
Abstract
The pathogenesis of NAFLD and NASH is hastened by a disturbance of adipose secretion, including decreased production of adiponectin and increased production of TNF-α, characteristic of obesity. NASH suppressive effects of the leptin are diminished by its pro-oxidant and fibrogenic properties. Resistin’s involvement in NASH is documented in rodent models, which may not accurately reflect NAFLD in humans. Vaspin, visfatin, apelin, nesfatin, omentin and chemerin require further study in patients with NAFLD and NASH.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Key Points
-
The pathogenesis of NAFLD and NASH is hastened by a disturbance of adipokine production.
-
Decreased production of adiponectin and increased production of TNF-α, characteristic of obesity, directly contribute to NASH.
-
NASH suppressive effects of leptin are diminished by the widespread leptin resistance, while its pro-oxidant and fibrogenic properties augment the progression of NAFLD.
-
Resistin’s involvement in NASH is documented in rodent models, which may not be applicable to the human disease of NAFLD.
-
Effects of vaspin, visfatin, apelin, nesfatin, omentin, and chemerin require further study in patients with NAFLD and NASH.
The Spectrum of Nonalcoholic Fatty Liver Disease (NAFLD)
Nonalcoholic fatty liver disease (NAFLD) represents a spectrum of clinicopathologic conditions characterized by significant lipid deposition in the liver parenchyma of patients who do not consume excessive amounts of alcohol [1, 2]. At one end of the NAFLD spectrum is steatosis alone (“simple steatosis”), and at the other end are nonalcoholic steatohepatitis (NASH), NASH-related cirrhosis and hepatocellular carcinoma. NASH is characterized by hepatic steatosis and by evidence for hepatocyte ballooning degeneration, lobular inflammation, and occasionally, Mallory hyaline or sinusoidal fibrosis [3]. Differential diagnosis between NASH and steatosis alone is important because of their differential risk for progression [3].
The impact of NAFLD relates to its prevalence and potential for progression. Estimates of the prevalence of NAFLD are high and are expected to increase with the global epidemic of obesity. According to the National Health and Nutrition Examination Surveys (NHANES) conducted between 1988 and 2008, the prevalence of nonalcoholic fatty liver disease (NAFLD) increased from 5.51 % to 9.84–11.01 % [4]. From 1988 to 1994, NAFLD accounted for 46.8 % of Chronic Liver Disease (CLD) cases; from 1994 to 2004 its prevalence increased to 62.84 %, and then to 75.1 % from 2005 to 2008 [4]. The prevalence of NAFLD is highest in populations with preexisting metabolic conditions such as obesity and type II diabetes (up to 50–90 %) [2, 5–7]. The prevalence of histologically confirmed NASH is estimated as 1.2–4 %. However, in patients with risk factors such as morbid obesity, prevalence of NASH is higher and is estimated as 20–47 % [5, 8, 9].
The progression of NAFLD and its subtypes can be estimated from historical cohort studies, population-based studies and studies reporting sequential liver biopsies. Importantly, patients with steatosis alone rarely progress to cirrhosis, while 10–25 % of those with biopsy proven NASH can progress to cirrhosis [1, 3, 8]. In fact, most patients with cryptogenic cirrhosis seem to have had “burned-out NASH” [11]. NASH-related cirrhosis can cause hepatocellular carcinoma (HCC) [10, 11]. The major risk factors for progression or hepatic fibrosis in NASH are the presence of type 2 diabetes, obesity, metabolic syndrome, as well as elevated aminotransferase and histologic features of ballooning degeneration of hepatocytes and Mallory’s hyaline [8, 12]. Additionally, patients with NAFLD have higher mortality rates than the general population, in particular, due to associated cardiovascular risks [13].
Definitive diagnosis of NASH can be made only by liver biopsy evaluated by strict pathologic criteria. Although many noninvasive biomarkers for NAFLD assessment have been developed [14–17], liver biopsy remains “the imperfect gold standard” for diagnosing NASH and staging the extent of fibrosis. Nevertheless, liver biopsy is expensive, associated with small but definite medical risks and can be flawed by sampling errors [18, 19]. Studies of the involvement of adipokines in the pathogenesis of NAFLD and NASH hold particular promise for the development of noninvasive diagnostic biomarkers for these conditions.
Several treatment strategies are currently in use, but no therapy has proven to be effective for NASH [9, 20]. Treatment strategies include modification of the clinical conditions associated with NASH such as type II diabetes mellitus, hyperlipidemia, and obesity [2, 9]. Specific therapeutic interventions that have been evaluated so far include weight reduction, the use of ursodeoxycholic acid (UDCA), clofibrate, betaine, N-acetylcysteine, gemfibrozil, atorvastatin, thiazolidinediones, nitro-aspirin, pentoxifylline, and vitamin E [2, 9, 20, 21]. Although some of the results are encouraging, none of these interventions have been approved by the FDA for preventing NASH progression. Importantly, commonly used bariatric procedures have an impact on liver histology; a number of studies have shown post-surgical improvement of steatosis with a few studies suggesting an increase in portal fibrosis [22].
Pathogenesis of NASH
In the past few years, a substantial body of knowledge on the pathogenesis of NASH has been accumulated. However, the factors involved in its progression from steatohepatitis to fibrosis and cirrhosis remain to be elucidated. The pathogenesis of NASH appears to be multifactorial. To acquire an insight into the relationship between triglyceride accumulation within the hepatocyte and the necroinflammation and fibrosis, the following hypotheses are being investigated: the influences of abnormal lipid metabolism and the production of reactive oxygen species, increased hepatic lipid peroxidation, activated fibrocytes, and abnormal patterns of cytokine production leading to liver cell injury and fibrosis [23].
The “two-hit hypothesis” of NASH suggests that the first “hit” is the accumulation of excessive fat in the hepatic parenchyma [2, 8, 24]. This first step has been linked to insulin resistance (IR), which is consistently observed in patients with NASH [23]. The evidence for IR in NAFLD comes from both animal and human studies. Animal models of NASH show insulin resistance [25] and the use of the insulin-sensitizing agent, metformin reverses hepatic steatosis [23]. Clinical features of the metabolic syndrome (obesity, diabetes mellitus, or hypertriglyceridemia) are commonly observed in NAFLD [1, 8]. Additionally, polycystic ovary syndrome (PCOS), an insulin resistance-associated condition, was identified as a risk factor for developing NAFLD [26]. Growing evidence suggests that patients with more “severe” forms of insulin resistance are at an even greater risk for progressive liver disease [8]. Importantly, insulin resistance in the adipose tissue plays a larger role in the severity of NAFLD as compared to liver or muscle IR [27].
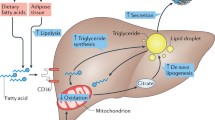
The second “hit” in the development of NASH could be multifactorial, involving fatty acid beta oxidation, oxidative stress, gut-derived endotoxins, pro-inflammatory cytokines, and adipokines. In fact, due to the multifactorial nature of the second “hit,” the “two-hit hypothesis” of NASH now is being expanded to “multi-hit model” (Fig. 17.1). Oxidative stress implies an imbalance between pro-oxidant and antioxidant processes. In the setting of NAFLD, oxidative stress can result from the induction of microsomal CYP2E1, H2O2 release from peroxisomal β-oxidation of fatty acids, cytokines released from activated inflammatory cells, or other unknown factors [23, 28]. This oxidative stress could potentially lead to peroxidation of membrane lipids resulting in the production of malondialdehyde and 4-hydroxynonenol, which in turn can induce the production of proinflammatory cytokines, stellate cell activation, and fibrogenesis, as well as direct hepatocyte damage [8, 23, 29]. The increase in oxidative stress observed in NAFLD hepatocytes may be linked to mitochondrial dysfunction, as mitochondria are a major potential source of reactive oxygen species (ROS) in living cells [29].

The “multi-hit” hypothesis of the pathogenesis of NASH
As noted, both the first and second hits in NASH may involve changes in circulating levels of various pro- and anti-inflammatory cytokines, including adipokines (Fig. 17.2). Although a role for cytokines such as TNF-α and IL-6 has been suggested for quite some time [30, 31], recent works center on the particular roles for adipokines in the pathogenesis of NASH [31].

Molecules involved in complex interplay between cells composing the adipose tissue and the liver
Adipose Tissue, Adipokines and NAFLD
White adipose tissue produces and releases a variety of proinflammatory and anti-inflammatory factors, including adipokines (leptin, adiponectin, resistin, apelin, omentin, vaspin, visfatin, chemerin, nesfatin, and others), cytokines (such as TNF-α, IL-6), and chemokines (such as monocyte chemoattractant protein 1, and others). In addition to adipocytes, white adipose tissue contains several other cell types including macrophages and monocytes. It is likely that macrophages are attracted into the adipose tissue by macrophage migration inhibitory factor (MIF), which is released by both preadipocytes and mature adipocytes in the amounts proportional to body mass index [32]. By producing several cytokines and adipokines, both adipocytes and other pro-inflammatory cells found within adipose tissue contribute to the increased systemic inflammation associated with obesity [33]. The exact contribution of each component of white adipose tissue in the “pro-inflammatory” state of obesity is not entirely clear. Over 90 % of the adipokines released from adipose tissue, except for adiponectin and leptin, originate from nonfat cells embedded in the extracellular matrix [34]. Additionally, most adipokines are also produced elsewhere in the body [35]. These data suggest that serum adipokine measurements in obese patients most likely reflect secretions by various cells, including, but not limited to, adipocytes.
Biologically active white adipose tissue plays an important role in metabolic syndrome and associated NAFLD. Although the role of proinflammatory cytokines (TNF-α, IL-6) in the pathogenesis of NASH is well documented, the role of adipokines in the pathogenesis of NAFLD is a recent observation. The exact source of adipokines in patients with NAFLD (e.g., adipocytes, macrophages, monocytes) remains unclear, because serum levels of adipokine probably reflect a “net” effect of the secretion from various cells in the body. Determining the exact role of adipokines in the pathogenesis of NAFLD is complicated by the interaction of adipokines with insulin resistance and obesity. Most studies have not controlled for these important confounders. This issue is critical for future research focused on elucidating pathways involved in the pathogenesis of NAFLD, NASH and its progression.
Adipokines in Experimental Models of NAFLD
Common experimental models of NAFLD include mice or rats that are fed high fat or high carbohydrate or methionine/choline deficient diets, or mice that exhibit one or another genetic deficiency, for example, leptin-deficient mice and rats. Additionally, many other animal models spontaneously develop steatosis, and some progress to steatohepatitis and cirrhosis.
Leptin-deficient ob/ob mice are obese, insulin resistant, hyperglycemic, and hyperlipidemic. Similar animal models such as db/db mice and fa/fa rats have homozygous loss-of-function due to a mutation in a leptin receptor encoding gene. All phenotypes (fa/fa rats, ob/ob and db/db mice) are essentially similar, except for hyperleptinemia, which is present in fa/fa and db/db animals. It is noteworthy that NAFLD occurs in both leptin-deficient and in hyperleptinemic animals with impaired leptin signaling. In leptin deficient animals, leptin restoration leads to NAFLD reversal [36]. Another important consideration in NAFLD development is the contribution of central versus peripheral leptin activity. To address this question, animals with tissue-specific leptin receptor knockouts were created. In contrast to the db/db and the neuron-specific knockout (KO) mice, the livers of hepatocyte-specific leptin receptor knockout animals were normal [37]. Unfortunately, the experimental approach does not exclude the possibility that fatty liver development is influenced by autocrinous leptin signaling in adipose tissue itself, or by endocrinous action of the leptin on cytokine releasing cells. So, the question of peripheral versus central involvement of leptin signaling in NAFLD development remains unanswered.
TNF-α serum levels increase in all animal models of NAFLD. However, the source of TNF-α (i.e., adipose tissue versus monocytes and macrophages) is not entirely clear. In NAFLD, TNF-α level may be influenced by the lipotoxic effects of excess fat. In fact, hepatic steatosis seems to increase oxidative stress and activation of the NF-κB pathway, leading in turn to an increase in TNF-α production. Once initiated, this vicious cycle of NF-κB/TNF-α becomes self-perpetuating, contributing to a “pro-inflammatory” state in patients with NASH [25]. Evidence supporting this concept is provided by anti-TNF-α treatment in ob/ob mice, which can improve liver histology, reduce hepatic total fatty acid content, and decrease serum alanine aminotransferase (ALT) levels. In addition, fatty acid beta-oxidation and uncoupling protein (UCP)-2 expression decreases with anti-TNF-α treatment in ob/ob mice, both of which suggest an improvement in oxidative stress and probably insulin resistance [38]. Similarly, metformin, which is known to inhibit hepatic expression of TNF-α and TNF-inducible factors, also seems to improve fatty liver disease [39]. This has led to the hypothesis that chronic exposure to TNF-α prompts accumulation of inflammatory cells in the liver parenchyma, thereby exposing hepatocytes to other damaging factors released by activated mononuclear cells.
In animal models, adiponectin decreases insulin resistance by decreasing triglyceride content in the muscle and liver tissue and by increasing the ability of sub-physiological levels of insulin to suppress glucose production by inhibiting hepatic gluconeogenic enzymes [40, 41]. This results from increased expression of molecules involved in both fatty-acid metabolism and energy dissipation in muscle tissue. It has been shown that mice chronically fed high-fat, ethanol containing food, have lower levels of adiponectin. Experimental replenishment of adiponectin dramatically alleviates hepatomegaly and steatosis in these animals, and attenuates inflammation by suppressing the hepatic production of TNF-α [42]. Similar effects are achieved in nonalcoholic ob/ob mice [42]. Experiments with adiponectin-knockout mice conducted by Kamada and coworkers demonstrated that adiponectin is capable of attenuating liver fibrosis that develops after carbon tetrachloride administration [43]. Adiponectin also alleviates lipopolysaccharide (LPS)-induced liver injury [44]. Suppressing local TNF-α production and signaling provides a protective effect in all cases of adiponectin-related decreases in the extent of acute liver injury.
Studies of the effects of resistin in animal models of NAFLD are substantially less relevant to human fatty liver disease due to functional differences between resistin encoding genes in animal models and humans. The expression of resistin mRNA in human adipose tissue and its serum content are substantially lower (1/250) than that in rodents. Moreover, the resistin-α encoding gene is absent in humans [45].
Adipokines in Patients with NAFLD
The following sections review the current clinical data on specific adipokines in NAFLD: adiponectin, resistin, leptin, and TNF-alpha, as well as several other soluble molecules.
Adiponectin
Adiponectin is the most frequently studied adipokine associated with NASH. Many authors have suggested that hypoadiponectinemia may contribute to the development of NASH in obese individuals. This hypothesis is supported by a study of 257 healthy individuals, which reports a negative correlation between adiponectin serum levels and two markers of liver injury, alanine aminotransferase (ALT) and gamma-glutamyltranspeptidase (GGT) before and after adjustment for sex, age, body mass index (BMI) and insulin resistance [46]. Pagano and coworkers showed that plasma adiponectin levels were significantly lower in NAFLD patients than in the matched controls (5.93 ± 0.45 versus 15.67 ± 1.60 ng/ml). However, there was no difference in adiponectin levels between patients with simple steatosis and those with NASH (6.16 ± 0.78 versus 5.69 ± 0.49 ng/ml) [47]. The authors reported an inverse correlation between adiponectin and homeostatic model assessment (HOMA) of insulin resistance (P = 0.008), but no correlation between adiponectin and serum transaminases or lipid values [47]. A second study confirmed the protective effect of adiponectin against the development of radiologically proven steatosis [48]. Finally, a study of 113 obese children confirmed the protective role of adiponectin against NAFLD in pediatric populations [49].
Despite increasing evidence supporting the association between hypoadiponectinemia and steatosis, the role of adiponectin in distinguishing NASH from simple steatosis remains controversial. A study by Bugianesi and colleagues related decreased levels of circulating adiponectin in NAFLD to hepatic insulin sensitivity and to the amount of hepatic fat content, but not to liver disease severity as measured by necroinflammation and fibrosis [50]. On the other hand, Hui and coworkers found that low serum adiponectin is associated with increased grades of hepatic necroinflammation independent of insulin resistance [51]. Our own observations suggest that higher serum adiponectin concentrations also protect against the progressive form of fatty liver disease, NASH (P = 0.024) [31]. Finally, a report by Musso and colleagues is of special interest [52]. This study of 25 nonobese, nondiabetic patients with biopsy-proven NASH showed that adiponectin was protective against histologically proven NASH (NASH 5,476 ± 344 versus matched controls 11,548 ± 836 ng/ml; P = 0.00001) [52]. Adiponectin was negatively correlated with the presence of necroinflammation (OR = 5.0; P = 0.009), and fibrosis (OR = 8.0; P = 0.003). On logistic regression controlling for all important confounders, hypoadiponectinemia remained an independent predictor of severe necroinflammation and of stage 3 fibrosis [51, 52]. In the same cohort of patients, the magnitude of postprandial lipemia was significantly higher in NASH than in controls and was related to fasting adiponectin (β = −0.78; P = 0.00003) [53]. Controls showed a significant increase in serum adiponectin in response to the fat load, whereas patients with NASH showed a slight decrease. Postprandial free fatty acids response correlated inversely with adiponectin response in both groups and independently predicted the severity of liver steatosis in NASH (β = 0.51; P = 0.031). In other words, the dynamic adiponectin response to an oral fat load is strikingly different in healthy subjects and patients with NASH and is related to the postprandial FFA response. These findings suggest that hypoadiponectinemia precedes the overt manifestation of diabetes and is linked to impairment of the postprandial lipid metabolism.
Circulating levels of adiponectin has a strong genetic component demonstrated by an additive genetic heritability of 46 % [54]. The regulation of serum adiponectin levels has been linked to regions on chromosome 5p (logarithm of odds [LOD] = 4.06) and 14q (LOD = 3.2) in a predominantly northern European population [54] and to chromosome 9p (LOD = 3.0) in Pima Indians [55]. In addition, four haplotype-tagging single-nucleotide polymorphisms (SNPs) have been identified at the adiponectin-encoding APM1 locus itself. One of them, +276G > T, is associated with serum adiponectin levels collected from nondiabetic, Caucasian individuals (P = 0.032). Individuals homozygous for the +276T allele have higher adiponectin levels than other subjects [56]. Individuals with an allelic combination of +45T and +276G (“TG” haplotype) have higher body weight (P = 0.03), waist circumference (P = 0.004), systolic (P = 0.01) and diastolic blood pressure (P = 0.003), total to HDL cholesterol ratio (P = 0.01), and insulin resistance as measured by HOMA scores (P = 0.003) as well as fasting serum glucose (P = 0.02) and serum insulin (P = 0.005) levels [57]. Subsequent studies of adiponectin polymorphisms in obese and diabetic subjects suggest similar trends. In NAFLD, the homozygous “GG” genotypes at positions −11377 and +45 were significantly more prevalent than in matched controls [58]. Moreover, the presence of the “G” allele at these positions was associated with a necroinflammatory grade [58].
It is important to note that adiponectin is secreted into the circulation as three oligomeric isoforms, including (low molecular weight, LMW), hexamer (middle molecular weight, MMW) and the high molecular weight (HMW) oligomeric complex. Obesity-related metabolic complications, including NAFLD, are especially tightly associated with lower concentrations of HMW adiponectin [59]. This observation is not incidental as HMW oligomer mediates the insulin-sensitizing effects of adiponectin on suppression of hepatic gluconeogenesis. In one of the studies, after adjustment for gender, age, and total body fat, the content of the fat in the liver and within the muscles is associated only with HMW adiponectin (r = −0.35, P = 0.012), but not with total-, MMW-, or LMW adiponectin [60]. Levels of HMW adiponectin negatively correlate with the expression of nuclear receptor peroxisome proliferator-activated receptors-γ (PPAR-γ) expression in the liver, a prosteatotic factor in fatty liver disease [61].
In addition to the studies focusing on serum adiponectin levels, some recent publications looked into the role of the adipokine receptors. The results are contradictory. Kaser and colleagues showed that immunostaining of the adiponectin receptor adipoRII as well as its mRNA expression level were significantly reduced in liver biopsies of patients with NASH compared to patients with simple steatosis, but found no differences in adipoRI mRNA expression between the two groups [62]. Similar finding were reported by Shimizu and colleagues [63]. On the other hand, Vuppalanci and colleagues reported an increase in the mRNA expression levels of adiponectin receptor AdipoRII in liver specimens of patients with NASH compared to normal liver tissue [64]. These investigators reported several other contradictory findings. The Kaser group reported adiponectin expression both in endothelial cells of portal vessels and in hepatic sinusoids [62], but Vuppalanci found no adiponectin mRNA expression in any of the liver samples studied [64]. The recent work of Carazo and co-authors corroborated findings of Vuppalanci in a larger group of morbidly obese patients (N = 60) showing that NAFLD progression is associated with increase in the hepatic expression of both adiponectin receptors [65]. Two other studies had not registered any difference in expression of adipoRI and adipoRII in NASH and steatosis only groups [66, 67].
In connection to NAFLD, the allelic states of the human adiponectin receptor encoding gene ADIPOR1 and ADIPOR2 have been studied as well. A longitudinal study showed that a common haplotype of −8503A and −1927C ADIPOR1 alleles were associated with higher liver fat at follow-up as determined by proton magnetic resonance spectroscopy (P = 0.02) compared with the haplotype consisting of −8503G and −1927T alleles. These observations were independent of basal measurements, sex, and baseline versus follow-up percentage of body fat [68]. In Northern European populations, the polymorphism of ADIPOR2 (rs767870) was significantly associated with liver fat content measured with (1)H-MRS after adjusting for age, gender, and BMI and related to serum gamma glutamyltransferase concentrations [69]. In subjects with diabetes Type II, the at-risk alleles for the common −64241T/G and +33447C/T SNPs in ADIPOR2 were associated with increased serum ALT and AST [70]. Both findings were confirmed by replication studies in larger cohorts.
Further studies of allelic states of adiponectin and its receptor in association with NASH and NAFLD are clearly warranted.
Resistin
Resistin has been implicated in the pathogenesis of obesity-mediated insulin resistance and Type II diabetes mellitus. In addition, resistin also appears to stimulate macrophage secretion of TNF-α and IL-12 to the same extent as lipopolysaccharides. Most likely, its proinflammatory action is an induction of the nuclear translocation of NF-κB transcription factor [71]. Both pro-inflammatory properties and association with insulin resistance suggest that resistin may play an important role in the pathogenesis of NASH. One study showed that plasma resistin concentrations in serum positively correlate with hepatic fat content (r = 0.66, P < 0.001) [72]. In pediatric NAFLD, hepatic progenitor cells express higher levels of resistin, and this increase is proportional to the degree of fibrosis (r = 0.432, P < 0.05) [73]. Another study of pediatric NAFLD indicated that serum resistin levels are lower in children with advanced liver steatosis (grade 3, N = 10) compared to patients with mild steatosis (grade 1–2, N = 23) [74].
However, a number of studies of adult NAFLD cohorts failed to see any NASH-related differences in plasma or serum resistin concentrations [31, 52]. The role of resistin in the pathogenesis of NAFLD requires further clarification.
Leptin
Leptin is release by adipocytes into circulation that transfers it to the central nervous system where it regulates food intake. However, leptin receptors encoded by the LEPR gene are expressed both centrally and peripherally, as they were found in many peripheral tissues. Human livers express high mRNA levels for both short and long isoforms of the leptin receptor Moreover, there is a trend toward lower levels of mRNA encoding the long form of the leptin receptor in hepatic tissue from patients with NASH as compared to those with steatosis only [75]. In human blood, the bioavailability for leptin is modulated by the so-called soluble leptin receptor (SLR), a product of ADAM10-dependent shedding of the extracellular domain of the leptin receptor [76]. This cleavage is enhanced by treatment with lipotoxic agents and apoptosis. On the other hand, when leptin levels and/or ER stress are high, the levels of SLR in the serum are reduced, which might reflect a decrease in the membrane expression of leptin receptors.
As the SLR seem to directly block leptin action, the profiling of the serum leptin levels might be difficult to interpret. So far, only two NAFLD-related studies profiled both leptin and SLR. Huang and co-authors found that enhanced release of leptin is accompanied by a decrease in SLR concentration, which suggests higher resistance of peripheral tissues towards the action of leptin [77]. Medici and co-authors found that the extent of steatotic changes in the liver could be predicted by the Free Leptin Index (FLI) calculated as the ratio of leptin to SLR, while levels of SLR specifically, were correlated to the stage of fibrosis [78].
Leptin contributes to the development of NASH in many ways. First, abnormalities in serum leptin or its receptor promote insulin resistance. Second, leptin-dependent changes in insulin signaling increase fatty acid influx into hepatocytes, promoting lipotoxicity [79]. In the later stages of pathogenesis of NASH, leptin enhances the systemic low-grade inflammation, thus providing the “second hit” that advances simple steatosis to steatohepatitis. In fact, levels of leptin are independently associated with that of C-reactive protein (CRP) after adjustment for age, gender, BMI, waist-to-hip ratio, smoking and alcohol consumption (F = 12.39, P = 0.0007) [80]. Additionally, leptin acts as a profibrogenic cytokine in several liver diseases. As a profibrogenic agent, leptin influence both endothelial and Kupffer cells [81].
It is important to remember that NAFLD is commonly seen in conjunction with lipodystrophy, a condition characterized by the partial or complete absence of adipose tissue and hypoleptinemia. In patients with congenital lipodystrophy, leptin administration improves insulin resistance and corrects liver steatosis and hepatocellular ballooning injury [82, 83]. It also corrects elevation of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels [84]. Interestingly, the degree of liver fibrosis in NASH patients treated with recombinant leptin remains unchanged [83].
Genetics-wise, polymorphisms in both leptin and leptin receptor encoding genes were reported to have an effect on NAFLD-related phenotypes. For intronic SNP rs6700896 located within an intron of the LEPR gene, significantly different allelic frequencies were reported between NAFLD with or without T2DM and non-steatotic controls [85]. Frequencies of mutant LepR polymorphism were also significantly associated with IR increments [85]. Similar finding were also reported by Lu and co-authors [86]. The study of Aller et al. showed a substantial influence for the Asparagine-656 variant on leptin receptors (Lys656Asn polymorphism within LEPR gene) with obesity-related, rather than NAFLD-related parameters [87]. Larger, better controlled studies of the polymorphisms of leptin and leptin receptor-encoding genes in NAFLD are warranted.
TNF-alpha
TNF-α is a proinflammatory cytokine that orchestrates the synthesis, secretion, and activity of other proinflammatory molecules. Macrophages are a major source of TNF-α in humans. TNF-α is also produced by many other tissues in response to various pathological processes such as infection, ischemia, and trauma. Adipocytic production of TNF-α is very low [88]. Nevertheless, an overall increase in the adipose mass usually leads to substantial, cumulative production of this cytokine that is achieved by stromavascular cells and macrophages infiltrating expanded adipose. Increased amounts of TNF-α released by excessive adipose tissue may contribute to the development of obesity-related NAFLD. Several studies have demonstrated that serum levels of TNF-α are significantly higher in patients with NASH than in healthy controls (see [30, 31] for review). A comprehensive study of TNF-α in steatohepatitis shows remarkable increases in the expression levels of mRNA encoding TNF-α in both hepatic and adipose tissues in NASH patients as compared to obese controls [89]. Similar mRNA increases were observed for the p55 receptor, but not for the p75 receptor of TNF-α. Furthermore, the degree of hepatic fibrosis correlated with TNF-α expression levels in adipose tissue and levels of mRNAs encoding p55 in the hepatic tissue [89]. Recent meta-analysis suggests that TNF-α gene promoter polymorphism at position −238 but not −308 might be a risk factor for NAFLD (GA/AA versus GG [odds ratio = 2.06, 95 % confidence interval = 1.58–2.69, P < 0.00001]) [90].
Additional indirect evidence of TNF-α involvement in NASH comes from a 12-month trial of pentoxifylline, a methylxanthine that can suppress both TNF-α at the level of mRNA accumulation and the bioactivity of its secreted form, possibly through the generation of intracellular cAMP. A study by Adams showed that both alanine and aspartate aminotransferase levels are significantly lower after 12 months of therapy compared to baseline, indicating a significant improvement in treated patients [91]. Another study examined 18 patients with histologically proven NASH who received pentoxifylline (400 mg three times per day) for 6 months. After 6 months of therapy, the mean AST and ALT improved significantly (P < 0.0001 and <0.0001, respectively). In fact, ALT levels normalized in 23 % of patients at month 1, 35 % at month 2, and 60 % at month 6 of treatment. The insulin resistance index also improved (P = 0.046) and the serum TNF-α was also reduced significantly after therapy (P = 0.011), while serum triglyceride, cholesterol, and body mass index (BMI) remained unchanged [92]. Both of these studies suggest a potential role for TNF-α in NASH as well as potential interventions targeting TNF-α. The most recent randomized intention-to-treat pentoxifylline trial (N = 26, pentoxifylline arm; N = 29, placebo arm) showed a decrease of ≥2 points in the NASH activity score (NAS) in 38.5 % of patients on PTX versus 13.8 % of those on placebo (P = 0.036); this effect was accompanied by a significant decrease in steatosis and lobular inflammation [93]. Despite the encouraging tone of these reports, recent systematic reviews show that pentoxifylline reduces AST and ALT levels and may improve liver histological scores in patients with NAFLD/NASH, but does not appear to affect TNF-α or other cytokine levels [94]. It is currently unclear whether the effects of pentoxifylline on NAFLD phenotypes are due to its suppression of TNF-α or to its non-TNF-α related anti-inflammatory properties.
Other Cytokines
Obesity is a chronic state of low-grade inflammation which predisposes obese individuals to both insulin resistance (IR) and NAFLD. A progressive infiltration of classically activated (M1) macrophages into obese adipose tissue leads to a release of proinflammatory cytokines. In lean individuals macrophages are in an alternatively activated (M2) state; these cells secrete IL-10, an anti-inflammatory cytokine, which may protect against inflammation. Differential activation of resident macrophages defines net inflammatory/anti-inflammatory balance of secreted peptides released by adipose. Additionally, overproduction of inflammatory cytokines in adipose could be explained by a decrease in the production of miRNAs that regulate their synthesis. Concerted decrease of mature miRNA levels has recently been reported in adipose of patients with advanced stages of NASH [95].
In obese subjects, both visceral and subcutaneous adipose tissues release potent pro-inflammatory cytokines interleukin-6 (IL-6) and interleukin-8 (IL-8) [34, 96]. In fact, explanted human adipose tissue releases even more IL-6 and IL-8 than adiponectin [97], probably due to an obesity-related increase in the TNF-α production involving p38 MAPK and NF-κB pathways [98]. Unfortunately, little is known about IL-6 and IL-8 in patients with NAFLD or NASH. Some studies showed that serum IL-6 and IL-8 levels in patients with NASH are significantly higher than the healthy controls [31, 99, 100]. On the other hand, IL-6 seems to have hepatoprotective effects [101]. In particular, it prevents sinusoidal endothelial cell damage and associated changes in hepatic microcirculation and decreases hepatocyte death [102]. Moreover, in vitro, IL-6 treatment improves the outcomes for patients with liver transplants for alcohol-related liver disease [102]. These contradictory data emphasize our incomplete understanding of the role of these cytokines in NAFLD.
Another significant focus of interest is on monocyte chemoattractant protein-1 (MCP-1)/CCL2. This potent chemoattractant is secreted mainly by macrophages known to infiltrate adipose tissue in obese humans. Levels of MCP-1 are elevated in both the adipose tissue and the plasma of obese mice [103]. Interestingly, MCP-1 deficiency in mice fed a high-fat diet decreases insulin resistance and hepatic steatosis whereas mice overexpressing MCP-1 in adipose tissue show increased insulin resistance and hepatic triglyceride levels [104]. In mouse models of acute and chronic hepatic injury, pharmacological inhibition of MCP-1 suppresses the infiltration of macrophages into the liver and intrahepatic production of proinflammatory cytokines [105]. In humans, data on the role of MCP-1 in NAFLD are sparse. MCP-1 serum levels are elevated in patients with ultrasound-diagnosed NAFLD and positively correlate with body-mass index and fasting glucose [106].
Recently, the presence and accumulation of the CD11c(+)CD1c(+) dendritic cells has been demonstrated in the adipose tissue of obese individuals [107]. These cells are capable of inducing the differentiation of IL17-producing type of T-helpers (Th17) that functionally oppose T(reg)-mediated responses. In turn, Th17 cells may infiltrate the liver and facilitate the transition from simple steatosis to steatohepatitis [108]. This line of thought is augmented by observations of Th17 cells acting synergistically with FFAs to induce IL-6 production in cultured hepatic cells and of an increase in the hepatic expression of Th17 cell-related genes encoding for retinoid-related orphan receptor gamma (ROR)γt, IL-17, IL-21, and IL-23 in NASH patients as compared to healthy controls [108]. Moreover, in mice, neutralization of IL17 with specific antibodies improved their resistance to LPS-induced liver injury as measured by lower serum alanine aminotransferase (ALT) levels and reduced inflammatory cell infiltrates in the liver [108]. These data suggest that Th17 cell expansion and hepatocyte-generated IL-6, particularly in the presence of FFAs, such as would occur in NAFLD, may contribute to a vicious cycle leading to increased levels of hepatic inflammation and steatosis. Targeting the balance between Th17 cells and T(regs) may lead to novel strategies in preventing NAFLD progression.
The Role of Adipokines in Promoting Hepatic Steatosis, Insulin Resistance Oxidative Stress, and Hepatic Fibrosis
As previously noted, hepatic steatosis, insulin resistance and oxidative stress all play critical roles in the pathogenesis of NAFLD, forming the basis of the “two-hit” or “multi-hit” hypotheses [23, 24]. The following paragraphs focuses on the potential role of adipokines in inducing hepatic steatosis, enhancing insulin resistance, and promoting oxidative stress or hepatic fibrosis.
Adipokines and Steatosis
Fat accumulation in hepatocytes may result from an increase in the delivery of FFAs to the liver, increased FA synthesis, decreased FA degradation, impaired triglyceride release from the liver, or a combination of these factors. The role of several key adipokines in each of these steps is described below.
Adiponectin as an Anti-steatotic Agent
One hypothesis linking low serum adiponectin to the development of NAFLD focuses on an increase in hepatic lipid retention, a consequence of adiponectin-dependent suppression of very-low-density lipoprotein (VLDL) synthesis, the chief route of hepatic lipid export. One of the rate-determining steps in hepatic VLDL production is the synthesis of apoB-100. The absolute synthesis rates of apoB-100 in patients with NASH are lower (31.5 ± 3.4 mg/kg/day) than in either obese (115.2 ± 7.2 mg/kg/day, P < 0.001) or lean non-NAFLD controls (82.4 ± 4.1 mg/kg/day, P = 0.002) [109]. In fact, plasma adiponectin concentrations are inversely associated with both VLDL-apoB-100 concentrations (r = −0.337) and VLDL-apoB-100 production rates (r = −0.373) [110]. Additionally, results reported by Ng and coworkers [111] indicate that lower than normal adiponectin levels may weaken its beneficial effects on the accumulation of triglycerides and on the concentration of fatty acids in skeletal muscle. Earlier work showed that adiponectin enhances fatty acid oxidation both in liver and muscle tissue through activation of acetyl CoA oxidase, carnitine palmitoyltransferase-1, and 5′-AMP activated protein kinase (AMPK) [112]. Adiponectin increases lipoprotein lipase (LPL) translocation to the cell surface where it could be released [113]; decreased serum adiponectin is associated with LPL deficiency and acts independently of systemic inflammation and/or insulin resistance [114, 115]. Seemingly, these data indicate that the decrease in serum adiponectin concentrations may stimulate the accumulation of fat in the liver by promoting LPL deficiency and subsequent increase in free fatty acid flux to the liver. However, there are other data suggesting that the logic of relationship between adiponectin and LPL levels may be inverted. In patients with loss-of-function LPL gene variants, plasma adiponectin concentrations are significantly lower than in matched controls [116]. In fact, in the LPL mutation group, lower levels of adiponectin explained a proportion of the variance in metabolic covariates and, after adjustments for anthropometrics, lipids, glucose and other factors, substantially contributed to risks of obesity-associated disorders [116].
Other researchers have suggested an alternative mechanism for the steatogenic effects of low levels of adiponectin. One mechanism involves an increase in FA synthesis and/or a decrease in FA degradation within the liver. Through PPARalpha, adiponectin stimulates the expression of carnitine palmitoyltransferase 1 (CPT1), a rate limiting enzyme involved in the transport of long-chain fatty acids into the mitochondrial matrix of liver cells [117]. At the same time, adiponectin decreases the activity of two key enzymes in the hepatic lipogenesis pathway, namely, acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) [117]. In transgenic mice that overexpress adiponectin via the aP2-promoter (ADNTg mice), lipogenic gene expression is reduced as well [118]. These data suggest that an increase in adiponectin concentrations should stimulate beta-oxidation of fatty acids in the liver, decreasing the intrahepatic lipid load, while low adiponectin levels should promote hepatic steatosis, further supporting the anti-steatotic properties of adiponectin.
Leptin as an Anti-steatotic Agent
Leptin protects against steatosis and lipotoxicity in non-adipose tissues, including the liver, but the molecular mechanisms underlying these effects are not fully understood. Most likely, the mechanism for this protection is peripheral. In cultured pancreatic islets, leptin lowers triglyceride content by increasing FFA oxidation and preventing its esterification [119]. A similar mechanism may be at work in the liver, as it expresses leptin receptors. In fact, tissue-specific over expression of wild-type leptin receptors in steatotic livers of leptin-receptor-null fa/fa rats reduces TG accumulation in the liver but not anywhere else [120].
Early studies demonstrated that, in hepatocytes, the stimulation of the long isoform of the leptin receptors provides IL-6-like signals, as it synergizes with IL-1 and TNF-alpha to activate STAT proteins and synthesize acute-phase plasma proteins [121]. In addition, leptin dramatically suppresses the expression of hepatic stearoyl-CoA desaturase-1 (SCD-1), the rate limiting enzyme in the biosynthesis of monounsaturated fats [122, 123]. SCD-1 suppression, in turn, supports resistance to both hepatic steatosis and obesity due to marked increase in energy expenditure. The proposed mechanisms for the metabolic effects mediated by leptin induced SCD-1 deficiency include the blocking of triglyceride synthesis and the export of VLDL [122, 124]. This, in turn, leads to a concomitant increase in the pool of saturated fatty acyl CoAs, which allosterically inhibits acetyl CoA carboxylase (ACC) and reduces the amount of malonyl CoA. As a result, inhibition of the mitochondrial carnityl palmitoyl shuttle system is relieved, stimulating the import and oxidation of fatty acids in mitochondria. Thus, leptin administration de-represses fatty acid oxidation, leading to increased fat burning [122]. Other proposed mechanisms of anti-steatotic effects of leptin involve increases in peroxisome proliferator-activated receptor-α (PPAR-α) signaling [125] and/or activity of AMP-activated protein kinase (AMPK) [126].
Beyond this, leptin seems to promote the elimination of plasma cholesterol by decreasing cholesterol biosynthesis. Cholesterol elimination is achieved by down regulating the hepatic activity of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, up-regulating the activities of both sterol 27-hydroxylase and cholesterol 7α-hydroxylase and diminishing the cholesterol fraction bound to VLDL by limiting triglyceride supply [127]. Lowered leptin signaling may even be responsible for the increased prevalence of cholesterol gallstones in obese patients as compared to the general population [128].
Because obesity is associated with leptin resistance, exogenous leptin administration does not diminish liver steatosis. The development of both central and peripheral leptin resistance depends on the liver. In animal models, chronic leptin treatment in leptin-naïve animals induces leptin receptors and subsequent increases in the serum concentration of the soluble leptin receptor protein (SLR). This increase occurs as a consequence of ectodomain shedding from the membrane-bound isoforms. SLR shedding sequesters leptin and prevents its productive interactions with the receptor. In this way, the hepatic circuit limits peripheral leptin activity all over the body. To reflect available leptin as opposed to any leptin, the free leptin index (FLI), calculated as the ratio of leptin to SLR, was developed. Studies correlating FLI and hepatic steatosis, however, contradict each other; some studies show that FLI scores are lower in steatosis as compared to non-NAFLD obese individuals [78], while others point at the opposite [77, 129].
Resistin as a Pro-steatotic Agent
In rodents, resistin is capable of influencing lipid metabolism. Adenovirus-mediated resistin over expression leads to an increase in plasma triglycerides in mice and rats [130, 131]. Loss of resistin ameliorates hyperlipidemia and hepatic steatosis in leptin-deficient mice [132]. In healthy men, serum resistin levels were found to negatively correlate with high-density lipoprotein cholesterol (HDL-C) levels [133]. When human hepatocytes are treated with resistin, at levels observed in human obesity, the secretion of apoB dramatically increases along with hepatocyte lipid content. These increases are due to the stimulation of de novo lipogenesis via the SREBP1 and SREBP2 pathways [134]. This suggests that higher than normal resistin levels, typically seen in obesity and type II diabetes, might contribute to the development of fatty liver through its dyslipidemic effects.
TNF-α as a Pro-steatotic Agent
The list of the pleiotropic effects of TNF-α includes changes in lipid metabolism. In the liver, TNF-alpha stimulates the expression of genes involved in de novo synthesis of FAs, while suppressing those responsible for FA oxidation. This activation is achieved through interactions with the insulin-Insig-Srebp signaling pathway. The direct consequences to steatogenesis include reduction of HDL-cholesterol, increase of LDL-cholesterol, the accumulation of potentially harmful precholesterol metabolites and the suppression of cholesterol elimination through bile acids [135]. It also enhances the secretion and metabolic processing of VLDLs [136].
Over the years, mice expressing T cell-targeted human TNF-α transgenes have served as an animal model for persistent low-grade exposure to TNF-α typical of morbid obesity. In these mice, hepatic triglyceride and cholesterol levels are increased, despite concomitant serum cholesterol lowering [137]. In addition, mitochondrial β-oxidation is inhibited in the livers of hTNFα-transgenic mice [137], as well as the activities of carnitine palmitoyltransferase-II (CPT-II) and mitochondrial HMG-CoA synthase [137]. Peroxisomal β-oxidation is also lowered [137]. These changes are probably mediated by low mRNA levels of peroxisome proliferator-activated receptors α and δ (PPARα and PPARδ) [137]. On the other hand, there is no increase in de novo fatty acid synthesis. In fact, both fatty acid synthase (FAS) activity and the gene expression of acetyl-CoA carboxylase 2 (ACC2) are reduced [137]. Therefore, liver-specific steatogenic effects of TNF-α are predominantly caused by lipid retention due to the suppression of fatty acid decomposition. It is important to mention that both the control and the transgenic mice in this study were fed ad libitum. Thus, this model does not allow us to determine whether these effects are direct consequences of TNF-α, peripheral consequences of elevated TNF-α levels, or secondary effects related to other TNF-α-mediated mechanisms such as appetite.
In addition to pronounced inhibition of mitochondrial and peroxysomal β-oxidation, TNF-α stimulates VLDL production in the liver [136] and inhibits the activity and mRNA expression of lipoprotein lipase in adipocytes [138]; these processes favor lipolysis in visceral and subcutaneous fat depots that contribute to the development of TNF-α dependent hypertriglyceridemia and associated NAFLD.
Adipokines and Insulin Resistance
There is a striking association between NASH and insulin resistance. As insulin resistance is thought to lead to the accumulation of triglycerides in the liver [139]; thus, any factor promoting a vicious cycle of insulin signaling can be steatogenic and factors counteracting insulin resistance can be protective against the development of NAFLD.
Adiponectin as Insulin Sensitizer
Hyperinsulinemia caused by insulin resistance increases fatty acid synthesis and impairs both mitochondrial β-oxidation and the export of triglycerides in multiple ways. Early studies indicate that adiponectin decreases insulin resistance by increasing oxidation of FAs, thereby reducing the triglyceride content in peripheral tissues [112]. Adiponectin also suppresses glucose production in the liver and enhances the hepatic action of insulin. These glucose-lowering effects of adiponectin require liver-specific activation of AMP-activated protein kinase (AMPK) [140], a central component of the protein kinase cascade that plays a key role in the regulation of energy control. It is tempting to speculate that the AMPK-mediated antiglycemic effects of adiponectin in the liver may play a role in the prevention of NAFLD, but this seems unlikely. Recent work indicates that short-term over expression of a constitutively active form of AMPK in the liver can lead to the development of steatosis even in the presence of lowered hepatic glycogen synthesis and circulating lipid levels [141]. Most likely, the NAFLD-like disorder in these animals develops from the hepatic accumulation of lipids released from adipose tissue in response to the relative scarcity of glucose. Therefore, additional stimulation of AMPK provided by a sudden increase of adiponectin (e.g., due to thiazolidinedione (TZD) treatment) may aggravate early stages of the steatogenic processes in the liver. This may also explain the infrequent but potentially serious hepatotoxic side effects of chronic administration of TZDs [142] and the pronounced exacerbation of hepatic steatosis in mice with polygenic obesity treated by rosiglitazone [143].
Insulin-sensitizing effects of adiponectin are not limited to AMPK signaling events. For example, in primary mouse hepatocytes, the absence of AMPK, or other components of the same signaling cascade did not prevent adiponectin from inhibiting glucose output or reducing gluconeogenic gene expression [144]. It is also important to note that adiponectin inhibits autophagy, while AMPK stimulates it, pointing at other signaling pathways sensitive to the presence of adiponectin. One potential contributor to AMPK-independent adiponecting response is newly discovered suppressor of glucose by autophagy (SOGA) that both inhibits autophagy and contributes to the inhibition of glucose production in hepatocytes [145].
Interestingly, various treatments successful at improving insulin response (thiazolidinediones (TZDs), n-3 polyunsaturated fatty acid (PUFA) supplementation) also stimulate adiponectin production [146].
Leptin as an Insulin Sensitizer
It is widely accepted that leptin exerts a systemic insulin-sensitizing effect. The interaction between the insulin and leptin signaling cascades in peripheral organs have been studied both in vitro and in vivo [147]. However, the results are inconsistent in different cell lines and the complete mechanism remains unclear. Most likely, these cross-cascade interactions involve down-regulation of the PKR-like endoplasmic reticulum (ER) kinase/eukaryotic translation inhibition factor 2α (PERK-eIF2α) arm of ER stress in liver, skeletal muscle, and adipose tissue [148]. One way of inhibiting the peripheral effects of leptin is through the feedback inhibition by SOCS3 via phosphorylation of Tyrosine 985 on its receptor [149]. When this tyrosine is mutated, hence, abrogating this inhibitory signaling, the insulin sensitivity is enhanced throughout the body via increased insulin action on the suppression of hepatic glucose production [150]. The liver is probably central to this mechanism, as some studies suggest that leptin selectively improves insulin receptor (IR) activation only in this organ, but not in the skeletal muscle or fat [151]. Unfortunately, the insulin-related branch of the leptin-dependent signaling in obese livers is profoundly suppressed [152]. Therefore, it is unlikely that therapeutic administration of leptin would alleviate liver steatosis through improved insulin sensitivity.
Resistin as an Inductor of Insulin Tolerance
Hyperresistinemia certainly contributes to impaired insulin sensitivity in obese rodents. In mice, resistin elimination reduces hepatic glucose production due to decreased gluconeogenic enzyme expression in the liver and to the activation of AMPK [153]. In humans, the situation is much more difficult to trace, because serum resistin levels are related to sex, age, testosterone and estradiol levels [154]. These fluctuations in resistin levels and the relatively low homology between resistin and resistin-like molecules in humans and rodents complicate the study of resistin involvement in the development of insulin resistance in the liver and NAFLD.
TNF-α Impairs Insulin Signaling
TNF-α alters systemic energy homeostasis in a way that closely resembles the insulin resistance phenotype. Mice with a complete knock-out of TNF-α signaling show significantly improved insulin sensitivity in both diet-induced and leptin-deficient obesity [155]. Molecular studies show that long-term exposure to TNF-α completely abolishes insulin-induced glycogen synthesis in hepatocytes [156]. TNF-α inhibits tyr phosphorylation of IRS-1 that promotes the transmission of the insulin signal, and stimulates ser phosphorylation instead, thus blunting peripheral insulin response [157, 158]. It was also shown that TNF-α-mediated insulin resistance of glucose uptake occurs through a MEK/Erk-dependent activation of CDK5 [159]. This compelling evidence demonstrates that abnormal production of TNF-α may predispose obese individuals to the development of insulin resistance and NAFLD.
Other Adipokines Influencing Insulin Resistance
The secretion of the extracellular form of nicotinamide phosphoribosyltransferase (NAMPT), also known as visfatin, is upregulated in obesity and has been shown to help in the regulation of glucose homeostasis. Visfatin regulates insulin secretion, insulin receptor phosphorylation and intracellular signaling and the expression of a number of beta-cell function-associated genes in the pancreas [160]. In rat livers, visfatin is strongly expressed, while in visceral fat its expression is significantly lower, and in subcutaneous adipose, it is undetectable. When visfatin was downregulated in rat hepatocytes by RNAi, a significantly decrease in glucose uptake after stimulation with insulin was observed, thus pointing at substantial autocrine effects on the sensitivity of liver cells to insulin action, possibly through its effects on NAD biosynthesis [161].
Another interesting adipokine with potential effects on the pathogenesis of NASH is vaspin, visceral adipose tissue-derived member of serine protease inhibitor (serpin) family [162]. Vaspin administration in obese mice fed with high-fat high-sucrose chow normalizes their serum glucose levels by reversing altered gene expression related to insulin resistance, including all other adipokines discussed above [162]. In humans, the role for vaspin in metabolic regulation is unclear at present. Serum vaspin concentrations display a circadian rhythm, along with a preprandial rise and postprandial fall, similar to that of ghrelin, while unscheduled meals lead to a decrease in vaspin levels [163]. The study in normoglycemic (N = 259) and diabetic Japanese patient (N = 275) showed that serum vaspin levels closely correlate with HOMA scores for insulin resistance rather than with BMI and other anthropometric parameters. Moreover, the minor (A) allele of SNP rs77060950 in the promoter region of the vaspin-encoding gene SERPINA12 appears to be closely linked to increased levels of serum vaspin and higher HOMA scores [164]. Additionally, insulin sensitivity was shown to be the strongest determinant of vaspin mRNA expression in human subcutaneous adipose [165]. These data point at vaspin as an important insulin sensitizer of adipocytic origin that may play an instrumental role in NAFLD.
Apelin inhibits glucose-stimulated insulin secretion both in vivo and in vitro by acting on its receptor, which is expressed in beta-cells of pancreatic islands [166]. The expression of apelin in fat cells and apelin plasma levels are largely increased in all the hyperinsulinemia-associated obese states of mice, independent of diet composition [167]. In obese patients, plasma apelin levels are also significantly higher than in normal controls. When apelin levels were studied in patients with NAFLD and healthy controls of the same gender, apelin levels correlated with and HOMA indexes positively (r = 0.4, P = 0.008). Importantly, adjustments for BMI and HOMA indices eliminated the differences in apelin concentrations between compared groups [168].
Chemerin, encoded by the retinoic acid receptor responder 2 gene RARRES2, exacerbates glucose intolerance, lowers serum insulin levels, and decreases tissue glucose uptake in obese/diabetic but not normoglycemic mice when administered exogenously [169]. The disruption of the chemokine-like receptor-1 (CMKLR1) gene that encodes the receptor for chemerin leads to glucose intolerance as evidenced by decreased glucose stimulated insulin secretion as well as decreased skeletal muscle and white adipose tissue glucose uptake [170]. Studies using hemerin-deficient murine islands and a chemerin-ablated β-cell line showed that chemerin regulates β-cell function via maintaining expression of MafA, a pivotal transcriptional factor that stimulates insulin gene promoter activity [171]. In adipocytes, chemerin potentiated insulin-stimulated glucose uptake concomitant with enhanced insulin signaling [172]. Intravenous administration of the chemerin analog in sheep led to a dramatic increase in the insulin levels and a drop in glucose levels, along with an immediate increase in the level of triglycerides [173]. In humans, receptors for chemerin, CMKLR1, could be detected in primary human hepatocytes (PHH), Kupffer cells, bile-duct cells, and hepatic stellate cells; its amounts were strongly induced by treatment with exogenous adiponectin [174].
It seems that the NUCB2-encoded nesfatin play important roles in both central and peripheral branches of the glucose homeostasis regulation. In the brain, it stimulates the PEPCK/InsR/IRS-1/AMPK/Akt/TORC2 pathway, thus contributing to increased peripheral and hepatic insulin sensitivity by decreasing gluconeogenesis and promoting hepatic glucose uptake in vivo [175]. In β-cells, nesfatin exerts a direct, glucose-dependent insulinotropic action [176, 177]. It acts by promoting Ca(2+) influx through L-type Ca(2+) channels independently of PKA and PLA(2) [177]. Levels of nesfatin are elevated in newly diagnosed type 2 diabetes patients and glucose intolerant subjects [178], but are decreased in patients with insulin resistance-associate polycystic ovary syndrome (PCOS) [179]. In line with the above, there is some evidence of the reduction of sensitivity to nesfatin in obese individuals, possibly due to the saturation of transporters [180].
Omentin is expressed in stromal vascular cells of the visceral adipose, but not in the fat cells per se [181]. In human adipocytes grown in vitro, omentin enhances insulin-stimulated glucose uptake, while not affecting basal influx of glucose [181]. In omental adipose tissue explants, both insulin and glucose significantly and dose-dependently decrease the secretion of omentin [182]. Fasting serum omentin levels negatively correlate with HOMA-IR scores and positively correlate with serum levels of adiponectin [183].
In serum samples of patients with NAFLD or with NAFLD-associated disorders, the concentration of these, newly emerging, adipokines are altered. The interplay between novel insulin-sensitizing and insulin suppressing soluble molecules may represent an important avenue for future studies of the pathogenesis of NAFLD and NASH.
Adipokines and Oxidative Stress
Several lines of evidence support the role of adipokines in the increased oxidative stress seen in patients with NASH. Most studies converge on CYP2E1, peroxisomal release of reactive oxygen species and mitochondrial dysfunction with high proton potential. Within the liver, the reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generated by the parenchymal cells, by activated Kupffer cells and by both resident and attracted inflammatory cells, which further mobilize cellular defense mechanisms and contribute to liver injury and necrosis. Here we will review ROS/RNS-promoting and antioxidant properties of adipokines, leaving out their cytokine-mediated proinflammatory/anti-inflammatory effects as these are reviewed in detail in other chapters.
Antioxidant Role of Adiponectin
The potential role of adiponectin in the prevention of oxidative stress in the liver lacks direct evidence. Nakanishi and coworkers presented the most compelling indirect evidence of such an influence [184]. The authors used an oral glucose test to study 259 Japanese Americans with normal glucose tolerance, impaired glucose tolerance, or diabetes. Concentrations of 8-iso-prostaglandin F(2-α) (isoprostane), a marker of oxidative stress, were measured in the urine and adiponectin concentrations were measured in the serum. Adiponectin levels negatively correlated with urinary isoprostane levels (adjusted for age, gender, and smoking status). This association was attenuated but remained significant after adjusting for waist-to-hip ratio, body mass index, percent body fat, C-reactive protein levels, glucose tolerance status, or HOMA scores [184].
A different line of evidence suggests a reverse causative relationship between oxidative stress and levels of adiponectin, such that the production of adiponectin might be suppressed in pro-oxidative conditions. For example, in differentiated murine adipocytes exposed to increasing concentrations of glucose oxidase, adiponectin mRNA expression is suppressed [185]. Moreover, a significant inverse correlation between the formation of 4-Hydroxynonenal (4-HNE), an important by-product of lipid peroxidation, and adiponectin secretion was observed. Additionally, 4-HNE inhibits adiponectin production when administered alone [185]. Researchers have proposed a direct transcriptional effect of 4-HNE on the adiponectin gene and/or effects mediated by NF-κB activation through the phosphorylation of Iκ [185].
On the other hand, there is a certainty that adiponectin exerts antioxidant effects in non-hepatic cell types. For example, it inhibits the generation of ROS in human glomerular mesangial cells treated with high concentrations of glucose; it also stimulates eNOS activity, which has additional protective effects [186]. Similar effects were observed in primary human phagocytes; these effects were invoked by full-length adiponectin only, while the globular fragment of adiponectin enhanced the production of ROS [187]. The list of other cellular types protected by full-size adiponectin from an excess of oxidative stress include: β-cells of the islets, endothelial cells, cardiomyocytes, and adipocytes. It is likely that in the human livers the HMW or full-size adiponectin is exerting similar effects, thus protecting from the progression of simple steatosis to NASH.
The most recent, intriguing observation on possible liver-specific anti-inflammatory function of adiponectin is that, in the liver cells, it stimulates the expression of UCP-2, the mitochondrial inner membrane transporter uncoupling protein 2 with hepatoprotective effects. A recent study of adiponectin knockout (ADN-KO) mice treated with adiponectin found that it stimulates mitochondrial superoxide production that, in turn, facilitates the transportation, stabilization and translation of UCP2 mRNA in nonparenchymal cells of the liver [188]. Livers of untreated AND-KO mice readily accumulate fat and have dysfunctional mitochondrial, while the replenishment of adiponectin restores the oxidative capability of the mitochondrial respiratory chain (MRC) complexes, thereby preventing the accumulation of lipid peroxidation products without a direct effect on mitochondrial biogenesis [189]. LPS induces elevation of TNFα and ALT levels in the livers of ADN-KO mice, but not in the livers of UCP2-KO mice [189]. These evidences suggest a UCP-2 dependent beneficial effect of adipose-derived adiponectin on the levels of oxidative stress in the liver (Fig. 17.3).

Mutual regulation of adiponectin and UCP2 across the liver and adipose tissue
Pro-oxidant Role of Leptin
Leptin inhibits antioxidant systems and enhances lipoperoxidation in the liver and other tissues. Administering leptin to experimental animals increases hepatic acetyl-coenzyme A carboxylase phosphorylation, fatty acid oxidation and ketogenesis [189] along with the hepatic levels of thiobarbituric acid reactive substances (TBARS), which are markers of lipoperoxidation [190]. On the other hand, this same treatment decreases antioxidant GSH levels and the activities of glutathione-S-transferases (GSTs), superoxide dismutase (SOD) and catalase [191]. These differences are more pronounced when hyperleptinemia is induced in alcohol-treated animals, suggesting that leptin may augment liver injury mediated by free radicals via other mechanisms [191].
Intravenous injections of leptin induce the release of nitric oxide (NO) by both inducible NO synthase (iNOS) and endothelial nitric oxide synthase (eNOS) [192]. When overexpressed eNOS remains uncoupled, it changes from a protective enzyme to a contributor to oxidative stress, thus adding up to a pro-oxidative environment [193]. Therefore, leptin induced stimulation of eNOS and iNOS may be a pro-oxidative event.
Leptin also upregulates CYP2E1 expression, a cytochrome P450 responsible for the oxidation of alcohol and production of activated oxygen species leading to oxidative stress. Proofs that leptin regulates CYP2E1 activity come from observations that livers of leptin-deficient mice express much lower levels of CYP2E1, while short-term leptin replacement completely reverses suppression of CYP2E1 [194]. Paradoxically, CYP2E1-dependent production of ROS inhibits apoptosis but accelerates necrosis, stimulated by polyunsaturated fatty acids [195]. This latter observation is consistent with the neuroinflammatory features seen in patients with NASH. Finally, higher hepatic CYP2E1 expression and activity have been frequently observed in the context of NAFLD (see [196] for review).
Pro-oxidant Role of Resistin
Despite the scarcity of observations showing that resistin directly influences oxidative stress, there is a body of evidence suggesting a pro-oxidative role for this adipokine. In porcine coronary arteries, resistin evokes an increase in the production of superoxide radical [197], while in rats, the long-term overexpression of resistin in cardiomyocytes in vivo results in larger amounts of TNFα release, along with increased phosphorylation of IκBα and intracellular ROS content [198]. It is of interest that individuals with high expression levels of NAD(P)H:quinone reductase (NQO), a prototypical phase II antioxidant enzyme, tend to have higher levels of resistin mRNA in their subcutaneous adipose tissue [199]. The positive effects of NQO are exercised only in individuals with at least one −180G allele that confers significant increase to the basal activity of the resistin promoter [199]. It is possible that antioxidant enzymes and oxidative stress-promoting resistin are co-regulated. Importantly, levels for both gene and protein expression of resistin are substantially higher in the liver than in the adipose [200]. Accordingly, it is important to find out wither locally produced resistin plays its pro-oxidative role within the liver parenchyma.
Pro-oxidant Role of TNF-α
The important role of TNF-α in the enhancement of ROS production, observed in steatotic livers, is certain. Key components of TNF signaling include sphingolipids, particularly ceramide, generated from acidic sphingomyelinase activation [201]. Mitochondria isolated from TNF-α-treated liver cells show a two- to threefold increase in the amount of ceramide when compared with mitochondria from untreated cells. Ceramide, in turn, influences the mitochondrial electron transport chain and evokes hydrogen peroxide overproduction [202], one of the most potent sources of oxidative damage. In addition, ceramide induces necrosis through the mitochondrial membrane permeability transition (MMPT) mechanism [203]. In mitochondria, treatment with TNF-α induces pronounced morphological changes, decreased mitochondrial membrane potential and reduced production of intracellular ATP [204]. These changes are accompanied by accumulation of significant amounts of reactive oxygen species (ROS) [204].
Some conditions sensitize liver cells to TNF-α-induced cell death; one of these is the depletion of reduced glutathione [205]. It is peculiar that chronic exposure to low levels of TNF-α, similar to that observed in NAFLD patients, profoundly modulates GSH metabolism. Reduced levels of catalase and glutathione peroxidase (reflecting impaired redox buffering capacity) were observed in the livers of mice with low, persistent expression of TNF-α in the T-cell compartment [206]. GSH depletion driven by TNF-α may further enhance oxidative stress, constituting a vicious circle augmenting parenchymal injury. Consistent with this notion, the oxidized/reduced glutathione ratio (GSSG/GSH) is increased in NASH patients, while superoxide dismutase, glutathione peroxidase and glutathione reductase activities remain unchanged [207].
Interestingly, TNF-α enhances UCP-2 expression in hepatocytes [208]. In these effects, the action of TNF-α is similar to that of adiponectin (see argument in respective subchapter). On the one hand, the up-regulation of UCP-2 may compromise cellular ATP levels and worsen liver damage by augmenting cell death; on the other hand, it may have a protective role by reducing ROS. Studies of adiponectin-dependent upregulation of UCP-2 [188, 189] point toward the second option. It is also possible that these two effects cancel each other out. The latter suggestion has been supported by the finding that serum alanine aminotransferase (ALT) levels and steatohepatitis scores in UCP-2−/− mice remains similar to those of wild-type controls, both in genetically and diet-induced obesity [209]. Little is known about the state of UCP-2 activity in NAFLD and NASH patients. There is only one study that covers hepatic expression of UCP-2 in humans; in this study, the staining intensity of TNF-α and UCP-2 were correlated with the severity of liver disease [210]. If the majority of NAFLD patients, indeed, overexpress UCP-2, as do NAFLD mice, and if this over expressed molecule remains functional, it seems that it is unable to protect against steatohepatitis, possibly due to the overwhelming effect of ROS that exceeds the compensatory potential of mitochondrial uncoupling. As mitochondrial uncoupling sensitizes cell to TNF-α induced death, it might be that this effect outweighs the simultaneous decrease in the ROS production.
Other Adipokines and Oxidative Stress
The effects of NAMPT/visfatin at the levels of oxidative stress are unclear at best. On the one hand, nicotinamide mononucleotide (NMN), a product of the NAMPT reaction and a key NAD(+) intermediate, restores sensitivity to glucose in diabetic mice through enhancement of hepatic insulin sensitivity and restoration of gene expression related to oxidative stress and inflammatory response [211]; On other hand, in both human primary pulmonary artery endothelial cells and A549 lung epithelial cells, the overexpression of visfatin affects intracellular ROS production in either the presence or absence of IL1-β. These effects are abrogated by visfatin-specific siRNA and by treatment with rotenone, a NADH oxidase complex I inhibitor [212]. In mouse models of ischemia and reperfusion in vivo, pharmacological inhibition of NAMPT with FK866 reduced both neutrophil infiltration and reactive oxygen species (ROS) generation within infarcted hearts. Sera from FK866-treated mice showed reduced circulating levels of the neutrophil chemoattractant, CXCL2, and impaired capacity to prime migration of these cells in vitro; these effects were reduced by either FK866, or sirtuin (SIRT) inhibitors, implying that visfatin stimulates the production of this chemokine [213]. In glomerular injury, often observed in diabetic patients, visfatin promotes the formation of lipid raft redox signaling platforms, which produces local oxidative stress resulting in the disruption of microtubular networks in glomerular endothelial cells and an increase in the glomerular permeability [214]. Similar effects are observed in coronary circulation, when visfatin-dependent lysosome-associated molecular trafficking and consequent ceramide accumulation in cell membrane mediates the assembly of NOX subunits and their activation, producing endothelial dysfunction [215]. The observations mentioned above might be explained by the pleiotropic action of visfatin. Being beneficial as an NMN-producing enzyme, visfatin may serve as a soluble signal detrimental to the oxidative state within the cells comprising a particular tissue. It is unclear whether hepatocytes or others cells of the liver are sensitive to the ROS-augmenting effects of visfatin.
Through its receptor APJ, apelin mediates oxidative stress in blood vessels, thus contributing to atherogenesis [216]; however, in cardiomyocytes, the same protein inhibits the hypertrophic response to oxidative stress in a dose-dependent manner through stimulation of the activity of catalase [217]. As pertains to NAFLD, treatment with apelin increases fatty acid oxidation and mitochondrial biogenesis in muscles of apelin-treated mice [218]. Apelin is highly expressed by stellate cells and its receptor is expressed in the parenchyma of the liver. Whether these insulin-sensitizing effects of apelin take place in the liver, or whether they increase oxidative stress in the liver, is currently unknown.
Studies of chemerin levels measured in portal venous (PVS), hepatic venous (HVS) and systemic venous (SVS) blood of patients with liver cirrhosis showed that its levels seem to be associated with inflammation rather than BMI, and that this adipokine is also released by the liver [219]. Serum chemerin concentrations are significantly higher in NAFLD patients as compared to healthy volunteers, and in NASH patients as compared to simple steatosis [220]. Almost nothing is known about the involvement of chemerin in the pathogenesis of NAFLD.
Treatment with omentin prevents TNF-α-induced COX-2 expression and subsequent vascular inflammation in human endothelial cells through stimulating eNOS [221]. Similar to chemerin, the influence of omentin levels on inflammatory features of NAFLD and NASH were never studied.
It is clear that thorough studies of inflammation-related effects of newly discovered adipokines are needed.
The Role of Adipokines in Hepatic Fibrosis
Liver fibrosis is a wound healing response that involves several cell types. It is characterized by inflammation, activation of matrix-producing cells, extracellular matrix (ECM) deposition and remodeling, and epithelial cell regeneration or an attempt thereof. The major matrix producing cells in the liver are hepatic stellate cells (HSC) that may undergo a phenotypic transition to myofibroblast-like cells that synthesize various ECM components. This fibrogenic response might result in cirrhosis in NASH livers and may instigate the progress to hepatocellular cancer and liver-related death. Despite a strong association between necroinflammatory activity and fibrosis on cross sectional studies, fibrosis progression in many NASH patients may still occur in the presence of an improvement in inflammation, ballooning, and steatosis [222].
Leptin as Fibrogenic Agent
The first evidence of the fibrogenic effects of leptin comes from studies of leptin deficient animals that are resistant to fibrosis resulting from long-term thioacetamide administration [223]. Ablation of leptin also alleviates profibrogenic effects of exposure to carbon tetrachloride [224, 225], Schistosoma mansoni infection [225], or as a consequence of steatohepatitis. Subsequent studies revealed a profound positive influence of leptin on TGF-β [223] and collagen I and III mRNA and protein production in HSCs [226]. Leptin effects on collagen production are at levels seen in obese individuals [226]. Leptin also augments the effect of TGF-β1 on collagen production [213].
The mechanism of leptin-dependent stimulation of collagen production includes the Janus kinase-phosphatidylinositol 3-kinase-Akt (JAKs-PI3K-Akt) pathway [227] and peroxide-dependent components coupled to the ERK1/2 and p38 pathways [228], thus opening the door to possible antioxidant therapy of liver fibrosis. Moreover, in HSCs, the same leptin-induced signaling leads to enhanced production of the tissue inhibitor of metalloproteinase TIMP-1 that suppresses collagen degradation [229]. To induce expression of the smooth muscle actin αSMA, leptin synergizes with IL-6 [230].
Taken together, these data indicate that leptin is a potent hepatic fibrosis promoter. In obese individuals, the increase in the leptin levels observed along with increasing fatty mass leads to persistent hyperleptinemia that may be a detriment to their livers through leptin-dependent profibrogenic effects. Observations in lipodystrophic patients treated with recombinant leptin support this conclusion. Despite significant improvements in hepatic steatosis, ballooning injury and NASH activity scores, there were no changes in hepatic fibrosis in patients treated with recombinant leptin [83, 231].
Adiponectin as Antifibrotic Agent
Kamada and co-authors revealed that adiponectin attenuates liver fibrosis in a carbon tetrachloride administration model [43]. In cultured hepatic stellate cells, adiponectin suppresses PDGF-induced proliferation and migration and attenuates the effects of TGF-β1 [43]. Another recent study demonstrates that adiponectin can inhibit proliferation and induce apoptosis in activated HSCs, but not in quiescent HSCs [232]. Local adiponectin production has been demonstrated in quiescent HSCs. Both AdipoR1 and AdipoR2 receptors are present in both quiescent and activated HSCs; however, AdipoR1 mRNA expression is reduced by 50 % in activated HSCs as compared to quiescent HSCs [232]. This data indicates that adiponectin is essential to either maintaining the quiescent phenotype of HSCs or is capable of reversing hepatic fibrosis by hampering the proliferation of activated HSCs and by inducing HSC apoptosis.
Importantly, in activated HSCs, adiponectin dampens the profibrogenic signaling through leptin receptor and prevents excess extracellular matrix production. Adiponectin-dependent suppression of the proliferation and the migration of HSCs is achieved through activation of AMPK [233, 234] and subsequent inhibition of leptin-mediated Stat3 phosphorylation and SOCS-3 binding to the leptin receptor [234, 235]. In addition, adiponectin-stimulated AMPK inhibits transforming growth factor (TGF)-β-induced fibrogenic properties of HSCs by regulating transcriptional coactivator p300 [236]. Adiponectin also stimulates PTP1B expression and activity, thus inhibiting JAK2/STAT3 signaling at multiple points [235].
Resistin as a Potential Fibrogenic Agent
Resistin has no known connection with hepatic fibrosis, except indirect evidence. In particular, resistin levels are associated with fibrosis severity in patients with chronic hepatitis B and C [237]. In liver cirrhosis, the plasma resistin levels are elevated as well [238]. On the other hand, resistin has been implicated in pulmonary fibrosis induced by bleomycin. In fact, microarray profiling has revealed a 17–25-fold induction of the RELM-alpha encoding gene FIZZ1 in the alveolar and airway epithelium of bleomycin treated rats. Co-cultures of FIZZ1-expressing epithelial cells and fibroblasts stimulate alpha-smooth muscle actin and type I collagen expression independently of transforming growth factor-beta. Similar effects were achieved in experiments transfecting a FIZZ1-expressing plasmid into fibroblasts [239]. Similar resistin-dependent responses might be produced in HSCs, if resistin indeed contributes to the development of NASH.
TNF-α and Fibrogenic Responses
There is direct evidence of the involvement of TNF-α in fibrogenic responses. When double knockout mice lacking both TNF receptors (TNFRDKO mice) are fed methionine- and choline-deficient (MCD) diets, they develop less pronounced liver steatosis than their wild-type counterparts [240]. The livers of these mice show significantly decreased numbers of recruited Kupffer cells, together with the extent of centrizonal fibrosis; stellate cell activation is also diminished [240]. Similar findings in TNFRp55 knock-out mice indicate that even partial suppression of TNF-α signaling can alleviate liver fibrosis [241]. Diet-driven induction of α1(I) collagen and TIMP-1 in TNFRDKO livers was also significantly lower, indicating TNF-α involvement in the stimulation of collagen deposition. Moreover, in primary cultures, TNF-α administration enhances TIMP-1 mRNA expression in activated hepatic stellate cells and suppresses apoptosis [241]. It seems that TNF-α increases the recruitment of Kupffer cells that, in turn, produce extra TNF-α and hasten fibrosis in either an autocrine or paracrine manner, thus contributing to NASH progression to cirrhosis. It seems that TNF regulates the profibrogenic effects of HSC through its binding to TNFR1, which is required for both HSC proliferation and MMP-9 expression. Both in vivo liver damage and fibrogenesis after bile-duct ligation were reduced in TNFR-DKO and TNFR1 knockout mice, compared to wild-type or TNFR2 knockout mice [242].
Other Adipokines and Fibrogenic Responses
Recent studies on rats have shown that apelin is overexpressed in activated HSCs and its receptor APJ—in the hepatic parenchyma of animals with cirrhosis [143]. The treatment of these animals with antagonist of APJ lead to the decrease in both grade of hepatic fibrosis and vessel density [243]. Similar effects of apelin signaling suppression were observed in rats treated with carbon tetrachloride [244]. Interestingly, the treatment with profibrogenic agent TNF-α stimulates the expression of apelin in cardiomyocytes and adipocytes [245, 246]. It is important to note that, in cardiovascular tissues, apelin prevents fibrosis by inhibition of the TGF-β-mediated expression of the myofibroblast marker α-SMA and the production of collagen. The prevention of collagen accumulation by apelin is mediated by a reduction in the activity of sphingosine kinase 1 (SphK1) [247]. Similarly, the decrease in the extent of the cardiovascular fibrosis and the levels of mRNA encoding plasminogen activator inhibitor type-1 (PAI-1) was observed in mice treated with both apelin and profibrotic peptide angiotensin II [248]. It is possible that the opposing effects of apelin on the liver and non-hepatic tissues are due to its differential regulation.
It seems that visfatin has at least some fibrogenic effects. In cardiac fibroblasts, visfatin stimulates collagen I and III production, procollagen I and III mRNA expression and protein production via p38MAPK, PI3K, and ERK 1/2 pathways in a dose- and time-dependent manner [249]. Importantly, in the livers of NAFLD patients, the expression levels of visfatin were significantly higher in patients with fibrosis (P = 0.036) and were positively correlated with the fibrosis stage (r = 0.52, P = 0.03), while no difference in intrahepatic visfatin levels were registered when patients with NASH were compared to those with simple steatosis [250]. These observations point at the necessity of further investigations of possible fibrotic effect of visfatin in the liver.
Conclusions
The past decade has produced a great deal of knowledge about the epidemiology and pathogenesis of NAFLD and NASH. It is increasingly clear that the development of NASH is a complex process involving multiple mechanisms including insulin-resistance, oxidative stress, abnormal free fatty acid metabolism, and inflammatory cytokines and adipokines. Increasing evidence indicates that the pathogenesis of NAFLD and NASH is hastened by a disturbance of adipocytic production of adipokines. Decreased production of adiponectin and increased production of TNF-α, characteristic of obesity, seem to contribute to all major NASH-related cellular processes (Table 17.1). Leptin, on the other hand, behaves as a “wolf in sheep’s clothing.” Its NASH suppressive effects are diminished by the widespread effects of leptin resistance, and it becomes potentially pro-oxidant and fibrogenic. Resistin’s involvement in NASH is documented in rodent models, which may not be applicable to the human disease of NAFLD. Therefore, the molecular effects of resistin in patients with NASH remain to be investigated. In addition, other adipokines that seem to be involved in insulin signaling, such as vaspin, visfatin, apelin, nesfatin, omentin and chemerin require further study in patients with NAFLD and NASH.
References
Falck-Ytter Y, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis. 2001;21(1):17–26. Review.
Younossi ZM, Diehl AM, Ong JP. Nonalcoholic fatty liver disease: an agenda for clinical research. Hepatology. 2002;35(4):746–52.
Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116(6):1413–9.
Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9(6):524–30.e1. quiz e60. Epub 2011 Mar 25.
Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34(3):274–85.
Suzuki A, Angulo P, Lymp J, St Sauver J, Muto A, Okada T, Lindor K. Chronological development of elevated aminotransferases in a nonalcoholic population. Hepatology. 2005;41(1):64–71.
Benedetti A. Attraction and growing interest for the fatty liver by the scientific associations. Eur Rev Med Pharmacol Sci. 2005;9(5):251–2.
Lam B, Younossi ZM. Treatment options for nonalcoholic fatty liver disease. Therap Adv Gastroenterol. 2010;3(2):121–37.
Ong JP, Younossi ZM. Approach to the diagnosis and treatment of nonalcoholic fatty liver disease. Clin Liver Dis. 2005;9(4):617–34. vi. Review.
Caldwell SH, Oelsner DH, Iezzoni JC, Hespenheide EE, Battle EH, Driscoll CJ. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology. 1999;29(3):664–9.
Yasui K, Hashimoto E, Tokushige K, Koike K, Shima T, Kanbara Y, Saibara T, Uto H, Takami S, Kawanaka M, Komorizono Y, Okanoue T, The Japan NASH Study Group. Clinical and pathological progression of non-alcoholic steatohepatitis to hepatocellular carcinoma. Hepatol Res. 2012;42(8):767–73.
Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, De Paolis P, Capussotti L, Salizzoni M, Rizzetto M. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123(1):134–40.
Bhatia LS, Curzen NP, Calder PC, Byrne CD. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur Heart J. 2012;33(10):1190–200.
Younossi ZM, Page S, Rafiq N, Birerdinc A, Stepanova M, Hossain N, Afendy A, Younoszai Z, Goodman Z, Baranova A. A biomarker panel for non-alcoholic steatohepatitis (NASH) and NASH-related fibrosis. Obes Surg. 2011;21(4):431–9.
Younossi ZM, Jarrar M, Nugent C, Randhawa M, Afendy M, Stepanova M, Rafiq N, Goodman Z, Chandhoke V, Baranova A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH). Obes Surg. 2008;18(11):1430–7.
Shen J, Chan HL, Wong GL, Choi PC, Chan AW, Chan HY, Chim AM, Yeung DK, Chan FK, Woo J, Yu J, Chu WC, Wong VW. Non-invasive diagnosis of non-alcoholic steatohepatitis by combined serum biomarkers. J Hepatol. 2012;56(6):1363–70.
Friedrich-Rust M, Romen D, Vermehren J, Kriener S, Sadet D, Herrmann E, Zeuzem S, Bojunga J. Acoustic radiation force impulse-imaging and transient elastography for non-invasive assessment of liver fibrosis and steatosis in NAFLD. Eur J Radiol. 2012;81(3):e325–31.
Ratziu V, Charlotte F, Heurtier A, Gombert S, Giral P, Bruckert E, Grimaldi A, Capron F, Poynard T, LIDO Study Group. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128(7):1898–906.
Arun J, Jhala N, Lazenby AJ, Clements R, Abrams GA. Influence of liver biopsy heterogeneity and diagnosis of nonalcoholic steatohepatitis in subjects undergoing gastric bypass. Obes Surg. 2007;17:155–61.
Torres DM, Williams CD, Harrison SA. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2012;10(8):837–58.
de Oliveira CP, Stefano JT, de Siqueira ER, Silva LS, de Campos Mazo DF, Lima VM, Furuya CK, Mello ES, Souza FG, Rabello F, Santos TE, Nogueira MA, Caldwell SH, Alves VA, Carrilho FJ. Combination of N-acetylcysteine and metformin improves histological steatosis and fibrosis in patients with non-alcoholic steatohepatitis. Hepatol Res. 2008;38(2):159–65.
Stephen S, Baranova A, Younossi ZM. Nonalcoholic fatty liver disease and bariatric surgery. Expert Rev Gastroenterol Hepatol. 2012;6(2):163–71.
Martel C, Esposti DD, Bouchet A, Brenner C, Lemoine A. Non-alcoholic steatohepatitis: new insights from OMICS studies. Curr Pharm Biotechnol. 2012;13(5):726–35.
Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21(1):27–41.
Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21(1):89–104.
Baranova A, Tran TP, Birerdinc A, Younossi ZM. Systematic review: association of polycystic ovary syndrome with metabolic syndrome and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2011;33(7):801–14.
Ortiz-Lopez C, Lomonaco R, Orsak B, Finch J, Chang Z, Kochunov VG, Hardies J, Cusi K. Prevalence of prediabetes and diabetes and metabolic profile of patients with nonalcoholic fatty liver disease (NAFLD). Diabetes Care. 2012;35(4):873–8.
Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K. Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet. 2011;26(1):30–46.
Serviddio G, Bellanti F, Vendemiale G, Altomare E. Mitochondrial dysfunction in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5(2):233–44.
Diehl AM. Tumor necrosis factor and its potential role in insulin resistance and nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8(3):619–38.
Jarrar MH, Baranova A, Collantes R, Ranard B, Stepanova M, Bennett C, Fang Y, Elariny H, Goodman Z, Chandhoke V, Younossi ZM. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;27(5):412–21.
Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115(5):911–9. quiz 920.
Lee YH, Pratley RE. The evolving role of inflammation in obesity and the metabolic syndrome. Curr Diab Rep. 2005;5(1):70–5.
Fain JN, Tichansky DS, Madan AK. Most of the interleukin 1 receptor antagonist, cathepsin S, macrophage migration inhibitory factor, nerve growth factor, and interleukin 18 release by explants of human adipose tissue is by the non-fat cells, not by the adipocytes. Metabolism. 2006;55(8):1113–21.
Wilkinson M, Brown R, Imran SA, Ur E. Adipokine gene expression in brain and pituitary gland. Neuroendocrinology. 2007;86(3):191–209.
Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, O’Rahilly S. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50(10):2199–202.
Nanji AA. Animal models of nonalcoholic fatty liver disease and steatohepatitis. Clin Liver Dis. 2004;8(3):559–74. ix.
Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song XY, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37(2):343–50.
Raso GM, Esposito E, Iacono A, Pacilio M, Cuzzocrea S, Canani RB, Calignano A, Meli R. Comparative therapeutic effects of metformin and vitamin E in a model of non-alcoholic steatohepatitis in the young rat. Eur J Pharmacol. 2009;604(1–3):125–31.
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7(8):947–53.
Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, Uchida S, Ito Y, Takakuwa K, Matsui J, Takata M, Eto K, Terauchi Y, Komeda K, Tsunoda M, Murakami K, Ohnishi Y, Naitoh T, Yamamura K, Ueyama Y, Froguel P, Kimura S, Nagai R, Kadowaki T. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278(4):2461–8.
Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112(1):91–100.
Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, Okamoto Y, Kihara S, Miyagawa J, Shinomura Y, Funahashi T, Matsuzawa Y. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125(6):1796–807.
Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, Yoshimatsu H. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology. 2004;40(1):177–84.
Yang RZ, Huang Q, Xu A, McLenithan JC, Eisen JA, Shuldiner AR, Alkan S, Gong DW. Comparative studies of resistin expression and phylogenomics in human and mouse. Biochem Biophys Res Commun. 2003;310(3):927–35. Erratum in: Biochem Biophys Res Commun. 2003 Dec 19;312(3):866. Eison Jonathan A [corrected to Eisen Jonathan A].
López-Bermejo A, Botas P, Funahashi T, Delgado E, Kihara S, Ricart W, Fernández-Real JM. Adiponectin, hepatocellular dysfunction and insulin sensitivity. Clin Endocrinol (Oxf). 2004;60(2):256–63.
Pagano C, Soardo G, Esposito W, Fallo F, Basan L, Donnini D, Federspil G, Sechi LA, Vettor R. Plasma adiponectin is decreased in nonalcoholic fatty liver disease. Eur J Endocrinol. 2005;152(1):113–8.
Mendez-Sanchez N, Chavez-Tapia NC, Villa AR, Sanchez-Lara K, Zamora-Valdes D, Ramos MH, Uribe M. Adiponectin as a protective factor in hepatic steatosis. World J Gastroenterol. 2005;11(12):1737–41.
Zou CC, Liang L, Hong F, Fu JF, Zhao ZY. Serum adiponectin, resistin levels and non-alcoholic fatty liver disease in obese children. Endocr J. 2005;52(5):519–24.
Bugianesi E, Pagotto U, Manini R, Vanni E, Gastaldelli A, de Iasio R, Gentilcore E, Natale S, Cassader M, Rizzetto M, Pasquali R, Marchesini G. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J Clin Endocrinol Metab. 2005;90(6):3498–504.
Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40(1):46–54.
Musso G, Gambino R, Biroli G, Carello M, Fagà E, Pacini G, De Michieli F, Cassader M, Durazzo M, Rizzetto M, Pagano G. Hypoadiponectinemia predicts the severity of hepatic fibrosis and pancreatic Beta-cell dysfunction in nondiabetic nonobese patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2005;100(11):2438–46.
Musso G, Gambino R, Durazzo M, Biroli G, Carello M, Fagà E, Pacini G, De Michieli F, Rabbione L, Premoli A, Cassader M, Pagano G. Adipokines in NASH: postprandial lipid metabolism as a link between adiponectin and liver disease. Hepatology. 2005;42(5):1175–83.
Comuzzie AG, Funahashi T, Sonnenberg G, Martin LJ, Jacob HJ, Black AE, Maas D, Takahashi M, Kihara S, Tanaka S, Matsuzawa Y, Blangero J, Cohen D, Kissebah A. The genetic basis of plasma variation in adiponectin, a global endophenotype for obesity and the metabolic syndrome. J Clin Endocrinol Metab. 2001;86(9):4321–5.
Lindsay RS, Funahashi T, Krakoff J, Matsuzawa Y, Tanaka S, Kobes S, Bennett PH, Tataranni PA, Knowler WC, Hanson RL. Genome-wide linkage analysis of serum adiponectin in the Pima Indian population. Diabetes. 2003;52(9):2419–25.
Menzaghi C, Ercolino T, Salvemini L, Coco A, Kim SH, Fini G, Doria A, Trischitta V. Multigenic control of serum adiponectin levels: evidence for a role of the APM1 gene and a locus on 14q13. Physiol Genomics. 2004;19(2):170–4. Epub 2004 Jul 13.
Menzaghi C, Ercolino T, Di Paola R, Berg AH, Warram JH, Scherer PE, Trischitta V, Doria A. A haplotype at the adiponectin locus is associated with obesity and other features of the insulin resistance syndrome. Diabetes. 2002;51(7):2306–12.
Gupta AC, Misra R, Sakhuja P, Singh Y, Basir SF, Sarin SK. Association of adiponectin gene functional polymorphisms (−11377C/G and +45T/G) with nonalcoholic fatty liver disease. Gene. 2012;496(1):63–7.
Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, et al. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem. 2004;279:12152–62.
Kantartzis K, Staiger H, Machann J, Schick F, Claussen CD, Machicao F, Fritsche A, Häring HU, Stefan N. Adiponectin oligomers and ectopic fat in liver and skeletal muscle in humans. Obesity (Silver Spring). 2009;17(2):390–2.
Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011;96(5):1424–30.
Kaser S, Moschen A, Cayon A, Kaser A, Crespo J, Pons-Romero F, Ebenbichler CF, Patsch JR, Tilg H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut. 2005;54(1):117–21.
Shimizu A, Takamura T, Matsuzawa N, Nakamura S, Nabemoto S, Takeshita Y, Misu H, Kurita S, Sakurai M, Yokoyama M, Zen Y, Sasaki M, Nakanuma Y, Kaneko S. Regulation of adiponectin receptor expression in human liver and a hepatocyte cell line. Metabolism. 2007;56(11):1478–85.
Vuppalanchi R, Marri S, Kolwankar D, Considine RV, Chalasani N. Is adiponectin involved in the pathogenesis of nonalcoholic steatohepatitis? A preliminary human study. J Clin Gastroenterol. 2005;39(3):237–42.
Carazo A, León J, Casado J, Gila A, Delgado S, Martín A, Sanjuan L, Caballero T, Muñoz JA, Quiles R, Ruiz-Extremera A, Alcázar LM, Salmerón J. Hepatic expression of adiponectin receptors increases with non-alcoholic fatty liver disease progression in morbid obesity in correlation with glutathione peroxidase. Obes Surg. 2011;21(4):492–500.
Ma H, Gomez V, Lu L, Yang X, Wu X, Xiao SY. Expression of adiponectin and its receptors in livers of morbidly obese patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2009;24(2):233–7.
Uribe M, Zamora-Valdés D, Moreno-Portillo M, Bermejo-Martínez L, Pichardo-Bahena R, Baptista-González HA, Ponciano-Rodríguez G, Uribe MH, Medina-Santillán R, Méndez-Sánchez N. Hepatic expression of ghrelin and adiponectin and their receptors in patients with nonalcoholic fatty liver disease. Ann Hepatol. 2008;7(1):67–71.
Stefan N, Machicao F, Staiger H, Machann J, Schick F, Tschritter O, Spieth C, Weigert C, Fritsche A, Stumvoll M, Häring HU. Polymorphisms in the gene encoding adiponectin receptor 1 are associated with insulin resistance and high liver fat. Diabetologia. 2005;48(11):2282–91.
Kotronen A, Yki-Järvinen H, Aminoff A, Bergholm R, Pietiläinen KH, Westerbacka J, Talmud PJ, Humphries SE, Hamsten A, Isomaa B, Groop L, Orho-Melander M, Ehrenborg E, Fisher RM. Genetic variation in the ADIPOR2 gene is associated with liver fat content and its surrogate markers in three independent cohorts. Eur J Endocrinol. 2009;160(4):593–602.
López-Bermejo A, Botas-Cervero P, Ortega-Delgado F, Delgado E, García-Gil MM, Funahashi T, Ricart W, Fernández-Real JM. Association of ADIPOR2 with liver function tests in type 2 diabetic subjects. Obesity (Silver Spring). 2008;16(10):2308–13.
Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun. 2005;334(4):1092–101.
Bajaj M, Suraamornkul S, Hardies LJ, Pratipanawatr T, DeFronzo RA. Plasma resistin concentration, hepatic fat content, and hepatic and peripheral insulin resistance in pioglitazone-treated type II diabetic patients. Int J Obes Relat Metab Disord. 2004;28(6):783–9.
Nobili V, Carpino G, Alisi A, Franchitto A, Alpini G, De Vito R, Onori P, Alvaro D, Gaudio E. Hepatic progenitor cells activation, fibrosis and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology. 2012;56(6):2142–53.
Lebensztejn DM, Wojtkowska M, Skiba E, Werpachowska I, Tobolczyk J, Kaczmarski M. Serum concentration of adiponectin, leptin and resistin in obese children with non-alcoholic fatty liver disease. Adv Med Sci. 2009;54(2):177–82.
Le D, Marks D, Lyle E, Corless CL, Diggs BS, Jobe BA, Kay T, Deveney CW, Wolfe BM, Roberts Jr CT, O’Rourke RW. Serum leptin levels, hepatic leptin receptor transcription, and clinical predictors of non-alcoholic steatohepatitis in obese bariatric surgery patients. Surg Endosc. 2007;21(9):1593–9.
Schaab M, Kausch H, Klammt J, Nowicki M, Anderegg U, Gebhardt R, Rose-John S, Scheller J, Thiery J, Kratzsch J. Novel regulatory mechanisms for generation of the soluble leptin receptor: implications for leptin action. PLoS One. 2012;7(4):e34787.
Huang XD, Fan Y, Zhang H, Wang P, Yuan JP, Li MJ, Zhan XY. Serum leptin and soluble leptin receptor in non-alcoholic fatty liver disease. World J Gastroenterol. 2008;14(18):2888–93.
Medici V, Ali MR, Seo S, Aoki CA, Rossaro L, Kim K, Fuller WD, Vidovszky TJ, Smith W, Jiang JX, Maganti K, Havel PJ, Kamboj A, Ramsamooj R, Török NJ. Increased soluble leptin receptor levels in morbidly obese patients with insulin resistance and nonalcoholic fatty liver disease. Obesity (Silver Spring). 2010;18(12):2268–73.
Ge F, Zhou S, Hu C, Lobdell IV H, Berk PD. Insulin- and leptin-regulated fatty acid uptake plays a key causal role in hepatic steatosis in mice with intact leptin signaling but not in ob/ob or db/db mice. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G855–66.
Shamsuzzaman AS, Winnicki M, Wolk R, Svatikova A, Phillips BG, Davison DE, Berger PB, Somers VK. Independent association between plasma leptin and C-reactive protein in healthy humans. Circulation. 2004;109(18):2181–5.
Wang J, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, Brigstock D, George J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology. 2009;137(2):713–23.
Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109(10):1345–50.
Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54(7):1994–2002.
Chan JL, Lutz K, Cochran E, Huang W, Peters Y, Weyer C, Gorden P. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr Pract. 2011;17(6):922–32.
Swellam M, Hamdy N. Association of nonalcoholic fatty liver disease with a single nucleotide polymorphism on the gene encoding leptin receptor. IUBMB Life. 2012;64(2):180–6.
Lu H, Sun J, Sun L, Shu X, Xu Y, Xie D. Polymorphism of human leptin receptor gene is associated with type 2 diabetic patients complicated with non-alcoholic fatty liver disease in China. J Gastroenterol Hepatol. 2009;24(2):228–32.
Aller R, De Luis DA, Izaola O, González Sagrado M, Conde R, Pacheco D, Velasco MC, Ovalle HF. Lys656Asn polymorphism of leptin receptor, leptin levels and insulin resistance in patients with non alcoholic fatty liver disease. Eur Rev Med Pharmacol Sci. 2012;16(3):335–41.
Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm. 2006;74:443–77.
Crespo J, Cayón A, Fernández-Gil P, Hernández-Guerra M, Mayorga M, Domínguez-Díez A, Fernández-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34(6):1158–63.
Wang JK, Feng ZW, Li YC, Li QY, Tao XY. Association of tumor necrosis factor-α gene promoter polymorphism at sites −308 and −238 with non-alcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. 2012;27(4):670–6.
Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99(12):2365–8.
Satapathy SK, Garg S, Chauhan R, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of tumor necrosis factor-alpha inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99(10):1946–52.
Zein CO, Yerian LM, Gogate P, Lopez R, Kirwan JP, Feldstein AE, McCullough AJ. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54(5):1610–9.
Li W, Zheng L, Sheng C, Cheng X, Qing L, Qu S. Systematic review on the treatment of pentoxifylline in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2011;10:49.
Estep M, Armistead D, Hossain N, Elarainy H, Goodman Z, Baranova A, Chandhoke V, Younossi ZM. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2010;32(3):487–97.
Fain JN. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediators Inflamm. 2010;2010:513948.
Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145(5):2273–82.
Fain JN, Bahouth SW, Madan AK. Involvement of multiple signaling pathways in the post-bariatric induction of IL-6 and IL-8 mRNA and release in human visceral adipose tissue. Biochem Pharmacol. 2005;69(9):1315–24.
Bahcecioglu IH, Yalniz M, Ataseven H, Ilhan N, Ozercan IH, Seckin D, Sahin K. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepatogastroenterology. 2005;52(65):1549–53.
Coulon S, Francque S, Colle I, Verrijken A, Blomme B, Heindryckx F, De Munter S, Prawitt J, Caron S, Staels B, Van Vlierberghe H, Van Gaal L, Geerts A. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine. 2012;59(2):442–9.
Teoh N, Field J, Farrell G. Interleukin-6 is a key mediator of the hepatoprotective and pro-proliferative effects of ischaemic preconditioning in mice. J Hepatol. 2006;45(1):20–7.
Gao B. Therapeutic potential of interleukin-6 in preventing obesity- and alcohol-associated fatty liver transplant failure. Alcohol. 2004;34(1):59–65.
Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2003;100:7265–70.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505.
Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, Trautwein C, Tacke F. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61(3):416–26.
Kirovski G, Dorn C, Huber H, Moleda L, Niessen C, Wobser H, Schacherer D, Buechler C, Wiest R, Hellerbrand C. Elevated systemic monocyte chemoattractrant protein-1 in hepatic steatosis without significant hepatic inflammation. Exp Mol Pathol. 2011;91(3):780–3.
Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, Blin-Wakkach C, Anty R, Iannelli A, Gugenheim J, Tran A, Bouloumié A, Gual P, Wakkach A. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61(9):2238–47.
Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, Han X, Peng Y, Chen X, Shen L, Qiu D, Li Z, Ma X. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. 2011;166(2):281–90.
Charlton M, Sreekumar R, Rasmussen D, Lindor K, Nair KS. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology. 2002;35(4):898–904.
Chan DC, Watts GF, Ooi EM, Chan DT, Wong AT, Barrett PH. Apolipoprotein A-II and adiponectin as determinants of very low-density lipoprotein apolipoprotein B-100 metabolism in nonobese men. Metabolism. 2011;60(10):1482–7.
Ng TW, Watts GF, Farvid MS, Chan DC, Barrett PH. Adipocytokines and VLDL metabolism: independent regulatory effects of adiponectin, insulin resistance, and fat compartments on VLDL apolipoprotein B-100 kinetics? Diabetes. 2005;54(3):795–802.
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–95. Epub 2002 Oct 7.
Ganguly R, Schram K, Fang X, Kim M, Rodrigues B, Thong FS, Sweeney G. Adiponectin increases LPL activity via RhoA/ROCK-mediated actin remodelling in adult rat cardiomyocytes. Endocrinology. 2011;152(1):247–54.
von Eynatten M, Schneider JG, Humpert PM, Rudofsky G, Schmidt N, Barosch P, Hamann A, Morcos M, Kreuzer J, Bierhaus A, Nawroth PP, Dugi KA. Decreased plasma lipoprotein lipase in hypoadiponectinemia: an association independent of systemic inflammation and insulin resistance. Diabetes Care. 2004;27(12):2925–9.
De Vries R, Wolffenbuttel BH, Sluiter WJ, van Tol A, Dullaart RP. Post-heparin plasma lipoprotein lipase, but not hepatic lipase activity, is related to plasma adiponectin in type 2 diabetic patients and healthy subjects. Clin Lab. 2005;51(7–8):403–9.
Loucif Y, Méthot J, Tremblay K, Brisson D, Gaudet D. Contribution of adiponectin to the cardiometabolic risk of postmenopausal women with loss-of-function lipoprotein lipase gene mutations. Menopause. 2011;18(5):558–62.
Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55(9):2562–70.
Shetty S, Ramos-Roman MA, Cho YR, Brown J, Plutzky J, Muise ES, Horton JD, Scherer PE, Parks EJ. Enhanced fatty acid flux triggered by adiponectin overexpression. Endocrinology. 2012;153(1):113–22.
Shimabukuro M, Koyama K, Chen G, Wang MY, Trieu F, Lee Y, Newgard CB, Unger RH. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci U S A. 1997;94(9):4637–41.
Lee Y, Wang MY, Kakuma T, Wang ZW, Babcock E, McCorkle K, Higa M, Zhou YT, Unger RH. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J Biol Chem. 2001;276(8):5629–35. Epub 2000 Nov 28.
Wang Y, Kuropatwinski KK, White DW, Hawley TS, Hawley RG, Tartaglia LA, Baumann H. Leptin receptor action in hepatic cells. J Biol Chem. 1997;272(26):16216–23.
Cohen P, Friedman JM. Leptin and the control of metabolism: role for stearoyl-CoA desaturase-1 (SCD-1). J Nutr. 2004;134(9):2455S–63. Review.
Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, Soukas AA, Kahn CR, Ntambi JM, Socci ND, Friedman JM. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113(3):414–24.
Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. 2000;275(39):30132–8.
Lee Y, Yu X, Gonzales F, Mangelsdorf DJ, Wang MY, Richardson C, Witters LA, Unger RH. PPAR alpha is necessary for the lipopenic action of hyperleptinemia on white adipose and liver tissue. Proc Natl Acad Sci U S A. 2002;99(18):11848–53. Epub 2002 Aug 23.
Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415(6869):339–43.
VanPatten S, Ranginani N, Shefer S, Nguyen LB, Rossetti L, Cohen DE. Impaired biliary lipid secretion in obese Zucker rats: leptin promotes hepatic cholesterol clearance. Am J Physiol Gastrointest Liver Physiol. 2001;281(2):G393–404.
Méndez-Sánchez N, Ponciano-Rodrigoez G, Chavez-Tapia N, Uribe M. Effects of leptin on biliary lipids: potential consequences for gallstone formation and therapy in obesity. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5(2):203–8.
Nobili V, Manco M, Ciampalini P, Diciommo V, Devito R, Piemonte F, Comparcola D, Guidi R, Marcellini M. Leptin, free leptin index, insulin resistance and liver fibrosis in children with non-alcoholic fatty liver disease. Eur J Endocrinol. 2006;155(5):735–43.
Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J Clin Invest. 2004;114(2):224–31.
Sato N, Kobayashi K, Inoguchi T, Sonoda N, Imamura M, Sekiguchi N, Nakashima N, Nawata H. Adenovirus-mediated high expression of resistin causes dyslipidemia in mice. Endocrinology. 2005;146(1):273–9. Epub 2004 Oct 7.
Singhal NS, Patel RT, Qi Y, Lee YS, Ahima RS. Loss of resistin ameliorates hyperlipidemia and hepatic steatosis in leptin-deficient mice. Am J Physiol Endocrinol Metab. 2008;295(2):E331–8.
Chen CC, Li TC, Li CI, Liu CS, Wang HJ, Lin CC. Serum resistin level among healthy subjects: relationship to anthropometric and metabolic parameters. Metabolism. 2005;54(4):471–5.
Costandi J, Melone M, Zhao A, Rashid S. Human resistin stimulates hepatic overproduction of atherogenic ApoB-containing lipoprotein particles by enhancing ApoB stability and impairing intracellular insulin signaling. Circ Res. 2011;108(6):727–42.
Fon Tacer K, Kuzman D, Seliskar M, Pompon D, Rozman D. TNF-alpha interferes with lipid homeostasis and activates acute and proatherogenic processes. Physiol Genomics. 2007;31(2):216–27.
Qin B, Anderson RA, Adeli K. Tumor necrosis factor-alpha directly stimulates the overproduction of hepatic apolipoprotein B100-containing VLDL via impairment of hepatic insulin signaling. Am J Physiol Gastrointest Liver Physiol. 2008;294(5):G1120–9.
Glosli H, Gudbrandsen OA, Mullen AJ, Halvorsen B, Røst TH, Wergedahl H, Prydz H, Aukrust P, Berge RK. Down-regulated expression of PPARalpha target genes, reduced fatty acid oxidation and altered fatty acid composition in the liver of mice transgenic for hTNFalpha. Biochim Biophys Acta. 2005;1734(3):235–46.
Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, Olefsky JM, Lodish HF. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes. 2002;51:3176–88.
Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16(17):1941–51.
Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond). 2008;32 Suppl 7:S13–8.
Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes. 2005;54(5):1331–9.
Parulkar AA, Pendergrass ML, Granda-Ayala R, Lee TR, Fonseca VA. Nonhypoglycemic effects of thiazolidinediones. Ann Intern Med. 2001;134(1):61–71. Review. Erratum in: Ann Intern Med 2001 Aug 21;135(4):307.
Watkins SM, Reifsnyder PR, Pan HJ, German JB, Leiter EH. Lipid metabolome-wide effects of the PPARgamma agonist rosiglitazone. J Lipid Res. 2002;43(11):1809–17.
Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest. 2011;121(6):2518–28.
Cowherd RB, Asmar MM, Alderman JM, Alderman EA, Garland AL, Busby WH, Bodnar WM, Rusyn I, Medoff BD, Tisch R, Mayer-Davis E, Swenberg JA, Zeisel SH, Combs TP. Adiponectin lowers glucose production by increasing SOGA. Am J Pathol. 2010;177(4):1936–45.
Tishinsky JM, Robinson LE, Dyck DJ. Insulin-sensitizing properties of adiponectin. Biochimie. 2012;94(10):2131–6.
Anubhuti, Arora S. Leptin and its metabolic interactions: an update. Diabetes Obes Metab. 2008;10(11):973–93.
Cummings BP, Bettaieb A, Graham JL, Stanhope KL, Dill R, Morton GJ, Haj FG, Havel PJ. Subcutaneous administration of leptin normalizes fasting plasma glucose in obese type 2 diabetic UCD-T2DM rats. Proc Natl Acad Sci U S A. 2011;108(35):14670–5.
Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers Jr MG. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275(51):40649–57.
Tom RZ, Sjögren RJ, Vieira E, Glund S, Iglesias-Gutiérrez E, Garcia-Roves PM, Myers Jr MG, Björnholm M. Increased hepatic insulin sensitivity in mice lacking inhibitory leptin receptor signals. Endocrinology. 2011;152(6):2237–46.
Lam NT, Lewis JT, Cheung AT, Luk CT, Tse J, Wang J, Bryer-Ash M, Kolls JK, Kieffer TJ. Leptin increases hepatic insulin sensitivity and protein tyrosine phosphatase 1B expression. Mol Endocrinol. 2004;18(6):1333–45.
Brabant G, Müller G, Horn R, Anderwald C, Roden M, Nave H. Hepatic leptin signaling in obesity. FASEB J. 2005;19(8):1048–50.
Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA. Regulation of fasted blood glucose by resistin. Science. 2004;303(5661):1195–8.
Gerber M, Boettner A, Seidel B, Lammert A, Bär J, Schuster E, Thiery J, Kiess W, Kratzsch J. Serum resistin levels of obese and lean children and adolescents: biochemical analysis and clinical relevance. J Clin Endocrinol Metab. 2005;90(8):4503–9. Epub 2005 May 31.
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389(6651):610–4.
Gupta D, Khandelwal RL. Modulation of insulin effects on phosphorylation of protein kinase B and glycogen synthesis by tumor necrosis factor-alpha in HepG2 cells. Biochim Biophys Acta. 2004;1671(1–3):51–8.
Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277(50):48115–21. Epub 2002 Sep 25.
Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord. 2003;27 Suppl 3:S53–5. Review.
Nohara A, Okada S, Ohshima K, Pessin JE, Mori M. Cyclin-dependent kinase-5 is a key molecule in tumor necrosis factor-α-induced insulin resistance. J Biol Chem. 2011;286(38):33457–65.
Brown JE, Onyango DJ, Ramanjaneya M, Conner AC, Patel ST, Dunmore SJ, Randeva HS. Visfatin regulates insulin secretion, insulin receptor signalling and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J Mol Endocrinol. 2010;44(3):171–8.
Skop V, Kontrová K, Zídek V, Pravenec M, Kazdová L, Mikulík K, Sajdok J, Zídková J. Autocrine effects of visfatin on hepatocyte sensitivity to insulin action. Physiol Res. 2010;59(4):615–8.
Hida K, Wada J, Eguchi J, Zhang H, Baba M, Seida A, Hashimoto I, Okada T, Yasuhara A, Nakatsuka A, Shikata K, Hourai S, Futami J, Watanabe E, Matsuki Y, Hiramatsu R, Akagi S, Makino H, Kanwar YS. Visceral adipose tissue-derived serine protease inhibitor: a unique insulin-sensitizing adipocytokine in obesity. Proc Natl Acad Sci U S A. 2005;102(30):10610–5. Epub 2005 Jul 19.
Jeong E, Youn BS, Kim DW, Kim EH, Park JW, Namkoong C, Jeong JY, Yoon SY, Park JY, Lee KU, Kim MS. Circadian rhythm of serum vaspin in healthy male volunteers: relation to meals. J Clin Endocrinol Metab. 2010;95(4):1869–75.
Teshigawara S, Wada J, Hida K, Nakatsuka A, Eguchi J, Murakami K, Kanzaki M, Inoue K, Terami T, Katayama A, Iseda I, Matsushita Y, Miyatake N, McDonald JF, Hotta K, Makino H. Serum vaspin concentrations are closely related to insulin resistance, and rs77060950 at SERPINA12 genetically defines distinct group with higher serum levels in Japanese population. J Clin Endocrinol Metab. 2012;97(7):E1202–7.
Klöting N, Berndt J, Kralisch S, Kovacs P, Fasshauer M, Schön MR, Stumvoll M, Blüher M. Vaspin gene expression in human adipose tissue: association with obesity and type 2 diabetes. Biochem Biophys Res Commun. 2006;339(1):430–6.
Sörhede Winzell M, Magnusson C, Ahrén B. The apj receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul Pept. 2005;131(1–3):12–7.
Boucher J, Masri B, Daviaud D, Gesta S, Guigné C, Mazzucotelli A, Castan-Laurell I, Tack I, Knibiehler B, Carpéné C, Audigier Y, Saulnier-Blache JS, Valet P. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146(4):1764–71. Epub 2005 Jan 27.
Ercin CN, Dogru T, Tapan S, Kara M, Haymana C, Karadurmus N, Karslioglu Y, Acikel C. Plasma apelin levels in subjects with nonalcoholic fatty liver disease. Metabolism. 2010;59(7):977–81.
Ernst MC, Issa M, Goralski KB, Sinal CJ. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology. 2010;151(5):1998–2007.
Ernst MC, Haidl ID, Zúñiga LA, Dranse HJ, Rourke JL, Zabel BA, Butcher EC, Sinal CJ. Disruption of the chemokine-like receptor-1 (CMKLR1) gene is associated with reduced adiposity and glucose intolerance. Endocrinology. 2012;153(2):672–82.
Takahashi M, Okimura Y, Iguchi G, Nishizawa H, Yamamoto M, Suda K, Kitazawa R, Fujimoto W, Takahashi K, Zolotaryov FN, Hong KS, Kiyonari H, Abe T, Kaji H, Kitazawa S, Kasuga M, Chihara K, Takahashi Y. Chemerin regulates β-cell function in mice. Sci Rep. 2011;1:123.
Takahashi M, Takahashi Y, Takahashi K, Zolotaryov FN, Hong KS, Kitazawa R, Iida K, Okimura Y, Kaji H, Kitazawa S, Kasuga M, Chihara K. Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett. 2008;582(5):573–8.
Suzuki Y, Song SH, Sato K, So KH, Ardiyanti A, Kitayama S, Hong YH, Lee SD, Choi KC, Hagino A, Katoh K, Roh SG. Chemerin analog regulates energy metabolism in sheep. Anim Sci J. 2012;83(3):263–7.
Wanninger J, Bauer S, Eisinger K, Weiss TS, Walter R, Hellerbrand C, Schäffler A, Higuchi A, Walsh K, Buechler C. Adiponectin upregulates hepatocyte CMKLR1 which is reduced in human fatty liver. Mol Cell Endocrinol. 2012;349(2):248–54.
Yang M, Zhang Z, Wang C, Li K, Li S, Boden G, Li L, Yang G. Nesfatin-1 action in the brain increases insulin sensitivity through Akt/AMPK/TORC2 pathway in diet-induced insulin resistance. Diabetes. 2012;61(8):1959–68.
Gonzalez R, Reingold BK, Gao X, Gaidhu MP, Tsushima RG, Unniappan S. Nesfatin-1 exerts a direct, glucose-dependent insulinotropic action on mouse islet β- and MIN6 cells. J Endocrinol. 2011;208(3):R9–16.
Nakata M, Manaka K, Yamamoto S, Mori M, Yada T. Nesfatin-1 enhances glucose-induced insulin secretion by promoting Ca(2+) influx through L-type channels in mouse islet β-cells. Endocr J. 2011;58(4):305–13.
Zhang Z, Li L, Yang M, Liu H, Boden G, Yang G. Increased plasma levels of nesfatin-1 in patients with newly diagnosed type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 2012;120(2):91–5.
Deniz R, Gurates B, Aydin S, Celik H, Sahin I, Baykus Y, Catak Z, Aksoy A, Citil C, Gungor S. Nesfatin-1 and other hormone alterations in polycystic ovary syndrome. Endocrine. 2012;42(3):694–9.
Tan BK, Hallschmid M, Kern W, Lehnert H, Randeva HS. Decreased cerebrospinal fluid/plasma ratio of the novel satiety molecule, nesfatin-1/NUCB-2, in obese humans: evidence of nesfatin-1/NUCB-2 resistance and implications for obesity treatment. J Clin Endocrinol Metab. 2011;96(4):E669–73.
Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, Shuldiner AR, Fried SK, McLenithan JC, Gong DW. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. 2006;290(6):E1253–61.
Tan BK, Adya R, Farhatullah S, Lewandowski KC, O’Hare P, Lehnert H, Randeva HS. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes. 2008;57(4):801–8.
Yan P, Liu D, Long M, Ren Y, Pang J, Li R. Changes of serum omentin levels and relationship between omentin and adiponectin concentrations in type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 2011;119(4):257–63.
Nakanishi S, Yamane K, Kamei N, Nojima H, Okubo M, Kohno N. A protective effect of adiponectin against oxidative stress in Japanese Americans: the association between adiponectin or leptin and urinary isoprostane. Metabolism. 2005;54(2):194–9.
Soares AF, Guichardant M, Cozzone D, Bernoud-Hubac N, Bouzaïdi-Tiali N, Lagarde M, Géloën A. Effects of oxidative stress on adiponectin secretion and lactate production in 3T3-L1 adipocytes. Free Radic Biol Med. 2005;38(7):882–9.
Yuan F, Li YN, Liu YH, Yi B, Tian JW, Liu FY. Adiponectin inhibits the generation of reactive oxygen species induced by high glucose and promotes endothelial NO synthase formation in human mesangial cells. Mol Med Rep. 2012;6(2):449–53.
Chedid P, Hurtado-Nedelec M, Marion-Gaber B, Bournier O, Hayem G, Gougerot-Pocidalo MA, Frystyk J, Flyvbjerg A, El Benna J, Marie JC. Adiponectin and its globular fragment differentially modulate the oxidative burst of primary human phagocytes. Am J Pathol. 2012;180(2):682–92.
Zhou M, Xu A, Tam PK, Lam KS, Huang B, Liang Y, Lee IK, Wu D, Wang Y. Upregulation of UCP2 by adiponectin: the involvement of mitochondrial superoxide and hnRNP K. PLoS One. 2012;7(2):e32349.
Huang W, Dedousis N, Bandi A, Lopaschuk GD, O’Doherty RM. Liver triglyceride secretion and lipid oxidative metabolism are rapidly altered by leptin in vivo. Endocrinology. 2006;147(3):1480–7.
Sailaja JB, Balasubramaniyan V, Nalini N. Effect of exogenous leptin administration on high fat diet induced oxidative stress. Pharmazie. 2004;59(6):475–9.
Balasubramaniyan V, Kalaivani Sailaja J, Nalini N. Role of leptin on alcohol-induced oxidative stress in Swiss mice. Pharmacol Res. 2003;47(3):211–6.
Oztay F, Kandil A, Gurel E, Ustunova S, Kapucu A, Balci H, Akgun-Dar K, Demirci C. The relationship between nitric oxide and leptin in the lung of rat with streptozotocin-induced diabetes. Cell Biochem Funct. 2008;26(2):162–71.
Förstermann U, Li H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol. 2011;164(2):213–23.
Leclercq IA, Field J, Enriquez A, Farrell GC, Robertson GR. Constitutive and inducible expression of hepatic CYP2E1 in leptin-deficient ob/ob mice. Biochem Biophys Res Commun. 2000;268(2):337–44.
Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42.
Aubert J, Begriche K, Knockaert L, Robin MA, Fromenty B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin Res Hepatol Gastroenterol. 2011;35(10):630–7.
Kougias P, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Adipocyte-derived cytokine resistin causes endothelial dysfunction of porcine coronary arteries. J Vasc Surg. 2005;41(4):691–8.
Chemaly ER, Hadri L, Zhang S, Kim M, Kohlbrenner E, Sheng J, Liang L, Chen J, K-Raman P, Hajjar RJ, Lebeche D. Long-term in vivo resistin overexpression induces myocardial dysfunction and remodeling in rats. J Mol Cell Cardiol. 2011;51(2):144–55.
Smith SR, Bai F, Charbonneau C, Janderová L, Argyropoulos G. A promoter genotype and oxidative stress potentially link resistin to human insulin resistance. Diabetes. 2003;52(7):1611–8.
Szalowska E, Elferink MG, Hoek A, Groothuis GM, Vonk RJ. Resistin is more abundant in liver than adipose tissue and is not up-regulated by lipopolysaccharide. J Clin Endocrinol Metab. 2009;94(8):3051–7.
Fernández-Checa JC. Alcohol-induced liver disease: when fat and oxidative stress meet. Ann Hepatol. 2003;2(2):69–75. Review.
García-Ruiz C, Colell A, Marí M, Morales A, Fernández-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272(17):11369–77.
Arora AS, Jones BJ, Patel TC, Bronk SF, Gores GJ. Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat. Hepatology. 1997;25(4):958–63.
Chen XH, Zhao YP, Xue M, Ji CB, Gao CL, Zhu JG, Qin DN, Kou CZ, Qin XH, Tong ML, Guo XR. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol Cell Endocrinol. 2010;328(1–2):63–9.
Nagai H, Matsumaru K, Feng G, Kaplowitz N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor-alpha-induced apoptosis in cultured mouse hepatocytes. Hepatology. 2002;36(1):55–64.
Glosli H, Tronstad KJ, Wergedal H, Müller F, Svardal A, Aukrust P, Berge RK, Prydz H. Human TNF-alpha in transgenic mice induces differential changes in redox status and glutathione-regulating enzymes. FASEB J. 2002;16(11):1450–2. Epub 2002 Jul 18.
Nobili V, Pastore A, Gaeta LM, Tozzi G, Comparcola D, Sartorelli MR, Marcellini M, Bertini E, Piemonte F. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin Chim Acta. 2005;355(1–2):105–11.
Cortez-Pinto H, Yang SQ, Lin HZ, Costa S, Hwang CS, Lane MD, Bagby G, Diehl AM. Bacterial lipopolysaccharide induces uncoupling protein-2 expression in hepatocytes by a tumor necrosis factor-alpha-dependent mechanism. Biochem Biophys Res Commun. 1998;251(1):313–9.
Baffy G, Zhang CY, Glickman JN, Lowell BB. Obesity-related fatty liver is unchanged in mice deficient for mitochondrial uncoupling protein 2. Hepatology. 2002;35(4):753–61.
Park JW, Jeong G, Kim SJ, Kim MK, Park SM. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol. 2007;22(4):491–7.
Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–36.
Zhang LQ, Adyshev DM, Singleton P, Li H, Cepeda J, Huang SY, Zou X, Verin AD, Tu J, Garcia JG, Ye SQ. Interactions between PBEF and oxidative stress proteins—a potential new mechanism underlying PBEF in the pathogenesis of acute lung injury. FEBS Lett. 2008;582(13):1802–8.
Montecucco F, Bauer I, Braunersreuther V, Bruzzone S, Akhmedov A, Lüscher TF, Speer T, Poggi A, Mannino E, Pelli G, Galan K, Bertolotto M, Lenglet S, Garuti A, Montessuit C, Lerch R, Pellieux C, Vuilleumier N, Dallegri F, Mage J, Sebastian C, Mostoslavsky R, Gayet-Ageron A, Patrone F, Mach F, Nencioni A. Inhibition of nicotinamide phosphoribosyltransferase reduces neutrophil-mediated injury in myocardial infarction. Antioxid Redox Signal. 2013;18(6):630–41.
Boini KM, Zhang C, Xia M, Han WQ, Brimson C, Poklis JL, Li PL. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys Acta. 2010;1801(12):1294–304.
Xia M, Zhang C, Boini KM, Thacker AM, Li PL. Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc Res. 2011;89(2):401–9.
Hashimoto T, Kihara M, Imai N, Yoshida S, Shimoyamada H, Yasuzaki H, Ishida J, Toya Y, Kiuchi Y, Hirawa N, Tamura K, Yazawa T, Kitamura H, Fukamizu A, Umemura S. Requirement of apelin-apelin receptor system for oxidative stress-linked atherosclerosis. Am J Pathol. 2007;171(5):1705–12.
Foussal C, Lairez O, Calise D, Pathak A, Guilbeau-Frugier C, Valet P, Parini A, Kunduzova O. Activation of catalase by apelin prevents oxidative stress-linked cardiac hypertrophy. FEBS Lett. 2010;584(11):2363–70.
Attané C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E, Guzmán-Ruiz R, Dray C, Bezaire V, Rancoule C, Kuba K, Ruiz-Gayo M, Levade T, Penninger J, Burcelin R, Pénicaud L, Valet P, Castan-Laurell I. Apelin treatment increases complete fatty acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin-resistant mice. Diabetes. 2012;61(2):310–20.
Weigert J, Neumeier M, Wanninger J, Filarsky M, Bauer S, Wiest R, Farkas S, Scherer MN, Schäffler A, Aslanidis C, Schölmerich J, Buechler C. Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin Endocrinol (Oxf). 2010;72(3):342–8.
Kukla M, Zwirska-Korczala K, Hartleb M, Waluga M, Chwist A, Kajor M, Ciupinska-Kajor M, Berdowska A, Wozniak-Grygiel E, Buldak R. Serum chemerin and vaspin in non-alcoholic fatty liver disease. Scand J Gastroenterol. 2010;45(2):235–42.
Yamawaki H, Kuramoto J, Kameshima S, Usui T, Okada M, Hara Y. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. 2011;408(2):339–43.
Ratziu V, Poynard T. NASH: a hidden and silent fibroser finally revealed? J Hepatol. 2005;42(1):12–4.
Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T, Sato N. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122(5):1399–410.
Lu B, Yu L, Li S, Si S, Zeng Y. Alleviation of CCl4-induced cirrhosis in rats by tetramethylpyrazine is associated with downregulation of leptin and TGF-beta1 pathway. Drug Chem Toxicol. 2010;33(3):310–5.
Potter JJ, Rennie-Tankesley L, Mezey E. Influence of leptin in the development of hepatic fibrosis produced in mice by Schistosoma mansoni infection and by chronic carbon tetrachloride administration. J Hepatol. 2003;38(3):281–8.
Choudhury J, Mirshahi F, Murthy KS, Yager DR, Sanyal AJ. Physiologic concentrations of leptin increase collagen production by non-immortalized human hepatic stellate cells. Metabolism. 2006;55(10):1317–22.
Niu L, Wang X, Li J, Huang Y, Yang Z, Chen F, Ni H, Jin Y, Lu X, Cao Q. Leptin stimulates alpha1(I) collagen expression in human hepatic stellate cells via the phosphatidylinositol 3-kinase/Akt signalling pathway. Liver Int. 2007;27(9):1265–72.
Cao Q, Mak KM, Lieber CS. Leptin enhances alpha1(I) collagen gene expression in LX-2 human hepatic stellate cells through JAK-mediated H2O2-dependent MAPK pathways. J Cell Biochem. 2006;97(1):188–97.
Cao Q, Mak KM, Ren C, Lieber CS. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279(6):4292–304. Epub 2003 Nov 18.
Liu Y, Brymora J, Zhang H, Smith B, Ramezani-Moghadam M, George J, Wang J. Leptin and acetaldehyde synergistically promotes αSMA expression in hepatic stellate cells by an interleukin 6-dependent mechanism. Alcohol Clin Exp Res. 2011;35(5):921–8.
Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, Kleiner DE, Gorden P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41(4):753–60.
Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166(6):1655–69. Erratum in: Am J Pathol. 2008 Dec;173(6):1929. Xu, Amin [corrected to Xu, Aimin].
Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47(2):677–85.
Handy JA, Saxena NK, Fu P, Lin S, Mells JE, Gupta NA, Anania FA. Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3). J Cell Biochem. 2010;110(5):1195–207.
Handy JA, Fu PP, Kumar P, Mells JE, Sharma S, Saxena NK, Anania FA. Adiponectin inhibits leptin signalling via multiple mechanisms to exert protective effects against hepatic fibrosis. Biochem J. 2011;440(3):385–95.
Lim JY, Oh MA, Kim WH, Sohn HY, Park SI. AMP-activated protein kinase inhibits TGF-β-induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. J Cell Physiol. 2012;227(3):1081–9.
Tsochatzis E, Papatheodoridis GV, Hadziyannis E, Georgiou A, Kafiri G, Tiniakos DG, Manesis EK, Archimandritis AJ. Serum adipokine levels in chronic liver diseases: association of resistin levels with fibrosis severity. Scand J Gastroenterol. 2008;43(9):1128–36.
Kakizaki S, Sohara N, Yamazaki Y, Horiguchi N, Kanda D, Kabeya K, Katakai K, Sato K, Takagi H, Mori M. Elevated plasma resistin concentrations in patients with liver cirrhosis. J Gastroenterol Hepatol. 2008;23(1):73–7.
Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, Phan SH. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol. 2004;164(4):1315–26.
Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, Han JY, Kato S, Shimoda M, Oike Y, Tomizawa M, Makino S, Ohkura T, Saito H, Kumagai N, Nagata H, Ishii H, Hibi T. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. 2006;55(3):415–24.
Kitamura K, Nakamoto Y, Akiyama M, Fujii C, Kondo T, Kobayashi K, Kaneko S, Mukaida N. Pathogenic roles of tumor necrosis factor receptor p55-mediated signals in dimethylnitrosamine-induced murine liver fibrosis. Lab Invest. 2002;82(5):571–83.
Tarrats N, Moles A, Morales A, García-Ruiz C, Fernández-Checa JC, Marí M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology. 2011;54(1):319–27.
Principe A, Melgar-Lesmes P, Fernández-Varo G, del Arbol LR, Ros J, Morales-Ruiz M, Bernardi M, Arroyo V, Jiménez W. The hepatic apelin system: a new therapeutic target for liver disease. Hepatology. 2008;48(4):1193–201.
Reichenbach V, Ros J, Fernández-Varo G, Casals G, Melgar-Lesmes P, Campos T, Makriyannis A, Morales-Ruiz M, Jiménez W. Prevention of fibrosis progression in CCl4-treated rats: role of the hepatic endocannabinoid and apelin systems. J Pharmacol Exp Ther. 2012;340(3):629–37.
Ronkainen VP, Ronkainen JJ, Hänninen SL, Leskinen H, Ruas JL, Pereira T, Poellinger L, Vuolteenaho O, Tavi P. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. FASEB J. 2007;21(8):1821–30.
Daviaud D, Boucher J, Gesta S, Dray C, Guigne C, Quilliot D, Ayav A, Ziegler O, Carpene C, Saulnier-Blache JS, Valet P, Castan-Laurell I. TNFalpha up-regulates apelin expression in human and mouse adipose tissue. FASEB J. 2006;20(9):1528–30.
Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau-Frugier C, Schaak S, Seguelas MH, Wanecq E, Valet P, Parini A, Kunduzova O. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J. 2012;33(18):2360–9.
Siddiquee K, Hampton J, Khan S, Zadory D, Gleaves L, Vaughan DE, Smith LH. Apelin protects against angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J Hypertens. 2011;29(4):724–31.
Yu XY, Qiao SB, Guan HS, Liu SW, Meng XM. Effects of visfatin on proliferation and collagen synthesis in rat cardiac fibroblasts. Horm Metab Res. 2010;42(7):507–13.
Kukla M, Ciupińska-Kajor M, Kajor M, Wyleżoł M, Żwirska-Korczala K, Hartleb M, Berdowska A, Mazur W. Liver visfatin expression in morbidly obese patients with nonalcoholic fatty liver disease undergoing bariatric surgery. Pol J Pathol. 2010;61(3):147–53.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Baranova, A., Birerdinc, A., Younossi, Z.M. (2014). Adipokines in Nonalcoholic Fatty Liver Disease. In: Fantuzzi, G., Braunschweig, C. (eds) Adipose Tissue and Adipokines in Health and Disease. Nutrition and Health. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-62703-770-9_17
Download citation
DOI: https://doi.org/10.1007/978-1-62703-770-9_17
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-62703-769-3
Online ISBN: 978-1-62703-770-9
eBook Packages: MedicineMedicine (R0)