphic cells with large, irregular nuclei and bizarre or multinucleated giant cells
2.2 Immunophenotype
The tumor cells of DLBCL express by definition pan B-cell markers such as CD19, CD20, or CD22. A number of other antigens may also be expressed on the membrane, in the cytoplasm, or in the nuclei of the tumor cells, including CD5 (5–10% of cases), CD10 (30%), or CD30 (10–20%). Reactivity for CD5 or CD10 does not necessarily indicate transformation from indolent lymphomas, and the vast majority of CD5- and/or CD10-positive DLBCL are de novo diagnoses. Additional markers include CD38, CD138, immunoglobulin (cytoplasmic), IRF4/MUM1, the GC-associated protein BCL6, and BCL2 [2]. Apart from correctly identifying the B-cell derivation of the tumor cells and rendering predictive information on CD20 expression (the target of anti-CD20 immunotherapy), various immunohistochemical algorithms have been constructed to assign DLBCL samples to the two main molecular classes identified by GEP analysis (discussed in the next section). The Ki67 proliferation index is generally high, and nuclear reactivity is often seen in 60–90% of tumor cells.
Of particular relevance is the immunohistochemical analysis of MYC and BCL2, because the simultaneous expression of these two proteins, either due to concurrent chromosomal translocations (double-hit lymphomas, 5–8% of cases) or independently of genetic lesions (so-called double-expressor lymphomas, accounting for 20–30% of cases), has been associated with inferior prognosis, although more recent prospective studies are starting to reveal comparable overall survival [9,10,11,12,13]. In the WHO update, “double-hit” or “triple-hit” lymphomas are included in the new category of high-grade B-cell lymphoma (see corresponding section for a detailed description), while dual expression of BCL2 and MYC without MYC/BCL2 chromosomal alterations is recognized as a negative prognostic indicator within DLBCL, NOS, but not as a separate category. The recommended cutoffs to define double-expressor DLBCLs are >40% for MYC expression and >50% for BCL2 expression (Fig. 2.2a, b) [1].

Double expressor lymphoma. (a) Immunohistochemical analysis of BCL2 expression, showing positivity in the cytoplasm of virtually all tumor cells. (b) The vast majority of the cells also express nuclear MYC protein
2.3 Cell of Origin and Gene Expression Profiling
The normal counterpart of DLBCL is a peripheral mature B-cell that has experienced the GC reaction, as demonstrated by the presence in these tumors of clonally rearranged, somatically hypermutated immunoglobulin genes [14].Seminal studies utilizing genome-wide transcriptional profiling approaches allowed the identification of at least two molecular subtypes of DLBCL, NOS, with an intermediate group that remains unclassifiable [5, 15]. The germinal center B-cell-like (GCB) DLBCL is characterized by a gene expression pattern that is more similar to that of a light zone (LZ) B cell, possibly recirculating toward the dark zone [15, 16]. Conversely, the activated B-cell-like (ABC) DLBCL is characterized by the expression of genes that are typically upregulated in a subset of GC LZ B cells poised to differentiate into plasma cells [15, 16]. Besides indicating the putative cell of origin (COO) of the disease, this classification identifies tumors with fundamentally different biological and genetic features, as well as clinical outcome, with ABC-DLBCL being generally less curable [5, 17, 18]. In the upcoming area of targeted therapies [19], the COO classification is, therefore, of important predictive impact in the management of DLBCL. Moreover, some association has been reported between COO subtype and cytomorphology, site of origin, and age at diagnosis [2, 5]. Particularly, centroblastic lymphomas are more often of GCB type, whereas immunoblastic tumors more often display an ABC-like GEP [5]. The tumor cells in specific entities such as PCNSL or DLBCL of the leg are more often of the ABC-type [20]. Finally, the prevalence of ABC-type tumors appears to rise with age [21].
The distinction in ABC- and GCB-DLBCL has now been officially incorporated into the updated WHO classification and is strongly recommended in the clinical practice [1]. Because genome-wide molecular profiling is not available in all laboratories, a number of immunohistochemical algorithms have been developed over the past decades in order to predict DLBCL molecular subtype on routine formalin-fixed paraffin-embedded (FFPE) material. Among these classifiers, the most widely used is the so-called Hans algorithm [22] that utilizes expression of CD10, BCL6, and IRF4/MUM1 to distinguish GC and non-GC DLBCL. However, other algorithms have been proposed, which employ additional markers such as GCET1, FOXP1, and LMO2 [23]. While the use of immunohistochemical algorithms is considered acceptable in the revised WHO classification, two major caveats have to be kept in mind: (1) these classifiers do not—at least formally—define an “unclassified” variant; (2) lack of reproducibility in the staining pattern and intensity has been frequently observed across different laboratories and institutions [24]. Indeed, the individual immunohistochemistry-based algorithms are far from yielding identical results, even when applied on the same case series [25]. In order to overcome these limits, several low-density GEP platforms have been recently developed, which can be applied to FFPE material and provided reproducible results comparable to conventional microarray-based GEP, thus showing promise as a routine diagnostic tool [26, 27].
2.4 Molecular Pathogenesis
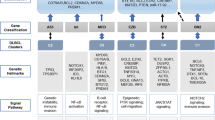
Over the past decade, the rapid expansion of next-generation sequencing technologies has significantly advanced our understanding of the molecular pathogenesis of DLBCL. These studies revealed a remarkable complexity in the DLBCL coding genome, which harbors between 30 and >100 lesions/case [28,29,30,31] and features approximately 150 candidate driver genes, as predicted in a recent study of over 500 cases [32]. Even greater complexity is expected to emerge as we start interrogating the noncoding portion of the DLBCL genome, including long noncoding RNAs, microRNAs, and promoter/enhancer elements that could be targeted by ASHM. Consistently, recent whole-genome sequencing studies uncovered clusters of mutations at enhancers, which may reflect the activity of the AID cytidine deaminase [33, 34]. While only a subset of the identified lesions has undergone detailed functional characterization to date, these studies revolutionized our knowledge of DLBCL biology, leading to the discovery of several potential targets for therapy. Here we will focus on selected genetic alterations that are more frequently observed and have been functionally dissected (Fig. 2.3).

Most common genetic lesions in DLBCL, NOS and PMBCL. Postulated cell of origin for germinal center B-cell-like (GCB) DLBCL, activated B-cell-like (ABC) DLBCL, and primary mediastinal B-cell lymphoma (PMBCL). The most common genetic alterations associated with these diseases (including those shared across different subtypes and those subtype-specific) are shown, with color codes indicating the subverted biological pathway. Blue textbox, loss-of-function; red, gain-of-function (Modified from Pasqualucci and Dalla-Favera, Semin Hematol, 2015) [18]
2.4.1 Genetic Lesions Found in Both GCB-DLBCL and ABC-DLBCL
Histone/chromatin modifier genes. A consistent theme in recent DLBCL sequencing studies has been the discovery of recurrent mutations in genes encoding for histone/chromatin modifiers, including methyltransferases, acetyltransferases, and linker histones [28,29,30]. Mutations of epigenetic modifiers (particularly, CREBBP and KMT2D) were shown to represent early events that are acquired by a common ancestral clone before final clonal expansion and, in the context of FL transformation, divergent evolution to FL or tFL [35,36,37,38], suggesting that these lesions contribute to the initial phases of lymphomagenesis.
Overall, ~30% of all DLBCL samples, with some enrichment for GCB-DLBCL, carry mutations and/or deletions inactivating the acetyltransferase genes CREBBP (25%) and EP300 (5%) [39]. These two highly conserved and ubiquitously expressed enzymes catalyze the addition of acetyl groups to the lysine residues of multiple histone and nonhistone proteins implicated in diverse signaling and developmental pathways [40]. Mutations include prototypical loss-of-function events that truncate the CREBBP/EP300 polypeptide, abolishing the histone acetyltransferase (HAT) domain, as well as amino acid changes that target the HAT domain, leading to diminished affinity for Acetyl-CoA and thus impaired enzymatic activity [39]. Of note, most of these events are heterozygous, and the residual wild-type allele is expressed, implicating a haploinsufficient tumor suppressor role. Indeed, reduced dosage of CREBBP/EP300 in mouse GC B cells was shown to cooperate with BCL2 deregulation to facilitate the development of FL [41]. Recent studies have started to unveil the mechanism by which these mutations contribute to neoplastic transformation. This involves in part the reduced ability of CREBBP to acetylate the oncoprotein BCL6, where acetylation serves as a negative modulator [42], and the tumor suppressor TP53, which, conversely, requires acetylation for its transcriptional activation [43, 44]. The balanced activity of these two proteins is fundamental for GC physiology and pathology and particularly for the regulation of DNA damage checkpoints during IG remodeling processes [45]; thus, one consequence of BCL6 activity overriding p53 would be an increased tolerance for DNA damage in the absence of proper apoptotic responses. CREBBP opposes the activity of BCL6 also by acetylating H3K27 residues at the promoter/enhancer sequences of most of its target genes, facilitating their transcription. This is particularly important for the commissioning of enhancers that need to be activated during the transition between GC and post-GC stages, when cells receive signals to down-regulate BCL6 and become plasma cells [41]. Reduced CREBBP acetyltransferase activity will impact on multiple biological programs that are critical to the normal GC reaction, with genes involved in GC exit being especially susceptible. The discovery of mutations in CREBBP and EP300 has clinical implications in view of available drugs (i.e., histone deacetylase inhibitors) that could provide therapeutic benefits by re-establishing physiologic acetylation levels in tumors carrying these alterations.
Inactivating mutations of KMT2D (also known as MLL2) are found in approximately one third of DLBCL cases and >80% of FL cases, representing one of the most frequent genetic aberrations associated with GC-derived malignancies [28, 29]. KMT2D is a member of the SET1 family of histone methyltransferases and controls epigenetic transcriptional regulation mostly by mono- and dimethylating the lysine 4 position of histone H3 (H3K4), a mark associated with active chromatin conformation [46]. In GC B cells, KMT2D occupies chromatin domains at enhancer and less frequently at promoter regions of genes that have established roles in B-cell physiology, including immune regulators and components of BCR and CD40 receptor-mediated signaling responses [47, 48].
KMT2D mutations impair the protein methyltransferase activity by either removing the C-terminal cluster of conserved domains, including the SET module (truncating mutations) [28, 39], or by introducing disruptive amino acid changes in the same portion of the protein (missense mutations) [48]. In analogy to the human data, conditional B-cell-specific deletion of KMT2D before GC formation leads to a significant increase in the GC B-cell population and to the establishment of a distinct transcriptional program enriched in anti-apoptotic and cell cycle regulatory genes [48]. While loss of KMT2D is insufficient to drive tumor formation, combined KMT2D deletion and BCL2 deregulation led to a significant increase in the development of lymphomas recapitulating the spectrum of phenotypes observed during human FL to DLBCL transition [48].
Collectively, the findings accumulated to date suggest that alterations in epigenetic modifiers may act differently from classical tumor suppressor genes and facilitate the initial stages of transformation by creating a permissive environment for the proliferation and survival of the cancer clone, in cooperation with other genetic lesions (e.g., BCL2 translocations).
Deregulation of BCL6 activity. BCL6 is a master regulator of the GC reaction [49, 50], and deregulation of its activity constitutes a major oncogenic mechanism exploited by DLBCL cells. The function of BCL6 is required in normal GC B cells to negatively modulate multiple biological programs that are critical for allowing the selection of cells with high-affinity antigen receptors and include the establishment of a proliferative phenotype, the attenuation of DNA damage sensing and replication checkpoints [45, 51,52,53], the protection from programed cell death [54, 55], and the block of terminal differentiation [56]. These programs are hijacked by DLBCL cells through multiple genetic aberrations that dysregulate BCL6 function either directly or indirectly (Fig. 2.4). Approximately 30% of cases—with a preference for ABC-DLBCL [57]—carry chromosomal translocations that juxtapose the intact coding domains of BCL6 to heterologous sequences derived from over 20 chromosomal partners (the IG heavy and light chain loci being most commonly involved) [58,59,60,61]. In an additional ~6% of cases, translocations cluster at an alternative breakpoint region and link BCL6 to distantly acting transcriptional regulatory elements [62]. The common feature of these promoters is a broader activity that extends past the GC stage [63], thus preventing the downregulation of BCL6 transcription necessary for differentiation into post-GC B cells.

Deregulation of BCL6 activity by genetic lesions in DLBCL. Recurrent genetic alterations deregulating the function of BCL6 in DLBCL, either directly or indirectly. Representative biological programs modulated by BCL6 in the GC and disrupted as a consequence of these lesions are shown at the bottom. Symbols depict loss-of-function and gain-of-function genetic alterations (Modified from Pasqualucci and Dalla-Favera, Semin Hematol, 2015) [18]
BCL6 is also altered by multiple somatically acquired mutations that are distributed within a ~2 kb region downstream to its TSS, encompassing the first noncoding exon (~75% of cases). While most of these events reflect the physiologic activity of the SHM process [64, 65], selected nucleotide changes within its untranslated first exon appear to be restricted to lymphoma cells, where they abrogate a negative autoregulatory feedback loop (10% of GCB-DLBCL cases) [66, 67] or prevent the IRF4-mediated BCL6 transcriptional repression induced by CD40 signaling in the GC LZ [68].
Besides genetic lesions directly affecting the BCL6 locus, several indirect mechanisms lead to deregulated BCL6 activity in DLBCL (Fig. 2.4). These include (1) inactivating mutations of CREBBP/EP300 (discussed above) [41, 42, 69]; (2) somatic mutations in the GC-specific transcription factor MEF2B [28, 70], which enhances BCL6 expression in ~15% of cases, by abrogating the binding to the corepressor CABIN1 (missense mutations in the N-terminal MEF/MAD domain) or by generating truncated proteins that lack phosphorylation- and sumoylation-mediated negative regulatory motifs located in the C-terminal portion of the protein (truncating mutations) [70, 71]; and (3) loss-of-function mutations and/or deletions of the ubiquitin adaptor protein FBXO11 (~5% of cases), leading to increased BCL6 protein stability [72]. In line with the genetic data, mouse models recapitulating these lesions develop clonal lymphoproliferative disorders mimicking various stages of the human disease [41, 73, 74]. Collectively, these data highlight BCL6 as an attractive therapeutic target for DLBCL, with several in vitro and preclinical studies showing promising results upon the use of small peptide inhibitors [75, 76].
Escape from immune surveillance. DLBCL cells have devised means to escape immune surveillance mechanisms, both cytotoxic T lymphocytes (CTL)-mediated and natural killer (NK) cell-mediated, by virtue of several genetic lesions. In approximately 60% of DLBCL clinical samples, the MHC class I complex is not expressed; this is explained in part by (1) biallelic inactivating mutations and/or focal homozygous deletions of the B2M gene, which encodes for β2-microglobulin and is required for assembly of the MHC-I complex on the cell surface (29% of cases), and (2) point mutations or genomic loss of the HLA A, B, and C loci [77]. In the remaining 30–45% of cases, lack of expression or aberrant cytoplasmic localization of the B2M/HLA-I protein complex is observed in the absence of such genetic lesions, suggesting additional mechanisms [78]. Interestingly, the loss of HLA-I expression is infrequently observed in other B-NHL types [77], but it is often associated with FL transformation [38, 79].
Nearly 20% of DLBCL cases carry disruptive point mutations and focal deletions inactivating CD58, the ligand for the CD2 receptor expressed on T cells and NK cells [80]. Notably, concurrent loss of HLA-I and CD58 expression is observed in over half of DLBCL samples, suggesting that lymphoma cells co-select these lesions to evade both arms of immune surveillance [78].
Finally, downregulation of MHC class II expression has been reported in 40–50% of samples [5, 81]. This phenotype involves at least in part the genetic inactivation of CIITA, the gene encoding for the MHC-II transactivator; CIITA was identified as a common target of ASHM in ~23% of DLBCL and can also be implicated in promiscuous chromosomal rearrangements (3% of cases) that were shown to either disrupt the gene or generate dominant-negative fusion proteins [82, 83]. Moreover, CIITA is a functional target of CREBBP, and reduced levels of MHC-II were observed in GC B cells from CREBBP conditional knockout mice, suggesting that CREBBP mutations could also contribute to its downregulated expression [41, 69, 84]. Reduced MHC-II levels were shown to correlate with poor outcome and may thus represent a relevant biomarker [5, 81].
Mutations of FOXO1. The transcription factor FOXO1 plays central roles during B-cell differentiation and, within the GC, is expressed specifically in the DZ, while its activity is downregulated in the LZ via the phosphatidylinositol 3-kinase (PI3K)/AKT and mTOR cascade. FOXO1 is an essential requirement for sustaining the DZ transcriptional program, in part by cooperating with BCL6. Accordingly, mice engineered to delete FOXO1 specifically in the GC form DZ-less structures [85, 86]. This circuit is disrupted in 8–10% of DLBCL cases that carry somatic mutations of this gene. FOXO1 mutations mainly cluster around a T24 phosphorylation site required for its AKT-mediated nuclear-cytoplasmic translocation and inactivation; other events substitute the initiating methionine of the FOXO1 polypeptide, leading to the synthesis of proteins that utilize a downstream in-frame start codon, and thus also lack the N-terminal nuclear export domain. These events were suggested to prevent the cytoplasmic translocation and inactivation of FOXO1 induced by PI3K signaling [87]. While mutations are preferentially enriched in GCB-DLBCL, aberrant nuclear localization of the FOXO1 protein is observed in both DLBCL phenotypic subgroups (unpublished), implicating a broader involvement for FOXO1 dysregulation in DLBCL pathogenesis. Clearly, additional studies will be needed to dissect the precise mechanism by which FOXO1 mutations contribute to lymphomagenesis, including a systematic examination of their consequences in the context of B cells and the analysis of larger cohorts to confirm the reported preferential enrichment of these mutations in patients with aggressive disease [87, 88].
2.4.2 Genetic Lesions Associated with GCB-DLBCL
Chromosomal translocations of BCL2. The BCL2 proto-oncogene encodes for the founding member of a protein family that governs commitment to programmed cell death at the mitochondrion [89]. BCL2 is also a direct target of BCL6, which binds to its promoter via the transcriptional co-activator Miz1 and prevents its expression in GC B cells, ensuring the maintenance of a default pro-apoptotic programme [55, 90]. This regulatory axis is disrupted in 30–40% of GCB-DLBCL due to the t(14;18) translocation, which brings the BCL2 coding exons under the control of potent regulatory elements from the IG locus, resulting in its ectopic expression [91]. As a consequence, BCL2 also becomes targeted by multiple AID-mediated somatic mutations, although the significance of these events remains controversial [90, 92]. In line with its ability to confer survival advantage, deregulation of BCL2 has been associated with an inferior outcome [93], particularly when coupled with MYC deregulation (see paragraph on “HGBL-DH”).
Chromosomal translocations of MYC. The MYC gene encodes for a sequence-specific transcription factor that acts as an amplifier of transcriptional programs associated with numerous biological functions, ranging from proliferation to cell growth, energy metabolism, differentiation, apoptosis [94], and DNA replication independently of its transcriptional activity [95]. In 10–14% of GCB-DLBCLs, the MYC protein is constitutively expressed due to chromosomal translocations that fuse its intact coding exons to the IG heavy or light chains loci [96,97,98,99]. Besides juxtaposing enhancer elements in close proximity of the MYC locus, a subset of these rearrangements abrogates regulatory sequences bound by BCL6 to negatively modulate its transcription in most GC cells [100]. The presence of MYC-IGH translocations has been linked with worse prognosis in DLBCL [101].
Mutations of the EZH2 methyltransferase. EZH2 encodes for the enzymatic component of the polycomb repressive complex-2 (PRC2), which is responsible for trimethylating the lysine 27 residue of histone H3 (H3K27me3) at the promoter/enhancer regions of silenced or poised genes [102,103,104]. In GC B cells, EZH2 facilitates the establishment of bivalent domains [105] in multiple genes that are critical for the termination of the GC reaction (IRF4, PRDM1) or for other GC programs (CDKN1A) [106, 107]. In line with these essential functions, conditional deletion of EZH2 in the mouse was sufficient to prevent GC formation and affinity maturation [106, 107].
Somatic heterozygous mutations of the EZH2 gene have been identified in up to 22% of GCB-DLBCL [108]. The vast majority of these events replace a single evolutionarily conserved residue (Y641) within the protein SET domain, leading to increased levels of H3K27me3 through altered catalytic specificity of EZH2 for its substrates [109,110,111]. Consistently, expression of the mutant EZH2Y641F allele in the mouse induces GC hyperplasia and, when combined with BCL2 deregulation, accelerates the development of mature B-cell lymphomas [106, 107]. Of note, selective small molecule inhibitors of EZH2 have shown specific activity in preclinical studies particularly in GCB-DLBCL [106, 112, 113], supporting their evaluation in early clinical trials. Indeed, a recent first-in-human phase II trial evaluating a highly selective EZH2 inhibitor showed promising results in patients with FL and, to less extent, relapsed/refractory DLBCL [114].
Mutations of the Gα13 pathway. The positioning and confinement of GC B cells within the B-cell follicle is regulated by the activity of two G-protein-coupled receptors that, among B-cell types, are expressed specifically in the GC: sphingosine-1-phosphate receptor 2 (S1PR2) [115] and the orphan purinergic receptor P2Y, G-protein-coupled 8 (P2RY8) [116]. These receptors recruit two closely related G proteins (Ga12 and Ga13) in response to lipid ligands and stimulate Rho activity via specific guanine nucleotide exchange factors (Rho-GEFs), ultimately suppressing pAKT signaling and cell migration. The importance of the Gα13 inhibitory circuit in the pathogenesis of GCB-DLBCLs is underscored by the presence of recurrent inactivating mutations in several of its components, including S1PR2, GNA13, and, more rarely, ARHGEF1 and P2RY8 (~20% of cases) (Fig. 2.5) [28, 30, 116]. Consistently, deletion of these genes in the mouse is associated with increased GC B-cell survival and disruption of the GC architecture, followed by dissemination of GC B cells to the lymph and bone marrow; with time, GNA13, ARHGEF1, and S1PR2 [117] knockout mice develop lymphomas exhibiting features of GCB-DLBCL [116]. Disruption of this pathway may thus contribute to malignant transformation by abrogating Gα13-mediated inhibitory signals to both cell migration and AKT signaling.

Disrupted signaling pathways in GCB-DLBCL. Genetic lesions preferentially associated with GCB-DLBCL include chromosomal translocations of BCL2 and/or MYC, which cause their ectopic expression in part by bypassing BCL6-mediated transcriptional repression; chromosomal translocations (15% of cases) and point mutations (10% of cases) of BCL6, leading to its deregulated expression; gain-of-function mutations of EZH2 (~20% of cases), which induce epigenetic remodeling in various lymphoma-relevant genes, such as CDKN1A, in part cooperating with BCL6. Additionally, loss of PTEN expression is observed in 55% of cases, as a consequence of genetic deletions (15%) and amplifications of miR17-92 (%), resulting in activation of the PI3K/Akt/mTOR signaling pathway
Mutations of TNFRSF14. TNFRSF14 is a member of the TNF receptor superfamily expressed in both T and B cells, which emanates inflammatory or inhibitory signals depending on its specificity for diverse ligands [118]. In 9–22% of DLBCL, almost exclusively of the GCB type, missense (~50%), nonsense (~40%), and frameshift (2.5%) mutations target the exons encoding for its ectodomain, leading to functional inactivation (Fig. 2.5). Additionally, TNFRSF14 is part of a genomic region that is frequently deleted on chromosome 1p36. A tumor suppressor function for this gene during DLBCL development was recently demonstrated in a BCL2-driven mouse model where silencing of Tnfrsf14 induced cell autonomous activation of B-cell proliferation and enhanced the development of GC-derived lymphomas [119]. Loss of TNFRSF14 contributes to tumorigenesis by inhibiting cell-cell interactions between TNFRSF14 and its ligand BTLA, thus inducing a supportive microenvironmental niche characterized by lymphoid stroma activation and increased recruitment of T follicular-helper cells. Consistent with this model, TNFRSF14 mutations and BTLA downregulation are largely mutually exclusive in FL, although no studies have investigated this aspect in DLBCL. Of note, administration of soluble HVEM to BTLA-expressing lymphoma cells in vitro was able to restore this circuit and to inhibit cell growth, while local production by modified CAR-T cells in vivo led to the significant reduction of established lymphomas [119].
Other Lesions. While the multitude of genetic alterations associated with DLBCL prevents their comprehensive and detailed description, loss of the tumor suppressor PTEN, amplifications of the REL gene, and mutations of SGK1 should be mentioned because of their frequency and/or potential therapeutic implications. Approximately 55% of GCB-DLBCL, as compared to 14% of ABC-DLBCL, lack expression of PTEN [120], due in part to the presence of mutually exclusive 10q chromosomal deletions and amplifications of the oncogenic miR-17-92 micro-RNA cluster (collectively, ~20% of patients) [17]. This pattern was inversely correlated with activation of the PI3K/AKT pathway and sensitivity to pharmacologic PI3K inhibition, uncovering potential therapeutic opportunities [120]. Also largely restricted to GCB-like DLBCL is the presence of chromosome 2p amplifications encompassing the REL gene, which encodes for a subunit of the NF-κB transcription complex. Finally, mutations of SGK1, presumably reflecting the aberrant activity of SHM, were found specifically in this phenotypic subgroup. The SGK1 genes encode for a PI3K-regulated kinase implicated in the control of FOXO transcription factors [121], NF-κB [122], and NOTCH signaling [123], but the functional consequences of these mutations remain unaddressed.
2.4.3 Genetic Lesions Associated with ABC-DLBCL
Alterations sustaining constitutive activation of the NF-κB signaling pathway. The genetic hallmark of ABC-DLBCL is the presence of multiple genetic aberrations that lead to the constitutive activation of the NF-κB signaling pathway, downstream of the BCR signaling cascade and the Toll-like receptor (TLR)/IL1R pathway (Fig. 2.6).

Disrupted signaling pathways in ABC-DLBCL. Multiple genetic alterations cluster around two main biological programs in ABC-DLBCL: constitutive activation of the NF-κB transcription complex and block of plasma cell differentiation. NF-κB activity can be induced by a variety of signals in GC LZ cells, including engagement of the BCR by the antigen, interaction of the CD40 receptor with the CD40L presented by T cells, and TLR activation, which all lead to the upregulated expression of its target genes (e.g., IRF4 and A20). IRF4, in turn, extinguishes the GC program by suppressing BCL6 and thus releasing the expression of the plasma cell master regulator PRDM1/BLIMP1. NF-κB responses are then terminated in part by a negative feedback loop that requires the activity of A20. Subversion of the BCR and NF-κB signaling pathways may fuel malignant transformation by hijacking the anti-apoptotic and pro-proliferative functions of NF-κB while blocking terminal B-cell differentiation through mutually exclusive alterations deregulating BCL6 or inactivation of BLIMP1 (Modified with permission from Pasqualucci and Dalla-Favera, Semin Hematol, 2015) [18]
Mutations in the BCR signaling pathway. Most ABC-DLBCL cells are characterized by a “chronic active” form of BCR signaling associated with the presence of clustered BCRs on the plasma membrane, similar to those formed in antigen-stimulated B cells. This signaling cascade requires CARD11 and is sustained in part by somatic mutations in the CD79A/B genes [124]. In particular, 20% of ABC-DLBCL patients harbor gain-of-function somatic variants in the immunoreceptor tyrosine-based activation motifs (ITAMs) of CD79B (or, rarely, CD79A) [124]. Most of these events replace the first tyrosine residue (Y196) in the cytoplasmic tail of the two BCR subunits and are thought to maintain BCR signaling chronically active by attenuating the phosphorylation and activation of the Lyn kinase, which is necessary for internalization of the surface BCR and serves as a negative feedback regulator.
In ~9% of ABC-DLBCL, activation of the BCR and NF-κB signaling cascade can be attributed to oncogenic mutations of CARD11 [125]. The CARD11 protein is a component of the CBM complex that is coordinately recruited to transduce signals emanating from the BCR [126]. Missense mutations typically target the CARD11 coiled-coil domain, facilitating the formation of cytosolic aggregates and the recruitment of downstream effector molecules, ultimately enhancing its ability to transactivate NF-κB target genes [125].
The genetic lesions discussed above maintain, yet do not initiate, chronic active BCR signaling, which instead was recently shown to require engagement by self-antigens [127]. The dependence of ABC-DLBCL from the BCR signaling pathway is underscored by the preferential sensitivity of these tumors to agents that inhibit Bruton’s tyrosine kinase (BTK), the molecule linking BCR to NF-κB, even in the absence of mutations targeting CD79A/B [128].
Mutations of the MYD88 gene. MYD88 encodes an adaptor molecule that mediates activation of NF-κB as well as type I interferon responses downstream the TLR signaling pathway [129]. Thirty percent of ABC-DLBCLs carry a hotspot L265P substitution within the protein TIR (Toll/IL1 receptor) domain, which was shown to induce the spontaneous assembly and activation of a protein complex containing the kinases IRAK1 and IRAK4, leading to engagement of the NF-κB signaling pathway [130]. MYD88-mutated DLBCL cases also show autocrine transcriptional activation of the JAK/STAT3 signaling cascade, another distinctive phenotype of ABC-DLBCL required for their survival [130, 131]. The significance of other MYD88 mutations found in both ABC- and GCB-DLBCL remains to be established. Importantly, MYD88-mutant DLBCL are not sensitive to BTK inhibition, but this treatment was exquisitely toxic to tumors carrying concurrent MYD88 and CD79A/B mutations, suggesting functional interaction between these two molecules [128].
Mutations of the TNFAIP3 gene. TNFAIP3 (also called A20) encodes a dual function ubiquitin-modification enzyme involved in the termination of NF-κB responses triggered by TLR and BCR stimulation. This protein removes K63-linked regulatory ubiquitins from a number of substrates via its OTU domain and subsequently conjugates K48-linked ubiquitins via its zinc finger domains, targeting these proteins for proteasomal degradation. In line with this, TNFAIP3 knockout mice show abnormally prolonged NF-κB responses associated with an overactive phenotype [132]. Almost one third of ABC-DLBCLs and fewer GCB-DLBCLs harbor biallelic truncating mutations and/or focal deletions that inactivate the function of TNFAIP3, leading to constitutively active NF-κB responses [133, 134]. A tumor suppressor role for this gene in ABC-DLBCL is supported by the observation that enforced expression of wild-type TNFAIP3 in TNFAIP3-null DLBCL cell lines results in cytoplasmic re-localization of the NF-κB complex and suppression of its activity, leading to apoptosis [133, 134].
At lower frequencies, a variety of other genes encoding for NF-κB positive and negative regulators have been found mutated in ABC-DLBCL, overall accounting for more than 50% of cases [133]. It should be considered that, since the BCR emanates signals to multiple downstream effectors besides canonical NF-κB (namely, PI3K, ERK, and NF-AT), the activation of these circuits could also represent vulnerabilities of the tumor cells. This may offer additional therapeutic opportunities based on the design of combinatorial strategies, as supported by the cooperative toxicity observed upon combined inhibition of NF-κB and PI3K [125].
Genetic lesions preventing terminal differentiation. A second pathogenic mechanism in ABC-DLBCL is blockade of post-GC differentiation. Indeed, these tumors derive from a GC B cell that has received signals to commit to plasma cell differentiation but is arrested at the plasma blast developmental stage due to a variety of genetic and epigenetic mechanisms abrogating the function of the plasma cell master regulator PRDM1/BLIMP1 (Fig. 2.6). In 25% of patients, both copies of the PRDM1 gene are lost owing to truncating mutations, loss-of-function missense mutations, and/or genomic deletions [135,136,137]. Another 25% of patients lack BLIMP1 expression as the result of direct transcriptional repression by constitutively active, translocated BCL6 alleles [135]. Alternative modes of inactivation could explain the absence of BLIMP1 in the remaining large fraction of cases, including epigenetic silencing or high expression of the ETS family factor SPIB. Consistent with this model, conditional ablation of PRDM1 in mouse GC B cells, either alone or in combination with a constitutively active IκB kinase, leads to the development of DLBCL with ABC-like phenotype [135, 138]. BCL6 rearrangements and PRDM1 inactivation are mutually exclusive [34, 35], supporting a complementary role for these lesions in fostering lymphomagenesis.
Other genetic lesions. Additional recurrent alterations include amplifications of the BCL2 locus, observed in more than one third of patients [5, 17], homozygous deletions or lack of expression of the CDKN2A/B tumor suppressor genes (30% of cases) [17], and gains or amplifications of a large region on chromosome 19q (27% of cases), which spans the SPIB locus and may contribute to the elevated expression of this protein observed in DLBCL [17].
3 High-Grade B-Cell Lymphomas
The fourth edition of the WHO classification of hematolymphoid tumors of 2008 has coined the provisional category of “B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL,” or so-called grey zone lymphoma [139]. These lymphomas were defined because of their intermediate morphological, immunohistochemical, and genetic features encompassing features of both DLBCL and Burkitt lymphoma. However, this category was poorly reproducible, and, owing to the fact that around 60% of greyzone lymphomas harbor double or triple hits involving MYC and BCL2 and/or BCL6, research had mainly focused on the double-/triple-hit subcategory. In the 2017 update of the fourth edition, this provisional category has been replaced by two new categories: high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (or HGBL-DH) and high-grade B-cell lymphoma, not otherwise specified (HGBL-NOS), which by definition lacks a double- or triple-hit constellation involving MYC but may be MYC-rearranged [1]. Most patients with HGBL will present at higher age and with aggressive and/or extensive disease, and bone marrow and extranodal involvements are common, as are elevated lactate dehydrogenase (LDH) levels.
3.1 High-Grade B-Cell Lymphoma with MYC and BCL2 and/or BCL6 Rearrangements (HGBL-DH)
The diagnosis of these tumors as a new WHO category relies exclusively on the presence of rearrangements involving both MYC and BCL2 and/or BCL6 (previously known as double-hit DLBCL). In contrast, DLBCL featuring high-level amplifications, copy number alterations, or elevated protein expression of MYC will not be included in this category. Therefore, by definition, HGBL-DH encompasses the following morphological cases:
-
Tumors with the morphology of DLBCL and a double (DH) or triple hit (TH) (Fig. 2.7a)
-
Tumors that, in the former taxonomy, would have been classified as “unclassifiable with features intermediate between DLBCL and BL” harboring dual rearrangements involving MYC (Fig. 2.7b)
-
Cases with blastoid cytomorphology and a double- or triple-hit genetic constitution (Fig. 2.7c)

(a) High-grade B-cell lymphoma, featuring large tumor cells of varying size and moderately pleomorphic nuclei equivalent to a DLBCL. (b) Double-hit DLBCL showing medium-sized cells with narrow cytoplasm and round, only slightly polymorphic nuclei. This tumor would have been classified as “B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL” in the 2008 WHO classification. (c) High-grade B-cell lymphoma with MYC/BCL2 translocation, transformed from FL. This tumor features blastoid cells with small to intermediate size and finely dispersed nuclear chromatin
One important new concept of the updated WHO classification is that DLBCLs with a DH or TH genetic constitution will be reclassified as HGBL-DH. Consequently, all DLBCL would be required to undergo FISH testing in order to identify and classify these cases. The update of the WHO classification, however, does not formally require FISH testing on all DLBCL; therefore, pathologists may preselect cases according to a variety of criteria in order to minimize the efforts inherent to extensive FISH testing.
With the probable exception of cases displaying immunoblastic morphology and cases with a higher number of tumor cells expressing nuclear MYC [140], HGBL—DH/TH are virtually indistinguishable from bona fide DLBCL upon morphology. Cases with blastoid cytomorphology have a lymphoblast-like appearance and, therefore, may require a differential diagnosis with B-cell lymphoblastic leukemia/lymphoma or the blastoid variant of mantle cell lymphoma. More frequently, cases transformed from preexisting or concurrent follicular lymphoma with secondary MYC rearrangement will have this appearance. The latter tumors are classified as transformed follicular lymphomas with dual MYC and BCL2 rearrangements and blastoid cytomorphology. Finally, tumors previously classified as greyzone lymphomas consist of predominantly medium-sized cells in between the size of Burkitt lymphoma and DLBCL cells, but featuring a more pleomorphic cytology than BL, and will occasionally show a starry-sky pattern [139]. Cases with a MYC and BCL6 double hit are almost exclusively of the GCB type, and, because of their particularly poor clinical outcome, they represent an unmet clinical need within this otherwise favorable prognostic category of DLBCL. Some clinico-pathological differences have been recorded between MYC-BCL2 and MYC-BCL6 DH tumors [141, 142].
3.2 High-Grade B-Cell Lymphoma Not Otherwise Specified (HGBL-NOS)
This new category mainly includes the former greyzone or high-grade B-cell lymphomas, unclassifiable, with features intermediate between DLBCL and BL, which lack a genetic DH or TH constellation. Morphologically, these cases are composed of predominantly medium-sized or blastoid cells with a narrow cytoplasmic rim, round nuclei slightly more pleomorphic than those of Burkitt lymphoma, a finely dispersed nuclear chromatin, and inconspicuous nucleoli, sometimes in combination with a starry-sky pattern (Fig. 2.8). HGBL-NOS often shows a GCB-like GEP, although some cases are of ABC-DLBCL type based on either GEP or immunohistochemical algorithms. By virtue of their definition, these tumors may carry isolated MYC, BCL2, or BCL6 rearrangements and may also show dual BCL2 and BCL6 rearrangements. It has to be noted that tumors showing the classical morphology of DLBCL and harboring isolated MYC rearrangements will continue to be diagnosed as DLBCL and do not fall into the HGBL-NOS category [1].

Bone marrow biopsy of a high-grade B-cell lymphoma, NOS, displaying diffuse infiltration by medium-sized blasts. Cytogenetic analysis in this case was positive for a MYC translocation, but no alterations of BCL2 or BCL6 were seen
4 Other Lymphomas of Large B Cells and Special Subtypes/Variants
4.1 T-Cell/Histiocyte-Rich Large B-Cell Lymphoma
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is defined as a DLBCL variant with few scattered tumor cells and an abundant reactive background consisting of T cells and histiocytes. The median age at presentation is in the sixth to seventh decade of life, but this variant may also be seen in children. There is a male predominance. THRLBCL is a primarily nodal disease and often presents with high-stage and bone marrow involvement. On morphology, there is complete effacement of the underlying architecture, with large lymphoid cells scattered in a predominantly reactive background of activated lymphocytes and histiocytes. The tumor cells in THRLBCL make up less than 10% of the whole cell population, without clusters or sheets (Fig. 2.9). Presence of an even focal component of NLPHL excludes de novo THRLBCL (see above). The cytomorphology of the tumor cells is centroblastic, immunoblastic, or LP or HRS cell-like. The tumor cells express pan B-cell markers, are negative for CD30 and CD15, and often co-express BCL6 and EMA. In the background, CD3-positive cells usually express CD8 and TIA1. THRLBCL shows clonally rearranged IG genes, and roughly one quarter of the cases has BCL2 rearrangements. EBV is negative in the vast majority of cases. In some cases, a biological overlap to progressed variants of nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) is suggested [143]. Malignant lymphomas with features of THRLBCL have been observed in progression of or as secondary lymphomas that develop after NLPHL. Diffuse variants of NLPHL may bear close similarity to THRLBCL, and therefore it is not clear whether predominantly diffuse NLPHL and THRLBCL may be representing a spectrum of the same disease or whether THRLBCL could represent transformation of NLPHL [144, 145]. Cases with T-cell/histiocyte-rich areas in NLPHL are designated as THRLBCL-like transformation of NLPHL in the revised fourth edition of the WHO classification.

T-cell/histiocyte-rich large B-cell-lymphoma (THRLBCL). In this tumor, rare large B blasts highlighted in a CD20 stain are seen in a background of small and slightly activated reactive T cells and histiocytes (a). Of importance, no small B cells are present. (b) H&E staining
4.2 Intravascular Large B-Cell Lymphoma
Intravascular large B-cell lymphoma (IVLBCL) is a DLBCL subtype in which blastic tumor cells are seen exclusively—or at least predominantly—within the lumina of small-sized blood vessels [146]. The lymphoma is rare, predominantly occurring in older patients with a peak in the seventh decade. IVLBCL can be seen in all organs, but most commonly in the CNS, skin, kidney, lung, and liver. Clinical symptoms are often nonspecific and related to the site of infiltration, such as cerebrovascular dysfunction. Involvement of the bone marrow is rare. A particular variant associated with hemophagocytic syndrome has been described in Asian patients.
Histologically, medium-sized to large lymphoma cells with varying cytological appearance are seen in often dilated lumens of small to intermediate-sized vessels (Fig. 2.10a, b). There may be secondary changes such as thrombosis or endothelial hyperplasia, causing infarction and hemorrhage. The tumor cells express pan B-cell markers and may be positive for CD5, CD10, and/or BCL6. Clonal IGH rearrangements are usually found. The majority of cases show a non-GCB immunophenotype, and no specific cytogenetic alteration has been defined so far [139].

Intravascular large B-cell lymphoma. (a) Hematoxylin-eosin (H&E) staining of a lung biopsy showing dense infiltrations of medium- to large-size tumor cells in distended capillary vessels of the alveolar septa. (b) CD20 staining of the same sample highlights the intravascular localization of the tumor cells
4.3 Plasmablastic Lymphoma
Plasmablastic lymphoma (PBL) is defined as a terminally differentiated B-cell neoplasia. Morphologically, it consists of immunoblastic or plasmablastic cells. The negativity of the tumor cells for the CD20 antigen is a hallmark defining feature [139]. PBL frequently occurs in adults with immunodeficiency states (most commonly HIV infection) but may also be seen in the setting of posttransplantation or autoimmune disorders. In addition, these tumors are also diagnosed in patients without any apparent immunodeficiency. PBL frequently arises in extranodal sites, mainly of the head and neck and often in the oral cavity, in which it was primarily described [147]. Other involved locations are the upper respiratory tract, gastrointestinal tract or soft tissues, skin, bone, lung, and lymph nodes. The latter localization is often characteristic in PBL occurring in a posttransplantation setting. Most patients are diagnosed in higher clinical stages (III/IV), but this is less apparent in patients without immunodeficiency. In general, the prognosis is poor.
PBL may demonstrate monomorphic cytological features or plasmacytoid differentiation. The common variant of PBL shows a diffuse proliferation of cohesively growing immunoblasts or plasmablasts, whereas in cases of plasmacytoid differentiation, the cells are rounder with more eccentric nuclei and abundant basophilic cytoplasm. In some cases, a starry-sky pattern may be seen, and there may be extensive necrosis. On immunophenotyping, the tumor cells are negative for CD45, CD20, and PAX5. Expression of CD79A is variable. Plasma cell-associated markers are regularly expressed (CD38, CD138, IRF4/MUM1, and others such as BLIMP1 and XBP1). EMA is frequently positive and CD56 expression may be seen in some cases. The proliferative index is high. BCL2 and BCL6 are usually not expressed [148]. EBV association may be seen in roughly 70% of cases, in most of the tumors with a latency type I pattern. HHV8 is always negative. Genetic analyses frequently show complex karyotypes, and MYC translocationsc to the IG genes have been seen in 50% of the cases [149]. These cells express the MYC protein, circumventing the BLIMP1-mediated suppression of its transcription usually encountered in plasma cells.
4.4 ALK-Positive Large B-Cell Lymphoma
ALK-positive large B-cell lymphoma is an uncommon subtype of DLBCL that shares with plasmablastic lymphoma its frequent immunoblastic-plasmablastic morphology and protein expression phenotype. In addition, ALK+ DLBCL strongly expresses the ALK protein. It occurs predominantly in young adults, but there is a broad age range at presentation. Generalized disease is common, and no obvious association with immunodeficiency states has been reported. The majority of patients have high-stage (III/IV) disease, and the tumor is aggressive with short 5-year overall survival.
The tumor cells feature an immunoblastic or plasmablastic appearance with vesicular nuclei and frequently a large central nucleolus (Fig. 2.11a). One characteristic architectural feature is a sinusoidal growth pattern in many cases, creating a differential diagnosis with carcinoma. CD20, CD79A, and PAX5 are usually negative, but the tumor cells express CD45, EMA, IRF4/MUM1, immunoglobulin light chain genes and often IgA. In addition, CD138, BLIMP1, and XBP1 are usually positive. CD30 is negative or only weakly expressed [150]. All cases are EBV- and HHV8-negative. By definition, the ALK kinase is expressed, usually in the cytoplasm with a granular pattern (Fig. 2.11b), and these cases frequently harbor ALK-CLTC translocations [150,151,152]. Tumors with the classical ALK-NPM translocation inferring nuclear and cytoplasmic expression of the ALK protein are rare. ALK activation leads to upregulation of the STAT3 pathway, and ALK-positive LBCL express high levels of phospho-STAT3. STAT3, in turn, upregulates MYC and also BLIMP1. Consistently, ALK-positive LBCLs also express the MYC protein, but do not harbor MYC translocations [153].

ALK-positive large B-cell lymphoma. (a) H&E staining shows large tumor cells reminiscent of immunoblasts with a certain cohesiveness featuring abundant cytoplasm and slightly eccentric nuclei. CD20 negativity of the tumor cells suggested plasmablastic differentiation. (b) ALK staining reveals positivity in the cytoplasm of the tumor cells with granular reactivity that is characteristic of an ALK-CLTC fusion
4.5 Primary Mediastinal (Thymic) Large B-Cell Lymphoma
Primary mediastinal (thymic) large B-cell lymphoma (PMBCL) is regarded as a distinct subtype of DLBCL with organotypic features. The tumor usually presents in the upper anterior mediastinum of predominantly young adults, with a median age of 35 years and a female to male ratio of 2:1. The tumor can invade into neighboring structures such as the thoracic wall, the pericardium, the pleura, and the lung. Symptoms are related to the often bulky mediastinal mass, such as superior vena cava obstruction, dyspnea, and chest pain [139]. More recently, cases with a gene expression profile of PMBCL have also been described outside the mediastinum [154].
On morphology, the tumor shows a diffuse infiltration of medium-sized to large blastic cells that have a variable cytological appearance (Fig. 2.12) [155]. Centroblastic, immunoblastic, anaplastic, or Hodgkin- and Reed-Sternberg-like cells may occur, and a clear cell morphology is not infrequent [156]. One hallmark feature of PMBCL is background sclerosis with delicate or coarse collagen fibers encompassing small groups of lymphoma cells in a way “compartmentalizing” the infiltrate. The immunophenotype, although not specific to PMBCL, is characteristic, frequently showing co-expression of CD30 and CD23, with pan B-cell markers and absent expression of MHC class I and II antigens [139]. Another feature characteristic of PMBCL is expression of the MAL protein [157]. PMBCL displays a unique GEP that is closer to that of classical Hodgkin lymphoma and reflects the origin from a thymic, GC-experienced B cell (with particular asteroid morphology) [158, 159].

Mediastinal (thymic) large B-cell lymphoma. This biopsy shows large blastic B cells diffusely infiltrating the mediastinal tissue. Note the slight fibrosis in the background
Molecular Pathogenesis. The core biology of PMBCL is defined by a constellation of somatic mutations, copy number changes, and genomic rearrangements that lead to deregulation of the JAK-STAT and NF-κB signaling pathways, as well as to acquired immune privilege [82, 160,161,162,163] (Fig. 2.3).
Constitutive activation of the JAK-STAT signaling pathway, a signature identified by gene expression profiling analysis and shared with classical Hodgkin lymphoma (cHL), is the result of both paracrine interleukin receptor-mediated signaling [164] and genetic alterations, including amplification of JAK2 (>50% of patients) [163, 165], deletions or inactivating mutations of the negative regulators SOCS1 (45% of cases) [166] and PTPN1 (22% of cases) [160], and mutations of STAT6. Amplification of chromosomal region 9q24 is a genetic hallmark of both PMBCL and cHL and spans multiple genes of possible pathogenic significance, in addition to JAK2, namely, CD274 (PDL1), PDCD1LG2 (PDL2), and JMJD2C. The latter encodes a demethylase that removes histone H3 lysine 9 trimethylation marks, interfering with the recruitment of HP1α and heterochromatin formation [167]. Interestingly, phosphorylation of JAK2 also contributes to an active chromatin conformation by phosphorylating both histone H3 and HP1α [168]. Thus, activation of JAK-STAT signaling may act synergistically with acquired immune privilege (through PDL1/2) and chromatin remodeling (through JMJD2C) in the pathogenesis of PMBCL [161]. SOCS1 mutations abrogate SOCS box function leading to hyperphosphorylation of JAK2 and STAT5 [166], while mutations of PTPN1, encoding a non-receptor member of the protein tyrosine phosphatases superfamily named PTP1B, impair its phosphatase activity causing hyperphosphorylation of STAT molecules. Consistent with these data, STAT6 phosphorylation has been suggested as a reliable marker for the differential diagnosis of PMBCL from other large cell lymphomas [169]. Finally, recurrent mutations of the transcription factor STAT6 are found in 36% of PMBCL cases, although the exact functional consequences of STAT6 mutations in the DNA binding domain are still a subject of debate.
Another major target of genetic alterations in PMBCL is the NF-κB signaling pathway [161], which is maintained constitutively active by a variety of somatic gene mutations and structural genomic changes. These include (1) copy number gains and amplifications of the REL gene (>50% of cases), correlated with nuclear localization of the REL protein [170]; (2) inactivating mutations of TNFAIP3 (36% of cases) [171]; and (3) amplification of the genomic regions encoding for the NF-κB regulators BCL10 (1p22) and MALT1 (18q21), observed in a subset of cases [172].
Finally, several genetic lesions impinge on the crosstalk between the tumor cells and the tumor microenvironment [173], leading to acquired immune privilege [82]. Among these lesions, the loss of MHC-II molecules is a prominent event that, analogous to DLBCL, NOS, has been linked to decreased infiltrating cytotoxic T cells and inferior outcome [5, 81]. Downregulation of surface MHC-II expression in these tumors is explained in part by allelic imbalances of the HLA-DR loci on chromosome 6p [174, 175] and by genomic breakpoints and mutations of the MHC class II transactivator gene CIITA (38% of samples) [83]. In addition, 20% of PMBCL carry recurrent genomic rearrangements involving the PDL1/PDL2 genes on the short arm of chromosome 9, which encode for inhibitors of T-cell responses. These rearrangements generate PDL1 and PDL2 gene fusions with various partner genes, leading to elevated protein expression, and may in part explain the unique features of PMBCL, which are characterized by a significant inflammatory infiltrate. The cumulative incidence of genetic alterations that interfere with the interaction between the lymphoma cells and the microenvironment supports a central role for escape from immunosurveillance mechanisms in the pathogenesis of these neoplasms. Importantly, the frequent co-amplification of JAK2 and PDL1/2 on chromosome 9p24 suggests that combination therapies with JAK-STAT pathway inhibitors and immune checkpoint inhibitors might have synergistic antitumor activity.
4.6 B-Cell Lymphoma, Unclassifiable, with Features Intermediate Between Diffuse Large B-Cell Lymphoma and Classical Hodgkin Lymphoma
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL (so-called mediastinal greyzone lymphoma, MGZL) still represents a peculiar tumor that shows features of both primary mediastinal large B-cell lymphoma and classical Hodgkin lymphoma. The clinical characteristics of the condition are predominantly determined by the site in which the tumor arises. Usually, there is a large mediastinal mass that can spread to supraclavicular lymph nodes and/or can invade neighboring structures per continuitatem, such as the lungs, the surrounding soft tissues, and even bone structures, but that rarely metastasizes to distant sites. Retrospective analyses have shown a poorer clinical outcome of MGZL in comparison with either CHL or PMBCL [176]. There are two more or less distinct morphologies that may be seen. One resembles that of CHL (frequently, of nodular sclerosis type), but, on immunohistochemistry, the B-cell program is greatly preserved as exemplified by strong expression of B-cell-associated antigens including CD20 and CD79a and also reactivity for the B-cell-specific transcription factors OCT-2 and BOB1, while expression of CD15 is usually absent. The other type of MGZL shows the morphology of large B-cell lymphoma with expression of B-cell markers, in which the tumor cells are also uniformly positive for CD30 and often CD15, or are EBV associated [177]. This tumor should be set apart from a composite lymphoma simultaneously showing classical Hodgkin lymphoma and primary large B-cell lymphoma in different areas of the same site or biopsy.
4.7 Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
Primary cutaneous DLBCL, leg type (PCDLBCL, leg type) is a special variant of diffuse large B-cell lymphoma with a characteristic clinical presentation, morphology, and immunophenotype [1]. It usually develops on the lower legs of predominantly female older adults. There is a diffuse infiltration of the skin and subcutaneous tissues by large cells, often centroblasts and immunoblasts, forming large cutaneous nodules [178]. The tumor cells express BCL2, IRF4/MUM1, and FOXP1, while CD10 and BCL6 are negative. A characteristic feature is strong cytoplasmic expression of IgM and MYC protein (without underlying MYC rearrangements). Leg-type DLBCL often displays an ABC-type (or non-GCB-type) expression profile. High-level amplifications of the BCL2 locus have been seen in two thirds of cases along with CDKN2A deletion or promoter methylation. Mutation analyses have revealed MYD88 L265 mutations in 60% of the cases and presence of mutations in genes characteristic of ABC-type DLBCL, such as CARD11, CD79B, und TNFAIP3 [179].
4.8 Primary Central Nervous System Lymphoma
Primary CNS lymphoma (PCNSL) is a malignant lymphoid neoplasm arising in the brain, spinal cord, or leptomeninges; intraocular lymphoma forms a subset of PCNSL. PCNSL are predominantly diagnosed as large B cell lymphomas [139] and can develop in either immunocompetent or immunosuppressed patients. The affected individuals are in general elderly adults with a slight male predominance while, among immunodeficient patients, HIV-infected individuals, with younger age and clear male predominance at presentation are commonly seen. Histology characteristically reveals a diffuse proliferation of blasts with perivascular growth and—often extensive—necrosis. The tumors are usually composed of immunoblasts or centroblasts. They express pan B-cell antigens, usually BCL2, IRF4/MUM1, and often BCL6 implying a non-GCB immunophenotype [180]. PCNS-DLBCL shares with other lymphomas arising at immune-privileged sites the characteristic downregulation of MHC class II antigens [181]. Virtually all cases of PCNS-DLBCL in immunosuppressed patients are positive for EBERs, and a subset of them also expresses LMP1.
At the molecular level, several lines of investigation support a role for both the NF-κB and JAK/STAT signaling pathways as mediators of pro-survival signals in this tumor type [182]: besides the presence of multiple genetic alterations in NF-κB regulators (see below), interleukin-4 is upregulated at the transcript and protein level within the vascular microenvironment; IL-10 (another first messenger in the JAK/STAT signaling) is increased in the vitreous and cerebrospinal fluid, and increased JAK1 transcripts are detected in the tumor cells [182]. Elevated IL-10 expression alongside activation of JAK/STAT signaling are consistent with aberrant activation of the MYD88 pathway.
Molecular Pathogenesis. The rarity of PCNS-DLBCL and the difficulty in obtaining material for investigational studies has hampered the molecular analysis of this DLBCL subtype. Nonetheless, a number of studies, including recent whole-exome sequencing efforts, have unveiled important genetic features associated with PCNS-DLBCL, including recurrent mutations in genes that are involved in NF-κB pathway activation (e.g., CARD11, CD79/B, and MYD88), cell cycle regulation (CCND3), and chromatin structure/histone modifications (CREBBP, KMT2D, ARID1A/B, SMARCA4, and SMARCA5) [183]. Of these, MYD88 mutations at the L265 hotspot represent the most common event, accounting for 38–50% of clinical cases [184, 185], followed by CD79B (approximately 20% of cases) [186]. PCNS-DLBCL also shows evidence of ASHM in multiple proto-oncogenes with established roles in B-cell development and differentiation, including PAX5, PIM1, c-MYC, and RhoH/TTF [187].
Moreover, a critical step in the molecular pathogenesis of PCNSL (and a biomarker of inferior clinical outcome) is inactivation of the CDKN2A/B genes, encoding for p14ARF and p16INK4a, by either homozygous deletion (40–50% of cases) or hypermethylation (15–30% of cases). BCL6 translocations are detected in 17% of tumors and have been associated with inferior overall survival especially when combined with 6q22 deletions [188]. Other potentially relevant genetic lesions in PCNS-DLBCL comprise recurrent copy number gains of MALT1 (a potential additional contributor to the aberrant activation of the NF-κB pathway in this disease) [189], chromosome 12 (particularly in the 12q region harboring STAT6, MDM2, and CDK4), and the long arms of chromosomes 1, 7, and 18 [182]; deletions of the short arm of chromosome 6 (which harbors the HLA loci) and chromosomes 17 and 18 [188, 190]; and deletions of 6q21-23, detected in 40–60% of cases. The latter encompasses several candidate tumor suppressor genes, among which PRDM1 [191], TNFAIP3 (A20), and PTPRK, a protein tyrosine phosphatase that participates in cell adhesion signaling events [192] may be most relevant. In contrast, mutations in the TP53 gene are observed in only a small proportion of PCNS-DLBCL specimens.
4.9 Epstein-Barr Virus (EBV)-Positive Diffuse Large B-Cell Lymphoma NOS
The update of the fourth edition of the WHO classification has renamed EBV-positive DLBCL because this lymphoma, called EBV-positive DLBCL of the elderly in the fourth edition of the classification (2008), can also be seen in younger patients [193]. The name “EBV-positive DLBCL, NOS” was adopted to emphasize the difference from other specific EBV-associated LBCL variants and subtypes. In elderly individuals EBV infection of the tumor cells is explained by an age-related waning of the immune system, whereas its association to DLBCL occurring in the younger age group is not clear [194]. The frequency of EBV-positive DLBCL increases with age and is higher in Japan and Latin America as compared to Europe. Extranodal disease is common, but lymph nodes are affected as well. In younger patients, nodal disease is the most common form of presentation. In both younger and elderly patients, the male gender predominates [195].
Generally, there is a diffuse effacement of the underlying architecture by diffuse infiltrates of blasts, in many cases with extensive necrosis (Fig. 2.13a). A monomorphic subtype characterized by tumor cells resembling centroblasts, immunoblasts, and/or Hodgkin-like cells has been described and set apart from cases where the infiltrate also harbors many reactive bystander cells such as lymphocytes, histiocytes, plasma cells, and eosinophils. Some cases show pleomorphic large cells with several nuclei mimicking Reed-Sternberg cells. The tumor cells usually express pan B-cell markers such as CD20, CD79A, and PAX5. In many cases, co-expression of CD30 and a non-GCB phenotype is common. By definition, in situ hybridization with EBV-encoded small RNAs (EBERs) demonstrates EBV infection in at least 50% of the tumor cells (Fig. 2.13b), although different and lower cutoffs have been used for its definition in the past. LMP1 is usually expressed, and EBNA2 may be positive, consistent with type II latency. As is the case in posttransplantation lymphoproliferative disease (PTLD), the tumor cells are frequently positive for PDL-1 [196], possibly leading to attenuated immune surveillance. In contrast with classical Hodgkin lymphoma, the tumor cells are usually CD15 negative.

EBV-positive DLBCL, NOS. (a) H&E staining of a lung tumor showing blastic tumor cells of varying size and shape, intermingled with some larger Hodgkin-like cells. Note focal necrotic areas. (b) EBER in situ hybridization discloses infection in more than 50% of the tumor cell nuclei by the EBV virus
EBV-positive DLBCL, NOS, must be differentiated from EBV-associated mucocutaneous ulcer because of the apparently indolent clinical course of the latter.
4.10 Primary Effusion Lymphoma
Primary effusion lymphoma (PEL) is a distinct clinico-pathological entity, the hallmark of which is malignant effusion in the pleural or peritoneal cavity or in the pericardium (Fig. 2.14a). Infection of the tumor cells by the Kaposi sarcoma herpes/human herpes virus 8 (KSHV/HHV8) (Fig. 2.14b) is characteristic [197, 198]. PEL is a disease of HIV-infected patients and, in this setting, it is also associated with EBV infection. Rare cases of HHV8-positive but EBV-negative cases have been described in immunocompetent patients [199]. PEL associated with HIV infection features malignant effusion without solid tumor infiltrations. Only rarely, there is tumor propagation into the lung, the mediastinum, or regional lymph nodes. On microscopy, centroblastic, immunoblastic, or anaplastic tumor cells are shed into body cavities. The tumor cells are positive for the pan leucocyte antigen (CD45) and activation-associated proteins like HLA-DR and CD30; B-cell specific antigens including immunoglobulins and PAX5 are not expressed. Rarely, aberrant co-expression of T-cell antigens is seen. Characteristically, a nuclear reactivity for the HHV8 latency-associated nuclear antigen (LANA) (Fig. 2.14b) and co-infection with EBV is observed. PEL-like effusion lymphomas without HHV8 may be seen in elderly patients mainly in the peritoneum [200]. Rarely, PEL may manifest as an extrapleural solid tumor without effusion [201]. Immunoglobulin genes are clonally rearranged. Classical cytogenetic analyses have revealed complex karyotypic alterations, but no recurrent aberration has been identified so far.

Primary effusion lymphoma (PEL). (a) Cytomorphological evaluation of a pleural effusion, disclosing large atypical cells with abundant cytoplasm, irregular nuclei, and prominent nucleoli. (b) Nuclear expression of the latency-associated nuclear antigen (LANA) proves infection of the tumor cells by the HHV8/KSHV virus
4.11 Diffuse Large B-Cell Lymphoma Associated with Chronic Inflammation
This DLBCL subtype arises in the context of usually long-standing chronic inflammation and involves body cavities or narrow spaces such as the joints. Pyothorax-associated lymphoma (PAL) is the classical form frequently arising in the pleural cavity in the setting of long-standing pyothorax. Most patients are in their fifth to eighth decade, and usually there is a long interval (>20–30 years) between the onset of chronic inflammation and the diagnosis of malignant lymphoma. Most patients present with localized disease [202, 203], and common sites are the pleural cavity, bone, and periarticular soft tissues, with possible invasion of adjacent structures. On histology, the infiltrating blasts are either of centroblastic or immunoblastic type. Immunophenotyping shows the expression of pan B-cell markers and usually a non-GCB expression profile with reactivity for IRF4/MUM1 and negativity for CD10 and BCL6. Occasionally, aberrant expression of T-cell markers may occur [204]. Common genetic features of PAL are MYC amplifications, TP53 mutations, and deletions of TNFAIP3/A20. Immunoglobulin genes are clonally rearranged [205, 206].
4.12 Lymphomatoid Granulomatosis
Lymphomatoid granulomatosis (LYG) is an angiocentric and angiodestructive lymphoproliferative disorder associated with EBV-positive B-cell blasts in a predominantly reactive background [207]. Most cases of LYG arise in healthy individuals, but the disease can also be seen in immune deficiencies of varying cause, especially iatrogenic immunosuppression. It is usually a disease of adulthood, and most cases occur in the fourth to sixth decade of life, with a male predominance. LYG mainly presents with pulmonary disease and can also manifest in the CNS, skin, liver, and kidneys. In contrast, lymph node, splenic, and bone marrow involvement is uncommon. Presenting clinical symptoms are unspecific. The radiologic hallmark of the disease is bilateral nodular infiltration of the lungs. Histology shows angiocentric and angiodestructive polymorphous infiltrates with lymphocytes, histiocytes, and large atypical lymphoid cells and tissue necrosis [208]. The EBV-associated B cells vary in size and correspond to lymphocytes, blasts, and/or HRS-like cells. Grading of LYG is based on the number of EBV-positive cells. Grade 1 lesions contain a minority of large atypical cells within a rich polymorphous background, and EBER in situ hybridization highlights fewer than five cells per HPF. In grade 3 lesions, the large atypical cells are readily seen and may form smaller or larger aggregates. In case of a uniform infiltration of EBV-positive blasts without the characteristic background of LYG, a diagnosis of EBV-associated DLBCL, NOS, is warranted. On immunophenotyping, the EBV-positive blasts express CD20, PAX5 or CD79a, and CD30 in many cases, but CD15 is negative. LYG frequently shows an EBV latency pattern III with positivity for LMP1, EBNA2, and EBER. Clonal IG rearrangements are usually seen in grades 2 and 3 [209].
4.13 HHV8-Positive Diffuse Large B-Cell Lymphoma, NOS
HHV8-positive DLBCL, NOS is a new disease category in the 2016 update of the WHO classification. It forms part of the spectrum of KSHV/HHV8-associated lymphoproliferative disorders and is considered a large B-cell lymphoma that frequently, but not invariably, arises in the background of multicentric Castleman’s disease (MCD) [210]. The KSHV/HHV8-positive plasmablasts are usually negative for CD45 and CD20, strongly express the KSHV/HHV8 latency-associated nuclear antigen (LANA), and show monoclonal cytoplasmic IgM/λ staining [211]. They often represent a tumor form of the KSHV-/HHV8-infected plasmablasts encountered in MCD in HIV-infected individuals.
5 Concluding Remarks
During the past decade, our understanding of the pathogenesis of DLBCL, particularly of its most common subtypes DLBCL NOS and PMBCL, has improved substantially, owing in part to the implementation of powerful genomic technologies. We have now identified a number of pathways that are recurrently dysregulated by genetic alterations in this disease and are emerging as central players in tumor development and maintenance. These lesions reveal vulnerabilities in the lymphoma cells, which are often uniquely associated with distinct lymphoma subtypes and could thus be exploited for patient stratification and for the design of more effective, possibly less toxic-targeted therapeutic approaches. Indeed, the information gained from these studies is already being translated into the development and clinical testing (or repositioning) of novel drugs/drug combinations directed against specifically disrupted programs in DLBCL. While these strategies are expected to impact the standard of care for this malignancy, the complexity of the involved pathways and the overall heterogeneity of the disease, which may also involve non-genetic mechanisms, underscores the need of precise patient stratification in order to identify sensitive and resistant cases.
References
Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90.
Swerdlow SH Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO classification of haematopoietic and lymphoid tissues. Revised 4th ed. Lyon: IARC; 2017.
Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumors of haematopoietic and lymphoid tissues. In: Bosman FT, Jaffe ES, Lakhani SR, Ohgaki H, editors. World Health Organization of tumor. Lyon: International Agency for Research on Cancer (IARC); 2008.
Jaffe ES, Arber DA, Campo E, Harris NL, Quintanilla-Fend L. Hematopathology. Philadelphia: Saunders/Elsevier; 2017.
Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47.
Engelhard M, Brittinger G, Huhn D, et al. Subclassification of diffuse large B-cell lymphomas according to the Kiel classification: distinction of centroblastic and immunoblastic lymphomas is a significant prognostic risk factor. Blood. 1997;89:2291–7.
Lennert K, Feller AC. High grade malignant lymphoma of B-cell type. Histopathology of non-Hodgkin’s lymphoma. Berlin: Springer; 1990.
Ott G, Ziepert M, Klapper W, et al. Immunoblastic morphology but not the immunohistochemical GCB/nonGCB classifier predicts outcome in diffuse large B-cell lymphoma in the RICOVER-60 trial of the DSHNHL. Blood. 2010;116:4916–25.
Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood. 2013;121:2253–63.
Johnson NA, Slack GW, Savage KJ, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3452–9.
Perry AM, Alvarado-Bernal Y, Laurini JA, et al. MYC and BCL2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with rituximab. Br J Haematol. 2014;165:382–91.
Petrella T, Copie-Bergman C, Briere J, et al. BCL2 expression but not MYC and BCL2 coexpression predicts survival in elderly patients with diffuse large B-cell lymphoma independently of cell of origin in the phase 3 LNH03-6B trial. Ann Oncol. 2017;28(5):1042–9.
Valera A, Lopez-Guillermo A, Cardesa-Salzmann T, et al. MYC protein expression and genetic alterations have prognostic impact in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Haematologica. 2013;98:1554–62.
Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med. 1999;341:1520–9.
Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11.
Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120:2240–8.
Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105:13520–5.
Pasqualucci L, Dalla-Favera R. The genetic landscape of diffuse large B-cell lymphoma. Semin Hematol. 2015;52:67–76.
Horn H, Staiger AM, Ott G. New targeted therapies for malignant lymphoma based on molecular heterogeneity. Expert Rev Hematol. 2017;10:39–51.
Rovira J, Karube K, Valera A, et al. MYD88 L265P mutations, but no other variants, identify a subpopulation of DLBCL patients of activated B-cell origin, extranodal involvement, and poor outcome. Clin Cancer Res. 2016;22:2755–64.
Paul U, Richter J, Stuhlmann-Laiesz C, et al. Advanced patient age at diagnosis of diffuse large B-cell lymphoma is associated with molecular characteristics including ABC-subtype and high expression of MYC. Leuk Lymphoma. 2018;59(5):1213–21.
Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–82.
Choi WW, Weisenburger DD, Greiner TC, et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res. 2009;15:5494–502.
Salles G, Dd J, Xie W, et al. Prognostic significance of immunohistochemical biomarkers in diffuse large B-cell lymphoma: a study from the Lunenburg Lymphoma Biomarker Consortium. Blood. 2011;117:7070–8.
Coutinho R, Clear AJ, Owen A, et al. Poor concordance among nine immunohistochemistry classifiers of cell-of-origin for diffuse large B-cell lymphoma: implications for therapeutic strategies. Clin Cancer Res. 2013;19:6686–95.
Scott DW, Mottok A, Ennishi D, et al. Prognostic significance of diffuse large B-cell lymphoma cell of origin determined by digital gene expression in formalin-fixed paraffin-embedded tissue biopsies. J Clin Oncol. 2015;33:2848–56.
Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123:1214–7.
Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303.
Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7.
Lohr JG, Stojanov P, Lawrence MS, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–84.
Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1398–403.
Reddy A, Zhang J, Davis NS, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171:481–94 e15.
Meng FL, Du Z, Federation A, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159:1538–48.
Qian J, Wang Q, Dose M, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159:1524–37.
Green MR, Gentles AJ, Nair RV, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604–11.
Green MR, Kihira S, Liu CL, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015;112:E1116–25.
Okosun J, Bodor C, Wang J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81.
Pasqualucci L, Khiabanian H, Fangazio M, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–40.
Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95.
Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–77.
Zhang J, Vlasevska S, Wells VA, et al. The CREBBP acetyltransferase is a haploinsufficient tumor suppressor in B-cell lymphoma. Cancer Discov. 2017;7:322–37.
Bereshchenko OR, Gu W, Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002;32:606–13.
Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606.
Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–26.
Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–9.
Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95.
Ortega-Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21:1199–208.
Zhang J, Dominguez-Sola D, Hussein S, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21:1190–8.
Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med. 2014;20:343–52.
Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. 2012;247:172–83.
Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054–60.
Ranuncolo SM, Polo JM, Dierov J, et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol. 2007;8:705–14.
Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol Dis. 2008;41:95–9.
Basso K, Saito M, Sumazin P, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115:975–84.
Ci W, Polo JM, Cerchietti L, et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood. 2009;113:5536–48.
Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004;173:1158–65.
Iqbal J, Greiner TC, Patel K, et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia. 2007;21:2332–43.
Ye BH, Lista F, Lo Coco F, et al. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science. 1993;262:747–50.
Ye BH, Rao PH, Chaganti RS, Dalla-Favera R. Cloning of bcl-6, the locus involved in chromosome translocations affecting band 3q27 in B-cell lymphoma. Cancer Res. 1993;53:2732–5.
Baron BW, Nucifora G, McCabe N, Espinosa R 3rd, Le Beau MM, McKeithan TW. Identification of the gene associated with the recurring chromosomal translocations t(3;14)(q27;q32) and t(3;22)(q27;q11) in B-cell lymphomas. Proc Natl Acad Sci U S A. 1993;90:5262–6.
Kerckaert JP, Deweindt C, Tilly H, Quief S, Lecocq G, Bastard C. LAZ3, a novel zinc-finger encoding gene, is disrupted by recurring chromosome 3q27 translocations in human lymphomas. Nat Genet. 1993;5:66–70.
Butler MP, Iida S, Capello D, et al. Alternative translocation breakpoint cluster region 5′ to BCL-6 in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2002;62:4089–94.
Ye BH, Chaganti S, Chang CC, et al. Chromosomal translocations cause deregulated BCL6 expression by promoter substitution in B cell lymphoma. EMBO J. 1995;14:6209–17.
Pasqualucci L, Migliazza A, Fracchiolla N, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci U S A. 1998;95:11816–21.
Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 1998;280:1750–2.
Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003;101:2914–23.
Wang X, Li Z, Naganuma A, Ye BH. Negative autoregulation of BCL-6 is bypassed by genetic alterations in diffuse large B cell lymphomas. Proc Natl Acad Sci U S A. 2002;99:15018–23.
Saito M, Gao J, Basso K, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12:280–92.
Jiang Y, Ortega-Molina A, Geng H, et al. CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discov. 2017;7:38–53.
Ying CY, Dominguez-Sola D, Fabi M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013;14:1084–92.
Brescia P, Schneider C, Holmes AB, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell. 2018;34:453–65.
Duan S, Cermak L, Pagan JK, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481:90–3.
Cattoretti G, Pasqualucci L, Ballon G, et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell. 2005;7:445–55.
Schneider C, Kon N, Amadori L, et al. FBXO11 inactivation leads to abnormal germinal-center formation and lymphoproliferative disease. Blood. 2016;128:660–6.
Cerchietti LC, Ghetu AF, Zhu X, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell. 2010;17:400–11.
Cerchietti LC, Lopes EC, Yang SN, et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat Med. 2009;15:1369–76.
Fangazio M, Dominguez-Sola D, Tabbo F, et al. Genetic mechanisms of immune escape in diffuse large B cell lymphoma. Blood. 2014;124:1692.
Challa-Malladi M, Lieu YK, Califano O, et al. Combined genetic inactivation of beta2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20:728–40.
Kridel R, Chan FC, Mottok A, et al. Histological transformation and progression in follicular lymphoma: a clonal evolution study. PLoS Med. 2016;13:e1002197.
Moingeon P, Chang HC, Wallner BP, Stebbins C, Frey AZ, Reinherz EL. CD2-mediated adhesion facilitates T lymphocyte antigen recognition function. Nature. 1989;339:312–4.
Rimsza LM, Roberts RA, Miller TP, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–8.
Mottok A, Woolcock B, Chan FC, et al. Genomic alterations in CIITA are frequent in primary mediastinal large B cell lymphoma and are associated with diminished MHC class II expression. Cell Rep. 2015;13:1418–31.
Steidl C, Shah SP, Woolcock BW, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471:377–81.
Hashwah H, Schmid CA, Kasser S, et al. Inactivation of CREBBP expands the germinal center B cell compartment, down-regulates MHCII expression and promotes DLBCL growth. Proc Natl Acad Sci U S A. 2017;114:9701–6.
Dominguez-Sola D, Kung J, Wells VA, et al. Role of FOXO1 in germinal center development and lymphomagenesis. Blood. 2014;124:3536.
Sander S, Chu VT, Yasuda T, et al. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity. 2015;43:1075–86.
Trinh DL, Scott DW, Morin RD, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013;121:3666–74.
Morin RD, Assouline S, Alcaide M, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22:2290–300.
Hata AN, Engelman JA, Faber AC. The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015;5:475–87.
Saito M, Novak U, Piovan E, et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:11294–9.
Iqbal J, Sanger WG, Horsman DE, et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am J Pathol. 2004;165:159–66.
Schuetz JM, Johnson NA, Morin RD, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–90.
Barrans SL, Evans PA, O’Connor SJ, et al. The t(14;18) is associated with germinal center-derived diffuse large B-cell lymphoma and is a strong predictor of outcome. Clin Cancer Res. 2003;9:2133–9.
Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4:a014357.
Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51.
Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7.
Dalla-Favera R, Martinotti S, Gallo RC, Erikson J, Croce CM. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science. 1983;219:963–7.
Ladanyi M, Offit K, Jhanwar SC, Filippa DA, Chaganti RS. MYC rearrangement and translocations involving band 8q24 in diffuse large cell lymphomas. Blood. 1991;77:1057–63.
Karube K, Campo E. MYC alterations in diffuse large B-cell lymphomas. Semin Hematol. 2015;52:97–106.
Dominguez-Sola D, Victora GD, Ying CY, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol. 2012;13:1083–91.
Copie-Bergman C, Cuillière-Dartigues P, Baia M, et al. MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: a GELA/LYSA study. Blood. 2015;126:2466–74. https://doi.org/10.1182/blood-2015-05-647602.
Mahmoudi T, Verrijzer CP. Chromatin silencing and activation by Polycomb and trithorax group proteins. Oncogene. 2001;20:3055–66.
Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–96.
Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43.
Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26.
Beguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92.
Caganova M, Carrisi C, Varano G, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013;123:5009–22.
Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5.
Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980–5.
Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–9.
McCabe MT, Graves AP, Ganji G, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A. 2012;109:2989–94.
Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12.
Morschhauser F, Salles GA, McKay P, et al. Interim report from a phase 2 multicenter study of tazemetostat, an EZH2 inhibitor, in patients with relapsed or refractory B-cell non-Hodgkin lymphomas. Hematol Oncol. 2017;35(suppl S2):24–5. https://doi.org/10.1002/hon.2437_3.
Green JA, Cyster JG. S1PR2 links germinal center confinement and growth regulation. Immunol Rev. 2012;247:36–51.
Muppidi JR, Schmitz R, Green JA, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516(7530):254–8.
Cattoretti G, Mandelbaum J, Lee N, et al. Targeted disruption of the S1P2 sphingosine 1-phosphate receptor gene leads to diffuse large B-cell lymphoma formation. Cancer Res. 2009;69:8686–92.
Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev. 2011;244:169–87.
Boice M, Salloum D, Mourcin F, et al. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T Cells. Cell. 2016;167:405–18 e13.
Pfeifer M, Grau M, Lenze D, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:12420–5.
Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol. 2001;21:952–65.
Tai DJ, Su CC, Ma YL, Lee EH. SGK1 phosphorylation of IkappaB Kinase alpha and p300 Up-regulates NF-kappaB activity and increases N-Methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem. 2009;284:4073–89.
Mo JS, Ann EJ, Yoon JH, et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) controls Notch1 signaling by downregulation of protein stability through Fbw7 ubiquitin ligase. J Cell Sci. 2011;124:100–12.
Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92.
Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–9.
Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–59.
Young RM, Wu T, Schmitz R, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A. 2015;112:13447–54.
Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–6.
Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–90.
Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9.
Lam LT, Wright G, Davis RE, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111:3701–13.
Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60.
Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21.
Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–6.
Mandelbaum J, Bhagat G, Tang H, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18:568–79.
Pasqualucci L, Compagno M, Houldsworth J, et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med. 2006;203:311–7.
Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. 2006;107:4090–100.
Calado DP, Zhang B, Srinivasan L, et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18:580–9.
Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoetic and lymphoid tissues. Lyon: IARC Press; 2008.
Kluk MJ, Ho C, Yu H, et al. MYC immunohistochemistry to identify MYC-driven B-cell lymphomas in clinical practice. Am J Clin Pathol. 2016;145:166–79.
Pillai RK, Sathanoori M, van Oss SB, Swerdlow SH. Double-hit B-cell lymphomas with BCL6 and MYC translocations are aggressive, frequently extranodal lymphomas distinct from BCL2 double-hit B-cell lymphomas. Am J Surg Pathol. 2013;37:323–32.
Turakhia SK, Hill BT, Dufresne SD, Nakashima MO, Cotta CV. Aggressive B-cell lymphomas with translocations involving BCL6 and MYC have distinct clinical-pathologic characteristics. Am J Clin Pathol. 2014;142:339–46.
Delabie J, Vandenberghe E, Kennes C, et al. Histiocyte-rich B-cell lymphoma. A distinct clinicopathologic entity possibly related to lymphocyte predominant Hodgkin’s disease, paragranuloma subtype. Am J Surg Pathol. 1992;16:37–48.
Boudova L, Torlakovic E, Delabie J, et al. Nodular lymphocyte-predominant Hodgkin lymphoma with nodules resembling T-cell/histiocyte-rich B-cell lymphoma: differential diagnosis between nodular lymphocyte-predominant Hodgkin lymphoma and T-cell/histiocyte-rich B-cell lymphoma. Blood. 2003;102:3753–8.
Hartmann S, Doring C, Jakobus C, et al. Nodular lymphocyte predominant Hodgkin lymphoma and T cell/histiocyte rich large B cell lymphoma—endpoints of a spectrum of one disease? PLoS One. 2013;8:e78812.
Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25:3168–73.
Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413–20.
Montes-Moreno S, Gonzalez-Medina AR, Rodriguez-Pinilla SM, et al. Aggressive large B-cell lymphoma with plasma cell differentiation: immunohistochemical characterization of plasmablastic lymphoma and diffuse large B-cell lymphoma with partial plasmablastic phenotype. Haematologica. 2010;95:1342–9.
Valera A, Balague O, Colomo L, et al. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am J Surg Pathol. 2010;34:1686–94.
Laurent C, Do C, Gascoyne RD, et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma: a rare clinicopathologic entity with poor prognosis. J Clin Oncol. 2009;27:4211–6.
Gascoyne RD, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood. 2003;102:2568–73.
Pd P, Baens M, van Krieken H, et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood. 2003;102:2638–41.
Valera A, Colomo L, Martinez A, et al. ALK-positive large B-cell lymphomas express a terminal B-cell differentiation program and activated STAT3 but lack MYC rearrangements. Mod Pathol. 2013;26:1329–37.
Yuan J, Wright G, Rosenwald A, et al. Identification of primary mediastinal large B-cell lymphoma at nonmediastinal sites by gene expression profiling. Am J Surg Pathol. 2015;39:1322–30.
Paulli M, Strater J, Gianelli U, et al. Mediastinal B-cell lymphoma: A study of its histomorphologic spectrum based on 109 cases. Hum Pathol. 1999;30:178–87.
Moller P, Lammler B, Eberlein-Gonska M, et al. Primary mediastinal clear cell lymphoma of B-cell type. Virchows Arch A Pathol Anat Histopathol. 1986;409:79–92.
Copie-Bergman C, Plonquet A, Alonso MA, et al. MAL expression in lymphoid cells: further evidence for MAL as a distinct molecular marker of primary mediastinal large B-cell lymphomas. Mod Pathol. 2002;15:1172–80.
Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–62.
Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 2003;102:3871–9.
Gunawardana J, Chan FC, Telenius A, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014;46:329–35.
Steidl C, Gascoyne RD. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood. 2011;118:2659–69.
Twa DD, Chan FC, Ben-Neriah S, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5.
Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77.
Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21.
Joos S, Otano-Joos MI, Ziegler S, et al. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood. 1996;87:1571–8.
Melzner I, Bucur AJ, Bruderlein S, et al. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood. 2005;105:2535–42.
Rui L, Emre NC, Kruhlak MJ, et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell. 2010;18:590–605.
Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–22.
Guiter C, Dusanter-Fourt I, Copie-Bergman C, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood. 2004;104:543–9.
Weniger MA, Gesk S, Ehrlich S, et al. Gains of REL in primary mediastinal B-cell lymphoma coincide with nuclear accumulation of REL protein. Genes Chromosomes Cancer. 2007;46:406–15.
Schmitz R, Hansmann ML, Bohle V, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–9.
Wessendorf S, Barth TF, Viardot A, et al. Further delineation of chromosomal consensus regions in primary mediastinal B-cell lymphomas: an analysis of 37 tumor samples using high-resolution genomic profiling (array-CGH). Leukemia. 2007;21:2463–9.
Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–34.
Rigaud G, Moore PS, Taruscio D, et al. Alteration of chromosome arm 6p is characteristic of primary mediastinal B-cell lymphoma, as identified by genome-wide allelotyping. Genes Chromosomes Cancer. 2001;31:191–5.
Roberts RA, Wright G, Rosenwald AR, et al. Loss of major histocompatibility class II gene and protein expression in primary mediastinal large B-cell lymphoma is highly coordinated and related to poor patient survival. Blood. 2006;108:311–8.
Wilson WH, Pittaluga S, Nicolae A, et al. A prospective study of mediastinal gray-zone lymphoma. Blood. 2014;124:1563–9.
Harris NL. Shades of gray between large B-cell lymphomas and Hodgkin lymphomas: differential diagnosis and biological implications. Mod Pathol. 2013;26(Suppl 1):S57–70.
Vermeer MH, Geelen FA, van Haselen CW, et al. Primary cutaneous large B-cell lymphomas of the legs. A distinct type of cutaneous B-cell lymphoma with an intermediate prognosis. Dutch Cutaneous Lymphoma Working Group. Arch Dermatol. 1996;132:1304–8.
Pham-Ledard A, Prochazkova-Carlotti M, Andrique L, et al. Multiple genetic alterations in primary cutaneous large B-cell lymphoma, leg type support a common lymphomagenesis with activated B-cell-like diffuse large B-cell lymphoma. Mod Pathol. 2014;27:402–11.
Camilleri-Broet S, Criniere E, Broet P, et al. A uniform activated B-cell-like immunophenotype might explain the poor prognosis of primary central nervous system lymphomas: analysis of 83 cases. Blood. 2006;107:190–6.
Riemersma SA, Jordanova ES, Schop RF, et al. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood. 2000;96:3569–77.
Rubenstein JL, Treseler P, O’Brien JM. Pathology and genetics of primary central nervous system and intraocular lymphoma. Hematol Oncol Clin North Am. 2005;19:705–17, vii.
Vater I, Montesinos-Rongen M, Schlesner M, et al. The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia. 2015;29:677–85.
Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011;122:791–2.
Gonzalez-Aguilar A, Idbaih A, Boisselier B, et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res. 2012;18:5203–11.
Montesinos-Rongen M, Schafer E, Siebert R, Deckert M. Genes regulating the B cell receptor pathway are recurrently mutated in primary central nervous system lymphoma. Acta Neuropathol. 2012;124:905–6.
Montesinos-Rongen M, Van Roost D, Schaller C, Wiestler OD, Deckert M. Primary diffuse large B-cell lymphomas of the central nervous system are targeted by aberrant somatic hypermutation. Blood. 2004;103:1869–75.
Cady FM, O’Neill BP, Law ME, et al. Del(6)(q22) and BCL6 rearrangements in primary CNS lymphoma are indicators of an aggressive clinical course. J Clin Oncol. 2008;26:4814–9.
Schwindt H, Vater I, Kreuz M, et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia. 2009;23:1875–84.
Harada K, Nishizaki T, Kubota H, Harada K, Suzuki M, Sasaki K. Distinct primary central nervous system lymphoma defined by comparative genomic hybridization and laser scanning cytometry. Cancer Genet Cytogenet. 2001;125:147–50.
Courts C, Montesinos-Rongen M, Brunn A, et al. Recurrent inactivation of the PRDM1 gene in primary central nervous system lymphoma. J Neuropathol Exp Neurol. 2008;67:720–7.
Nakamura M, Kishi M, Sakaki T, et al. Novel tumor suppressor loci on 6q22-23 in primary central nervous system lymphomas. Cancer Res. 2003;63:737–41.
Nicolae A, Pittaluga S, Abdullah S, et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood. 2015;126:863–72.
Sansoni P, Vescovini R, Fagnoni F, et al. The immune system in extreme longevity. Exp Gerontol. 2008;43:61–5.
Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124–32.
Chen BJ, Chapuy B, Ouyang J, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19:3462–73.
Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–91.
Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood. 1996;88:645–56.
Said JW, Tasaka T, Takeuchi S, et al. Primary effusion lymphoma in women: report of two cases of Kaposi’s sarcoma herpes virus-associated effusion-based lymphoma in human immunodeficiency virus-negative women. Blood. 1996;88:3124–8.
Hermine O, Michel M, Buzyn-Veil A, Gessain A. Body-cavity-based lymphoma in an HIV-seronegative patient without Kaposi’s sarcoma-associated herpesvirus-like DNA sequences. N Engl J Med. 1996;334:272–3.
Chadburn A, Hyjek E, Mathew S, Cesarman E, Said J, Knowles DM. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am J Surg Pathol. 2004;28:1401–16.
Nakatsuka S, Yao M, Hoshida Y, Yamamoto S, Iuchi K, Aozasa K. Pyothorax-associated lymphoma: a review of 106 cases. J Clin Oncol. 2002;20:4255–60.
Narimatsu H, Ota Y, Kami M, et al. Clinicopathological features of pyothorax-associated lymphoma; a retrospective survey involving 98 patients. Ann Oncol. 2007;18:122–8.
Petitjean B, Jardin F, Joly B, et al. Pyothorax-associated lymphoma: a peculiar clinicopathologic entity derived from B cells at late stage of differentiation and with occasional aberrant dual B- and T-cell phenotype. Am J Surg Pathol. 2002;26:724–32.
Ando M, Sato Y, Takata K, et al. A20 (TNFAIP3) deletion in Epstein-Barr virus-associated lymphoproliferative disorders/lymphomas. PLoS One. 2013;8:e56741.
Yamato H, Ohshima K, Suzumiya J, Kikuchi M. Evidence for local immunosuppression and demonstration of c-myc amplification in pyothorax-associated lymphoma. Histopathology. 2001;39:163–71.
Guinee D, Jaffe E, Kingma D, et al. Pulmonary lymphomatoid granulomatosis. Evidence for a proliferation of Epstein-Barr virus infected B-lymphocytes with a prominent T-cell component and vasculitis. Am J Surg Pathol. 1994;18:753–64.
Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:e35–48.
Roschewski M, Wilson WH. Lymphomatoid granulomatosis. Cancer J. 2012;18:469–74.
Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95:1406–12.
Du MQ, Liu H, Diss TC, et al. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood. 2001;97:2130–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pasqualucci, L., Ott, G. (2019). Pathology and Molecular Pathogenesis of DLBCL and Related Entities. In: Lenz, G., Salles, G. (eds) Aggressive Lymphomas. Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-00362-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-00362-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00361-6
Online ISBN: 978-3-030-00362-3
eBook Packages: MedicineMedicine (R0)