
Abstract
Neuroendocrine tumors (NETs) of the digestive tract are usually slow-growing neoplasms carrying an overall favorable prognosis. Even if most of patients have metastatic or locally advanced tumors, surgery, from resection to transplantation, remains the only potential curative option for these patients and should always be considered. Nevertheless, because of the very few randomized controlled trials available, the optimal place of surgery within a global treatment strategy remains controversial.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Neuroendocrine tumors
- Surgical resection
- Primary tumor
- Liver metastases
- Debulking surgery
- Transplantation
- Surgical strategy
Introduction
Neuroendocrine tumors (NETs) of the digestive tract NET are usually slow-growing neoplasms carrying an overall favorable prognosis compared with their adenocarcinoma counterpart. These fascinating tumors are best treated with a multimodal management including oncologists, radiologists, pathologists, and surgeons. Despite an extensive scientific literature on these tumors, high-level evidence are sparse and the optimal treatment for patients with NET remains controversial [1] and should be tailored according to tumor’s characteristics, especially their site, grade, and relation to genetic syndromes. Surgical resection definitely has a cornerstone role within a global treatment strategy. Nevertheless, if there are some recent large randomized controlled trials for medical treatment for metastatic NET, there is, to date, no randomized trial concerning surgical management or comparing different treatment strategies. Overall, surgery, from resection to transplantation, remains the only potential curative option for these patients and should always be considered, even in the presence of synchronous metastases or locally advanced tumors [2–18].
We herein discuss the role of surgical resection in the treatment for NET of the digestive tract.
Preoperative Workup: The Surgeon’s Perspective
From the surgeon’s perspective, preoperative workup aims:
-
To exclude genetic syndromes such as multiple endocrine neoplasia type 1 (MEN 1), von Hippel–Lindau’s disease, or neurofibromatosis type 1. Indeed, these inherited diseases call for a specific preoperative workup, management, as well as postoperative follow-up.
-
To characterize the primary tumor, i.e., evaluate its local extension and relationship to adjacent organs, stage the regional (node) and distant (liver, bone, lung) disease extension and assess its secreting status.
-
To assess, when possible, the natural history of NET, which is highly variable, depending of tumor location, secretion, size, Tumor, Node, Metastasis (TNM) status, differentiation, and grade. It is important to remember that most of them are slowly growing neoplasms.
-
And, finally, to estimate the benefit–risk balance of surgery for a specific patient in order to tailor the management.
This preoperative workup is best multimodal including clinical examination, biological tests, and tumor markers [such as chromogranin A, urine 5-hydroxyindoleacetic acid (5-HIAA) or specific secreted hormones (gastrin, glucagon, somatostatine, etc.)], and various imaging modalities including computed tomography (CT), magnetic resonance imaging (MRI), and nuclear imaging. High-quality CT scan, including three phases spiral multidetector CT acquisitions with contiguously reconstructed sections, as well as dedicated liver MRI sequence are required, especially for liver metastasis detection [19]. In this setting, high sensitivity of diffusion-weighted MRI appears to be a useful tool [20]. Enterography (by CT or MRI) can be extremely helpful in detecting small intestinal lesions, especially when liver metastases are discovered but no primary tumor is found through other tests [21, 22]. In this specific condition, colonoscopy can be useful in searching for colorectal and terminal ileus neoplasms as well. Technique of nuclear imaging should be chosen according to tumor size, location, and characteristics: for all NET, 111In-somatostatin receptor scintigraphy; for pancreatic, NET 68GA-DOTATOC PET; for aggressive NET, 18F-FDG-PETs, and for small bowel NET, 18F-DOPA-PET, even though there is, at the present time, no clear consensus on the choice of tracer [23–26]. Endoscopic ultrasound is a very accurate tool to detect small pancreatic NET, especially sporadic insulinoma [27], and to stage gastroduodenal and rectal NET.
Histoprognostic Classification
Several classifications, recently updated, have been used for NET [28]. The World Health Organization (WHO) recently published an update on its classification for neuroendocrine tumor (Table 6.1) [29]. This histological classification divides neuroendocrine tumor into grades, according to mitotic rate and proliferative index (Ki-67 assessed by immunohistological staining). This classification is the one that now should be used in addition to the seventh TNM UICC stage [30]. If the value of the new 2010 WHO classification has been recently confirmed [31], it raises question especially regarding G3 tumors. Indeed, tumors with a mitotic count and/or Ki-67 above 20 % can have well- or poorly differentiated features, and consequently very different biological behavior, requiring different therapeutic strategy.
Aim of Surgery
Surgery aims either to provide/improve local control of disease burden, or to stop the natural course of the disease and, above all, definitively cure the patient. Treatment strategy and surgical indications are highly variable according to tumor site, genetic origin, local and regional extension, and biological behavior of the tumor.
Despite lack of high-level evidence such as randomized controlled trials, surgical strategies for NET are now better defined. Most of these recommendations are detailed in the European Neuroendocrine Tumor Society (ENETS) guidelines recently published and available online (www.enets.org). If some surgical indications are consensual, surgery should also sometimes be avoided.
Overall, surgical resection is rarely an “oncological emergency,” and a temporary “wait and see” policy is often acceptable to better assess the tumor natural history. If some surgical indications are consensual, surgery should also sometimes be avoided. Considering the rarity of such disease, surgery needs to be discussed on a case-by-case basis in a multidisciplinary neuroendocrine tumor board.
When Surgery is Required
First, it is noteworthy that surgery is the single most effective therapy for NET. Whatever abdominal procedure is planned, cholecystectomy should always be performed during primary resection of the tumor [32]. This is justified by the risk of gallstone-related complications or acute cholecystitis if patients are later treated with somatostatin analogs or liver arterial embolization [33, 34].
Some surgical indications are consensual, because of the clear benefit on long-term outcome.
Gastric Neuroendocrine Tumor
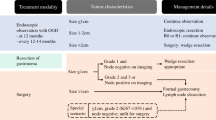
Gastric NETs represent less than 10 % of digestive NET. They can be due to chronically elevated gastrin (ECLomas) because of achlorydia from atrophic fundic gastritis (type 1, the most frequent) or to gastrin tumoral secretion (type 2, associated with Zollinger–Ellison Syndrome—ZES) (Table 6.2). Patients with type 1 or type 2 gastric NET above 1 cm with deep gastric parietal wall invasion and/or positive margins after endoscopic resection should undergo surgical resection [35–37]. It is important to recognize the low malignancy risk of type 1 lesions. These lesions can be treated either by local resection or antrectomy (by open or laparoscopic approach) [38], and this latter anatomical resection can, in theory, suppress the source of gastrin and decrease recurrence rate [35]. Overall, total gastrectomy should be avoided when possible, and its indications are limited to widely diffuse and large or malignant lesions. Whatever procedure performed, endoscopic surveillance is recommended thereafter [39].
Regarding rare sporadic primary gastric NET (type 3), they carry an overall dismal prognosis and should undergo curative-intent (R0) resection, by formal gastrectomy according to tumor location with regional lymphadenectomy, similar to gastric adenocarcinoma.
Duodenopancreatic Neuroendocrine Tumor
Insulinomas are the most common functioning endocrine neoplasms of the pancreas, frequently presenting with nonspecific symptoms due to hypoglycemia, such as weakness, confusion, headaches, sweating, tremors, palpitation, and visual disturbances. They are most of the time sporadic and benign, with less than 5 % of them being associated with MEN 1 and less than 10 % being malignant (i.e., with distant or nodal metastasis) [40, 41]. Accurate preoperative localization and characterization (including endoscopic ultrasonography (EUS) [42, 43], triphasic CT scan, or MRI [44]) are mandatory in order to tailor surgical treatment and to avoid blind distal pancreatectomy as it used to be recommended. Usually, allow adequate tumor localization and can avoid invasive exams such as intra-arterial calcium stimulation with hepatic venous sampling [44, 45]. Additionally, EUS can accurately assess relationship between the tumor and the main pancreatic duct. Surgery is the treatment of choice with an overall cure rate close to 100 % for benign lesions, if complete resection is achieved [46]. Procedures should be performed by an experienced team in pancreatic surgery and can be either performed laparoscopically or through open approach [41, 47]. After additional intraoperative localization of the tumor by palpation and ultrasonography, the lesion can be either enucleated or resected by standard pancreatectomy such as pancreaticoduodenectomy or distal pancreatectomy. Whenever possible, parenchyma-sparing resection [48] including central pancreatectomy or enucleation should be preferred because of a better long-term exocrine and endocrine function. Enucleation is best indicated for small benign lesions located in the head of the pancreas and far enough, i.e., 2–3 mm, from the main pancreatic duct [49]. Interestingly, preoperative EUS can accurately help the surgeon to choose the adequate surgical procedure assessing relationship between the tumor and the main pancreatic duct.
In patients with MEN 1, insulinoma can be multiple in about 10 % of patients and one should keep in mind of it in order to locate all lesions, pre and/or intraoperatively, thus avoiding blind pancreatectomy.
Treatment for other rare duodenopancreatic secreting lesions is in first line surgical resection [50], as for sporadic gastrinoma, glucagonoma, or vipoma. Most of these lesions are malignant, and standard pancreatectomy with formal lymphadenectomy is required. Regarding duodenal gastrinoma that even when malignant, often grows slowly, routine duodenotomy during surgical exploration usually allows accurate identification, and consequently, local resection can be performed. It is important to note that local lymphadenectomy should be systematically performed in order to decrease recurrence rate. Nodal extension of disease can occur in about 45 % of both duodenal and pancreatic gastrinomas [51].
Nonsecreting tumors began to be more incidentally diagnosed because of the widespread use of cross-sectional imaging, representing up to 75 % in recent surgical series [52, 53]. Their natural history is heterogeneous and difficult to assess during preoperative workup, and whether an incidental finding is associated with improved prognosis is still a matter of debate [52, 53]. Nevertheless, size being an important prognostic factor, surgical resection with regional lymphadenectomy is required for lesions above 2 cm [52]. Indeed, in this setting, the risk to develop metastatic disease during the follow-up is above 10 % [54].
Small Bowel Neuroendocrine Tumor
They represent from 25--40 % [55–57] of gastrointestinal NET. They mostly occur after 60 years of age, can be multiple in up to 40 % of cases, and they present with carcinoid syndrome in about a quarter of patients [55, 57]. Their prognosis is not as good as for other gastrointestinal tumors justifying an aggressive surgical management. However, the overall survival is around 60 % and can be up to 85 % in cases with curative resection [17, 58]. If possible, primary should be localized before surgery, using double balloon enteroscopy, capsule endoscopy, CT/MRI enterography as well as cross-sectional, and nuclear imaging such as 18F-DOPA-PET.
Even small and asymptomatic lesions need to undergo surgery because lesion size does not correlate with biological behavior, and they can be associated with node or liver metastases [15]. Small bowel lesions need to go through segmental resection with a formal wide lymphadenectomy including all gross metastatic nodes even when they are located around the superior mesenteric artery origin. Nevertheless, a special attention must be paid to avoid large resections leading to small bowel syndrome. Surgery for the primary tumor should always be considered even in the presence of metastatic disease, since the primary lesion can be responsible for local complications such as intussusception, small bowel obstruction, or ischemia. Additionally, also some lesions can present with important peritumoral fibrosis involving the mesentery root and the retroperitoneum, leading to occlusion, hydronephrosis, and chronic pain. Studies showed better results among patients who had at least their primary tumor resected along with nodal resection, with a better disease-free and overall survival [15, 17]. Since abdominal complications remain one of the major causes of death, we believe that in the setting of unresectable lesion, 90 % cytoreductive surgery can be considered, if a long enough small bowel can be conserved [56]. Surgery is at best performed after medical control of carcinoid syndrome, if present, by somatostatin analogs. Preoperatively, a special attention must be paid to carcinoid heart disease assessment in case of patient with carcinoid syndrome. Up to now, laparoscopic approach in this setting has been poorly studied, but we believe that open approach should be preferred in order to explore the full length of small bowel and to achieve a large lymphadenectomy.
Despite complete surgical resection of small bowel carcinoids, recurrence can occur in about 30–40 % of cases [16, 59]. Liver recurrence is best treated with resection in the case it is possible, which occurs usually in less than 20 %. Otherwise, other modalities of local treatment such as chemoembolization and pure embolization can be used as well as systemic therapy, from somatostatin analogs to chemotherapy.
Appendiceal Neuroendocrine Tumor
Appendiceal NET is currently diagnosed incidentally after appendectomy. They represent about a third of all gastrointestinal endocrine tumors and are the most benign of carcinoids. Although the overall 5-year survival is around 85 %, size is the most important prognostic factor. Tumors below 1 cm are cured by appendectomy alone, without need for any additional treatment. For tumors above 2 cm, a right colectomy is needed [60], due to the risk of lymph node extension. Between 1 and 2 cm, the risk-to-benefit ratio needs to be determined on a case-by-case basis.
Colonic and Rectal Neuroendocrine Tumor
These rare tumors are often incidentally diagnosed during colonoscopy and are nonfunctioning in most of the cases.
For well-differentiated colonic tumors, colonic resection with standard oncologic criteria should be performed in a similar way to adenocarcinoma, because of the poor 5-year prognosis of these tumors, between 40 and 70 %.
For well-differentiated rectal tumors below 1 cm, developed within the submucosal layer (T1) and without nodal involvement on preoperative EUS and MRI, local resection, either endoscopical or surgical is appropriate. For tumors above 2 cm, standard oncologic anterior resection is required. Between 1 and 2 cm, the risk-to-benefit ratio needs to be determined on a case-by-case basis.
Concerning endoscopic resection, endoscopic submucosal resection has replaced standard polypectomy as the preferred technique for tumors below 1 cm, and newer techniques such as submucosal resection with band ligation or endoscopic submucosal dissection are likely to be associated with less residual disease [61]. Submucosal resection with band ligation has the advantages of being easier to perform, demanding a shorter procedure time and having better negative margin rates; thus, it may be considered the treatment of choice for small rectal carcinoid tumors [62, 63].
Neuroendocrine Tumor Within a Genetic Background
NET can be associated with multiple endocrine neoplasia type 1, or less frequently with von Hippel–Lindau’s disease, von Recklinghausen’s disease, or tuberous sclerosis (Table 6.3) [64].
Regarding MEN 1, tumors such as nonfunctioning tumors above 2 cm [65], glucagonoma, vipoma, somatostatinoma, or GRFoma should undergo standard resection, since malignant NET is one of the main determinants of long-term survival in this disease. Insulinoma [66] represents a formal indication of resection because of its potentially harmful secretion (Table 6.4) [67].
In VHL disease, pancreatic NETs are usually nonfunctioning and have a better prognosis than sporadic nonfunctioning pancreatic NETS (metastatic disease in 10–20 vs. 60–90 %), and patients are usually younger about 30 years [68–70]. Tumors can also be multiple in about 20 % and locate throughout the pancreas. Usually, nonsurgical management is recommended for those patients, but criteria predicting poor prognosis have been described, being: tumor size ≥3 cm, tumor doubling time ≤500 days, and mutation in exon 3. In the case that more than two of these criteria are present, surgery is to be considered. Otherwise, surveillance with CT/MRI can be advocated [68, 69]. Patients with VHL should be screened for pancreatic lesions starting at 12 years of age and resecting PNETs should be considered if the patient is having an exploratory laparotomy for another manifestation of VHL [70].
NETs related to NF-1 are usually somatostatinomas frequently located around the papilla. These tumors metastasize in one-third to half of cases irrespective of primary tumor size. Although some experts advocate local excision for tumors smaller than 2 cm and surgical excision for tumors larger than 2 cm, as many as 70 % of surgical cases are associated with regional or liver metastasis, an aggressive surgical approach is best indicated in the form of pancreaticoduodenectomy [70–73].
Tuberous sclerosis-related pancreatic NETs are very rare (about 10 cases reported in the literature), and pancreatic NET in these patients can be both nonfunctional and functional. Because of the possible malignant potential of these tumors, surgical resection should be considered [51, 70, 74].
When Surgery Should be Avoided
Surgery should only be avoided after a complete clinical, biological, and imaging workup and discussion in a multidisciplinary neuroendocrine tumor board. Workup should include complete clinical examination, static and dynamic biological tests adapted to tumor origin and secretion. Imaging workup should at least include thoraco-abdominal CT scan, to assess tumor burden. When liver is involved, because of the frequent numerous small metastases [75], a liver MRI should be added routinely. If in doubt, nuclear imaging should be performed to better assess tumor biology (18F-FDG-PET CT) or tumor burden (111In-somatostatin receptor scintigraphy, 68GA-DOTATOC PET, or 18F-DOPA-PET).
Gastric Neuroendocrine Tumor
In patients with gastric NET associated with chronic atrophic gastritis (type 1) or with Zollinger–Ellison syndrome with multiple endocrine neoplasia type 1 (type 2), tumors under 1 cm can undergo endoscopic resection and follow-up with yearly endoscopic surveillance (every 6–12 months) with gastric biopsies is necessary [35–37], because of their very low risk of lymph node extent.
High-grade and Poorly Differentiated Tumors
These tumors are characterized by a mitotic count over 20 per high-power field and/or a Ki-67 index over 20 % and a poorly differentiated pathologic pattern. These tumors have a specific and very aggressive biological behavior totally different from G1 and G2 tumors. Platin-based chemotherapy is the first line treatment for these tumors. Surgical indications are very limited and can be considered for very localized tumors and/or tumors well controlled under chemotherapy.
When Surgery Should be Discussed
Metastastic Tumor to the Liver
The presence of metastases, most commonly located in the liver, is a major adverse prognostic factor [76–78] and affects up to 75 % of patients. Metastases are often bilobar, numerous, and synchronous. Despite a lack of high-level evidence, complete (R0) resection is the only hope for cure in these patients, and an aggressive surgical approach—including resection of metastatic liver disease—is widely accepted [2–18]. Nevertheless, intra- and extrahepatic recurrences are frequent, and a careful patient selection is needed. Patient selection is based on operative risk and general status, but mainly on tumor biological behavior. Surgery should be only discussed for patients with well-differentiated tumor (G1 and G2), at low risk of postoperative mortality and with no extrahepatic disease, when R0 resection is technically doable. On a technical point of view, two-step hepatectomy or intraoperative ablation enables complete resection with low mortality in patients with bilobar liver metastases, with a 5-year overall survival above 90 % and a 5-year disease-free survival around 50 % [79]. It is important to note that metastases are sometimes far more numerous than what is previously suggested by imaging [75].
Unresectability is defined as the technical impossibility to achieve R0 resection: metastases involving the right or left hepatic pedicle and abutting the contralateral pedicle, or involving or abutting the vena cava, or involving two hepatic veins and abutting the third one, or lesions that would leave <25 % of functional liver after resection. It is important to note that unresectability should be only defined par an experienced team including a hepatobiliary surgeon. Whether, in this setting, debulking surgery is justified remains highly controversial and should be limited to highly selected cases if at least 90 % of the tumor could be resected.
A recent meta-analysis demonstrated that surgery was superior to chemoembolization or other treatment modalities (non surgical) in the treatment for liver metastases of NET, with a significant longer survival [80].
Orthotopic liver transplantation has been proposed as an alternative option. Indeed, liver metastases are one of the main causes of dead, are usually confined to the liver for a long time, and can be responsible of debilitating symptoms.
Main criteria for transplantation are the absence of extrahepatic disease, a low Ki-67, and symptomatic disease refractory to previous therapies. In a French multicentric study, overall 5-year survival was 50 % and poor prognostic factors included upper-abdominal exenteration, primary tumor in duodenum or pancreas and hepatomegaly [81]. A review including 150 transplanted patients extracted from the UNOS database [82] demonstrated a long-term survival similar to that of patients with hepatocellular carcinoma, especially in patients with stabilized disease. Additionally, a recent European multicentric study of 213 patients showed an overall survival of up to 60 % after liver transplantation and suggested hepatomegaly, concurrent resections and age over 45 years as poor outcome predictors in the more recent cases [83]. Nguyen et al. also showed 5-year survival rate of near 60 % after 2002 and introduction of the Model for End-Stage Liver Disease (MELD) criteria [84]. Máthé et al. [85] found that age over 55 years and simultaneous pancreatic resection were poor prognostic factors in a study with 89 patients (Table 6.5).
In conclusion, in highly selected patients with unresectable liver metastasis, liver transplantation is a valid option that should be considered and discussed on a case-by-case basis. But still, optimal time for the transplant, if during stable or progressive disease, remains unclear [83].
Small Nonsecreting Pancreatic Neuroendocrine Tumor and Incidentaloma
Whether NET discovered as incidentalomas carry a better prognosis remains controversial [52, 53, 86]. If operative management is mandatory for symptomatic lesions, nonoperative management can be discussed on a case-by-case basis according to the individual risk-to-benefit evaluation. Since tumor size correlates with malignancy [52], for lesions below 2 cm and especially for those below 1 cm requiring aggressive surgery such as pancreaticoduodenectomy because of their anatomical localization, surveillance should be considered as an option. In this setting, preoperative assessment of grade or malignancy remains difficult. Fine needle aspiration cytology could estimate the Ki-67 value preoperatively, but its accuracy has been assessed in small series only [87, 88]. New imaging modalities, such as perfusion CT [89], diffusion-weighted MRI [90], or nuclear medicine assessment, including 18-FDG-PET [91], are currently in development and could help to identify tumors with a more aggressive behavior, which will need to be resected.
Appendiceal Neuroendocrine Tumor
Surgical indications should be discussed on a case-by-case basis for tumors between 1 and 2 cm, and patients are usually warranted a right hemicolectomy according to the presence of deep mesoappendiceal invasion (>3 mm), positive lymph node, positive margin, microscopic lymphatic, or venous invasion.
Neuroendocrine Tumor Within a Genetic Background
In patients with MEN 1, the situation is controversial regarding small, i.e., below 2 cm, often multiple nonfunctioning tumors [65]. A better understanding of their natural history shows most of the time-indolent growth, and regular follow-up can be advocated. Resection can be discussed when tumor is growing, especially over 2 cm.
If insulinoma should always be resected, the situation regarding gastrinoma is more complex. Treatment for gastrinomas in patients with MEN 1 has evolved with the development of highly efficient drugs such as proton pumps inhibitor to treat gastric acid hypersecretion. Gastrinomas are present in about half of patients with MEN 1, and their management has long been controversial [51]. If diagnosis is most of the time easily done, tumor localization is more challenging. Gastrinomas are often small, multiple, associated with metastatic lymph node and located in the duodenum in about 80 % of cases, but hard to accurately localized even with duodenotomy and intraoperative endoscopy. Local resection or duodenectomy is unlikely to cure patients, whom, however, have a long life expectancy. Pancreaticoduodenectomy has been advocated by some [92] with encouraging results, but is not recommended by most of the teams because of the postoperative mortality of this procedure and its long-term side effects for a slow-growing neoplasm. Overall, resection could be recommended for lesions above 2 cm because of a higher risk of aggressive tumor growth and liver metastatic disease [65]. It is important to take into account that pancreatic surgery should only be done after surgical treatment for hyperparathyroidism and should include local lymphadenectomy.
Conclusion and Perspectives
NET represents a wide range of neoplasms with various biological behaviors. Treatment strategy pursued for each patient needs to be individualized and discussed in a multidisciplinary tumor board specialized in neuroendocrine tumor. Surgery represents the only chance for cure and should always be discussed, even in the setting of advanced or metastatic disease.
References
Gurusamy KS, Ramamoorthy R, Sharma D et al (2009) Liver resection versus other treatments for neuroendocrine tumours in patients with resectable liver metastases. Cochrane Database Syst Rev CD007060
Cho CS, Labow DM, Tang L et al (2008) Histologic grade is correlated with outcome after resection of hepatic neuroendocrine neoplasms. Cancer 113:126–134
Sarmiento JM, Heywood G, Rubin J et al (2003) Surgical treatment of neuroendocrine metastases to the liver: a plea for resection to increase survival. J Am Coll Surg 197:29–37
McKenzie S, Lee W, Artinyan A et al (2010) Surgical resection and multidisciplinary care for primary and metastatic pancreatic islet cell carcinomas. J Gastrointest Surg 14:1796–1803
Elias D, Lasser P, Ducreux M et al (2003) Liver resection (and associated extrahepatic resections) for metastatic well-differentiated endocrine tumors: a 15-year single center prospective study. Surgery 133:375–382
Osborne DA, Zervos EE, Strosberg J et al (2006) Improved outcome with cytoreduction versus embolization for symptomatic hepatic metastases of carcinoid and neuroendocrine tumors. Ann Surg Oncol 13:572–581
Sarmiento JM, Que FG (2003) Hepatic surgery for metastases from neuroendocrine tumors. Surg Oncol Clin N Am 12:231–242
Fendrich V, Langer P, Celik I et al (2006) An aggressive surgical approach leads to long-term survival in patients with pancreatic endocrine tumors. Ann Surg 244:845–851; discussion 852–843
Schurr PG, Strate T, Rese K et al (2007) Aggressive surgery improves long-term survival in neuroendocrine pancreatic tumors: an institutional experience. Ann Surg 245:273–281
Que FG, Nagorney DM, Batts KP et al (1995) Hepatic resection for metastatic neuroendocrine carcinomas. Am J Surg 169:36–42; discussion 42–33
Hill JS, McPhee JT, McDade TP et al (2009) Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer 115:741–751
Chu QD, Hill HC, Douglass HO Jr et al (2002) Predictive factors associated with long-term survival in patients with neuroendocrine tumors of the pancreas. Ann Surg Oncol 9:855–862
Grfnbech JE, Søreide O, Bergan A (1992) The role of respective surgery in the treatment of the carcinoid syndrome. Scand J Gastroenterol 27:433–437
Han SL, Cheng J, Zhou HZ et al (2010) Surgically treated primary malignant tumor of small bowel: a clinical analysis. World J Gastroenterol 16:1527–1532
Hellman P, Lundström T, Öhrvall U, Eriksson B, Skogseid B, Öberg K et al (2002) Effect of surgery on the outcome of midgut carcinoid disease with lymph node and liver metastases. World J Surg 26:991–997
Landerholm K, Zar N, Andersson RE et al (2011) Survival and prognostic factors in patients with small bowel carcinoid tumour Brit J Surg 98:1617–1624
Nave H, Mossinger E, Feist H et al (2001) Surgery as primary treatment in patients with liver metastases from carcinoid tumors: a retrospective, unicentric study over 13 years. Surgery 129:170–175
Søreide O, Berstad T, Bakka A et al (1992) Surgical treatment as a principle in patients with advanced abdominal carcinoid tumors. Surgery 111:48–54
Dromain C, de Baere T, Lumbroso J et al (2005) Detection of liver metastases from endocrine tumors: a prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. J Clin Oncol 23:70–78
d’Assignies G, Fina P, Bruno O, Vullierme MP, Tubach F, Paradis V, Sauvanet A, Ruszniewski P, Vilgrain V (2013) High sensitivity of diffusion-weighted MRI for the detection of liver metastases from neuroendocrine tumors compared with T2-weighted and dynamic gadolinium-enhanced MRI, using surgical and histological findings as a standard of reference. Radiology Mar 2013
Hakim FA, Alexander JA, Huprich JE et al (2011) CT-enterography may identify small bowel tumors not detected by capsule endoscopy: eight years experience at Mayo Clinic Rochester. Dig Dis Sci 56(10):2914–2919
Masselli G, Polettini E, Casciani E et al (2009) Small-bowel neoplasms: prospective evaluation of MR enteroclysis. Radiology 251(3):743–750
Schiesser M, Veit-Haibach P, Muller MK et al (2010) Value of combined 6-[18F] fluorodihydroxyphenylalanine PET/CT for imaging of neuroendocrine tumours. Br J Surg 97:691–697
Abgral R, Leboulleux S, Deandreis D et al (2011) Performance of (18) fluorodeoxyglucose-positron emission tomography and somatostatin receptor scintigraphy for high Ki67 (>/=10%) well-differentiated endocrine carcinoma staging. J Clin Endocrinol Metab 96:665–671
Dudczak R, Traub-Weidinger T (2010) PET and PET/CT in endocrine tumours. Eur J Radiol 73:481–493
Masui T, Doi R, Ito T et al (2010) Diagnostic value of (18)F-fluorodeoxyglucose positron emission tomography for pancreatic neuroendocrine tumors with reference to the World Health Organization classification. Oncol Lett 1(1):155–159
McLean A (2004) Endoscopic ultrasound in the detection of pancreatic islet cell tumours. Cancer Imaging 4:84–91
Kloppel G, Rindi G, Perren A et al (2010) The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: a statement. Virchows Arch 456:595–597
Rindi G, Arnold R, Bosman F et al (2010) Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman F, Carneiro F, Hruban R et al (eds) WHO classification of tumours of the digestive system, international agency for research on cancer 2010
Soblin L, Gospodarowicz M, Wittekind C (2009) TNM classification of malignant tumours, 7th edn. Wiley, New York
Dolcetta-Capuzzo A, Villa V, Albarello L et al (2012) Gastroenteric neuroendocrine neoplasms classification: comparison of prognostic models. Cancer 119(1):36–44
Norlen O, Hessman O, Stalberg P et al (2010) Prophylactic cholecystectomy in midgut carcinoid patients. World J Surg 34:1361–1367
Wagnetz U, Jaskolka J, Yang P et al (2010) Acute ischemic cholecystitis after transarterial chemoembolization of hepatocellular carcinoma: incidence and clinical outcome. J Comput Assist Tomogr 34:348–353
Chua HK, Sondenaa K, Tsiotos GG et al (2004) Concurrent vs. staged colectomy and hepatectomy for primary colorectal cancer with synchronous hepatic metastases. Dis Colon Rectum 47:1310–1316
Borch K, Ahren B, Ahlman H et al (2005) Gastric carcinoids: biologic behavior and prognosis after differentiated treatment in relation to type. Ann Surg 242:64–73
Kulke MH (2003) Neuroendocrine tumours: clinical presentation and management of localized disease. Cancer Treat Rev 29(5):363–370
Akerström G, Hellman P (2007) Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab 21(1):87–109
Kim BS, Oh ST, Yook JH et al (2010) Typical carcinoids and neuroendocrine carcinomas of the stomach: differing clinical courses and prognoses. Am J Surg 200:328–333
Modlin IM, Lye KD, Kidd M (2003) Carcinoid tumors of the stomach. Surg Oncol 12:153–172
Service FJ, McMahon MM, O’Brien PC et al (1991) Functioning insulinoma–incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 66:711–719
Crippa S, Zerbi A, Boninsegna L et al (2012) Surgical management of insulinomas: short- and long-term outcomes after enucleations and pancreatic resections. Arch Surg 147:261–266
Nesje LB, Varhaug JE, Husebye ES et al (2002) Endoscopic ultrasonography for preoperative diagnosis and localization of insulinomas. Scand J Gastroenterol 37:732–737
McLean AM, Fairclough PD (2005) Endoscopic ultrasound in the localisation of pancreatic islet cell tumours. Best Pract Res Clin Endocrinol Metab 19:177–193
Daneshvar K, Grenacher L, Mehrabi A et al (2011) Preoperative tumor studies using MRI or CT in patients with clinically suspected insulinoma. Pancreatology 11:487–494
Zhang T, Mu Y, Qu L et al (2012) Accurate combined preoperative localization of insulinomas aid the choice for enucleation: a single institution experience over 25 years. Hepatogastroenterology 59:1282–1285
Nikfarjam M, Warshaw AL, Axelrod L et al (2008) Improved contemporary surgical management of insulinomas: a 25-year experience at the Massachusetts general hospital. Ann Surg 247:165–172
Hu M, Zhao G, Luo Y et al (2011) Laparoscopic versus open treatment for benign pancreatic insulinomas: an analysis of 89 cases. Surg Endosc 25:3831–3837
Crippa S, Boninsegna L, Partelli S et al (2010) Parenchyma-sparing resections for pancreatic neoplasms. J Hepatobiliary Pancreat Sci 17:782–787
Crippa S, Bassi C, Salvia R et al (2007) Enucleation of pancreatic neoplasms. Br J Surg 94:1254–1259
O’Toole D, Salazar R, Falconi M et al (2006) Rare functioning pancreatic endocrine tumors. Neuroendocrinology 84:189–195
Jensen RT et al (2008) Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 113(S7):1807–1843
Bettini R, Partelli S, Boninsegna L et al (2011) Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery 150:75–82
Haynes AB, Deshpande V, Ingkakul T et al (2011) Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors: short-term and long-term patient outcomes. Arch Surg 146:534–538
Kuvibidila S, Baliga BS, Gardner R et al (2005) Differential effects of hydroxyurea and zileuton on interleukin-13 secretion by activated murine spleen cells: implication on the expression of vascular cell adhesion molecule-1 and vasoocclusion in sickle cell anemia. Cytokine 30:213–218
Eriksson B, Kloppel G, Krenning E et al (2008) Consensus guidelines for the management of patients with digestive neuroendocrine tumors–well-differentiated jejunal-ileal tumor/carcinoma. Neuroendocrinology 87:8–19
Modlin IM, Lye KD, Kidd M (2003) A 5-decade analysis of 13,715 carcinoid tumors. Cancer 97:934–959
Bilimoria KY, Bentrem DJ, Wayne JD et al (2009) Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg 249:63–71
Le Roux C, Lombard-Bohas C, Delmas C et al (2011) Groupe d’étude des Tumeurs Endocrines (GTE). Relapse factors for ileal neuroendocrine tumours after curative surgery: a retrospective French multicentre study. Dig Liver Dis 43(10):828–833
Moertel CG, Weiland LH, Nagorney DM et al (1987) Carcinoid tumor of the appendix: treatment and prognosis. N Engl J Med 317:1699–1701
Mandair D, Caplin ME (2012) Colonic and rectal NET’s. Best Pract Res Clin Gastroenterol 26(6):775–789
Choi CW, Kang DH, Kim HW et al (2013) Comparison of endoscopic resection therapies for rectal carcinoid tumor: endoscopic submucosal dissection versus endoscopic mucosal resection using band ligation. J Clin Gastroenterol 47(5):432–436
Lee SH, Park SJ, Kim HH et al (2012) Endoscopic resection for rectal carcinoid tumors: comparison of polypectomy and endoscopic submucosal resection with band ligation. Clin Endosc 45(1):89–94
Chen et al (2012) Molecular pathology of pancreatic neuroendocrine tumors. J Gastrointest Oncol 3(3):182–188
Triponez F, Goudet P, Dosseh D et al (2006) Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg 30:654–662; discussion 663–654
O’Riordain DS, O’Brien T, van Heerden JA et al (1994) Surgical management of insulinoma associated with multiple endocrine neoplasia type I. World J Surg 18:488–493; discussion 493–484
de Wilde RF, Edil BH, Hruban RH et al (2012) Well-differentiated pancreatic neuroendocrine tumors: from genetics to therapy. Nat Rev Gastroenterol Hepatol 9(4):199–208
Tamura K, Nishimori I, Ito T et al (2010) Diagnosis and management of pancreatic neuroendocrine tumor in von Hippel–Lindau disease. World J Gastroenterol 16(36):4515–4518
Blansfield JA, Choyke L, Morita SY et al (2007) Clinical, genetic and radiographic analysis of 108 patients with von Hippel–Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 142:814–818; discussion 818.e1–818.e2
Landry CS, Waguespack SG, Perrier ND (2009) Surgical management of nonmultiple endocrine neoplasia endocrinopathies: state-of-the-art review. Surg Clin N Am 89:1069–1089
De Palma GD, Masone S, Siciliano S et al (2010) Endocrine carcinoma of the major papilla: report of two cases and review of the literature. Surg Oncol 19(4):235–242
Klein A, Clemens J, Cameron J (1989) Periampullary neoplasms in von Recklinghausen’s disease. Surgery 106(5):815–819
Makhlouf HR, Burke AP, Sobin LH (1999) Carcinoid tumors of the ampulla of Vater: a comparison with duodenal carcinoid tumors. Cancer 85(6):1241e9
Lodish MB, Stratakis CA (2010) Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract Res Clin Endocrinol Metab 24(3):439–449
Elias D, Lefevre JH, Duvillard P et al (2010) Hepatic metastases from neuroendocrine tumors with a “thin slice” pathological examination: they are many more than you think. Ann Surg 251:307–310
Panzuto F, Nasoni S, Falconi M et al (2005) Prognostic factors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer 12:1083–1092
Bilimoria KY, Talamonti MS, Tomlinson JS et al (2008) Prognostic score predicting survival after resection of pancreatic neuroendocrine tumors: analysis of 3851 patients. Ann Surg 247:490–500
Modlin IM, Sandor A (1997) An analysis of 8305 cases of carcinoid tumors. Cancer 79:813–829
Kianmanesh R, Sauvanet A, Hentic O et al (2008) Two-step surgery for synchronous bilobar liver metastases from digestive endocrine tumors: a safe approach for radical resection. Ann Surg 247:659–665
Bacchetti S, Bertozzi S, Londero AP et al (2013) Surgical treatment and survival in patients with liver metastases from neuroendocrine tumors: a meta-analysis of observational studies. Int J Hepatol 2013:235040
Le Treut YP, Gregoire E, Belghiti J et al (2008) Predictors of long-term survival after liver transplantation for metastatic endocrine tumors: an 85-case French multicentric report. Am J Transplant 8:1205–1213
Gedaly R, Daily MF, Davenport D et al (2011) Liver transplantation for the treatment of liver metastases from neuroendocrine tumors: an analysis of the UNOS database. Arch Surg 146:953–958
Le Treut YP et al (2013) Liver transplantation for neuroendocrine tumors in Europe-results and trends in patient selection: a 213-case European liver transplant registry study. Ann Surg 257(5):807–815
Nguyen NT, Harring TR, Goss JA, O’Mahony CA (2011) Neuroendocrine liver metastases and orthotopic liver transplantation: the US experience. Int J Hepatol 2011:742890
Lahat G, Ben Haim M, Nachmany I et al (2009) Pancreatic incidentalomas: high rate of potentially malignant tumors. J Am Coll Surg 209:313–319
Piani C, Franchi GM, Cappelletti C et al (2008) Cytological Ki-67 in pancreatic endocrine tumours: an opportunity for pre-operative grading. Endocr Relat Cancer 15:175–181
Figueiredo FA, Giovannini M, Monges G et al (2009) EUS-FNA predicts 5-year survival in pancreatic endocrine tumors. Gastrointest Endosc 70:907–914
d’Assignies G, Couvelard A, Bahrami S et al (2009) Pancreatic endocrine tumors: tumor blood flow assessed with perfusion CT reflects angiogenesis and correlates with prognostic factors. Radiology 250:407–416
Wang Y, Chen ZE, Yaghmai V et al (2011) Diffusion-weighted MR imaging in pancreatic endocrine tumors correlated with histopathologic characteristics. J Magn Reson Imaging 33:1071–1079
Garin E, Le Jeune F, Devillers A et al (2009) Predictive value of 18F-FDG PET and somatostatin receptor scintigraphy in patients with metastatic endocrine tumors. J Nucl Med 50:858–864
Tonelli F, Fratini G, Nesi G et al (2006) Pancreatectomy in multiple endocrine neoplasia type 1-related gastrinomas and pancreatic endocrine neoplasias. Ann Surg 244:61–70
Máthé Z, Tagkalos E, Paul A et al (2011) Liver transplantation for hepatic metastases of neuroendocrine pancreatic tumors: a survival-based analysis. Transplantation 91:575–582
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag France
About this chapter
Cite this chapter
Belghiti, J., Gaujoux, S., Figueiredo, M., Fuks, D., Sauvanet, A. (2014). Place of Surgical Resection in the Treatment Strategy for Gastrointestinal Neuroendocrine Tumors. In: Raymond, E., Faivre, S., Ruszniewski, P. (eds) Management of Neuroendocrine Tumors of the Pancreas and Digestive Tract. Springer, Paris. https://doi.org/10.1007/978-2-8178-0430-9_6
Download citation
DOI: https://doi.org/10.1007/978-2-8178-0430-9_6
Published:
Publisher Name: Springer, Paris
Print ISBN: 978-2-8178-0429-3
Online ISBN: 978-2-8178-0430-9
eBook Packages: MedicineMedicine (R0)