
Abstract
Focal development of an increasing number of renal cysts with age is a hallmark of autosomal dominant polycystic kidney disease (ADPKD), leading to the distortion of normal kidney architecture and, ultimately, ESRD in a majority of patients. Mutations of two genes (i.e., PKD1 and PKD2) account for most of the cases. Clinically, a wide spectrum of cystic disease variability is displayed in ADPKD in part due to strong effects from the gene locus (i.e., PKD1 vs. PKD2) and alleles (i.e., truncating vs. non-truncating PKD1 mutations). Presymptomatic screening of subjects at risk for ADPKD is commonly performed by conventional ultrasonography, which is inexpensive and widely available. Age-dependent ultrasound criteria have been derived for PKD1 and subsequently refined (a.k.a. unified criteria) for evaluation of at-risk subjects with unknown gene type. However, reduced diagnostic sensitivity by conventional ultrasound renders disease exclusion in younger at-risk subjects not possible until 40 years of age. More recently, magnetic resonance imaging (MRI) has been shown to greatly improve imaging-based diagnostic performance in ADPKD. Specifically, “more than a total of 10 renal cysts” can be regarded as sufficient for diagnosis while “less than a total of 5 renal cysts,” sufficient for disease exclusion in at-risk subjects between 16 and 40 years of age. These criteria will greatly facilitate the evaluation of at-risk subjects who wish to be considered as living kidney donors. High-resolution ultrasound can provide excellent diagnostic performance, rivaling that of MRI, but is both center- and operator-dependent. The diagnosis of subjects suspected to have ADPKD without a definitive family history can be challenging and will often require molecular genetic testing.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common Mendelian genetic kidney disease, affecting 1 in 500–1000 births worldwide [1]. Focal development of an increasing number of renal cysts with age is a hallmark of ADPKD, leading to the distortion of normal kidney architecture and, ultimately, end-stage renal disease (ESRD) in a majority of patients [2]. Mutations of two genes, PKD1 and PKD2 , have been, respectively, implicated for the disease in 85% and 15% of linkage-characterized European families [3]. However, a more recent population-based study has reported a higher prevalence of PKD2 of 26% [4]. Disease severity and progression is highly variable in ADPKD in part due to strong effects from the gene locus and alleles [5,6,7,8,9,10,11]. Adjusted for age, patients with PKD1 have more renal cysts, larger kidneys, and earlier onset of ESRD than patients with PKD2 (mean age at ESRD: 53.4 vs. 72.7 years, respectively) [7, 8]. More recent studies have further demonstrated a significant allelic effect in PKD1 with milder renal disease associated with non-truncating mutations and severe disease, with truncating mutations [9,10,11]. Marked renal disease variability within families has been well documented in ADPKD and suggests a modifier effect from genetic and environmental factors [12, 13].
Imaging-based testing is commonly performed for presymptomatic screening or clinical diagnosis of ADPKD. Conventional ultrasound, which is inexpensive and widely available, is the most common modality used for presymptomatic screening of ADPKD. As simple cysts occur with increasing age in the general [14] and hospital patient [15] population, age-dependent ultrasound diagnostic criteria have been established for PKD1 [16] and subsequently refined and extended for evaluation of at-risk subjects of unknown gene type (a.k.a. “unified diagnostic criteria ”) [17]. More recently, highly sensitive and specific criteria based on magnetic resonance imaging (MRI) have also been derived for earlier diagnosis and exclusion of ADPKD in younger at-risk subjects [18]. In general, presymptomatic screening of at-risk children (under 18 years of age) cannot be recommended at this time based on the potential risks including adverse psychological consequences and denial of future insurance coverage. Additionally, there is currently no evidence that clinical management in this setting would improve outcomes. The possible implications of all diagnostic screening should be discussed beforehand, and the results clearly explained to the test subjects. Blood pressure assessment should be performed routinely in at-risk subjects of all age groups whether or not they undergo the screening.
It is important to note that the diagnostic criteria derived for US and MRI are applicable only to test subjects with a definitive family history of ADPKD who are born with a 50% risk of disease. By contrast, the pretest probability of subjects without a positive family history is that of the population risk (i.e., 1 in 500–1000); thus, the above criteria may not be valid. Moreover, the possibility of other genetic and nongenetic causes of PKD needs to be considered in the latter setting [2]. In subjects without a positive family history, it is useful to screen their parents and older first-degree relatives with ultrasound as mild disease associated with a PKD2 or non-truncating PKD1 mutation may not be apparent, especially in small families [2]. If one or both parents are deceased, review of their medical record for prior renal imaging results may be helpful. The documentation of at least one affected first-degree relative with bilaterally enlarged kidneys and numerous cysts is sufficient evidence to support the use of the imaging-based diagnostic criteria. On the other hand, the presence of several cysts without kidney enlargement in an elderly relative may be due to simple cysts and should not be considered as sufficient evidence for a positive family history.
Diagnosis of ADPKD by Conventional Ultrasound
The diagnosis of ADPKD is generally straightforward in patients with symptomatic disease and a positive family history. For presymptomatic screening of at-risk subjects, age-dependent criteria based on conventional ultrasound have been derived for PKD1 [16]. However, the utility of these criteria is limited by the unknown gene type of most test subjects in the clinical setting and the negative impact on diagnostic performance by the milder cases associated with PKD2. To overcome these limitations, Pei et al. derived age-dependent diagnostic criteria from a cohort of 948 at-risk subjects by comparing their molecular genetic results (for the presence or absence of disease) and ultrasound findings, using a simulated case mix of 85–15% for PKD1 and PKD2, respectively [17]. Based on the “unified criteria” derived from this study (Table 7.1), the presence of “a total of three or more renal cysts” for at-risk subjects aged 15–39 years and “two cysts or more in each kidney” for at-risk subjects aged 40–59 years can be considered as sufficient for diagnosis. Conversely, the “absence of any renal cyst” can be considered sufficient for disease exclusion only in at-risk subjects aged 40 years or older. It should be noted that a resolution of approximately 1 cm or more is required for conventional ultrasound to detect most of the renal cysts. Thus, the validity of the unified criteria should not be extrapolated to high-resolution ultrasound using modern scanners, which has imaging resolution enabling routine detection of very small renal cysts of 2–3 mm. It is also clear that reduced diagnostic sensitivity (e.g., ~81% for unknown gene type) is a limitation of conventional ultrasound rendering early diagnosis of at-risk subjects with milder disease suboptimal. A corollary is that the “absence of any renal cyst” cannot be used as definitive evidence for disease exclusion in younger at-risk subjects of unknown gene type, where their negative predictive values are ~91% and 98%, under 30 and between 30 and 39 years of age, respectively.
Diagnosis of ADPKD by MRI and High-Resolution Ultrasound
Given that younger subjects at risk for ADPKD are increasingly being evaluated as potential living kidney donors [19, 20], there is a need to develop highly sensitive and specific tests for disease exclusion. While molecular diagnostics may be used here for disease exclusion, it is expensive and time-consuming and may not provide a definitive diagnosis in up to one third of cases [21]. With increased resolution for detecting very small cysts by magnetic resonance imaging (MRI) and contrast-enhanced computed tomography (CT), many transplant centers have routinely included one of these imaging modalities in their evaluation of subjects at risk of ADPKD. However, until recently, validated diagnostic criteria for these modalities were lacking [19, 20]. Thus, Huang et al. empirically proposed that CT or MRI be used for screening in this setting, and if there are at least three unilateral or bilateral renal cysts, that the kidney donation be deferred pending the results of molecular genetic testing [19]. On the other hand, Niaudet proposes that CT or MRI be used for detection of small renal cysts in at-risk subjects under 30 years of age and, if the scan is negative, molecular genetic testing be considered for disease exclusion [20].
With improved resolution to detect smaller cysts, there is a general concern that MRI or CT may also detect more simple renal cysts. However, until recently, there was no data on the prevalence of simple renal cysts detected by these imaging modalities in the heathy general population. Instead, a widely quoted study by Nascimento et al. [15] provides a retrospective survey of the prevalence of renal cysts by MRI in 528 patients with various non-specified medical conditions from a large university hospital in the United States. In this study, 31% (i.e., 11/35) and 51% (i.e., 97/190) of the patients aged 18–29 years and 30–44 years, respectively, had at least one renal cyst, and 11% (i.e., 4/35) and 12% (i.e., 23/190) of the patients from the two respective age strata had a total of three or more renal cysts. However, chronic renal disease (including polycystic kidney disease), if not manifested as renal atrophy, was not excluded in the study. Furthermore, molecular genetic analysis was not employed to ascertain whether those patients with renal cysts had ADPKD. Thus, the prevalence estimates of renal cysts provided by this study should not be taken to reflect those from a healthy general population.
The Toronto Radiological Imaging Study of Polycystic kidney disease (TRISP) has recently shown that MRI and high-resolution ultrasound provide improved performance for early diagnosis and exclusion of ADPKD compared to conventional ultrasound [18]. In this study, we prospectively enrolled 126 subjects at risk for ADPKD aged 16–40 years to undergo renal MRI and high-resolution ultrasound as well as comprehensive mutation screening of PKD1 and PKD2 (to define the disease status). Concurrently, 46 age-matched healthy controls without a family history of ADPKD also underwent the same imaging protocol to provide specificity data. Using these imaging modalities, we were able to detect very small renal cysts down to 3 mm in diameter. Despite increased sensitivity for detecting very small cysts by MRI, we only found 1/82 unaffected subject aged 16–40 years with more than three renal cysts. Thus, simple renal cysts in this healthy population remain few in number despite enhanced resolution for detecting very small cysts. By contrast, all but one (i.e., 72/73) of our genetically affected subjects had a total of more than 20 renal cysts (left panel, Fig. 7.1). These two features together enable MRI to provide highly discriminant diagnostics for ADPKD. Thus, the presence of “a total of more than 10 renal cysts” (with both positive predictive value and sensitivity of 100%) can be considered as sufficient for diagnosis in a subject at risk of ADPKD. Conversely, “less than a total of 10 renal cysts” (with both negative predictive value and specificity of 100%) can be considered as sufficient for disease exclusion. For evaluation of living kidney donors, among whom the clinical agenda is disease exclusion with high certainty, we recommend using “less than a total of 5 renal cysts” (with negative predictive value of 100% and specificity of 98.3%) as a more stringent criterion. Pending future studies demonstrating diagnostic equivalence between the two modalities, we do not recommend extrapolating the MRI-specific criteria to contrast-enhanced CT.

Distribution of total renal cyst counts by disease status: MRI (left panel) versus high-resolution US (right panel). Both MRI and HR-US provided highly discriminant diagnostics for ADPKD; however, HR-US is both operator- and center-dependent and may be affected by the body habitus of the test subject (Adapted from Pei et al. JASN; [18]). *TOR190.1 is a genetically affected subject with a BMI of 35.6 kg/m2 who had more than 20 renal cysts by MRI but no cyst detectable by a suboptimal US. #TOR31.2 is a subject with six renal cysts on US and ten renal cysts on MRI; he did not carry the familial PKD2 mutation and was considered as unaffected. **TOR404.1 and TOR208.5 were both unaffected but had three renal cysts by US
Overall, we found improved diagnostic performance by high-resolution ultrasound compared to conventional ultrasound [18]. Specifically, we noted a significant increase in sensitivity with a small decrease in specificity across all the criteria tested, likely due to increased imaging resolution in detecting cysts as small as 3 mm with the modern scanners and experienced operators attuned to detection of small cysts. Using the unified criterion of “a total of three or more renal cysts” in at-risk subjects under 30 years of age, we found a significant increase in sensitivity (i.e., 97.3% from 81.7%) with a small decrease in positive predictive value (i.e., 100–97.3%). To minimize false-positive cases with high-resolution ultrasound, a more stringent criterion such as “two or more cysts in each kidney” (with positive predictive value of 100%) should be used. Conversely, the “absence of any renal cyst” by high-resolution ultrasound may be considered sufficient for disease exclusion in at-risk subjects aged 30–40 years, but not younger. An important caveat is that a suboptimal scan (e.g., due to body habitus) should be interpreted as indeterminate, and this point was well illustrated by patient TOR190.1 (right panel, Fig. 7.1). Another limitation of ultrasound is that its diagnostic performance is both operator- and center-dependent, reflecting differences such as imaging resolution of the scanners and experience of the technicians/radiologists. Thus, current availability of ultrasound scanners with different imaging resolution has important implication for diagnostic testing. Specifically, the diagnostic criteria we derived here should be applicable to experienced centers using high-resolution ultrasound; otherwise, the unified criteria [17] should be used for centers that utilize conventional ultrasound. While conventional ultrasound will continue to be the first-line test for presymptomatic screening of ADPKD, MRI or high-resolution ultrasound will be useful for diagnostic clarification in cases with equivocal results and for disease exclusion in at-risk subjects being evaluated as potential kidney donors. Moreover, MRI also provides an assessment of renal cystic burden and disease severity in affected subjects (Fig. 7.2).

T2-weighted MRI images showing a spectrum of autosomal dominant polycystic kidney disease. (a) 26-year-old female with a protein-truncating PKD1 mutation (TKV = 890 ml), (b) 22-year-old female with a protein-truncating PKD2 mutation (TKV = 610 ml), (c) 26-year-old male with a hypomorphic PKD1 mutation (TKV = 310 ml), (d) 42-year-old male with protein-truncating PKD 1 mutation (TKV = 1280 ml), (e) 40-year-old male with a complete PKD2 deletion (TKV = 470 ml), and (f) 43-year-old male with a hypomorphic PKD1 mutation (TKV = 470 ml). Each small unit of the ruler denotes 1 cm
Diagnosis of ADPKD Without a Positive Family History
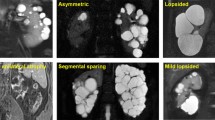
The absence of a positive family history in 15–28% of patients with suspected ADPKD poses a diagnostic challenge [2, 22, 23]. This problem is well illustrated by our findings in the Toronto Genetic Epidemiology Study of PKD (TGESP) in which 28% (58/209) of our probands did not report a family history of ADPKD [22]. In this subgroup of patients, we reviewed their parental medical records and screened all available parents by renal imaging. We found that 15.3% (32/209) of them had de novo PKD, 2% (4/209) had a positive family history in retrospect due to mild disease from non-truncating PKD1 mutations in one of their parents, and 10.5% (22/209) had an indeterminate family history due to missing parental information. Of interest, no pathogenic PKD1 and PKD2 mutation could be identified in ~16% (34/209) of our patients with atypical (i.e. asymmetric, focal, or unilateral) polycystic kidney disease on renal imaging. In one family with both an affected mother (proband) and daughter, we were able to prove that the mother was a somatic mosaic harboring a frameshift PKD1 mutation. Somatic mosaicism of a dominantly inherited disease such as ADPKD results from a pathogenic somatic mutation affecting one of the pluripotent progenitor cells during early embryo development [24]. The hallmark of somatic mosaicism is the presence of two distinct cell populations (i.e., one with and one without the pathogenic mutation) in the affected subject whose disease is typically focal, milder, and variably expressed due to dilution of the mutant gene dosage at one or more disease tissues [25]. This diagnosis should be considered in patients with asymmetric polycystic kidney disease of de novo onset (see molecular genetic testing).
In the absence of a family history of ADPKD, other causes of renal cystic diseases need to be considered in the differential diagnosis (Table 7.2). Autosomal dominant polycystic liver disease (ADPLD) [MIM 174050] is caused by mutations in at least two genes that are distinct from PKD1 and PKD2 [26]. In the classical form, ADPLD presents as a late-onset disease with liver cysts. However, patients with ADPLD may have a few renal cysts which can be confused as mild ADPKD. By contrast, patients with autosomal recessive polycystic kidney disease (ARPKD) [MIM 263200] can present with enlarged echogenic kidneys in the prenatal period as an incidental finding detected by screening ultrasound. The presence of oligohydramnios and respiratory failure at the perinatal period or congenital hepatic fibrosis and renal failure during childhood heralds more severe disease typically associated with two protein-truncating mutations [27]. Two other syndromic forms of PKD due to tuberous sclerosis complex (TSC) [MIM 191100] [28] and Von Hippel-Lindau (VHL) syndrome [MIM 193300] [29] should be easily recognized by their extrarenal clinical features (Table 7.2). However, when present atypically they can mimic ADPKD. Of note, the characteristic renal angiomyolipomas or skin lesions may be absent in up to 30% of patients with mild disease, some of whom may harbor TSC somatic mosaicism [28]. Glomerulocystic kidney disease [MIM 137920] due to hepatocyte nuclear factor 1-beta (HNF-1B) gene mutations can mimic ADPKD. However, the association of renal cysts with urogenital abnormalities, maturity onset diabetes mellitus, as well as chronic renal insufficiency that is discordant with the cystic disease burden (i.e., renal insufficiency without enlarged cystic kidneys) should raise a suspicion for this disorder [30]. Autosomal dominant medullary cystic kidney disease [OMIM 174000 and 603,860] typically presents as chronic renal insufficiency with few or no renal cysts and may be associated with hyperuricemia and gout [31, 32]. Other non-syndromic causes of PKD including simple renal cysts, acquired renal cystic disease, medullary sponge kidney [33], and somatic mosaicism of ADPKD should also be considered in patients without a family history of ADPKD. Examples of both syndromic and non-syndromic forms of PKD are shown in Fig. 7.3.

Examples of other cystic disorders. (a) Acquired cystic kidney disease in a 63-year-old female with end-stage renal failure due to diabetes mellitus, (b) simple renal cysts in a 61-year-old-male with normal eGFR and negative PKD1 and PKD2 mutation screen, (c) parapelvic cysts (arrows) in a 64-year-old-male, (d) unilateral ureteropelvic junction (UPJ) obstruction in a 44-year-old-male with markedly distended left renal collecting system and abrupt transition at UPJ (arrow), (e) multiple renal cysts (arrows) and renal cell carcinomas (RCCs) (arrowheads) in a 44-year-old male with Von-Hippel Lindau disease and a previous left nephrectomy for RCC, and (f) kidney enlargement from numerous cysts (asterisks) and angiomyolipomas (arrows) in a 32-year-old female with tuberous sclerosis complex. All figures are T2-weighted MRI images except for panels (d) and (f) which are from contrast-enhanced CT scans
An Integrated Approach for Diagnosis of Renal Cysts
We present in Fig. 7.4 an integrated approach for the evaluation of patients with renal cysts. The first step in the evaluation is a detailed family history including renal disease severity (i.e., age at ESRD) in older affected family members. For at-risk subjects with a definitive family history of ADPKD, the unified criteria based on conventional ultrasound can be used for both diagnosis and disease exclusion (Table 7.1). Specifically, the presence of “a total of three or more renal cysts” for at-risk subjects aged 15–39 years and “two cysts or more in each kidney” for at-risk subjects aged 40–59 years can be considered as sufficient for diagnosis. Conversely, the “absence of any renal cyst” can be considered sufficient for disease exclusion only in at-risk subjects aged 40 years or older. In addition, the absence of any renal cyst by 30 years of age (“Ravine criterion”) can be used for disease exclusion in at-risk subjects from families with one or more affected member who developed ESRD before 50 years of age or known truncating PKD1 mutations [4]. In at-risk subjects with equivocal results, MRI may be used for further diagnostic clarification [18]. Specifically, the presence of “a total of more than 10 renal cysts” can be considered as sufficient for diagnosis and “less than a total of 5 renal cysts” for disease exclusion with high stringency. In patients without an apparent family history of ADPKD, review of medical record or ultrasound screening of other older first-degree relatives may uncover one or more affected but undiagnosed members with mild disease due to PKD2 or non-truncating PKD1 mutations. In the absence of a definitive family history of ADPKD, the differential diagnosis will need to broaden to include other syndromic and non-syndromic causes of renal cysts (Table 7.2) with molecular genetic testing often indicated for diagnostic clarification .

An integrated approach for evaluation of renal cysts detected by conventional ultrasound (US). The documentation of a positive family history is a critical step allowing the use of age-dependent imaging-based criteria for both diagnosis and exclusion of ADPKD. In the absence of a family history, the differential diagnosis needs to include other syndromic and non-syndromic causes of renal cysts. *See Table 7.2 for differential diagnosis of atypical polycystic kidney disease; **at-risk subjects from families with known protein-truncating PKD1 mutations may use the Ravine criteria for earlier disease exclusion; MRI magnetic resonance imaging, HR-US high-resolution ultrasound
References
Harris PC, Torres V. Polycystic kidney disease. Annu Rev Med. 2009;60:321–37.
Pei Y, Watnick T. Diagnosis and screening of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:140–52.
Peters D, Sandkuijl L. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128–39.
Barua M, Cil O, Paterson AD, Wang KW, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol. 2009;20:1833–8.
Dicks E, Ravani P, Langman D, et al. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population and family-based cohort followed for 22 years. Clin J Am Soc Nephrol. 2006;1:710–7.
Magistroni R, He N, Wang KR, et al. Genotype-renal function correlation in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2003;14:1164–74.
Hateboer N, Dijk MA V, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353:103–7.
Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17:3013–9.
Rossetti S, Kubly VJ, Consugar MB, et al. Incomplete penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–55.
Pei Y, Lan Z, Wang KR, et al. Attenuated renal disease severity associated with a missense PKD1 mutation. Kidney Int. 2012;81:412–7.
Cornec-Le Gall E, Audrezet M-P, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–13.
Paterson AD, Magistroni R, He N, et al. Progressive loss of renal function is a heritable trait in type 1 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2005;16:755–62.
Liu Q-X, Shi S, Senthilnathan S, et al. Genetic variation of DKK3 may modify renal disease severity in PKD1. J Am Soc Nephrol. 2010;21:1510–20.
Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626–9.
Nascimento AB, Mitchell DG, Zhang XM, et al. Rapid MR imaging detection of renal cysts: age-based standards. Radiology. 2001;221:628–32.
Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–7.
Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205–12.
Pei Y, Hwang YH, Conklin J, et al. Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015;26:746–53.
Huang E, Samaniego-Picota M, McCune T, et al. DNA testing for live kidney donors at risk for autosomal dominant polycystic kidney disease. Transplantation. 2009;87:133–7.
Niaudet P. Living donor kidney transplantation in patients with hereditary nephropathies. Nat Rev Nephrol. 2010;6:736–43.
Rossetti S, Consugar M, Chapman A, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–60.
Iliuta I-A, Kalatharan V, Wang KR, et al. Polycystic kidney disease without an apparent family history. J Am Soc Nephrol. 2017;28:2768–76.
Reed B, McFann K, Kimberling WJ, et al. Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. Am J Kidney Dis. 2008;52:1042–50.
Youssoufian H, Pyeritz R. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–58.
Gottlieb B, Beitel L, Trifiro M. Somatic mosaicism and variability expressivity. Trends Genet. 2001;17:79–82.
Gevers TJ, Drenth JP. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:101–8.
Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease. Medicine (Baltimore). 2006;85:1–21.
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–68.
Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67.
Ulinski T, Lescure S, Beaufils S, et al. Renal phenotypes related to HNF1B mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17:497–503.
Heidet L, Decramer S, Pawtowski A, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal disease. Clin J Am Soc Nephrol. 2010;5:1079–90.
Ekici A, Hackenbeck T, Moriniere V, et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney Int. 2014;86:589–99.
Fabris A, Lupo A, Ferraro P, et al. Familial clustering of medullary sponge kidney is autosomal dominant with reduced pnentrance and variable expressivity. Kidney Int. 2013;83:272–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media, LLC, part of Springer Nature
About this chapter

Cite this chapter
Hwang, YH., Barua, M., McNaught, A., Khalili, K., Pei, Y. (2018). Imaging-Based Diagnosis of Autosomal Dominant Polycystic Kidney Disease. In: Cowley, Jr., B., Bissler, J. (eds) Polycystic Kidney Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7784-0_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7784-0_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-7782-6
Online ISBN: 978-1-4939-7784-0
eBook Packages: MedicineMedicine (R0)