
Abstract
Graft-versus-host disease (GVHD) refers to a constellation of adverse immune responses resulting in tissue destruction following hematopoietic stem cell or solid organ transplantation. Through a complex network of priming and activation events, immune-competent T cells residing in the transplanted tissue (the graft) become stimulated, migrate into target organs, and mediate immune destruction of the recipient’s healthy tissue (the host). Paradoxically, this immune activation can also eradicate residual leukemic cells, when hematopoietic stem cell transplantation occurs in the context of hematological malignancies, resulting in a beneficial graft-versus-leukemia (GVL) effect. The Notch family of transmembrane receptors functions in many aspects of immune responses, including those that mediate GVHD. Here we will review the complex nature of GVHD and how Notch signaling may play a prominent role during the initiation and progression of the disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Notch
- Graft-versus-host disease
- GVHD
- Graft-versus-leukemia
- GVL
- Immune destruction
- Hematopoietic stem cell transplantation
- Dll4
- M1 macrophages
- Chemokine receptors
- Toll-like receptors
7.1 Overview of Notch Signaling
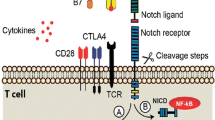
Signaling mediated by Notch receptors is essential to many varied cell processes. Excellent reviews discuss Notch signaling in embryonic tissue and organ development [108], fetal hematopoiesis [13, 104], intestinal cell homeostasis [25], T cell maturation in the thymus [53, 61], and the regulation of numerous immune responses in the periphery [6, 8, 30, 80, 88]. As depicted in Fig. 7.1, in mammals the Notch family of signaling proteins is comprised of four transmembrane receptors (Notch1–4) and five signal-initiating ligands that belong to two distinct groups: Jagged ligands (Jag1,2) and Delta-like ligands (Dll1,3,4). Notch receptors undergo a series of posttranslational events that ultimately result in a mature, non-covalently linked heterodimer that is inserted in the plasma membrane of the signal-receiving cell [31]. Upon engaging a cognate ligand, often displayed on the surface of nearby signal-sending cells, Notch receptors undergo conformational change that allows membrane proximal cleavage at the S2 site by ADAM proteases [24]. The extracellular domain of Notch receptors is transendocytosed by the signal-sending cell, through a process that initiates signaling within the ligand-bearing cell [98]. Removal of the extracellular domain leaves only a short transmembrane “stub” protruding from the cell surface, generating a structural substrate for the enzymatic actions of gamma secretase. Gamma secretase, a multiprotein complex comprised of presenilins 1 and 2, APH, and nicastrin, cleaves Notch receptors at the S3 site, liberating an active, intracellular form of Notch (NIC), capable of translocating to the nucleus to regulate downstream targets [27]. Within the context of canonical signaling, NIC is recruited to preformed, DNA-bound complexes containing its canonical nuclear binding partner CSL/RBPj. Notch binding to CSL/RBPj displaces transcriptional repressors, which are subsequently replaced by transcriptional activators, and allows for gene transcription [56]. In addition to canonical signaling, noncanonical signaling (i.e., CSL/RBPj-independent) and ligand-independent signaling have also been described [10, 73, 78, 135].

Overview of Notch signaling. Ligand-mediated Notch signaling is initiated when a ligand of the Delta-like (Dll 1,3,4) or Jagged (Jag 1,2) family, present on the surface of a nearby signal-sending cell, binds to a Notch receptor expressed on a signal-receiving cell. Following cleavage of the extracellular domain of Notch by an ADAM protease, the Notch extracellular domain is transendocytosed by the signal-sending cell, together with the bound Notch ligand. Notch ligands can be recycled and re-expressed on the surface of the signal-sending cell. In the signal-receiving cell, following processing by ADAM protease, transmembrane-tethered Notch is cleaved just proximal to the cell membrane to release intracellular domain (ICD) of Notch, which is transcriptionally active. Notch ICD associates with its canonical DNA-binding partner, CSL, which itself resides on DNA in repressor complexes that prohibit transcription of canonical gene targets. Upon association with CSL, additional coactivators including mastermind-like (MAML) and p300 are recruited to the NotchICD-CSL complex, activating its transcriptional capabilities. Transcription of some Notch-regulated genes can proceed in the absence of CSL and is referred to as noncanonical Notch signaling
7.2 Pathophysiology of GVHD
Hematopoietic stem cell transfer (HSCT) provides the means for full hematopoietic reconstitution following myeloablative therapy commonly used to treat hematologic malignancies, solid tumors, or certain immune-mediated bone marrow failure diseases, such as aplastic anemia [26, 99, 118]. Graft-versus-host disease (GVHD) refers to a constellation of adverse sequelae following HSCT, whereby immunocompetent cells present in the stem cell graft are activated and damage host tissues [109]. Studies suggest GVHD can affect greater than 40% of patients following HSCT, and, as such, it remains a significant barrier to the broader use of HSCT in the clinic. GVHD is classified as acute or chronic, based on how soon after HSCT symptoms appear. Symptoms presenting within the first 100 days post-HSCT are attributed to acute GVHD, while those occurring later than 100 days constitute chronic GVHD. Preventing acute GVHD is thought to decrease the likelihood of chronic GVHD, which also appears to have autoimmune underpinnings.
In cases of hematologic malignancy or transplant in the setting of solid tumors, conventional conditioning in preparation of HSCT is intended to reduce tumor burden and neutralize host immune responses, so as to prevent HSC rejection [67]. However, pre-transplant conditioning can also create the environment for host tissue to be targeted by HSCT-derived T cells, resulting in GVHD [121]. The pathophysiology of GVHD can be divided into three stages that represent a continuum of disease (Fig. 7.2). During the initiation phase, conditioning regimens cause the release of endogenous bacterial lipopolysaccharides (LPS) and damage host cells, resulting in inflammatory cytokine release [43]. The second, inductive phase begins when host, and possibly donor, antigen-presenting cells (APCs) are activated in response to potent LPS stimulation in the presence of pro-inflammatory cytokines, which serves to enhance their ability to present alloantigens to donor CD4 and CD8 T cells [41]. This stage progresses into the effector phase as immunocompetent T cells, resident within the stem cell graft, proliferate and differentiate after encountering major or minor histocompatibility antigens that are mismatched between donor and host. Tissue destruction is mediated during the effector phase of GVHD, which manifests when distinct subsets of differentiated T cells, directed by the chemokine receptors they express, infiltrate and damage specific host tissues [57].

Stages of graft-versus-host disease. Graft-versus-host disease (GVHD) is initiated as the result of host conditioning regimens, especially those that involve total body irradiation, prior to hematopoietic stem cell transplantation. Tissue damaged by pre-transplant conditioning causes the release of lipopolysaccharide (LPS) from gut bacteria, together with induction of pro-inflammatory cytokine secretion. Host/donor antigen-presenting cells (APC) and host macrophages (MΦ) are activated and primed to present alloantigen to donor T cells. Activated, differentiated donor T cells increase expression of pro-inflammatory cytokines and chemokine receptors, migrate to target tissues, and mediate tissue destruction. Regulatory T cells may negatively regulate T cell activation and differentiation, minimizing GVHD pathology
Organ involvement in GVHD can vary and may be linked to the intensity of the conditioning regimen [72, 95]. The major organs targeted for immune attack include the gut, liver, skin, and lungs. Increasingly, evidence suggests distinct populations of T helper (Th) cells mediate organ-specific destruction, likely as a combined result of the cytokines they produce together with the repertoire of chemokine receptors they express [39, 60]. To this end, Th type 1 cells (Th1) appear to be important in GVHD affecting the gut and liver, although these cells are thought to mediate, to some degree, immune attack against all of these tissues. Th2 and Th17 cells exacerbate immune-mediated damage to the skin and lungs, but Th2 cells also can contribute substantially to gut GVHD [131]. Chemokine-directed migration of Th cells has also been implicated in organ-specific trafficking. CCR9 and CCR5 are important in migration to the gut [7, 100]. CCR6 mediates targeting to the skin, while CXCR3 is critical for GVHD of the liver, gut, and skin [40, 120]. Finally, CCR2, CCR4, and CCR6 all can contribute to Th cell trafficking to the lungs [131].
For patients undergoing HSCT following myeloablative therapy for hematologic malignancies, such as leukemia or lymphoma, the allo-specific responses that result in GVHD can also provide a benefit to HSCT recipients through an immune attack directed against residual leukemic cells. This “graft-versus-leukemia” (GVL) or “graft-versus-tumor” (GVT) effect is neither well understood nor well defined, but studies do suggest it can be separated from the detrimental GVHD responses commonly observed [55, 64, 65, 133]. To preserve GVL/GVT, while selectively abrogating GVHD, is the ultimate therapeutic goal of researchers in this field.
This review will focus on the various stages of GVHD initiation and progression: activation of APCs, differentiation of the major Th subsets and secretion of their signature cytokines, as well as expression of chemokine receptors that direct tissue-specific infiltration, and how Notch signaling may be implicated in each of these.
7.3 Notch Receptors and Ligands During the Initiation and Induction Phase of GVHD
Acute GVHD occurs within 100 days in the post-transplant period and culminates in extensive tissue destruction characterized by apoptosis. Its development is absolutely dependent on the presence and function of alloreactive T cells in the donor inoculum and is closely tied to the curative GVT/GVL effect [64, 65]. However, following HSCT , tissue injury and inflammation, defined by pro-inflammatory cytokine release, are initiated by the conditioning regimen. Both the severity and the incidence of GVHD following HSCT have been associated with the intensity of the conditioning regimen, especially when total body irradiation (TBI) is included [42]. During the initiation phase of GVHD, pro-inflammatory cytokines, together with lipopolysaccharide (LPS) , released as a result of conditioning-induced gut damage, converge to craft the inflammatory environment. Subsequent activation of host antigen-presenting cells (APC) , including conventional dendritic cells (cDC) and macrophages, occurs during the inductive phase.
The requirement for host APCs to induce GVHD is complicated and controversial. While host APCs may not be required for GVHD to ensue [63], host DCs are sufficient to activate donor T cells [28, 64, 65] and initiate GVHD [58]. Toll receptor 4 (TLR4) is a major pattern recognition receptor capable of responding to LPS, and TLR4 expression on host cDCs exacerbates GVHD [16, 137]. Furthermore, cDC exposure to LPS also enhances expression of the Notch ligand Delta-like 4 (Dll4), likely through a TLR4-mediated process (Fig. 7.3 (1); [68, 111]). Seminal work by Flavell’s group suggested engagement of specific Notch ligands can influence T helper cell differentiation, with ligands of the Delta-like family (Dll1,3,4) promoting T helper type 1 responses and those of the Jagged family (Jag1,2) facilitating T helper type 2 differentiation [3]. In the context of GVHD, Dll4 expression on host cDCs has been shown to drive allogeneic T cell responses, including increased production of IFNγ and IL-17, both in vitro and in vivo (Fig. 7.3 (2); [75, 81, 85]).This study coincided with elegant work by Maillard’s group showing that administering antibodies specific for Dll1 and Dll4 in a mouse model of GVHD, early after induction, provided durable protection against disease pathology without significantly sacrificing the beneficial effects of GVL/GVT [114]. These studies not only highlight the contribution of Notch signaling to the induction of GVHD during the very early priming stages, their results suggest that inhibiting the signaling capacity of specific Notch ligands represents a viable therapeutic strategy to prevent GVHD.

Potential for Notch involvement in graft-versus-host disease. Notch signaling may mediate numerous aspects of GVHD. (1) Toll receptor (TLR) signaling in response to lipopolysaccharide (LPS) release following pre-transplant conditioning upregulates Dll ligands on host/donor APCs; (2) Dll4 expression on donor dendritic cells increases IFNγ and IL-17 secretion by Th1 and Th17 cells, respectively; (3) Notch1 upregulation in activated macrophages (MΦ) promotes a pro-inflammatory “M1” phenotype; (4) Notch receptors play an important role in reinforcing differentiated phenotypes of T helper (Th) CD4 cells, including Th1, Th2, and Th17 cells. (5) Notch signaling may play an important role in the development of induced regulatory T cells (iTregs); (6) Notch signaling may regulate chemokine receptor expression to direct T cell trafficking to GVHD target organs. Please refer to the text for expanded description of Notch signaling at each of these potential stages of GVHD induction and progression
The contribution of macrophages, both of host and donor origin, appears to be equally complex, especially in terms of Notch signaling. Macrophages can be polarized toward “M1” or “M2” phenotypes, and a critical involvement of Notch in this process has been described [110, 122, 129]. TLR4 signaling in macrophages increases Notch1 expression which, in turn, promotes their differentiation into pro-inflammatory M1 cells, through interaction with its canonical DNA-binding partner, CSL/RbpJ [45, 128]. Some reports show M1 macrophages exhibit increased capacity for antigen presentation, as well as increased secretion of pro-inflammatory TNF, IL-6, IL-12, and IFNγ, all through Notch1-mediated signaling cascades (Fig. 7.3 (3); [15, 82, 124, 125]). In a model of atherosclerosis, macrophage exposure to LPS was also shown to increase Dll4 expression via TLR4 signaling and promoted an M1 phenotype [34]. Thus, it is attractive to speculate that the Dll4-specific antibodies used in Maillard’s studies may target M1 macrophages, as well as effectively targeting Dll4+ cDCs, although additional studies are needed to confirm this. Nonetheless, M1 macrophages can accumulate in the skin to mediate cutaneous GVHD, where the number of infiltrating M1 macrophages is directly correlated with the severity of skin pathology [86], and reducing M1 differentiation post-HSCT transfer using dexamethasone palmitate can also mitigate acute damage to the skin [87], although its effects on Notch receptors and ligands were not evaluated.
The expansion and activity of host-derived M2 macrophages can also provide protection against GVHD-mediated tissue destruction, and inhibiting Notch1 signaling in macrophages can induce an M2 cell fate [110]. CSF-1 promotes the M2 phenotype in macrophages, with reduced secretion of pro-inflammatory cytokines, suggesting an increase in M2 macrophages may provide protection from GVHD. This hypothesis was supported in a mouse model by administering CSF-1 prior to GVHD induction. CSF-1 greatly expanded the host M2 macrophage pool, decreased the expansion of donor T cells, and reduced the severity of acute GVHD [38]. Conversely, donor-derived macrophages appear to require CSF-1 to mediate chronic GVHD, since depleting macrophages using an antibody directed against the CSF-1 receptor or transferring macrophages from csf1 deficient donors resulted in markedly reduced skin and lung GVHD, due to decreased IL-17 production [2].
Collectively, these data suggest that lingering, donor-derived M2 macrophages may contribute to tissue destruction characteristic of chronic GVHD. These confounding data underscore the complexity of temporal GVHD pathology, the diversity of cell types involved, and the potentially conflicting contributions of Notch signaling in acute vs chronic GVHD. Additional studies that specifically address Notch signaling in chronic GVHD will be imperative to clarify these discrepancies.
7.4 Notch, T Helper Cells, and the Effector Phase of GVHD
As the continuum of acute GVHD progresses into the effector phase, activated APCs are now suited to prime immunocompetent donor T cells to differentiate into various T helper (Th) subsets and expand effector CD8 T (Teff) cells. CD4 Th cells, along with CD8 cytolytic T cells (CTL), will infiltrate target organs to mediate tissue damage (Fig. 7.3 (4)).
In response to antigen stimulation, mature T cells integrate myriad external signals derived from the T cell receptor, co-stimulation, and cytokines present in the microenvironment to initiate the genetic programming that will result in their full activation, expansion, and subsequent differentiation [107]. Th type 1 (Th1), Th2, Th17, Th9, and Th22 cells have all been described based on the unique combinations of cytokines that drive their differentiation, the master transcription factor they upregulate, and the signature profile of cytokines they produce. Accumulating evidence suggests that individual Notch receptors and ligands critically influence Th cell differentiation, both in healthy and aberrant immune responses, including in those that mediate GVHD [130].
Notch signaling in donor CD4 and CD8 T cells augments their activation and expansion and regulates expression of CD25, the high-affinity subunit of the IL-2 receptor [1, 92], supporting the notion that Notch signaling likely acts as a signal amplifier [29]. Engagement of Notch receptors on T helper cells by cognate ligands expressed on APCs was initially thought to instruct T helper cell differentiation: exposure to Dll family ligands promoted a Th1 phenotype, while binding to Jag family ligands directed a Th2 cell fate [3]. A closer examination of the conditions under which T helper cells adopt a specific, differentiated state now supports the notion that Notch signaling provides an unbiased amplification signal to helper T cells, allowing them to proliferate and reinforce a T helper phenotype (i.e., cytokine secretion) acquired as a result of response to the extracellular cytokine milieu [11, 90]. This perspective helps to reconcile the pleiotropic requirements for Notch activation in Th1, Th2, Th17, and induced regulatory T cell (iTreg) differentiation, with different ligands augmenting, rather than inducing Th signature cytokine secretion. Furthermore, redundant functions for some Notch receptors have been described, suggesting strategies aiming to target specific Notch receptors in the management of GVHD may prove ineffective [9].
7.5 Th1 Cells in GVHD
GVHD that manifests in the gut and liver is driven by a strong Th1 response [93] although IFNγ, a signature Th1 cytokine, appears to contribute to tissue damage in nearly all organs affected by GVHD. T cells that adopt a Th1 cell fate exhibit increased and sustained expression of the master transcriptional regulator, T-bet. Th1 cells secrete IFNγ which, together with IL-12, serves to reinforce the Th1 phenotype. First described by Szabo et al. in 2000, T-bet is a T cell-specific transcription factor that is absolutely required for Th1 differentiation [112]. In GVHD, T-bet is also an important determinant of disease pathology. GVHD induction using T-bet-deficient T cells resulted in significantly reduced tissue damage in the gut and liver, compared to disease induction using wild-type or IFNγ-deficient T cells, and regardless of whether the model used was mismatched for major or minor histocompatibility antigens [33]. In this study, although loss of T-bet enhanced Th17 cell differentiation, the Th17 cells were less effective at inducing GVHD. Of note, GVL/GVT effects were also compromised but could be restored following neutralization of IL-17 in hosts. By contrast, selectively inhibiting Th17 development with low-dose halofuginone in a mouse model of GVHD exacerbated Th1-mediated pathology in the liver and gut, although pulmonary GVHD was attenuated with reduced Th17 differentiation [22, 23, 93]. Moreover, in a prospective study of patients with cutaneous GVHD, an increased number of IFNγ-producing Th1, but not Th17, cells were found in skin lesions of patients with acute GVHD, compared to those without GVHD, suggesting that Th1 cells can also mediate skin-associated tissue damage [17].
The gene encoding T-bet, Tbx21, is a direct transcriptional target of Notch1, and inhibiting Notch signaling greatly impairs both the differentiation of Th1 cells and the production of pro-inflammatory cytokines [77, 92, 101]. Canonical Notch signaling in T cells can be compromised when a dominant negative form of a key transcriptional coactivator mastermind-like (DNMAML) is expressed [70]. In the context of GVHD, inducing disease by transferring DNMAML T cells from C57BL/6 donor mice into Balb/c recipients attenuates disease pathology and impairs IFNγ secretion, although T-bet expression remained intact [105, 136]. However, a subsequent report demonstrated that T-bet expression was independent of CSL/Rbpj signaling and, rather, was regulated via a noncanonical signaling pathway [29]. In light of these combined studies, it is likely Notch signaling contributes significantly to Th1-mediated pathology in GVHD, through its regulation of canonical and noncanonical transcriptional targets.
7.6 Th17 Cells in GVHD
Th17 cells are an additional subset of differentiated T cells capable of migrating into the gut, lung, and skin tissues where they can mediate tissue destruction [74, 133]. Th17 cells require RORγt for their terminal differentiation, and this process is facilitated by the presence of IL-6 and TGFβ [51, 71]. Plasticity between Th1 and Th17 phenotypes has been demonstrated, suggesting that within the appropriate constellation of signaling modifiers, especially those conveyed by the cytokine milieu, Th1 cells can adopt features of Th17 cells and vice versa [18, 37, 69]. Within the complex progression of GVHD, it has been shown that even when the Th1-predominant, IFNγ, and the Th17-signature, IL-17, cytokines are present, the kinetics of their accumulation in target tissues differ. In an MHC minor antigen-mismatched model of GVHD, IFNγ secretion by CD4 and CD8 T cells preceded IL-17 production in the liver and lung, as well as in the spleen and mesenteric lymph nodes. This finding suggests that, in this model of GVHD, Th1 cell differentiation occurred prior to that of Th17 cells [49]. A recent study using a mouse model of colitis suggests that, in the presence of TGFβ, Th1 cells can be converted to IFNγ+IL-17+ CD4 T cells, which exacerbate gut pathology [37]. Whether or not a similar conversion can or does occur in the context of gut GVHD remains to be examined. Th1 and Th17 cells share a common cytokine subunit: the IL-12/IL23 p40 subunit. Each of these cytokines contributes to stabilizing their respective associated Th phenotype: IL-12 will reinforce Th1 polarization, while IL-23 increases Th17 cell fate stability. Although neutralization of the common p40 subunit can attenuate GVHD in a mouse model [126], it is not known, definitively, if signaling through this common subunit influences the interconversion of these Th cell subsets in response to the complex array of cytokines in the microenvironment.
Overall, the question of how extensively Th17 cells contribute to GVHD pathology remains unanswered [47, 119]. It is clear that Th17 cells can be found in target tissues during GVHD progression, and disease severity can be attenuated when T cells lacking RORγt are used in a model of CD4 T cell-mediated GVHD. The resulting decreased levels of TNFα in the gut, liver, and lung were thought to be significant factors in the reduced pathology noted [138]. Interestingly, in a model of spontaneous type I diabetes, TNFα could be induced in a Th17-dependent manner [62, 66]. Whether or not differentiated Th17 cells act to sustain TNFα secretion produced during the induction and/or initiation phases of GVHD will require further investigation. In a separate report, in vitro-generated Th17 cells were found to be sufficient but not required to induce GVHD, and their presence also correlated with higher levels of TNFα [47]. However, RORγt-deficient CD4 T cells were also capable of inducing lethal GVHD in this model, suggesting that, although Th17 cells may contribute significantly to GVHD progression, additional subsets of Th cells also are critically important.
One important observation of the study by Iclozan et al. was that Th17 cells showed extensive expansion, in vivo, and were highly resistant to activation-induced cell death (AICD). Additional work has demonstrated that human Th17 cells constitute a long-lived memory T cell population [59]. Under various pathological conditions, these cells were shown to be highly proliferative, in vivo, resistant to AICD, and preserved their ability to further differentiate into functional Th1 and iTreg cells, although they retained their capacity for IL-17 secretion. Notably, survival of these human Th17 cells was attributed to a Notch-dependent pathway, downstream of HIF1α, since overexpression of NICD could rescue a significant percentage of Th17 cells from the apoptosis induced when HIFα signaling was blocked. These data add to our understanding of Notch signaling in Th17 cell differentiation. Dll4-Notch signaling between DCs and T cells, in the presence of the Th17-polarizing cytokines, IL-6 and TGFβ, resulted in higher percentages of Th17 cells than those polarized under identical conditions in the absence of Notch signaling [85]. Although not expressly examined in their studies, it is also possible that the robust protection from GVHD induction observed by Maillard’s group during Dll4-blockade may include reduced differentiation of Th17 cells and the concomitant benefit that provides.
7.7 Th2 Cells in GVHD
A detailed study conducted by Zeng’s group would suggest that some of the complexities of GVHD may be attributed to cross-regulation among Th cell subsets which, in turn, influences the severity and organ specificity of the associated pathology [131]. Using a major MHC antigen-mismatched model of GVHD (C57BL/6 donor into Balb/c recipient), they showed that transplanting IFNγ-deficient T cells augmented both Th17- and Th2-mediated tissue destruction in the lung and skin. By contrast, inducing GVHD using T cells that were doubly deficient for IFNγ and IL-17 led to a massive upregulation and infiltration of Th2 cells into the lungs, resulting in idiopathic pneumonia syndrome (IPS). Thirteen days after GVHD induction, there was a nearly 16-fold increase in the number of CD4 Th2 cells in the lungs of mice that received doubly deficient T cells, compared to mice whose disease was induced using wild-type CD4 T cells. Mice that received IFNγ-only deficient cells had a nearly tenfold increase in Th2 and Th17 cells in the lung, compared to mice receiving wild-type T cells. This study further showed that induction of the immune-modulating ligand, B7-H1, in host lung epithelium required IFNγ for its upregulation, and absence of its expression was a compounding factor leading to IPS. In a complementary study by Yu et al. [133] that employed the same model of GVHD, transferring donor T cells that were doubly deficient for the transcription factors, T-bet and RORγt, resulted in overall decreased pathology in the gut, liver, and lung; however, significant increases in IL-4 and IL-5 expression 5 days after GVHD induction were also noted in the lungs of recipient animals [133]. These conclusions were further supported using a model of LPS inhalation-induced acute pulmonary GVHD in the context of HSCT. Transferring T-bet-deficient T cells in this model resulted in more severe lung pathology and pulmonary fibrosis, together with increased IL-13, IL-17, and Th17 cells, suggesting that, in the absence of cross regulation by IFNγ, Th17 cells can contribute significantly to lung GVHD [35].
As with other Th subsets, a role for Notch signaling in Th2 cell differentiation has also been described [4]. Using a model of Trichuris muris infection, Pear’s group demonstrated a requirement for canonical, MAML-mediated Notch signaling to generate the protective, in vivo Th2 response necessary to clear the parasitic infection [115].
Furthermore, while TCF and β-catenin, components of the Wnt signaling pathway, initiate early, IL-4-independent GATA3 expression via their binding to the Gata3-1b enhancer, it would appear that Notch binding to the Gata3-1a enhancer leads to sustained Th2 cell differentiation by providing synergistic upregulation of IL-4 [32, 132]. Thus, as with the other Th cell subsets discussed, and which are critically important in mediating GVHD, Notch is also an important regulator of Th2 cell differentiation [5].
7.8 Regulatory T Cells in GVHD
In addition to Th cells , distinct subsets of immunosuppressive regulatory T (Treg) cells also have been identified, including naturally occurring (nTreg) and inducible (iTreg) regulatory T cells that can express either CD4 or CD8 [69, 83, 106]. To date, Tregs exhibiting a CD4+ CD25+FoxP3+ phenotype are perhaps the most well-studied. However, unlike their Th and Teff cell counterparts which drive GVHD pathology, Treg cells have been shown to attenuate the severity of acute GVHD, mitigate the extent of tissue involvement, and prevent chronic GVHD altogether [76, 116].
Naïve T cells will adopt a Treg phenotype when they differentiate in the presence of TGFβ and IL-2 and are characterized by increased expression of the master transcriptional regulator FoxP3. In vivo, the Treg phenotype is considered to be unstable, with reports of interconversion of Tregs to effector T cells in the presence of high levels of inflammatory cytokines, especially IL-6 and IL-17 [54, 127].
Extensive and impressive progress has been made in defining the protective effects of Tregs and their mechanisms of action, both in murine and human studies. Functional Treg suppression has been reported to be faulty in patients with acute GVHD [14]. This may be due to the observed lower surface expression of CCR5 and CXCR3 on donor Tregs, resulting in impaired trafficking to target organs [117]. Studies also suggest that donor-derived, or third-party, Tregs provide far more potent immunosuppression than host Tregs when used as an immunotherapy [94]. Human trials using ex vivo-expanded nTregs as a potential prophylaxis for GVHD were recently concluded [19].
The encouraging results showed that, when administered at the time of HSCT, only 9% of patients who received expanded Tregs exhibited grade II–IV acute GVHD at 100 days post-transplant, compared to a control cohort in which 45% showed symptoms of GVHD, grade II or higher.
Patients in both cohorts received identical conditioning regimens prior to HSCT. Furthermore, while approximately 14% of the non-Treg-treated patients showed signs of chronic GVHD at 1 year post-HSCT, none of the patients who received the ex vivo-expanded Tregs exhibited chronic GVHD. The impressive results of this trial may be due to the co-administration of Tregs at the time of HSC transfer. In complementary studies, Negrin’s group utilized a minor antigen MHC-mismatched model to show that the immunosuppressive capacity of transferred Tregs, both donor-derived and third party, is most effective early after their transplantation and suggests that the timing of administering Tregs is a critical factor to be considered [97]. A small study using donor-derived or third-party expanded Tregs in the treatment of established cases of chronic and acute GVHD were less positive [113], lending support to Negrin’s findings.
There is a preponderance of evidence supporting a positive role for Notch signaling in iTreg differentiation and which further support the influence of specific Notch ligands in this process (Fig. 7.3 (5)). Notch signaling may generate Tregs through both direct and indirect mechanisms. In an early report, APCs engineered to overexpress Jag1 (serrate1) could induce a regulatory phenotype in co-cultured CD4 T cells, and these cells maintained antigen-specific suppressive activity when transferred into naïve hosts prior to immune challenge [44]. Two subsequent studies revealed Notch1 and TGFβ can cooperate to induce FoxP3 expression, and this process may involve canonical Notch signaling via CSL/Rbpj binding to the FOXP3 promoter [89, 91, 103]. Notch3 has also been implicated in Foxp3 transcription during nTreg generation, and this has been shown to proceed through an NF-κB-dependent pathway [12, 20]. Furthermore, it was demonstrated that human memory CD4 T cells could adopt a Treg phenotype, including expression of FoxP3 and upregulation of TGFβ, when exposed to Dll1 during in vitro culture [84].
Indirect involvement of Notch signaling in Treg-mediated immunosuppression has also been demonstrated. Co-incubating CD4+CD25- T cells with Dll4 or Jag1 increased their responsiveness to Treg-mediated suppression by upregulating TGFβRII expression [46]. Additionally, Notch signaling is also important for production of the immunosuppressive cytokine, IL-10, in T cells, and this is enhanced when CD4 T cells are co-cultured with Dll4-expressing plasmacytoid DCs [50, 102]. Elegant work by Sarin’s group showed that differential localization of Notch1 mediated Treg survival through a membrane-associated NICD/Rictor complex, which was stabilized by interactions with Dll1. This protection was lost when NICD was targeted to the nucleus [96]. Recent work, however, challenges the notion that Notch signaling positively regulates Treg induction [21]. The results of this study showed that when NICD was overexpressed in FoxP3+ Tregs, the Treg phenotype was destabilized, resulting in an autoimmune lymphoproliferative manifestation. Canonical Notch signaling mediated this phenomenon, since loss of Rbpj restored the number and frequency of Tregs and protected them from apoptosis in a model of GVHD generated by major histocompatibility antigen mismatch. Reconciling these seemingly disparate data will be important to informing therapeutic strategies aimed at limiting Notch signaling in GVHD.
7.9 Concluding Remarks and Future Directions
Much has been learned about the constellation of alloimmune responses that can be generated after HSCT and that result in the complex pathology of GVHD. However, much less is known about the mechanisms of the beneficial GVT responses that may also come into play following HSCT . Indeed, details regarding this phenomenon are only recently emerging and suggest that factors responsible for mediating this effect will be as complex as those that drive GVHD. It is also likely that multiple cellular subsets contribute to GVT; therefore, approaches that target multiple cell populations may ultimately prove to be the most successful.
Effective ex vivo expansion and delivery of donor-derived nTregs or in vivo generation of donor-derived iTregs represent viable approaches to attenuating GVDH, although the effect of increased numbers of Tregs on GVT has not been well-characterized. One caveat to this approach is the demonstrated capacity for Tregs to be converted to Th1 or Th17 cells in the context of a robust pro-inflammatory environment and will need to be considered when designing Treg-based therapies. As discussed above, the degree to which Notch signaling positively or negatively regulates Treg development also has not been fully elucidated. Thus, approaches that aim to attenuate GVHD by manipulating Notch signaling may need to be precise and acutely applied.
Manipulating chemokine receptors, which play a vital role in T cell trafficking, may be one means of achieving selective organ targeting to reduce Teff cell infiltration or to increase trafficking of Tregs to specific organs. CXCR3, CCR5, and CCR9 have been identified as chemokine receptors that facilitate T cell migration, and gut infiltration of CCR9+ CD4 T cells is associated with tissue destruction [7, 134]. CXCR3 has been shown to be a transcriptional target of T-bet [48, 52], which itself is regulated by Notch1 [77, 101]. Furthermore, CCR5 and CCR9 have both been demonstrated to be regulated by Notch1 in T-ALL [79]; however, whether Notch also primes Teff cells for gut infiltration in GVHD is not yet known (Fig. 7.3 (6)). Interestingly, CCR9 also mediates the migration of plasmacytoid DCs (pDC) to the intestines [123], and CCR9+ pDCs show an immature phenotype with a high capacity for suppressing GVHD by reducing the number of IL-17 producing cells and increasing the number of FoxP3+ Treg cells [36]. Further investigation is needed to determine if Notch signaling regulates CCR9 expression in pDC, as it does in CD4+ T cells.
Therapeutic modalities for the treatment and prevention GVHD that aim to limit Notch signaling represent an exciting and active area of investigation. Very early reports suggest that, at least in animal models, this is a promising approach that may serve both to attenuate tissue damage and preserve beneficial GVT effects. Current research in therapies that range from delivering ligand-specific neutralizing antibodies to developing γ-secretase inhibitors designed to selectively modulate Notch signaling, to expanded use of Treg cells as immune modulators, makes for an impressive, and hopefully successful, array of approaches to tackle a complex and complicated disease.
References
Adler, S. H., Chiffoleau, E., Xu, L., Dalton, N. M., Burg, J. M., Wells, A. D., Wolfe, M. S., Turka, L. A., & Pear, W. S. (2003). Notch signaling augments T cell responsiveness by enhancing CD25 expression. Journal of Immunology, 171, 2896–2903.
Alexander, K. A., Flynn, R., Lineburg, K. E., Kuns, R. D., Teal, B. E., Olver, S. D., Lor, M., Raffelt, N. C., Koyama, M., Leveque, L., Le Texier, L., Melino, M., Markey, K. A., Varelias, A., Engwerda, C., Serody, J. S., Janela, B., Ginhoux, F., Clouston, A. D., Blazar, B. R., Hill, G. R., & MacDonald, K. P. (2014). CSF-1- dependant donor-derived macrophages mediate chronic graft-versus-host disease. The Journal of Clinical Investigation, 124, 4266–4280. https://doi.org/10.1172/JCI75935.
Amsen, D., Blander, J. M., Lee, G. R., Tanigaki, K., Honjo, T., & Flavell, R. A. (2004). Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell, 117, 515–526.
Amsen, D., Antov, A., Jankovic, D., Sher, A., Radtke, F., Souabni, A., Busslinger, M., McCright, B., Gridley, T., & Flavell, R. A. (2007). Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity, 27, 89–99.
Amsen, D., Spilianakis, C. G., & Flavell, R. A. (2009). How are T(H)1 and T(H)2 effector cells made? Current Opinion in Immunology, 21, 153–160. https://doi.org/10.1016/j.coi.2009.03.010.
Amsen, D., Helbig, C., & Backer, R. A. (2015). Notch in T cell differentiation: All things considered. Trends in Immunology, 36, 802–814. https://doi.org/10.1016/j.it.2015.10.007.
Aoyama, K., Saha, A., Tolar, J., Riddle, M. J., Veenstra, R. G., Taylor, P. A., Blomhoff, R., Panoskaltsis Mortari, A., Klebanoff, C. A., Socié, G., Munn, D. H., Murphy, W. J., Serody, J. S., Fulton, L. M., Teshima, T., Chandraratna, R. A., Dmitrovsky, E., Guo, Y., Noelle, R. J., & Blazar, B. R. (2013). Inhibiting retinoic acid signaling ameliorates graft-versus-host disease by modifying T-cell differentiation and intestinal migration. Blood, 122, 2125–2134. https://doi.org/10.1182/blood-2012-11-470252.
Aster, J. C. (2014). In brief: Notch signalling in health and disease. The Journal of Pathology, 232, 1–3. https://doi.org/10.1002/path.4291.
Auderset, F., Schuster, S., Coutaz, M., Koch, U., Desgranges, F., Merck, E., MacDonald, H. R., Radtke, F., & Tacchini-Cottier, F. (2012). Redundant Notch1 and Notch2 signaling is necessary for IFNγ secretion by T helper 1 cells during infection with Leishmania major. PLoS Pathogens, 8, e1002560. https://doi.org/10.1371/journal.ppat.1002560.
Ayaz, F., & Osborne, B. A. (2014). Non-canonical notch signaling in cancer and immunity. Frontiers in Oncology, 4, 345. https://doi.org/10.3389/fonc.2014.00345.
Bailis, W., Yashiro-Ohtani, Y., Fang, T. C., Hatton, R. D., Weaver, C. T., Artis, D., & Pear, W. S. (2013). Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity, 39, 148–159. https://doi.org/10.1016/j.immuni.2013.07.006.
Barbarulo, A., Grazioli, P., Campese, A. F., Bellavia, D., Di Mario, G., Pelullo, M., Ciuffetta, A., Colantoni, S., Vacca, A., Frati, L., Gulino, A., Felli, M. P., & Screpanti, I. (2011). Notch3 and canonical NF- kappaB signaling pathways cooperatively regulate Foxp3 transcription. Journal of Immunology, 186, 6199–6206. https://doi.org/10.4049/jimmunol.1002136.
Bigas, A., Guiu, J., & Gama-Norton, L. (2013). Notch and Wnt signaling in the emergence of hematopoietic stem cells. Blood Cells, Molecules & Diseases, 51, 264–270. https://doi.org/10.1016/j.bcmd.2013.07.005.
Beres, A., Komorowski, R., Mihara, M., & Drobyski, W. R. (2011). Instability of FOXP3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clinical Cancer Research, 17, 3969–3983. https://doi.org/10.1158/1078-0432.CCR-10-3347.
Boonyatecha, N., Sangphech, N., Wongchana, W., Kueanjinda, P., & Palaga, T. (2012). Involvement of Notch signaling pathway in regulating IL-12 expression via c-Rel in activated macrophages. Molecular Immunology, 51, 255–262. https://doi.org/10.1016/j.molimm.2012.03.017.
Brennan, T. V., Lin, L., Huang, X., Cardona, D. M., Li, Z., Dredge, K., Chao, N. J., & Yang, Y. (2012). Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood, 120, 2899–2908. https://doi.org/10.1182/blood-2011-07-368720.
Broady, R., Yu, J., Chow, V., Tantiworawit, A., Kang, C., Berg, K., Martinka, M., Ghoreishi, M., Dutz, J., & Levings, M. K. (2010). Cutaneous GVHD is associated with the expansion of tissue-localized Th1 and not Th17 cells. Blood, 116, 5748–5751. https://doi.org/10.1182/blood-2010-07-295436.
Brown, C. C., Esterhazy, D., Sarde, A., London, M., Pullabhatla, V., Osma-Garcia, I., Al-Bader, R., Ortiz, C., Elgueta, R., Arno, M., de Rinaldis, E., Mucida, D., Lord, G. M., & Noelle, R. J. (2015). Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity, 42, 499–511. https://doi.org/10.1016/j.immuni.2015.02.003.
Brunstein, C. G., Miller, J. S., McKenna, D. H., Hippen, K. L., DeFor, T. E., Sumstad, D., Curtsinger, J., Verneris, M. R., ML, M. M., Levine, B. L., Riley, J. I., June, C. H., Le, C., Weisdorf, D., McGlave, P. B., Blazar, B. R., & Wagner, J. E. (2015). Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile and clinical effect. Journal of Immunology, 195, 347–355. https://doi.org/10.4049/jimmunol.1402861.
Campese, A. F., Grazioli, P., Colantoni, S., Anastasi, E., Mecarozzi, M., Checquolo, S., De Luca, G., Bellavia, D., Frati, L., Gulino, A., & Screpanti, I. (2009). Notch3 and pTalpha/pre-TCR sustain the in vivo function of naturally occurring regulatory T cells. International Immunology, 21, 727–743. https://doi.org/10.1093/intimm/dxp042.
Charbonnier, L. M., Wang, S., Georgiev, P., Sefik, E., & Chatila, T. A. (2015). Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. Nature Immunology, 16, 1162–1173. https://doi.org/10.1038/ni.3288.
Cheng, H., Tian, J., Li, Z., Zeng, L., Pan, B., Song, G., Chen, W., & Xu, K. (2012). TH17 cells are critical for skin-specific pathological injury in acute graft-versus-host disease. Transplantation Proceedings, 44, 1412–1418. https://doi.org/10.1016/j.transproceed.2011.12.078.
Cheng, H., Tian, J., Zeng, L., Pan, B., Li, Z., Song, G., Chen, W., & Xu, K. (2012). Halofugine prevents cutaneous graft versus host disease by suppression of Th17 differentiation. Hematology, 17, 261–267. https://doi.org/10.1179/1607845412Y.0000000016.
Christian, L. M. (2012). The ADAM family: Insights into Notch proteolysis. Fly (Austin)., 6, 30–34. https://doi.org/10.4161/fly.18823.
Demitrack, E. S., & Samuelson, L. C. (2016). Notch regulation of gastrointestinal stem cells. The Journal of Physiology, 594, 4791–4803. https://doi.org/10.1113/JP271667.
Dietz, A. C., Lucchini, G., Samarasinghe, S., & Pulsipher, M. A. (2016). Evolving hematopoietic stem cell transplantation strategies in severe aplastic anemia. Current Opinion in Pediatrics, 28, 3–11. https://doi.org/10.1097/MOP.0000000000000299.
Duggan, S. P., & McCarthy, J. V. (2016). Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cellular Signalling, 28, 1–11. https://doi.org/10.1016/j.cellsig.2015.10.006.
Duffner, U. A., Maeda, Y., Cooke, K. R., Reddy, P., Ordemann, R., Liu, C., Ferrara, J. L., & Teshima, T. (2004). Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. Journal of Immunology, 172, 7393–7398.
Dongre, A., Surampudi, L., Lawlor, R. G., Fauq, A. H., Miele, L., Golde, T. E., Minter, L. M., & Osborne, B. A. (2014). Non-canonical Notch signaling drives activation and differentiation of peripheral CD4(+) T cells. Frontiers in Immunology, 5, 54. https://doi.org/10.3389/fimmu.2014.00054.
Ebens, C. L., & Maillard, I. (2013). Notch signaling in hematopoietic cell transplantation and T cell alloimmunity. Blood Reviews, 27, 269–277. https://doi.org/10.1016/j.blre.2013.08.001.
Ehebauer, M., Hayward, P., & Martinez-Arias, A. (2006). Notch signaling pathway. Science’s STKE, 2006(364), cm7.
Fang, T. C., Yashiro-Ohtani, Y., Del Bianco, C., Knoblock, D. M., Blacklow, S. C., & Pear, W. S. (2007). Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity, 27, 100–110.
Fu, J., Wang, D., Yu, Y., Heinrichs, J., Wu, Y., Schutt, S., Kaosaard, K., Liu, C., Haarberg, K., Bastian, D., McDonald, D. G., Anasetti, C., & Yu, X. Z. (2015). T-bet is critical for the development of acute graft- versus-host disease through controlling T cell differentiation and function. Journal of Immunology, 194, 388–397. https://doi.org/10.4049/jimmunol.1401618.
Fung, E., Tang, S. M., Canner, J. P., Morishige, K., Arboleda-Velasquez, J. F., Cardoso, A. A., Carlesso, N., Aster, J. C., & Aikawa, M. (2007). Delta-like 4 induces notch signaling in macrophages: Implications for inflammation. Circulation, 115, 2948–2956.
Gowdy, K. M., Nugent, J. L., Martinu, T., Potts, E., Snyder, L. D., Foster, W. M., & Palmer, S. M. (2011). Protective role of T-bet and Th1 cytokines in pulmonary graft-versus-host disease and peribronchiolar fibrosis. American Journal of Respiratory Cell and Molecular Biology, 46, 249–256. https://doi.org/10.1165/rcmb.2011-0131OC.
Hadeiba, H., Sato, T., Habtezion, A., Oderup, C., Pan, J., & Butcher, E. C. (2008). CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nature Immunology, 9, 1253–1260. https://doi.org/10.1038/ni.1658.
Harbour, S. N., Maynard, C. L., Zindl, C. L., Schoeb, T. R., & Weaver, C. T. (2015). Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proceedings of the National Academy of Sciences of the United States of America, 112, 7061–7066. https://doi.org/10.1073/pnas.1415675112.
Hashimoto, D., Chow, A., Greter, M., Saenger, Y., Kwan, W. H., Leboeuf, M., Ginhoux, F., Ochando, J. C., Kunisaki, Y., van Rooijen, N., Liu, C., Teshima, T., Heeger, P. S., Stanley, E. R., Frenette, P. S., & Merad, M. (2011). Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. The Journal of Experimental Medicine, 208, 1069–1082. https://doi.org/10.1084/jem.20101709.
Hayashida, J. N., Nakamura, S., Toyoshima, T., Moriyama, M., Sasaki, M., Kawamura, E., Ohyama, Y., Kumamaru, W., & Shirasuna, K. (2013). Possible involvement of cytokines, chemokines and chemokine receptors in the initiation and progression of chronic GVHD. Bone Marrow Transplantation, 48, 115–123. https://doi.org/10.1038/bmt.2012.100.
He, S., Cao, Q., Qiu, Y., Mi, J., Zhang, J. Z., Jin, M., Ge, H., Emerson, S. G., Zhang, Y., & Zhang, Y. (2008). A new approach to the blocking of alloreactive T cell-mediated graft-versus-host disease by in vivo administration of anti-CXCR3 neutralizing antibody. Journal of Immunology, 181, 7581–7592.
Heidegger, S., van den Brink, M. R., Haas, T., & Poeck, H. (2014). The role of pattern-recognition receptors in graft-versus-host disease and graft-versus-leukemia after allogeneic stem cell transplantation. Frontiers in Immunology, 5, 337. https://doi.org/10.3389/fimmu.2014.00337.
Hill, G. R., Crawford, J. M., Cooke, K. R., Brinson, Y. S., Pan, L., & Ferrara, J. L. (1997). Total body irradiation and acute graft-versus-host disease: The role of gastrointestinal damage and inflammatory cytokines. Blood, 90, 3204–3213.
Holler, E., Landfried, K., Meier, J., Hausmann, M., & Rogler, G. (2013). The role of bacteria and pattern recognition receptors in GVHD. International Journal of Inflammation, 2010, 814326. https://doi.org/10.4061/2010/814326.
Hoyne, G. F., Le Roux, I., Corsin-Jimenez, M., Tan, K., Dunne, J., Forsyth, L. M., Dallman, M. J., Owen, M. J., Ish-Horowicz, D., & Lamb, J. R. (2000). Serrate1-induced notch signalling regulates the decision between immunity and tolerance made by peripheral CD4(+) T cells. International Immunology, 12, 177–185.
Hu, X., Chung, A. Y., Wu, I., Foldi, J., Chen, J., Ji, J. D., Tateya, T., Kang, Y. J., Han, J., Gessler, M., Kageyama, R., & Ivashkiv, L. B. (2008). Integrated regulation of Toll-like receptor responses by Notch and interferon-gamma pathways. Immunity, 29, 691–703. https://doi.org/10.1016/j.immuni.2008.08.016.
Hue, S., Kared, H., Mehwish, Y., Mouhamad, S., Balbo, M., & Levy, Y. (2012). Notch activation on effector T cells increases their sensitivity to Treg cell-mediated suppression through upregulation of TGF-βRII expression. European Journal of Immunology, 42, 1796–1803. https://doi.org/10.1002/eji.201142330.
Iclozan, C., Yu, Y., Liu, C., Liang, Y., Yi, T., Anasetti, C., & Yu, X. Z. (2009). T helper17 cells are sufficient but not necessary to induce acute graft-versus-host disease. Biology of Blood and Marrow Transplantation, 16, 170–178. https://doi.org/10.1016/j.bbmt.2009.09.023.
Imanguli, M. M., Swaim, W. D., League, S. C., Gress, R. E., Pavletic, S. Z., & Hakim, F. T. (2009). Increased T- bet+ cytotoxic effectors and type I interferon-mediated processes in chronic graft-versus-host disease of the oral mucosa. Blood, 113, 3620–3630. https://doi.org/10.1182/blood-2008-07-168351.
Ju, J. M., Lee, H., Oh, K., Lee, D. S., & Choi, E. Y. (2014). Kinetics of IFN-γ and IL-17 production by CD4 and CD8 T cells during acute graft-versus-host disease. Immune Network, 14, 89–99. https://doi.org/10.4110/in.2014.14.2.89.
Kassner, N., Krueger, M., Yagita, H., Dzionek, A., Hutloff, A., Kroczek, R., Scheffold, A., & Rutz, S. (2010). Cutting edge: Plasmacytoid dendritic cells induce IL-10 production in T cells via the Delta-like-4/Notch axis. Journal of Immunology, 184, 550–554. https://doi.org/10.4049/jimmunol.0903152.
Kimura, A., Naka, T., & Kishimoto, T. (2007). IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proceedings of the National Academy of Sciences of the United States of America, 104, 12099–12104.
Koch, M. A., Tucker-Heard, G., Perdue, N. R., Killebrew, J. R., Urdahl, K. B., & Campbell, D. J. (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nature Immunology, 10, 595–602. https://doi.org/10.1038/ni.1731.
Koch, U., & Radtke, F. (2011). Mechanisms of T cell development and transformation. Annual Review of Cell and Developmental Biology, 27, 539–562. https://doi.org/10.1146/annurev-cellbio-092910-154008.
Komatsu, N., Okamoto, K., Sawa, S., Nakashima, T., Oh-hora, M., Kodama, T., Tanaka, S., Bluestone, J. A., & Takayanagi, H. (2014). Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nature Medicine, 20, 62–68. https://doi.org/10.1038/nm.3432.
Kotsiou, E., & Davies, J. K. (2013). New ways to separate graft-versus-host disease and graft-versus- tumour effects after allogeneic haematopoietic stem cell transplantation. British Journal of Haematology, 160, 133–145. https://doi.org/10.1111/bjh.12115.
Kovall, R. A. (2007). Structures of CSL, Notch and Mastermind proteins: Piecing together an active transcription complex. Current Opinion in Structural Biology, 17, 117–127.
Koyama, M., Cheong, M., Markey, K. A., Gartlan, K. H., Kuns, R. D., Locke, K. R., Lineburg, K. E., Teal, B. E., Leveque-El Mouttie, L., Bunting, M. D., Vuckovic, S., Zhang, P., Teng, M. W., Varelias, A., Tey, S. K., Wockner, L. F., Engwerda, C. R., Smyth, M. J., Belz, G. T., McColl, S. R., MacDonald, K. P., & Hill, G. R. (2015). Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. The Journal of Experimental Medicine, 212, 1303–1321. https://doi.org/10.1084/jem.20150329.
Koyama, M., Hashimoto, D., Aoyama, K., Matsuoka, K., Karube, K., Niiro, H., Harada, M., Tanimoto, M., Akashi, K., & Teshima, T. (2009). Plasmacytoid dendritic cells prime alloreactive T cells to mediate graft-versus-host disease as antigen-presenting cells. Blood, 113, 2088–2095. https://doi.org/10.1182/blood-2008-07-168609.
Kryczek, I., Zhao, E., Liu, Y., Wang, Y., Vatan, L., Szeliga, W., Moyer, J., Klimczak, A., Lange, A., & Zou, W. (2011). Human TH17 cells are long-lived effector memory cells. Science Translational Medicine, 3, 104ra100. https://doi.org/10.1126/scitranslmed.3002949.
Lai, H. Y., Chou, T. Y., Tzeng, C. H., & Lee, O. K. (2012). Cytokine profiles in various graft-versus- host disease target organs following hematopoietic stem cell transplantation. Cell Transplantation, 21, 2033–2045. https://doi.org/10.3727/096368912X653110.
Laky, K., & Fowlkes, B. J. (2008). Notch signaling in CD4 and CD8 T cell development. Current Opinion in Immunology, 20, 197–202. https://doi.org/10.1016/j.coi.2008.03.004.
Li, C. R., Mueller, E. E., & Bradley, L. M. (2014). Islet antigen-specific Th17 cells can induce TNF-α- dependent autoimmune diabetes. Journal of Immunology, 192, 1425–1432. https://doi.org/10.4049/jimmunol.1301742.
Li, H., Demetris, A. J., McNiff, J., Matte-Martone, C., Tan, H. S., Rothstein, D. M., & Lakkis, F. G. (2012). Shlomchik WD (2012) Profound depletion of host conventional dendritic cells, plasmacytoid dendritic cells, and B cells does not prevent graft-versus-host disease induction. Journal of Immunology, 188(8), 3804–3811. https://doi.org/10.4049/jimmunol.1102795.
Li, J. M., Giver, C. R., Lu, Y., Hossain, M. S., Akhtari, M., & Waller, E. K. (2009). Separating graft-versus- leukemia from graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Immunotherapy, 1, 599–621.
Li, N., Chen, Y., He, W., Yi, T., Zhao, D., Zhang, C., Lin, C. L., Todorov, I., Kandeel, F., Forman, S., & Zeng, D. (2009). Anti-CD3 preconditioning separates GVL from GVHD via modulating host dendritic cell and donor T-cell migration in recipients conditioned with TBI. Blood, 113, 953–962. https://doi.org/10.1182/blood-2008-06-165522.
Li, S., Xie, Q., Zeng, Y., Zou, C., Liu, X., Wu, S., Deng, H., Xu, Y., Li, X. C., & Dai, Z. (2014). A naturally occurring CD8(+)CD122(+) T-cell subset as a memory-like Treg family. Cellular & Molecular Immunology, 11, 326–331. https://doi.org/10.1038/cmi.2014.25.
Lin, X., Lu, Z. G., Song, C. Y., Huang, Y. X., Guo, K. Y., Deng, L., Tu, S. F., He, Y. Z., Xu, J. H., Long, H., & Wu, B. Y. (2015). Long-term outcome of HLA-haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion based on an FBCA conditioning regimen for hematologic malignancies. Bone Marrow Transplantation, 50, 1092–1097. https://doi.org/10.1038/bmt.2015.108.
Liotta, F., Frosali, F., Querci, V., Mantei, A., Filì, L., Maggi, L., Mazzinghi, B., Angeli, R., Ronconi, E., Santarlasci, V., Biagioli, T., Lasagni, L., Ballerini, C., Parronchi, P., Scheffold, A., Cosmi, L., Maggi, E., Romagnani, S., & Annunziato, F. (2008). Human immature myeloid dendritic cells trigger a TH2- polarizing program via Jagged-1/Notch interaction. The Journal of Allergy and Clinical Immunology, 121, 1000–1005.e8. https://doi.org/10.1016/j.jaci.2008.01.004.
Liu, H. P., Cao, A. T., Feng, T., Li, Q., Zhang, W., Yao, S., Dann, S. M., Elson, C. O., & Cong, Y. (2015). TGF-β converts Th1 cells into Th17 cells through stimulation of Runx1 expression. European Journal of Immunology, 45, 1010–1018. https://doi.org/10.1002/eji.201444726.
Maillard, I., Weng, A. P., Carpenter, A. C., Rodriguez, C. G., Sai, H., Xu, L., Allman, D., Aster, J. C., & Pear, W. S. (2004). Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood, 104, 1696–1702.
Mangan, P. R., Harrington, L. E., O’Quinn, D. B., Helms, W. S., Bullard, D. C., Elson, C. O., Hatton, R. D., Wahl, S. M., Schoeb, T. R., & Weaver, C. T. (2006). Transforming growth factor-beta induces development of the T(H)17 lineage. Nature, 441, 231–234.
Mapara, M. Y., Leng, C., Kim, Y. M., Bronson, R., Lokshin, A., Luster, A., & Sykes, M. (2006). Expression of chemokines in GVHD target organs is influenced by conditioning and genetic factors and amplified by GVHR. Biology of Blood and Marrow Transplantation, 12, 623–634.
Martinez Arias, A., Zecchini, V., & Brennan, K. (2002). CSL-independent Notch signalling: A checkpoint in cell fate decisions during development? Current Opinion in Genetics & Development, 12, 524–533.
Mauermann, N., Burian, J., von Garnier, C., Dirnhofer, S., Germano, D., Schuett, C., Tamm, M., Bingisser, R., Eriksson, U., & Hunziker, L. (2008). Interferon-gamma regulates idiopathic pneumonia syndrome, a Th17+CD4+ T-cell-mediated graft-versus-host disease. American Journal of Respiratory and Critical Care Medicine, 178, 379–388. https://doi.org/10.1164/rccm.200711-1648OC.
Meng, L., Bai, Z., He, S., Mochizuki, K., Liu, Y., Purushe, J., Sun, H., Wang, J., Yagita, H., Mineishi, S., Fung, H., Yanik, G. A., Caricchio, R., Fan, X., Crisalli, L. M., Hexner, E. O., Reshef, R., Zhang, Y., & Zhang, Y. (2016). The Notch ligand DLL4 defines a capability of human dendritic cells in regulating Th1 and Th17 differentiation. Journal of Immunology, 196(3), 1070–1080. https://doi.org/10.4049/jimmunol.1501310. Epub 2015 Dec 28.
Michael, M., Shimoni, A., & Nagler, A. (2013). Regulatory T cells in allogeneic stem cell transplantation. Clinical & Developmental Immunology, 2013, 608951. https://doi.org/10.1155/2013/608951.
Minter, L. M., Turley, D. M., Das, P., Shin, H. M., Joshi, I., Lawlor, R. G., Cho, O. H., Palaga, T., Gottipati, S., Telfer, J. C., Kostura, L., Fauq, A. H., Simpson, K., Such, K. A., Miele, L., Golde, T. E., Miller, S. D., & Osborne, B. A. (2005). Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nature Immunology, 6, 680–688.
Minter, L. M., & Osborne, B. A. (2012). Canonical and non-canonical Notch signaling in CD4+ T cells. Current Topics in Microbiology and Immunology, 360, 99–114. https://doi.org/10.1007/82_2012_233.
Mirandola, L., Chiriva-Internati, M., Montagna, D., Locatelli, F., Zecca, M., Ranzani, M., Basile, A., Locati, M., Cobos, E., Kast, W. M., Asselta, R., Paraboschi, E. M., Comi, P., & Chiaramonte, R. (2012). Notch1 regulates chemotaxis and proliferation by controlling the CC-chemokine receptors 5 and 9 in T cell acute lymphoblastic leukaemia. The Journal of Pathology, 226, 713–722. https://doi.org/10.1002/path.3015.
Mochizuki, K., He, S., & Zhang, Y. (2011). Notch and inflammatory T-cell response: New developments and challenges. Immunotherapy, 3, 1353–1366. https://doi.org/10.2217/imt.11.126.
Mochizuki, K., Xie, F., He, S., Tong, Q., Liu, Y., Mochizuki, I., Guo, Y., Kato, K., Yagita, H., Mineishi, S., & Zhang, Y. (2013). Delta-like ligand 4 identifies a previously uncharacterized population of inflammatory dendritic cells that plays important roles in eliciting allogeneic T cell responses in mice. Journal of Immunology, 190, 3772–3782. https://doi.org/10.4049/jimmunol.1202820.
Monsalve, E., Pérez, M. A., Rubio, A., Ruiz-Hidalgo, M. J., Baladrón, V., García-Ramírez, J. J., Gómez, J. C., Laborda, J., & Díaz-Guerra, M. J. (2006). Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. Journal of Immunology, 176, 5362–5373.
Morikawa, H., & Sakaguchi, S. (2014). Genetic and epigenetic basis of Treg cell development and function: From a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunological Reviews, 259, 192–205. https://doi.org/10.1111/imr.12174.
Mota, C., Nunes-Silva, V., Pires, A. R., Matoso, P., Victorino, R. M., Sousa, A. E., & Caramalho, I. (2014). Delta-like 1-mediated Notch signaling enhances the in vitro conversion of human memory CD4 T cells intoFOXP3-expressing regulatory T cells. Journal of Immunology, 193, 5854–5862. https://doi.org/10.4049/jimmunol.1400198.
Mukherjee, S., Schaller, M. A., Neupane, R., Kunkel, S. L., & Lukacs, N. W. (2009). Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. Journal of Immunology, 182, 7381–7388. https://doi.org/10.4049/jimmunol.0804322.
Nishiwaki, S., Terakura, S., Ito, M., Goto, T., Seto, A., Watanabe, K., Yanagisawa, M., Imahashi, N., Tsukamoto, S., Shimba, M., Ozawa, Y., & Miyamura, K. (2009). Impact of macrophage infiltration of skin lesions on survival after allogeneic stem cell transplantation: A clue to refractory graft- versus-host disease. Blood, 114, 3113–3116. https://doi.org/10.1182/blood-2009-03-209635.
Nishiwaki, S., Nakayama, T., Murata, M., Nishida, T., Terakura, S., Saito, S., Kato, T., Mizuno, H., Imahashi, N., Seto, A., Ozawa, Y., Miyamura, K., Ito, M., Takeshita, K., Kato, H., Toyokuni, S., Nagao, K., Ueda, R., & Naoe, T. (2014). Dexamethasone palmitate ameliorates macrophages-rich graft-versus- host disease by inhibiting macrophage functions. PLoS One, 9, e96252. https://doi.org/10.1371/journal.pone.0096252.
Osborne, B. A., & Minter, L. M. (2007). Notch signalling during peripheral T-cell activation and differentiation. Nature Reviews. Immunology, 7, 64–75.
Ostroukhova, M., Qi, Z., Oriss, T. B., Dixon-McCarthy, B., Ray, P., & Ray, A. (2006). Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta. The Journal of Clinical Investigation, 116, 996–1004.
Ong, C. T., Sedy, J. R., Murphy, K. M., & Kopan, R. (2008). Notch and presenilin regulate cellular expansion and cytokine secretion but cannot instruct Th1/Th2 fate acquisition. PLoS One, 3, e2823. https://doi.org/10.1371/journal.pone.0002823.
Ou-Yang, H. F., Zhang, H. W., Wu, C. G., Zhang, P., Zhang, J., Li, J. C., Hou, L. H., He, F., Ti, X. Y., Song, L. Q., Zhang, S. Z., Feng, L., Qi, H. W., & Han, H. (2009). Notch signaling regulates the FOXP3 promoter through RBP-J- and Hes1-dependent mechanisms. Molecular and Cellular Biochemistry, 320, 109–114. https://doi.org/10.1007/s11010-008-9912-4.
Palaga, T., Miele, L., Golde, T. E., & Osborne, B. A. (2003). TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. Journal of Immunology, 171, 3019–3024.
Pan, B., Zhang, Y., Sun, Y., Cheng, H., Wu, Y., Song, G., Chen, W., Zeng, L., & Xu, K. (2014). Deviated balance between Th1 and Th17 cells exacerbates acute graft-versus-host disease in mice. Cytokine, 68, 69–75. https://doi.org/10.1016/j.cyto.2014.04.002.
Parmar, S., Liu, X., Tung, S. S., Robinson, S. N., Rodriguez, G., Cooper, L. J., Yang, H., Shah, N., Yang, H., Konopleva, M., Molldrem, J. J., Garcia-Manero, G., Najjar, A., Yvon, E., McNiece, I., Rezvani, K., Savoldo, B., Bollard, C. M., & Shpall, E. J. (2015). Third-party umbilical cord blood-derived regulatory T cells prevent xenogenic graft-versus-host disease. Blood. pii: blood-2015-06-653667. [Epub ahead of print].
Pasquini, M. C. (2008). Impact of graft-versus-host disease on survival. Best Practice & Research. Clinical Haematology, 21, 193–204. https://doi.org/10.1016/j.beha.2008.02.011.
Perumalsamy, L. R., Marcel, N., Kulkarni, S., Radtke, F., & Sarin, A. (2012). Distinct spatial and molecular features of notch pathway assembly in regulatory T cells. Science Signaling, 5, ra53. https://doi.org/10.1126/scisignal.2002859.
Pierini, A., Colonna, L., Alvarez, M., Schneidawind, D., Nishikii, H., Baker, J., Pan, Y., Florek, M., Kim, B. S., & Negrin, R. S. (2014). Donor requirements for regulatory T cell suppression of murine graft- versus-host disease. Cytotherapy, 16, 90–100. https://doi.org/10.1016/j.jcyt.2013.07.009.
Pratt, E. B., Wentzell, J. S., Maxson, J. E., Courter, L., Hazelett, D., & Christian, J. L. (2011). The cell giveth and the cell taketh away: An overview of Notch pathway activation by endocytic trafficking of ligands and receptors. Acta Histochemica, 113, 248–255. https://doi.org/10.1016/j.acthis.2010.01.006.
Ratajczak, M. Z., & Suszynska, M. (2016). Emerging strategies to enhance homing and engraftment of hematopoietic stem cells. Stem Cell Reviews, 12, 121–128. https://doi.org/10.1007/s12015-015-9625-5.
Reshef, R., Luger, S. M., Hexner, E. O., Loren, A. W., Frey, N. V., Nasta, S. D., Goldstein, S. C., Stadtmauer, E. A., Smith, J., Bailey, S., Mick, R., Heitjan, D. F., Emerson, S. G., Hoxie, J. A., Vonderheide, R. H., & Porter, D. L. (2012). Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. The New England Journal of Medicine, 367, 135–145. https://doi.org/10.1056/NEJMoa1201248.
Roderick, J. E., Gonzalez-Perez, G., Kuksin, C. A., Dongre, A., Roberts, E. R., Srinivasan, J., Andrzejewski, C., Jr., Fauq, A. H., Golde, T. E., Miele, L., & Minter, L. M. (2013). Therapeutic targeting of NOTCH signaling ameliorates immune-mediated bone marrow failure of aplastic anemia. The Journal of Experimental Medicine, 210, 1311–1329. https://doi.org/10.1084/jem.20112615.
Rutz, S., Janke, M., Kassner, N., Hohnstein, T., Krueger, M., & Scheffold, A. (2008). Notch regulates IL- 10 production by T helper 1 cells. Proceedings of the National Academy of Sciences of the United States of America, 105, 3497–3502. https://doi.org/10.1073/pnas.0712102105.
Samon, J. B., Champhekar, A., Minter, L. M., Telfer, J. C., Miele, L., Fauq, A., Das, P., Golde, T. E., & Osborne, B. A. (2008). Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood, 112, 1813–1821. https://doi.org/10.1182/blood-2008-03-144980.
Sandy, A. R., Jones, M., & Maillard, I. (2012). Notch signaling and development of the hematopoietic system. Advances in Experimental Medicine and Biology, 727, 71–88. https://doi.org/10.1007/978-1-4614-0899-4_6.
Sandy, A. R., Chung, J., Toubai, T., Shan, G. T., Tran, I. T., Friedman, A., Blackwell, T. S., Reddy, P., King, P. D., & Maillard, I. (2013). T cell-specific notch inhibition blocks graft-versus-host disease by inducing a hyporesponsive program in alloreactive CD4+ and CD8+ T cells. Journal of Immunology, 190, 5818–5828. https://doi.org/10.4049/jimmunol.1203452.
Schmitt, E. G., & Williams, C. B. (2013). Generation and function of induced regulatory T cells. Frontiers in Immunology, 4, 152. https://doi.org/10.3389/fimmu.2013.00152. eCollection 2013.
Schmitt, N., & Ueno, H. (2015). Regulation of human helper T cell subset differentiation by cytokines. Current Opinion in Immunology, 34, 130–136. https://doi.org/10.1016/j.coi.2015.03.007.
Schwanbeck, R. (2015). The role of epigenetic mechanisms in Notch signaling during development. Journal of Cellular Physiology, 230, 969–981. https://doi.org/10.1002/jcp.24851.
Shlomchik, W. D. (2007). Graft-versus-host disease. Nature Reviews. Immunology, 7, 340–352.
Singla, R. D., Wang, J., & Singla, D. K. (2014). Regulation of Notch 1 signaling in THP-1 cells enhances M2 macrophage differentiation. American Journal of Physiology. Heart and Circulatory Physiology, 307, H1634–H1642. https://doi.org/10.1152/ajpheart.00896.2013.
Skokos, D., & Nussenzweig, M. C. (2007). CD8− DCs induce IL-12–independent Th1 differentiation through Delta 4 Notch-like ligand in response to bacterial LPS. The Journal of Experimental Medicine, 204, 1525–1531. https://doi.org/10.1084/jem.20062305.
Szabo, S. J., Kim, S. T., Costa, G. L., Zhang, X., Fathman, C. G., & Glimcher, L. H. (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell, 100, 655–669.
Theil, A., Tuve, S., Oelschlägel, U., Maiwald, A., Döhler, D., Oßmann, D., Zenkel, A., Wilhelm, C., Middeke, J. M., Shayegi, N., Trautmann-Grill, K., von Bonin, M., Platzbecker, U., Ehninger, G., Bonifacio, E., & Bornhäuser, M. (2015). Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy, 17, 473–486. https://doi.org/10.1016/j.jcyt.2014.11.005.
Tran, I. T., Sandy, A. R., Carulli, A. J., Ebens, C., Chung, J., Shan, G. T., Radojcic, V., Friedman, A., Gridley, T., Shelton, A., Reddy, P., Samuelson, L. C., Yan, M., Siebel, C. W., & Maillard, I. (2013). Blockade of individual Notch ligands and receptors controls graft-versus-host disease. The Journal of Clinical Investigation, 123, 1590–1604.
Tu, L., Fang, T. C., Artis, D., Shestova, O., Pross, S. E., Maillard, I., & Pear, W. S. (2005). Notch signaling is an important regulator of type 2 immunity. The Journal of Experimental Medicine, 202, 1037–1042.
Ukena, S. N., Grosse, J., Mischak-Weissinger, E., Buchholz, S., Stadler, M., Ganser, A., & Franzke, A. (2011). Acute but not chronic graft-versus-host disease is associated with a reduction of circulating CD4(+)CD25 (high)CD127 (low/-) regulatory T cells. Annals of Hematology, 90, 213–218. https://doi.org/10.1007/s00277-010-1068-0. (a).
Ukena, S. N., Velaga, S., Geffers, R., Grosse, J., Baron, U., Buchholz, S., Stadler, M., Bruder, D., Ganser, A., & Franzke, A. (2012). Human regulatory T cells in allogeneic stem cell transplantation. Blood, 118, e82–e92. https://doi.org/10.1182/blood-2011-05-352708. (b).
van den Brink, M. R., Velardi, E., & Perales, M. A. (2015). Immune reconstitution following stem cell transplantation. Hematology. American Society of Hematology. Education Program, 2015, 215–219. https://doi.org/10.1182/asheducation-2015.1.215.
van der Waart, A. B., van der Velden, W. J., Blijlevens, N. M., & Dolstra, H. (2014). Targeting the IL17 pathway for the prevention of graft-versus-host disease. Biology of Blood and Marrow Transplantation, 20, 752–759. https://doi.org/10.1016/j.bbmt.2014.02.007.
Varona, R., Cadenas, V., Gómez, L., Martínez-A, C., & Márquez, G. (2005). CCR6 regulates CD4+ T- cell-mediated graft-versus-host disease responses. Blood, 106, 18–26.
Vianello, F., Cannella, L., Coe, D., Chai, J. G., Golshayan, D., Marelli-Berg, F. M., & Dazzi, F. (2013). Enhanced and aberrant T cell trafficking following total body irradiation: a gateway to graft- versus-host disease? British Journal of Haematology, 162, 808–818. https://doi.org/10.1111/bjh.12472.
Wang, Y. C., He, F., Feng, F., Liu, X. W., Dong, G. Y., Qin, H. Y., Hu, X. B., Zheng, M. H., Liang, L., Feng, L., Liang, Y. M., & Han, H. (2010). Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Research, 70, 4840–4849. https://doi.org/10.1158/0008-5472.CAN-10-0269.
Wendland, M., Czeloth, N., Mach, N., Malissen, B., Kremmer, E., Pabst, O., & Förster, R. (2007). CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proceedings of the National Academy of Sciences of the United States of America, 104, 6347–6352.
Wongchana, W., & Palaga, T. (2012). Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cellular & Molecular Immunology, 9, 155–162. https://doi.org/10.1038/cmi.2011.36.
Wongchana, W., Lawlor, R. G., Osborne, B. A., & Palaga, T. (2015). Impact of Notch1 deletion in macrophages on proinflammatory cytokine production and the outcome of experimental autoimmune encephalomyelitis. Journal of Immunology, 195, 5337–5346. https://doi.org/10.4049/jimmunol.1401770.
Wu, Y., Bastian, D., Schutt, S., Nguyen, H., Fu, J., Heinrichs, J., Xia, C., & Yu, X. Z. (2015). Essential role of interleukin-12/23p40 in the development of graft-versus-host disease in mice. Biology of Blood and Marrow Transplantation, 21, 1195–1204. https://doi.org/10.1016/j.bbmt.2015.03.016.
Xiao, S., Jin, H., Korn, T., Liu, S. M., Oukka, M., Lim, B., & Kuchroo, V. K. (2008). Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. Journal of Immunology, 181, 2277–2284.
Xu, H., Zhu, J., Smith, S., Foldi, J., Zhao, B., Chung, A. Y., Outtz, H., Kitajewski, J., Shi, C., Weber, S., Saftig, P., Li, Y., Ozato, K., Blobel, C. P., Ivashkiv, L. B., & Hu, X. (2012). Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nature Immunology, 13, 642–650. https://doi.org/10.1038/ni.2304.
Xu, J., Chi, F., Guo, T., Punj, V., Lee, W. N., French, S. W., & Tsukamoto, H. (2015). NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. The Journal of Clinical Investigation, 125, 1579–1590. https://doi.org/10.1172/JCI76468.
Yamane, H., & Paul, W. E. (2013). Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunological Reviews, 252, 12–23. https://doi.org/10.1111/imr.12032.
Yi, T., Chen, Y., Wang, L., Du, G., Huang, D., Zhao, D., Johnston, H., Young, J., Todorov, I., Umetsu, D. T., Chen, L., Iwakura, Y., Kandeel, F., Forman, S., & Zeng, D. (2009). Reciprocal differentiation and tissue- specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood, 114, 3101–3112. https://doi.org/10.1182/blood-2009-05-219402.
Yu, Q., Sharma, A., Oh, S. Y., Moon, H. G., Hossain, M. Z., Salay, T. M., Leeds, K. E., Du, H., Wu, B., Waterman, M. L., Zhu, Z., & Sen, J. M. (2009). T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nature Immunology, 10, 992–999. https://doi.org/10.1038/ni.1762.
Yu, Y., Wang, D., Liu, C., Kaosaard, K., Semple, K., Anasetti, C., & Yu, X. Z. (2011). Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORγt in mice. Blood, 118, 5011–5020. https://doi.org/10.1182/blood-2011-03-340315.
Yuan, J., Ren, H. Y., Shi, Y. J., & Liu, W. (2015). Prophylaxis of acute graft-versus-host disease by CCR5 blockade combined with cyclosporine A in a murine model. Inflammation Research, 64, 137–144. https://doi.org/10.1007/s00011-014-0793-6.
Zeng, C., Xing, R., Liu, J., & Xing, F. (2016). Role of CSL-dependent and independent Notch signaling pathways in cell apoptosis. Apoptosis, 21, 1–12. [Epub ahead of print].
Zhang, Y., Sandy, A. R., Wang, J., Radojcic, V., Shan, G. T., Tran, I. T., Friedman, A., Kato, K., He, S., Cui, S., Hexner, E., Frank, D. M., Emerson, S. G., Pear, W. S., & Maillard, I. (2011). Notch signaling is a critical regulator of allogeneic CD4+ T-cell responses mediating graft-versus-host disease. Blood, 117, 299–308. https://doi.org/10.1182/blood-2010-03-271940.
Zhao, Y., Liu, Q., Yang, L., He, D., Wang, L., Tian, J., Li, Y., Zi, F., Bao, H., Yang, Y., Zheng, Y., Shi, J., Xue, X., & Cai, Z. (2013). TLR4 inactivation protects from graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Cellular & Molecular Immunology, 10, 165–175. https://doi.org/10.1038/cmi.2012.58.
Fulton, L. M., Carlson, M. J., Coghill, J. M., Ott, L. E., West, M. L., Panoskaltsis-Mortari, A., Littman, D. R., Blazar, B. R., & Serody, J. S. (2012). Attenuation of Acute Graft-versus-Host Disease in the Absence of the Transcription Factor RORÂ t. The Journal of Immunology, 189(4):1765–1772. https://doi.org/10.4049/jimmunol.1200858. Epub 2012 Jul 9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media, LLC, part of Springer Nature
About this chapter

Cite this chapter
Minter, L.M. (2018). Notch Signaling in Graft-Versus-Host Disease. In: Miele, L., Artavanis-Tsakonas, S. (eds) Targeting Notch in Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-8859-4_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-8859-4_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-8857-0
Online ISBN: 978-1-4939-8859-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)