
Abstract
Testicular germ cell tumors (TGCTs) lie at the intersection of cancer and developmental biology. These tumors arise from defects in germ cell development, pluripotent primordial germ cells that fail to develop into normal male gametes. To understand the developmental defects that allow these tumors to form, we must study the developmental biology surrounding embryonic germ cell development, specifically during sex specification. Fortunately, excellent mouse models are available that recapitulate the pathology of the human disease. In this chapter, we focus on what has been learned by studying embryonic germ cell development in the 129/Sv inbred mouse model, and how this model is contributing to the study of human TGCTs.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Pathogenesis of Human Testicular Germ Cell Tumors
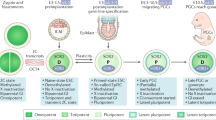
TGCTs represent three of the five types of germ cell tumors, as described by Oosterhuis and Looijenga (2005) and recognized by the World Health Organization (Ulbright et al. 2016): (Type I ) the teratomas and yolk-sac tumors of newborns and infants; (Type II ) the seminomatous and non-seminomatous tumors of adolescents and young adults; and (Type III ) the spermatocytic seminomas of the elderly (Oosterhuis et al. 1997; Looijenga and Oosterhuis 2002). These three types of TGCTs are classified based on chromosomal constitution and genomic imprinting, in addition to epidemiology and clinical presentation. Type I infantile germ cell tumors and Type III spermatocytic tumors are rare, with incidences of 0.2–0.3 per million and 0.4 per million individuals for each respective age group (Carriere et al. 2007; Ulbright et al. 2016). Type II TGCTs are the most frequent type of solid tumor diagnosed in Caucasian males 20–40 years of age in industrialized nations, with incidences in the range of 6–11 per 100,000 individuals; however, Type II TGCT incidences are much lower among non-whites in developing countries (Ulbright et al. 2016).
1.1 Cell of Origin and Pathology of Human TGCTs
All type I germ cell tumors are proposed to arise from primordial germ cells (PGCs) that have undergone immediate transformation into pluripotent embryonal carcinoma cells (ECCs) and clinically manifest before puberty. These germ cell tumors most often arise in the gonads but can also develop extragonadally, and are histologically classified as teratomas, yolk sac tumors, embryonal carcinomas, and mixed tumors, such as teratocarcinomas (teratomas with ECC elements; Fig. 10.1) (Oosterhuis et al. 1997; Oosterhuis and Looijenga 2005). In teratomas all three germinal layers are present (endoderm, mesoderm, and ectoderm). Additionally, teratomas may be composed exclusively of well differentiated, mature tissues that are typically benign, or have immature, fetal-like tissues that can be highly malignant (Ulbright et al. 2016). The most common testicular neoplasm in children are yolk sac tumors, which contain tissues that recapitulate the yolk sac, allantois, and other extra-embryonic lineages (Kaplan et al. 1988).

Cells of origin and pathogenesis of Type I and II testicular germ cell tumors (TGCTs). Both Type I and Type II TGCTS arise during embryogenesis from primordial germ cells (PGCs). In Type I TGCTs, embryonic germ cells are proposed to directly transform into pluripotent embryonal carcinoma cells (ECCs) , which form pure ECC tumors or differentiate to form teratomas or yolk sac tumors. Testicular teratomas in 129 inbred mice are also proposed to arise from gonocytes that directly transform into ECCs. However, evidence from both human and mouse studies suggest that a transient germ cell neoplasia in situ (GCNIS) -like state may occur prior to ECC formation. In Type II TGCTs, GCNIS precursor cells have been identified as the cell-of-origin. These cells give rise to either seminomatous or non-seminomatous tumors. Non-seminomas are the result of reprogramming of GCNIS, or potentially seminomatous tumor cells, into pluripotent ECCs, which form pure ECC tumors or differentiate to form teratoma, yolk sac tumor, or choriocarcinoma. Non-seminomas are generally found in mixed tumors (i.e. seminoma and non-seminoma components), however, pure forms are also observed. Of the type II TGCTs, 50% are pure seminomas and 30% are non-seminomas, with the remaining percentage of tumors a mix of seminoma and non-seminoma (Horwich et al. 2006). Percentages for Type I TGCTs and Type II Non-seminomas were collected from several references (Krag Jacobsen et al. 1984; Mostofi et al. 1988; Howlader et al. 2012; Ulbright et al. 2016)
Type II TGCTs arise from a precursor lesion, termed germ cell neoplasia in situ (GCNIS) of the seminiferous tubules (Skakkebaek 1978; Ulbright et al. 2016) (Fig. 10.1). GCNIS cells are positive for the kit receptor (KIT) (Rajpert-De Meyts and Skakkebaek 1994) and the pluripotency factor POU5F1 (OCT4) (Palumbo et al. 2002). However, GCNIS cells appear to not express all factors necessary to establish a pluripotent state.
Although GCNIS originates during embryogenesis, Type II TGCTs clinically manifest at or after puberty and are histologically subclassified as seminomas, non-seminomas, or tumors of mixed seminoma and non-seminomas components. The default pathway of Type II TGCTs is hypothesized to be from GCNIS towards the development of a seminoma, which consists of undifferentiated, KIT/OCT4-positive cells morphologically similar to GCNIS . The development of a non-seminoma requires activation (reprogramming) of pluripotency in either a GCNIS cell or a seminoma cell to establish ECCs (Oosterhuis and Looijenga 2005). Non-seminomas are found as pure tumor types (teratomas, embryonal carcinomas , yolk sac tumors, and choriocarcinomas) or as mixed tumor types, either as mixed non-seminoma (including teratocarcinomas) or as mixed seminoma and non-seminoma (Bahrami et al. 2007). Pure form embryonal carcinomas comprise only 2–10%, while more than 80% of mixed tumors have embryonal carcinoma as a component (Mostofi et al. 1988). Yolk sac tumors in adults are more often seen in mixed non-seminomas, occurring in about 40% of non-seminomas (Ulbright et al. 2016). Pure choriocarcinoma represents less than 1% (0.19%) of TGCTs; choriocarcinoma is also found mixed with other germ cell tumor elements in 8% of TGCTs (Krag Jacobsen et al. 1984).
Genomic imprinting studies suggest Type I and Type II TGCTs originate from PGCs at different stages of development (van Gurp et al. 1994; Ross et al. 1999; Bussey et al. 2001; Schneider et al. 2001). Type I teratomas and yolk sac tumors of infants show a slightly different pattern of genomic imprinting (Ross et al. 1999; Schneider et al. 2001), supporting the model that these tumors originate from an earlier stage of germ cell development than Type II TGCTs. Based on genomic imprinting patterns Type I teratomas and yolk sac tumors have been postulated to originate from an early PGC that has retained biparental epigenetic marks (Oosterhuis et al. 1997; Oosterhuis and Looijenga 2005). Type II TGCTs are most likely derived from a PGC blocked or delayed in maturation and with erased genomic imprinting (Oosterhuis and Looijenga 2005). Changes in DNA methylation is a hallmark of most cancers, and like developing PGCs, changes in DNA methylation status may contribute to genome instability to promote transformation to ECCs . Interestingly, a significant difference in genome methylation has been reported between seminomas (hypomethylated) and non-seminomas (hypermethylated) (Gillis et al. 1997; Smiraglia et al. 2002). The difference in methylation status could reflect the pluripotent potential of each subtype and the capacity of the non-seminomas to mimic embryonal and extra-embryonal development. Gene expression patterns of OCT4 and X-inactivation status in TGCTs support this theory (Looijenga et al. 1997; Palumbo et al. 2002).
There is conflicting data as to whether GCNIS exists in both Type I and Type II TGCTs. Several groups have reported that GCNIS is not observed in Type I TGCTs and therefore is not a precursor of these tumors; transformed PGCs progress directly to ECCs (Koide et al. 1987; Manivel et al. 1988, 1989; Soosay et al. 1991). Other studies provide evidence that TGCTs of infants and young men share a common precursor, and have documented GCNIS in Type I infantile teratomas (Stamp et al. 1993; Stamp and Jacobsen 1995). It is possible that GCNIS is a transient state of tumor progression in Type I TGCTs and is more difficult to observe clinically in fully developed tumors (Fig. 10.1). The transient state model is similar to that proposed for transition of seminomas to non-seminomas, where a clinically manifested seminoma stage may not be observed (de Jong et al. 1990).
Type III spermatocytic tumors display paternal patterning of genomic imprinting, and therefore most likely do not have an embryonic origin but instead develop from spermatocytes (Looijenga et al. 2006, 2007). This chapter focuses on the pathogenesis and genetics of Type I and Type II TGCTs, which initiate during embryogenesis. A review on Type III spermatocytic tumors is available (Looijenga et al. 1994).
1.2 Comparisons of Cell of Origin and Pathology of TGCTs in 129 Inbred Mice to Human TGCTs
Considering the embryonic origins of Type I and II of TGCTs, animal models are critical for the study of tumor initiation and pathogenesis. The 129/Sv inbred strain of mice has a spontaneous TGCT incidence between 5 and 10%. These spontaneous tumors closely resemble human Type I infantile teratomas and share many pathological characteristics with adult Type II non-seminomas. However, an animal model that fully recapitulates all of Type II TGCT pathology has not been established. In this chapter, we focus on what has been learned by studying embryonic germ cell development in the 129/Sv inbred mouse model, and how this model is contributing to the study of human Type I and II TGCTs.
TGCTs in 129/Sv mice are first evident microscopically at E15 as foci of EC cells, and macroscopically at 3–4 weeks after birth. (Stevens 1962, 1967a, b; Vos et al. 1990; Rodriguez et al. 1992; Looijenga et al. 1998). Tumors evolve in tissue type from being mainly comprised of EC cells in embryonic and neonatal mice. Shortly after birth, tumors will contain both differentiated and embryonal tissues, comprising a teratocarcinoma (Pierce et al. 1967; Matin et al. 1998). Most adult germ cell tumors in mice are benign teratomas, however ECCs can persist, as evident by the ability to transplant primary tumor cells to the testis of adult mice to form new tumors (Stevens 1958, 1981). Seminal studies from Leroy Stevens demonstrated the transplantability of genital ridges from E12.5129/Sv mice into adult testes to form teratomas, demonstrating the PGC as the originating cell of teratomas in mice (Stevens 1967b). Curiously, ECCs morphologically resemble totipotent cells of normal embryos (Pierce et al. 1967) and have similar developmental potential (Kleinsmith and Pierce 1964). ECCs are also similar to ES cells obtained from the inner cell mass of blastocysts of normal preimplantation mouse embryos; depending on the microenvironment ECCs can participate in normal mouse development (Martin 1981). Blastocysts injected with in vivo passaged ECCs give rise to chimeric offspring, demonstrating that ECCs can revert to aspects of normal development (Martin 1981; Rossant and Papaioannou 1984; Chadalavada et al. 2007).
It has been suggested that GCNIS is not the TGCT precursor lesion in 129/Sv mice, and similar to Type I infantile teratomas, TGCTs in mice also develop directly from the transformation of PGCs to ECCs (around E15) (Walt et al. 1993). However, as suggested from the human Type I TGCTs data, GCNIS in mice may be a transient stage that is not observed in developed tumors. Curiously, atypical gonocytes resembling GCNIS of humans have been observed in mice (Stevens and Bunker 1964; Walt et al. 1993). These abnormal cells have been disregarded as GCNIS because they are present in both TGCT susceptible (129/Sv) and nonsusceptible mouse strains, and in the nontumorigenic grafts of experimentally induced TGCT studies. However, the pro-survival, anti-apoptotic environment of the 129/Sv background may be required for tumor progression beyond GCNIS , which will be discussed in detail later.
2 Human TGCT Chemoresistance
TGCTs are highly treatable (>95% cure rate) by surgery, radiation, and platinum-based (e.g. cisplatin) chemotherapy, which induces apoptosis through DNA damage (Bosl and Motzer 1997; Horwich et al. 2006). However, there are limited treatment options for patients that demonstrate platinum resistance, a group for whom the long-term survival rate decreases to 10–15% (Mayer et al. 2003; Horwich et al. 2006; Nitzsche et al. 2012). Several hypotheses have been proposed to explain both the exceptionally high sensitivity of TGCTs to platinum-based therapy and the evolution of resistance in a small subset of tumors (Litchfield et al. 2016). One of the most convincing models for TGCT hypersensitivity to platinum is active (or even upregulated) TP53 mediating apoptotic responses to DNA damage (Gutekunst et al. 2011). Unlike most solid tumors, mutations in TP53 are extremely rare in TGCTs (Litchfield et al. 2016; Taylor-Weiner et al. 2016). The mechanisms driving platinum resistance remain unclear and are likely to involve genetic and epigenetic changes. Targeted analysis of mutational hotspots have identified chemoresistant-specific mutations in PIK3CA, AKT1, RAS, and FGFR3 in a subset of tumors (Feldman et al. 2014). Moreover, whole exome sequencing of two treatment resistant TGCTs identified mutations in the DNA repair gene XRCC2, suggesting that activation of DNA repair pathways and the corresponding suppression of apoptosis induced by DNA damage may induce chemoresistance (Litchfield et al. 2015c). Finally, CCND1 overexpression in TGCTs, as well as other tumor types, has been associated with cisplatin resistance (Noel et al. 2010). The role of CCND1 in promoting cell cycle progression and suppression of apoptosis has been proposed to mediate resistance (Zhou et al. 2009).
Curiously, a recent analysis of teratomas and transformed carcinomas that developed chemoresistance revealed a loss of pluripotency marker expression (NANOG and POU5F1), suggesting that tumor differentiation drives resistance (Taylor-Weiner et al. 2016). In agreement with these genetic findings, in vitro studies employing human EC cell lines demonstrated that retinoic acid-induced differentiation, and the resulting loss of NANOG and POU5F1 expression, increased cisplatin resistance (Abada and Howell 2014). Moreover, cisplatin alone was sufficient to reduce pluripotency gene expression and induce resistance to itself. Importantly, this same study demonstrated that enforced expression of NANOG can suppress cisplatin resistance. How differentiation facilitates chemoresistance has not been directly tested. However, the link between differentiation and resistance to apoptosis in pluripotent cell types has been proposed as the underlying mechanism for resistance (Abada and Howell 2014). Importantly, these findings may explain the overall sensitivity of undifferentiated seminomas and resistance of differentiated non-seminomas to systemic therapy (Oosterhuis and Looijenga 2005).
3 Genetic Contributions to TGCTs
3.1 Genetic Susceptibility and Human Genome-Wide Association Studies
There is a strong genetic component of human TGCTs, indicated by a high familial index (Lindelof and Eklund 2001) and significantly elevated relative risk of sons and brothers of affected individuals (Heimdal et al. 1996; Bromen et al. 2004; Chia et al. 2009). The heritability of TGCTs is third highest among all cancers, with genetic effects accounting for nearly 50% of risk (Heimdal et al. 1997; Czene et al. 2002; Litchfield et al. 2015d). Traditional genetic studies of candidate gene approaches and linkage analysis have been hampered by the genetic complexity of TGCT development and the lack of multigenerational pedigrees with affected individuals (Rapley et al. 2000; Nathanson et al. 2005; Crockford et al. 2006). In contrast, recent genome-wide association studies (GWAS ) have identified approximately 25 genomic intervals (loci) associated with TGCT risk (Kanetsky et al. 2009, 2011; Rapley et al. 2009; Turnbull et al. 2010; Kratz et al. 2011; Poynter et al. 2012; Andreassen et al. 2013; Chung et al. 2013; Ruark et al. 2013; Litchfield et al. 2015a). These loci have provided considerable new insights into testicular germ cell tumorigenesis, implicating genes involved in PGC specification and/or sex differentiation (DAZL, PRDM14, HPGDS DMRT1, and ZFPM1), including the KIT-KITLG signaling pathway (KITLG, SPRY4, BAK1, GAB2), and genes involved in microtubule assembly (TEX14, CENPE, PMF1, and MAD1L1), DNA repair (RAD51C and RFWD3), apoptosis (BAK1, CLPTM1L, and GSPT1) and telomerase regulation (TERT, ATF7IP, and PITX1) (Litchfield et al. 2015a). The strong genetic component to TGCTs is underlined by the per-allele odds ratios (ORs) for TGCT susceptibility loci, which are often in excess of 2.5, among the highest reported in GWAS of any cancer type (Chanock 2009). The TGCT-associated SNP rs995030 (in KITLG) has the strongest effect of all common SNPs for which a statistically significant association with a cancer phenotype has been reported (Welter et al. 2014). Notably, TGCT susceptibility loci interact in an additive rather than epistatic manner and are predominantly dominant; SNP variants identified often represent the common allele in the population.
Historically, none of the loci identified in GWAS show significant differences in effect on tumor risk when comparing Type II seminomas and non-seminomas (Kanetsky et al. 2009; Rapley et al. 2009; Rapley and Nathanson 2010; Turnbull et al. 2010; Ruark et al. 2013; Litchfield et al. 2015b). The absence of a difference between seminoma and non-seminoma is rather remarkable, as additional GWAS are conducted, sample sizes are now sufficiently powered to detect a difference in these two subgroups (Litchfield et al. 2015b). Follow-up studies investigating risk alleles have not yet identified associations with histological subtype (Karlsson et al. 2013). However, the absence of difference between seminoma and non-seminoma for assessing risk with GWAS TGCT loci is not surprising, considering both tumor types arise from the same cell of origin. Additionally, at least 10–15% of TGCT tumors identified are of mixed pathology (Gori et al. 2005; Horwich et al. 2006) and bilateral and familial cases do not show evidence of clustering within histological subtype or display histological similarity greater than that expected by chance (Forman et al. 1992; Mai et al. 2010). Despite the diversity in Type II TGCT subtypes, these findings provide further evidence that there is relative uniformity and complexity in the genetics of susceptibility. The genetic similarity between TGCT histological subtypes can most likely be attributed to the germ cell origin of TGCTs and early pathogenesis of the disease.
3.2 Genetic Susceptibility in 129/Sv Mice
The development of spontaneous TGCTs in 129/Sv mice but not in other inbred mouse strains denotes the complex genetic component of TGCT susceptibility. Classic genetic approaches, such as segregating crosses between 129/Sv and other strains, have failed to identify susceptibility loci in 129 mice, due to the complex genetic interactions required for tumor initiation (Matin et al. 1999; Muller et al. 2000; Anderson et al. 2009; Zhu and Matin 2014). In segregating crosses between 129/Sv and other strains, only 1 affected male was found among more than 11,000 progeny tested, which is consistent with as many as 15 different genes that interact to control TGCT susceptibility (Stevens and Mackensen 1961; Stevens 1967a, 1981; Matin et al. 1998, 1999; Jiang and Nadeau 2001). The low frequency of affected males (0.01%) in the segregating population precludes analysis of TGCT susceptibility with standard genetic approaches. However, specific genetic mutations introduced on the 129/Sv background have been shown to modify TGCT susceptibility (Table 10.1). A modifier gene, unlike a susceptibility gene, is not required or sufficient to induce a phenotype, but instead interacts with susceptibility genes to alter the penetrance of a phenotype (Heaney and Nadeau 2008). All of the genetic variants listed are at least partially dependent on the 129/Sv background to modify TGCT incidence, and will not cause TGCTs when congenic on other inbred mouse backgrounds. Modifier genes allow researchers to explore the genetic basis for susceptibility and provide avenues to characterize the genes and pathways involved in tumorigenesis.
The 129/Sv inbred strain has been used as a model of Type I TGCTs, considering the similarities in tumor emergence, pathology, and the seeming lack of GCNIS . However, parallels can be drawn between the spontaneous tumors observed in mice and Type II non-seminomas. It is interesting to consider the susceptibility genes KITLG and DMRT1 identified in human GWAS susceptibility loci were first discovered to contribute to TGCT susceptibility in 129/Sv mice (Heaney et al. 2008; Krentz et al. 2009). Additional studies in mice might be able to shed light to the initial commonality and the eventual dichotomy in the evolution of Type I and Type II TGCTs.
3.3 Chromosomal Abnormalities, Fusion Genes, and Single Nucleotide Variants in Type I and Type II TGCTs
As mentioned previously, human Type II TGCT GWAS have identified several susceptibility loci that harbor genes with roles in microtubule assembly, attachment of chromosomes to spindle microtubules, and alignment of chromosomes at the metaphase plate (Litchfield et al. 2015b). Telomerase function and DNA damage repair genes have also been identified in susceptibility loci of human TGCT GWAS (Turnbull et al. 2010; Kanetsky et al. 2011; Chung et al. 2013; Ruark et al. 2013). Together these observations implicate correct chromosomal segregation and destabilization of the genome in TGCT pathogenesis and may explain the karyotype evolution characteristic of TGCT progression.
A number of chromosomal abnormalities have been identified in different Type I and Type II TGCTs (Kraggerud et al. 2002; von Eyben 2004). Intriguingly, a pattern begins to emerge while studying the different abnormalities identified between TGCT types and subtypes. Foremost, Type I infantile teratomas (and coincidently teratomas in 129 mice) are nearly diploid (Kommoss et al. 1990; Hoffner et al. 1994; Silver et al. 1994; Stock et al. 1994; Bussey et al. 1999, 2001; Mostert et al. 2000; Schneider et al. 2001). However, Type I yolk sac tumors are aneuploid and have chromosomal abnormalities distinct from Type II TGCTs (Hoffner et al. 1994; Silver et al. 1994; Stock et al. 1994; Bussey et al. 1999; Mostert et al. 2000). Chromosomal aberrations often seen specifically in Type I yolk sac tumors include overrepresentation of regions of chromosomes 1, 12, 20, and 22, and an underrepresentation of parts of chromosomes 1, 4, and 6 (Mostert et al. 2000; Schneider et al. 2001). However, the contribution of these chromosomal abnormalities to tumor initiation and progression are not known.
Ploidy of Type II seminomas and non-seminomas progresses from tetraploid (in GCNIS ), to hypertriploid (in seminomas) and finally hypotriploid (in non-seminomas) (Oosterhuis et al. 1989; de Jong et al. 1990; Vos et al. 1990; de Graaff et al. 1992; Bosl and Motzer 1997; von Eyben 2004). This karyotype evolution is consistent with a model of multipolar cell division starting from a tetraploid tumor stem cell population (Frigyesi et al. 2004). In both seminomas and non-seminomas, loss of chromosomes 4, 5, 11, 13, 18, and Y, and gain of chromosomes 7, 8, 12, and X are observed (Castedo et al. 1989; Rodriguez et al. 1993; van Echten 1995; Ottesen et al. 1997; Summersgill et al. 1998; Looijenga et al. 2000; Kraggerud et al. 2002). However, gains in chromosomes 15 and 22 are more specific to seminomas, whereas gain of chromosome 17 and loss of chromosome 10 have been more closely associated with non-seminomas, suggesting that particular chromosome losses or gains may be involved in establishing Type II TGCT subtypes (Kraggerud et al. 2002). Of all the chromosomal abnormalities observed in Type II TGCTs, gains in a region of chromosome 12 (12p) may be the most important to TGCT progression, with at least one study showing that ~70% of TGCTs harbor 12p amplifications (Litchfield et al. 2015c; Taylor-Weiner et al. 2016). The vast majority of these amplifications are due to one or more copies of isochromosome 12p (i(12p) ) (Atkin and Baker 1983; Looijenga et al. 2003c). Importantly, most studies have demonstrated that premalignant GCNIS with no adjacent invasive tumor does not harbor 12p amplifications, in particular i(12p); however, malignant GCNIS has been identified with 12p amplifications (Summersgill et al. 2001; Ottesen et al. 2003). Therefore, i(12p) is not required for TGCT initiation, but plays an important role in the transition from a noninvasive to invasive phenotype. Of note, 12p harbors several candidate genes (e.g. KRAS, NANOG, and STELLAR) whose roles as oncogenes and pluripotency/stem cell regulators may promote tumorigenesis when overrepresented (Oosterhuis and Looijenga 2005).
Evidence for smaller somatic mutations such as fusion genes or transcripts , single nucleotide variants (SNVs) , and interval deletions contributing to TGCT pathogenesis is beginning to emerge. Hoff et al. used next-generation RNA sequencing to analyzed human EC cell lines and nonmalignant ES cell line controls for fusion genes or aberrant fusion transcripts (Hoff et al. 2016). Eight novel fusion transcripts and one gene with alternative promoter usage were identified in the EC cell lines. Intriguingly, four of the nine transcripts were found to be recurrently expressed in primary Type II TGCTs, including GCNIS and EC tumor stem cells, suggesting putative roles as driver mutations. However, whether these fusions contribute to germ cell transformation (tumor initiation), disease progression/metastasis, or both remains to be determined. Litchfield et al. (2015c) employed whole-exome sequencing of 42 Type II TGCTs and matched normal blood samples to identify somatic SNV and copy number driver mutations. Reoccurring SNV mutations were observed in only two genes (KIT, 14% of TGCTs and CDC27, 11.9% of TGCTs). KIT mutations were concentrated in seminomas (31%), which has been previously reported in studies utilizing targeted sequencing (Kemmer et al. 2004; McIntyre et al. 2005). Overall, nonsynonymous mutation rates were found to be low compared to other cancers. Two previously undescribed, reoccurring amplifications involving FSIP2 and a region of the X chromosome were also discovered with both occurring in 15% of TGCTs. However, copy number gain in chromosome region 12p was by far the most common amplification observed (71% of TGCTs). This dataset was subsequently used to determine whether somatic mutations reoccur in genes within four TGCT susceptibility loci associated with inherited risk (Litchfield et al. 2015a). Only reoccurring events within the susceptibility locus on chromosome 11 (deletions encompassing GAB2 and USP35) were observed (7% of the TGCTs). Therefore, even though TGCT susceptibility loci are important determinants of inherited risk, somatic mutations within these loci appear to be rare.
Importantly, the whole-exome sequencing studies by Litchfield et al. were powered to detect recurrent mutations having a tumor-associated frequency greater than 15% (84% power) (Litchfield et al. 2015c). Therefore, it is unlikely that additional high frequency driver mutations exist in TGCTs other than KIT SNVs or chromosome 12p amplifications. Two additional whole-exome sequencing studies (Brabrand et al. 2015; Cutcutache et al. 2015), using smaller sample sizes, also found a low incidence of nonsynonymous mutations in TGCTs with little evidence for high frequency driver mutations. Therefore, most somatic mutations observed in TGCTs are either passengers of the tumorigenic process or oncogenic factors in only a small subset of TGCTs. Importantly, these mutations are likely to be drivers of TGCT progression rather than initiation, as they most likely occur after the tumor stem cell population is established. TGCT data are currently under analysis for The Cancer Genome Atlas (TCGA) project (Chin et al. 2011). Once analysis of whole-exome, whole-genome, copy number variation (CNV), and microarray data from a larger cohort of patient tumor and control samples is complete, the mutational landscape of TGCTs will become more apparent.
Based on the strong heritable component, the evolving genome, and the low somatic mutation rates observed in TGCTs, a model of tumor susceptibility (initiation) and progression is beginning to emerge. TGCT initiation appears to be primarily caused by genetic factors inherited through the germline (i.e. common SNP variants) working in combination possibly with environmental factors, which predisposes embryonic germ cell transformation into ECCs . Once ECCs are established, the TGCT genome can evolve over time, gaining or losing chromosomal components and accumulating SNVs and gene/transcript fusions. Together these somatic mutations influence disease progression (e.g. development of a specific tumor subtype) and metastasis. Such a model can explain the higher incidence of aneuploidy and somatic mutations observed in post-pubertal Type II TGCTs compared to infantile Type I TGCTs. Type II TGCTs may simply accumulate more genetic abnormalities during the latency period between tumor stem cell development in the embryo and tumor expansion after puberty. Importantly, inherited genetic factors within TGCT susceptibility loci harbor genes associated with chromosome segregation and DNA repair. These risk alleles may be the cause of the evolving karyotypes and infrequent somatic mutations of TGCTs. Importantly, this tumor initiation/progression model contrasts those for spontaneous cancers in other tissues, such as the colon (Davies et al. 2005), in which accumulation of somatic mutations are the primary drivers of tumor initiation and progression, and inherited genetic factors modulate disease risk and severity.
The evolving model of tumor susceptibility has been largely supported by data uncovered in GWAS. These studies provide ample opportunity for identifying risk loci, but fail to provide an avenue for validation. As mentioned previously, two genes first characterized as modifiers of TGCT incidence in 129/Sv mice, Kitl and Dmrt1 (Heaney et al. 2008; Krentz et al. 2009), were also later identified as susceptibility loci in human GWAS (Kanetsky et al. 2009, 2011). This overlap highlights the similarity between mouse and human TGCT genetic susceptibility and pathogenesis, and suggests that additional genes in susceptibility loci identified in human TGCT GWAS may also be modeled as modifiers of TGCT incidence in mice. The ultimate goal of mouse models of human disease is to translate genetic alterations in mice to identical alterations in humans. To study tumor development in mice, a basic understanding of the normal developmental biology is critical to place in context the defects leading to tumorigenesis. We will present an overview of germ cell development, and then discuss the key disruptions in male germ cell development that contribute to testicular germ cell tumorigenesis.
4 Mouse Germ Cell Development and the Embryonic Origins of Tumorigenesis
For reference, Fig. 10.2 summarizes the main developmental time points and key gene expression patterns associated with mouse germ cell development. Additionally, time points associated with published deficiencies associated with tumor formation and the potential formative windows for TGCT development are also illustrated.

(a) Schematic of the development of mouse PGCs through the four major phases: Specification, migration, licensing, and sex specification. ExE, extra-embryonic ectoderm; Epi, epiblast; Al, allantois. (b) Temporal expression patterns of key genes and developmental events involved in specification and epigenetic reprogramming of mouse PGCs. The green bars represent the expression of indicated genes associated with PGC specification; orange bars represent the expression of the chromatin modifiers and methylation status; light blue bars represent the expression of pluripotency genes; pink and dark blue bars indicate female and male specific events, respectively. Figure adapted from several references (Saitou et al. 2012; Bustamante-Marin et al. 2013; Moshfegh et al. 2016; Saitou and Miyauchi 2016), data as revealed by immunohistochemistry and other methods (Seki et al. 2005, 2007; Hajkova et al. 2008; Popp et al. 2010). Extensive remodeling of additional histone modifications occurs in the genital ridges at around E11.5, duringFig. 10.2 (continued) the rapid genome demethylation (Hajkova et al. 2008; Hajkova et al. 2010). Interestingly, Oct4 (Pou5f1) has continuous RNA expression even beyond E15.5, while Sox2 and Nanog are reactivated during GC specification and downregulated again starting at E14.5. However, OCT4 protein is not detected by immunofluorescence in gonocytes by E15.5 (Western et al. 2010). Blimp1 (Prdm1), PR domain containing 1, with ZNF domain; Prdm14, PR domain containing 14; Dppa3 (Stella), developmental pluripotency-associated 3. (c) Expression patterns of key genes in XY germ cells during male sex specification. Purple bars indicates concomitant expression in both sexes, dark blue bars, male, and pink bars, female. Licensing of PGCs by Dazl is a key event (Lin and Page 2005; Gill et al. 2011) for upregulation of genes involved in the male specification pathway, such as Nanos2 (Tsuda et al. 2003; Suzuki and Saga 2008) and Dnmt3l (Bourc'his et al. 2001), or female specification pathway (Stra8, Rec8) (Menke et al. 2003; Koubova et al. 2014). Other factors produced from somatic cells serve to initiate expression of key genes for sex specification, such as Fgf9 expression in males and Wnt4 in females, starting at E11.5 (Lin and Capel 2015). Somatic signaling from the Tgfβ pathway, including Activin, serves to initiate mitotic arrest and induces male fate in XY germ cells through p38 MAPK and SMAD2 (Miles et al. 2012; Wu et al. 2013, 2015). The black boxes with red outline indicate published defects observed in TGCT-susceptible gonocytes during sex specification (Kimura et al. 2003; Krentz et al. 2009; Cook et al. 2011; Heaney et al. 2012; Lanza et al. 2016); see text for in-depth discussion. (d) Postulated windows during which defects could accumulate to initiate tumorigenesis at E15.5. For example, defects during migration are known to occur, through the presence of extragonadal tumors observed in human neonates and children. Kit and Kitl are critical components to the migration of PGCs from the primary streak through the hindgut to the genital ridge (Mahakali Zama et al. 2005; Kunwar et al. 2006). GWAS have implicated several variants in or around KITLG (Kanetsky et al. 2009; Rapley et al. 2009; Kratz et al. 2011). Epigenetic abnormalities could occur during PGC migration or pre-sex specification, which could result in the misexpression of genes during the wrong developmental window. Altered microenvironments within the gonad during male specification, such as inappropriate exposure to meiosis-promoting factors or insufficient expression of meiosis-inhibiting factors, could also provide avenues for TGCT initiating events
4.1 Primordial Germ Cells
PGCs originate from the proximal epiblast cells of the mouse at embryonic day (E) 6.5, in response to bone morphogenetic protein (BMP)4 signaling from the extra-embryonic ectoderm at ~E6.0 (Lawson et al. 1999; McLaren 2000; Surani 2001). BMP4 pathway signaling induces the expression of Fragilis, defining the portion of embryonic mesoderm with germ cell competence (Saitou et al. 2002). During PGC specification, the majority of epiblast cells are being pushed towards somatic fates and losing pluripotency (Kurimoto et al. 2008). High Fragilis-expressing cells induce Stella, to repress Homeobox gene expression and differentiate themselves from their somatic neighbors (Saitou et al. 2002). However, subsequent studies have demonstrated that neither Stella nor Fragilis are required for PGC specification (Payer et al. 2003; Lange et al. 2008; Saitou 2009).
BMP4 signaling also induces the expression of two transcriptional regulators, Prdm1 (also known as Blimp1, PR domain containing 1, with ZNF domain) and Prdm14, in the most proximal epiblasts at ~E6.25 and E6.5, respectively. BLIMP1- and PRDM14-positive cells progress to form a cluster of ~40 alkaline phosphatase (AP)-positive PGCs at the base of the incipient allantois at ~E7.25 (Ginsburg et al. 1990; Ohinata et al. 2005, 2009; Vincent et al. 2005; Yamaji et al. 2008). Blimp1 is exclusively expressed in founder PGCs and required for PGC specification (Ohinata et al. 2005; Vincent et al. 2005). BLIMP1 and PRDM14 work in concert to achieve repression of the somatic program, genome-wide epigenetic reprograming, and re-acquisition of potential pluripotency in PGCs (Saitou et al. 2008; Yamaji et al. 2008; Saitou 2009).
Epigenetic reprogramming in newly specified PGCs goes beyond suppression of the somatic program by BLIMP1 and PRDM14. In early PGCs, histone methylation markers and de novo methylases Dnmt3b, Dnmt3a and Uhrf1 are transcriptionally repressed (Kurimoto et al. 2008; Sasaki and Matsui 2008). Changes in genome-wide DNA methylation, removal of histone H3 lysine9 dimethylation (H3K9me2), and acquisition of high levels of tri-methylation of H3K27 (H3K27me3) occur just prior and continue throughout PGC migration, processes that might be crucial for the maintenance of potency in the germline (Seki et al. 2005). During migration, PGC methylation sites at imprinted loci are maintained (Hajkova et al. 2002; Lee et al. 2002), however, other studies have identified a heterogeneous “reprogramming” in a cell-by-cell manner (Hajkova et al. 2002; Lee et al. 2002; Lane et al. 2003; Seki et al. 2007; Hajkova et al. 2008). After arriving at the genital ridge PGCs undergo rapid genome-wide demethylation to cause reactivation of the inactivated X-chromosome in females, imprinted loci no longer retain methylation marks, and most transposable elements are demethylated by E13.5 (Surani 2001; Hajkova et al. 2002; Li 2002; McLaren 2003; Hayashi and Surani 2009). Extensive reviews on epigenetic reprogramming in PGC specification and maintenance have been published (Surani et al. 2007; Sasaki and Matsui 2008; Saitou et al. 2012).
Pluripotency is maintained in PGCs through approximately E13.5 (Yamaguchi et al. 2005; Western et al. 2010), which is evident from the ability to generate embryonal germ cells (EGCs) and ES cells from PGCs in vitro (Matsui et al. 1992; Pesce and Scholer 2000). At E12.5 PGCs can be cultured to generate alkaline phosphatase-positive, specific embryonic antigen 1 (SSEA1)-positive cells resembling undifferentiated embryonic stem cells that can be transplanted to form teratomas in nude mice (Matsui et al. 1992). Critical to the maintenance of pluripotency PGCs express the gene Pou5f1 (Oct4), which is expressed exclusively in PGCs starting at E7.5 through spermatogenesis up to the onset of spermatogenic differentiation (Scholer 1991; Pesce et al. 1998; Pesce and Scholer 2000). BMP4 signaling also controls the activity of the Oct4 distal enhancer in founder PGCs during germ cell specification (Yeom et al. 1996; McLaren 1999; Pesce and Scholer 2000). Oct4 expression is also necessary for survival of migrating PGCs in later stage embryos (Kehler et al. 2004).
4.2 PGC Migration
Beginning around E10.5–E11.5, PGCs complete their migration along the midline through the hindgut to arrive at the genital ridge (McLaren 2000). While migrating, the PGCs greatly increase in number by proliferating, relying on the c-kit/stem cell factor signal transduction pathway for continued proliferation and migratory guidance to the genital ridge (Matsui et al. 1990; Sutton 2000). Correspondingly, embryos homozygous for mutations in genes coding for either the receptor (W) or the ligand (Steel) are deficient in PGCs (McLaren 2000). By E11.5, the genital ridge is clearly defined from the mesonephros, thereby preventing any subsequent migration .
4.3 Sex Specification and the Mitotic : Meiotic Switch
After arriving in the genital ridge, PGCs continue proliferating for the next 2–3 days to expand the germ cell pool, going from tens to hundreds at their inception in the primary streak to over 12,000 PGCs per colonized gonad at E13.5 (Mintz and Russell 1957; Tam and Snow 1981). It is during this time period that germ cells, now termed gonocytes or oogonia, commit to either a male or female fate (respectively) and enter G1/G0 mitotic arrest or initiate meiosis (the mitotic:meiotic switch), respectively (McLaren 1984). These sex-specific developmental events are controlled by cues from the somatic environment. In normal development, Sry expression in somatic cells at E10.5 causes the upregulation of Fgf9 and Sox9 to signal XY PCGs to begin suppression of the female pathway (Kim et al. 2006; Sekido and Lovell-Badge 2008). Somatic cells increase proliferation during this peak of Sry expression (Hacker et al. 1995). Blocking this somatic proliferation disrupts the male pathway of development (Schmahl et al. 2000; Schmahl and Capel 2003) and inhibits the survival of the XY PGCs by preventing the eventual enclosure of germ cells and somatic cells in specific germ cell compartments (Byskov 1986). This chapter does not go into the differentiation of supporting somatic cells outside the context of male germ cell specification. For more information, excellent reviews have been published on cell fate commitment during sex determination (Park and Jameson 2005; Lin and Capel 2015).
FGF9 signaling from somatic cells induces the upregulation of the Nodal/Activin pathway in PGCs around E12.5 (Spiller et al. 2012; Wu et al. 2013). Activins and Nodal are members of the transforming growth factor β (TGFβ) superfamily of morphogens (Oshimori and Fuchs 2012). TGFβ family members play important roles in gonadal development in both sexes (Munsterberg and Lovell-Badge 1991; Yi et al. 2001; Nicholls et al. 2009; Moreno et al. 2010; Mendis et al. 2011). Activins and Nodal, with co-receptor CRIPTO, can signal through the same receptors and effectors to regulate transcription (Pauklin and Vallier 2015). In gonocytes co-expression of Cripto and Nodal generate a positive feedback loop to sustain NODAL signaling and expression of downstream targets Lefty1 and Lefty2, which display peak expression at E13.5 (Spiller et al. 2013). Expression of Nodal and Cripto at this time serves to maintain pluripotency in the XY germ cell population, and in the absence of NODAL/CRIPTO signaling , pluripotency potential of the gonocytes is reduced (Spiller et al. 2012). FGF9 signaling from Sertoli cells to gonocytes also helps to transiently maintain expression of pluripotency genes Oct4 and Sox2, prior to mitotic arrest (Bowles et al. 2010). Nodal has previously been shown to maintain Nanog expression in early embryos and pluripotent cells (Mesnard et al. 2006; Vallier et al. 2009). Recent studies have also indicated a role of NODAL signaling, and its activation of SMAD2 and p38 MAPK pathways, in promoting male differentiation through the induction of Nanos2 expression (Wu et al. 2013). In the absence of NODAL/Activin signaling, XY germ cells will enter meiosis (Souquet et al. 2012; Miles et al. 2013). TGFβ and Activin signaling are required to ensure correct mitotic arrest in XY germ cells (Moreno et al. 2010; Mendis et al. 2011; Miles et al. 2013; Wu et al. 2015).
Germ cells activate the G1-S phase cell cycle checkpoint in a gradual and unsynchronized manner due to a shift in the expression of positive and negative regulators of the G1-S phase transition. Prior to entry into mitotic arrest, germ cells express cyclins E1 and E2 (CCNE1/2) and cyclin D3 (CCND3), which form complexes with cyclin dependent kinases 2 (CDK2) and 4 or 6 (CDK4/6), respectively (Western et al. 2008). These cyclin-CDK complexes hyperphosphorylate (inactivate) retinoblastoma protein 1 (pRB1), leading to de-repression of E2F transcription factors and activation of genes required for progression into S phase (Deshpande et al. 2005; Western et al. 2008; Spiller et al. 2010). Cyclin D-CDK4/6 complexes also promote the G1-S transition through the sequestration of cyclin E-CDK2 inhibitors p27KIP1 (CDKN1B) and p21CIP1 (CDKN1A) (Deshpande et al. 2005). Mitotic arrest is initiated around E13.5 through decreases in expression of CCNE1/2 and CCND3 and increases in expression of p27KIP1 and cyclin D-CDK4/6 inhibitors p15INK4B (CDKN2B) and p16INK4A (CDKN2A), which result in hypophosphorylation (activation) of pRB1, suppression of E2F transcriptional activity, and gonocyte transition into G1/G0 arrest (Western et al. 2008). These negative regulators of the cell cycle, in addition to TGFβ/Activin signaling and Prostaglandin D2 signaling (Moniot et al. 2014) ensure the proper mitotic arrest in male gonocytes. Additional factors or a master regulator may be in control of the switch between mitotic arrest and meiosis (Adamah et al. 2006; Feng et al. 2014).
In addition to proper signaling to initiate mitotic arrest, checks and balances exist in the male gonad to prevent premature meiosis. Sertoli cell differentiation and the establishment of testis cords are critical to prevent XY germ cells from entering meiosis (Byskov 1978). If a male genital ridge is disaggregated and reaggregated at E11.5, testis cords do not develop and all the PGCs enter the oogenesis pathway (McLaren and Southee 1997; Adams and McLaren 2002). These studies investigating sex specification in the mouse ovary and testis postulated a “meiosis inducing substance” to be responsible for the initiation of meiosis, which was subsequently blocked in the testis around E12.5–E13.5, when germ cells commit to a sex specific pathway (McLaren and Southee 1997). Subsequent studies would identify this meiosis-inducing substance as retinoic acid, which normally induces genes such as Stra8 to initiate meiosis in the XX fetal ovary (Bowles et al. 2006; Koubova et al. 2006; Koubova et al. 2014) and is sufficient to initiate meiosis in the fetal testis ex vivo (Trautmann et al. 2008). An extensive review on the influence of retinoic acid in sex specification of embryonic germ cells was presented by Bowles and Koopman (2010).
Exposure to retinoic acid is a tightly regulated process in the developing mouse gonad, critical for the mitotic:meiotic switch in gonocytes between E13.5 and E15.5. During this time period, female oogonia are exposed to a wave of retinoic acid to induce expression of Stra8, which mirrors the anterior-to-posterior wave of retinoic acid and initiates meiotic differentiation that lasts for 4 days (E12.5–E16.5) (Menke et al. 2003). During the same developmental period in male gonocytes, CYP26B1 degrades retinoic acid, thereby blocking STRA8 expression to prevent the initiation of meiosis (Menke and Page 2002; Bowles et al. 2006; Koubova et al. 2006; Vernet et al. 2006; MacLean et al. 2007). To further inhibit Stra8 expression as CYP26B1 levels begin to decline and to promote the male germ cell differentiation program, Nanos2 expression is activated in male gonocytes at E14.5 (Suzuki and Saga 2008; Barrios et al. 2010; Bowles et al. 2010). NANOS2 represses meiosis and the female differentiation pathway in embryonic male germ cells independent of the expression of Nanos3 (Suzuki et al. 2007; Suzuki and Saga 2008; Barrios et al. 2010). NANOS proteins are evolutionary conserved RNA-binding proteins, involved in post-transcriptional RNA metabolism via their binding to target mRNAs in germ cells (Kadyrova et al. 2007). Interestingly, Nanos2 is required for normal male germ cell differentiation, as evident by the lack of rescue of male differentiation gene expression at E15.5 in Nanos2/Stra8 double knockout mice (Saba et al. 2014a). These data suggest that Nanos2 plays larger role in male germ cell development than inhibiting retinoic acid.
By E15.5 male gonocytes should have committed to mitotic arrest and should be expressing genes associated with male germ cell differentiation (e.g., Nanos2, Dnmt3l, Piwil4/Miwi2, Tdrd9, and Mili) (Shovlin et al. 2007; Aravin et al. 2008; Shoji et al. 2009). Following initiation of mitotic arrest at E13.5, male gonocytes normally downregulate expression of pluripotency factors (e.g. Oct4, Nanog, and Sox2) (Pesce et al. 1998; Avilion et al. 2003; Yamaguchi et al. 2005; Western et al. 2010). Accordingly, downregulation of Nodal expression is observed starting at E14.5 (Spiller et al. 2012), which supports the role of Nodal expression in transiently maintaining pluripotency. From the misregulation of mitotic arrest and the failure to downregulate pluripotency, male gonocytes in the 129 mouse strain are susceptible to develop into TGCTs. To note, misregulation of mitotic arrest and retention of pluripotency have also been proposed to cause TGCTs in humans (Palumbo et al. 2002; Looijenga et al. 2003b; Rajpert-De Meyts et al. 2004; Spiller et al. 2012).
5 TGC Tumorigenesis in the 129 Inbred Strain of Mice : Dysfunctional Germ Cell Development
5.1 Failure to Enter into Mitotic Arrest
Delayed entry into G1/G0 mitotic arrest has been linked with susceptibility to teratoma formation in the 129/Sv inbred strain of mice (Stevens 1964, 1967a; Noguchi and Stevens 1982; Matin et al. 1998). Many of the genetic modifiers that increase 129/Sv tumor incidence of 129/Sv characterize this failure to enter mitotic arrest as sustained proliferation through the mitotic:meiotic switch to E15.5: Noguchi and Stevens identified in genital ridge grafting experiments that sub-strains of 129/Sv with increased incidence of teratoma formation also exhibited a longer period of proliferation compared to 129/Sv sub-strains with lower teratoma frequency (Noguchi and Stevens 1982). Populations of germ cells in males of the 129-Chr19MOLF/Ei (M19) chromosome substitution strain, which has a tenfold tumor incidence compared to wild-type 129/Sv mice (Matin et al. 1999), still proliferate at E15.5, as evident by KI67 expression (Heaney et al. 2012). Mice with homozygous deletions of Trp53 and Pten also exhibit increased TGCT incidence, which underlies the role of cell cycle control in TGCT tumorigenesis (Harvey et al. 1993; Kimura et al. 2003; Western 2009). Importantly, several other genes that regulate male germ cell entry into mitotic arrest may influence TGCT susceptibility. Mice deficient for Dazl, a gene critical for germ cell development and survival of XY germ cells, have a few surviving germ cells that display sustained proliferation and retained pluripotency (Lin and Page 2005). Of note, DAZL is located near one of the TGCT risk loci identified in human TGCT GWAS. Mice deficient for genes involved in prostaglandin D2 synthesis, Ptgds and Hpgds, show increased proliferation of gonocytes at E13.5 through E15.5, compared to wild-type controls (Moniot et al. 2014). HPDGS is also located near a human TGCT GWAS risk locus. Therefore, in both mice and humans, a pro-proliferative germ cell program appears to be a central component of TGCT initiation.
Germ cell entry into G1/G0 mitotic arrest during the mitotic:meiotic switch is dependent on coordinated alterations in the expression of positive and negative regulators of the G1-S phase transition (Western et al. 2008). During embryogenesis, D-type cyclin expression is primarily restricted to CCND3 in male germ cells (Beumer et al. 2000; Western et al. 2008). Curiously, even though its expression decreases during the mitotic:meiotic switch, CCND3 protein persisted in FVB gonocytes through at least E17.5, a time point at which these cells are quiescent. Thus, expression of negative regulators of the G1-S transition must be sufficient to counteract residual D-type cyclin expression to induce G1/S mitotic arrest (Beumer et al. 2000). A sufficient increase in the ratio of positive to negative regulators of G1-S cell cycle progression during the mitotic:meiotic switch might tip the balance toward proliferation rather than mitotic arrest. It has been previously demonstrated that Ccnd1 expression levels are significantly higher in TGCT-susceptible gonocytes (Heaney et al. 2012); Ccnd1 expression is normally restricted to differentiating postnatal spermatogonia (Beumer et al. 2000). Furthermore, more recent data has revealed that Ccnd1 is the only G1-S phase cyclin upregulated in TGCT-susceptible gonocytes at E15.5, the time point at which ECCs are first evident (Lanza et al. 2016). Ccnd1-deficiency permitted TGCT-susceptible gonocytes to activate the G1-S cell cycle checkpoint and induce G1/G0 mitotic arrest in a more developmental stage-appropriate manner, as evident by phospho-pRB1 and KI67 immunostaining of E14.5 and E15.5 gonocytes. Thus, cyclin D1 appears to be the G1-S phase cyclin delaying mitotic arrest of TGCT-susceptible gonocytes. While Ccnd1-deficiency was not sufficient to prevent tumor initiation in TGCT-susceptible gonocytes, the misexpression of CCND1 in embryonic germ cells represents a severe consequence of the larger developmental defect present in 129/Sv mice, which permits spontaneous germ cell tumorigenesis.
Mutations of other genetic modifiers of the 129/Sv background, in addition to delayed mitotic entry, also display alterations in genes controlling the G1-S checkpoint. Dnd1 Ter/Ter mutants on a 129/Sv background fail to express the negative regulators of the cell cycle p27 KIP1 and p21 CIP1 at E14.5, while expression of both proteins is detected in wild-type littermates (Western et al. 2008, 2011; Cook et al. 2011). DND1 has been postulated to promote translation of P27KIP1, in addition to NANOS2, which would directly link Dnd1 to male differentiation and cell cycle control (Western 2009; Cook et al. 2011). Dmrt1 null mutants on the 129/Sv background have decreased expression of the negative regulators of the cell cycle, p18 INK4c and p19 iNK4d, and DMRT1 has been shown to bind to the promoter of p19 INK4d in E13.5 testes (Krentz et al. 2009). Therefore, misregulation of entry into mitotic arrest is affected in multiple modifiers of TGCT incidence, highlighting this checkpoint as a critical step in tumorigenesis.
5.2 Failure to Repress Pluripotency
Retention of pluripotency has been shown to play an important role in TGCT initiation. In normal PGCs of both TGCT-resistant and susceptible mice, pluripotency is maintained through E13.5, as evident by the expression of NANOG and other pluripotency genes (Heaney et al. 2012). In TGCT-susceptible mice, gonocytes that fail to enter mitotic arrest continue to express pluripotency factors through the transition to ECCs (Kimura et al. 2003; Krentz et al. 2009; Cook et al. 2011). TGCT-susceptible gonocytes have significantly increased expression levels of NANOG at E15.5 (Heaney et al. 2012). Importantly, signaling pathways involving OCT4 and NANOG have been implicated in TGCT initiation in humans (Looijenga et al. 2003a; Clark et al. 2004; Oosterhuis and Looijenga 2005).
The germ cell specification gene DAZL has been shown to regulate pluripotency in both mouse and human. Forced overexpression of Dazl in ES cells, in the absense of LIF, promotes germ cell differentation and in germ cells meiotic induction; Dazl deficiency results in germ cell apoptosis and infertility (Lin and Page 2005; Kee et al. 2009; Yu et al. 2009; Medrano et al. 2012). Another gene involved in regulating pluripotency in mouse embryonic germ cells, PRDM14, has also been shown to regulate the expression of OCT4, NANOG, and SOX2 in human embryonic stem cells (Tsuneyoshi et al. 2008; Chia et al. 2010). Both DAZL and PRDM14 have been identified in LD with risk loci from human TGCT GWAS (Ruark et al. 2013).
The maintenance of pluripotency in germ cells has also been linked to tumor susceptibility in mice. The transcription factor Dmrt1 (doublesex and mab-3 related) has been shown to control pluripotency by regulating transcription of several genes, including Sox2 (Krentz et al. 2013), and its deficiency is sufficient to induce tumors on the 129/Sv inbred background (Krentz et al. 2009). Studies have also demonstrated a direct relationship of the maintenance of pluripotency to active NODAL/CRIPTO signaling, and an overexpression of Nodal signaling components in TGCTs (Spiller et al. 2012, 2013). In normal development, NODAL/Activin signaling directly affects pluripotency, male differentiation, and entry into meiosis in XY germ cells (Mendis et al. 2011; Souquet et al. 2012; Spiller et al. 2012; Miles et al. 2013; Wu et al. 2013). Decreased NODAL/CRIPTO signaling leads to significant increases expression of male differentiation makers, such as p15 INK4b and Dnmt3l (Spiller et al. 2012). Notably, CRIPTO was first identified in a human EC cell line (Ciccodicola et al. 1989), and NODAL signaling components are overexpressed in human TGCTs (Spiller et al. 2012).
It has not been determined whether retention of pluripotency is regulated independently of failed mitotic arrest in TGCT-susceptible gonocytes. The tumor initiation capacity of ECCs is dependent upon their pluripotent capacity (Gidekel et al. 2003). Retention of both proliferation and pluripotency therefore appear to be necessary for germ cell transformation into ECCs. There are varying data in normal gonocytes as to whether mitotic arrest and downregulation of pluripotency in gonocytes are linked or independently regulated. Miles et al. previously demonstrated that Activin signaling from somatic cells and autocrine NODAL signaling induces gonocytes to enter into mitotic arrest and transiently maintain pluripotency, respectively (Miles et al. 2013). Separate studies showed that subsequent loss of NODAL expression by gonocytes facilitates suppression of pluripotency (Spiller et al. 2012). Thus, aspects of mitotic arrest and suppression of pluripotency are independently regulated.
Importantly, retention of pluripotency is, at least in part, dependent on the misexpression of genes that promote G1-S cell cycle progression, such as Ccnd1. Ccnd1-deficiency suppressed TGCT-susceptible gonocyte pluripotency during the mitotic:meiotic switch (Lanza et al. 2016). In both ES and ECCs , rapid transition through G1 into S phase facilitates the maintenance of pluripotency (Filipczyk et al. 2007; Singh and Dalton 2009). A short G1 and long S phase promotes the euchromatic state of chromatin and suppresses differentiation, which preferentially occurs during the G1 phase in pluripotent cells (Mummery et al. 1987; Jonk et al. 1992; Herrera et al. 1996). Moreover, recent data demonstrate that pRB1 directly binds to the regulatory regions the core components of pluripotency (Oct4, Nanog, Sox2) and suppresses their expression (Kareta et al. 2015). Therefore, regulation of the cell cycle may contribute to retention of pluripotency observed in TGCT-susceptible gonocytes.
5.3 Pro-survival, Anti-apoptotic Microenvironment for Aberrant Germ Cell Proliferation
By E15.5 all male gonocytes should have entered mitotic quiescence, to eventually reinitiate proliferation and differentiate to form the spermatogonial lineage after birth (McLaren 1984). As previously discussed, TGCT susceptibility is only observed in mice on the 129 inbred background. To date, it is unclear as to why TGC tumorigenesis is possible on the 129 strain, but not other inbred mouse strains. This sensitivity issue has been addressed using the Ter mutation in Dnd1 (dead end homolog 1) on both 129/Sv and non-TGCT susceptible strains. The increase in the occurrence of teratomas in 129 mice caused by the Ter mutation was first reported in 1973 (Stevens 1973). Intriguingly, the Ter mutation also causes a dramatic loss of germ cells in both sexes in all genetic backgrounds, which led eventually researchers to identify Ter as a nonsense mutation in the gene Dnd1 (Asada et al. 1994; Youngren et al. 2005). The loss of germ cells in Dnd1 Ter/Ter mice is consistent with the early defect in germ cell specification (Noguchi et al. 1996).
It has been postulated that more efficient cell death pathways might protect certain strains by eliminating errant germ cells prior to tumor initiation (Bustamante-Marin et al. 2013). Apoptosis of fetal germ cells through a BAX-dependent mechanism has been postulated in the absence of teratomas in C57BL/6 mice with mutations in known 129 susceptibility genes (Cook et al. 2009). To test this hypothesis, a mutation in the pro-apoptotic gene Bax was introduced into mice of several genetic backgrounds carrying the Dnd1 Ter mutation. Bax-deficient mice had partial rescue of the germ cell loss phenotype in all strains (Cook et al. 2009) and a high incidence of teratomas was detected in double mutant Dnd1 Ter/Ter , Bax −/− and Dnd1 Ter/Ter , Bax −/+ mice on mixed genetic backgrounds, where teratomas were not seen in the absence of the Bax mutation. However, on a pure C57BL/6 background where ~50% of germ cells were rescued, no teratomas were seen, even in double mutants (Cook et al. 2011). These data underlie the complex control needed in the regulation of apoptosis in male germ cell development.
The anti-apoptosis phenotype is involved in the pathology of human TGCTs as well. Human GWAS have identified a susceptibility locus in humans that falls within an intron of the gene BAK1 (BCL2-antagonist/killer 1). BAK1 promotes apoptosis by antagonizing the apoptosis repressor activity of BCL2 and other anti-apoptotic proteins (Yan et al. 2000; Rapley et al. 2009). Therefore, a direct link between TGCT susceptibility in mouse and humans can be established by the apparent need to establish a pro-survival environment for transformed germ cells to evolve into TGCTs.
Additional studies in mice sampling gene expression differences in E14.5 XY gonocytes identified cell cycle regulators, apoptotic pathways, and tumor suppressors to be among the genes enriched in C57BL/6 compared to 129/Sv (Cook et al. 2011). These findings suggest that increased expression of factors that promote cell cycle arrest or apoptotic pathways prior to mitotic arrest in gonocytes may be sufficient to prevent teratoma formation, even in the presence of mutations that promote the transformation of germ cells. A better understanding of the genetic basis for the pro-survival phenotype in 129/Sv tumor susceptibility versus the apoptosis-driven C57BL/6 TGCT resistance could lead to the identification of additional genetic factors/modifiers that contribute to the developmental defects in 129/Sv mice that permit testicular germ cell tumorigenesis.
5.4 Altered Epigenetic States
As previously described, Blimp1, Prdm14, and a third transcriptional regulator, Tfap2c (also known as AP2γ or Tcfap2c), specify PGCs by inducing DNA demethylation and histone remodeling, repressing the somatic cell program, and establishing a naïve pluripotent expression profile (Saitou and Yamaji 2010). Expression of Blimp1 ceases at E11.0 as PGCs migrate into the gonad, whereas expression of Prdm14 and Tfap2c continues through the mitotic:meiotic switch (E13.5–14.5) (Yamaji et al. 2008; Weber et al. 2010). Blimp1, Prdm14, and Tfap2c expression is observed during comparable developmental periods in humans (Saitou and Yamaji 2010). Genetic experiments indicate that each factor is essential for germ cell specification (Saitou et al. 2003; Yamaji et al. 2008; Weber et al. 2010). Additionally, Prdm14 suppress early somatic tissue specification genes (e.g. Fgfr1 and Fgfr2), DNA methyltransferases (e.g. Dnmt3a and Dnmrt3b), and mediators of G1-S phase transition (e.g. Ccnd1), and activates germ cell specification genes (e.g. Nanos3, Dmrt1, & Tfap2c) and mitotic arrest factors (e.g. Dnd1) (Yamaji et al. 2008; Grabole et al. 2013; Magnusdottir et al. 2013; Yamaji et al. 2013). PRDM14 has also been identified in linkage disequilibrium with risk loci from human TGCT GWAS (Ruark et al. 2013). A recent study showed that Tfap2c haploinsufficiency increases TGCT incidence in 129/Sv mice by tenfold (Schemmer et al. 2013). Historically, Tfap2c haploinsufficiency does not cause TGCTs in C57Bl/6 mice (Werling and Schorle 2002; Weber et al. 2010). These results suggest that haploinsufficiency for Tfap2c has phenotypic consequences only in the context of 129 developmental defects that cause TGCT initiation.
Genome-wide demethylation of 5-methylcytosine sites occurs during normal PGC development. Interestingly, expression of de novo Dnmt3A and Dnmt3L methyltransferases are required for germ cell viability; Male mice that lack Dnmt3L are viable but sterile, with a complete absence of germ cells in adult males (Bourc'his et al. 2001). Additionally, Dnmt3L is required for normal imprinting of male germ cells, and normal male meiosis, but is not expressed in spermatocytes (Bourc'his and Bestor 2004). There is some evidence that there are strain-specific differences in the establishment of new methylation imprints, 129/Sv mice have been shown to establish new imprints more slowly than C57BL/6 mice (Davis et al. 2000; Durcova-Hills et al. 2006). While it remains to be elucidated whether the methylation states merely reflects the pluripotency of TGCTs or are part of the changes leading to tumorigenesis, it is interesting to consider epigenetic changes contributing to TGCT formation .
6 Remaining Questions
TGCTs are highly treatable with platinum-based chemotherapy . However, current treatment regimens cause long-term side effects including hearing loss, cardiovascular disease, cognitive impairment, and infertility (Horwich et al. 2006; Oldenburg et al. 2007b; Kraggerud et al. 2013). Moreover, long-term prognosis markedly worsens as the disease progresses with the potential for metastasis and there are limited alternative treatment options for patients that demonstrate platinum resistance. Thus, improvements in risk assessment, screening, and alternative treatment options remain important and the social, emotional, and medical costs remain high. Over the last decade, significant improvements in our understanding of the developmental origins, inherited risk factors, and somatic mutations that contribute to TGCT initiation and progression have been made. However, despite these advances several important questions remain to be answered regarding TGCT pathogenesis. The answers to these questions will not only provide us with a clearer understanding of the basic biology and genetics of TGCTs, but also may provide targets for more efficacious screening and treatment paradigms.
What are the genetic risk factors for chemoresistance and the morbidities of platinum treatment? As previously discussed, a subset of TGCTs are resistant to platinum-based chemotherapy agents and the long-term outlook for individuals with these tumors is bleak. By contrast, for those patients whose tumors do respond to treatment, there are significant long-term survivorship issues (associated morbidities) resulting from platinum-based treatment (Singhera et al. 2012; Bujan et al. 2013; de Haas et al. 2013). Although evidence is beginning to emerge for inherited genetic risk factors and somatic mutations that determine chemoresistance (Litchfield et al. 2016; Taylor-Weiner et al. 2016) and predisposition to treatment morbidities (Peters et al. 2000; Oldenburg et al. 2007a), much still remains to be learned.
How does genomic instability start? Although the karyotypes of developed TGCTs have been well characterized, the origin and progression of chromosomal abnormalities is unclear. What promotes nuclear instability, when do chromosomal abnormalities first evolve, and how do karyotypic abnormalities contribute to tumor progression remain unanswered questions. Unfortunately, it is difficult to study human TGCTs during the early stages of tumorigenesis; such studies are possible in mice.
Observations from investigations using both human tissue samples from TGCTs and ECC lines have led researchers to suggest that the polyploidization observed in GCNIS and TGCT (Atkin and Baker 1983; Kraggerud et al. 2002; Skotheim et al. 2002; Adamah et al. 2006; Rajpert-De Meyts 2006; Rajpert-de Meyts and Hoei-Hansen 2007) might be a result of confused meiosis signaling (Adamah et al. 2006; Jorgensen et al. 2013). Additionally, Jorgensen et al. hypothesize that germ cells with highly expressed genes located on 12p and 17q (human) that are frequently amplified in TGCT, especially in non-seminomas, could be among the genetic abnormalities that escape normal DNA repair checkpoints. GCNIS cells and TGCT cells do not complete meiosis, which could be the result of concurrent expression of CYP26B1 and NANOS2 in addition to the high expression of pluripotency factors. Again, germ cells may be responding to the conflicting signals present in the surrounding microenvironment after ensuing genomic instability, as postulated in the mouse (Bustamante-Marin et al. 2013).
Sexual disorders in humans, conditions that often blur morphological differences between testis and ovary (Hughes et al. 2006), increase the risk of an individual developing TGCTs (Muller et al. 1985; Looijenga et al. 2010; Pleskacova et al. 2010; Cools et al. 2011; Jorgensen et al. 2015). In the gonads of patients with sexual development disorders, immature germ cells persist as the supporting niche is not able to provide the appropriate environment for germ cell development. Based on evidence in mice, under-development of the somatic niche in the testis has also been associated with increased frequency of GCNIS (Skakkebaek et al. 2001; Hoei-Hansen et al. 2003).
The dysregulation of the mitotic:meiotic switch and inappropriate exposure of male gonocytes to retinoic acid has influenced researchers to screen for GCNIS. These studies utilize human tissue samples from adult testes and testicular tumors, and diagnostic biopsies from young boys with sex chromosome aneuploidy to monitor for GCNIS. Several human studies have also demonstrated the role of retinoic acid and its contribution to the disruption of meiosis regulation in the progress towards TGCT development (Childs et al. 2011; Jorgensen et al. 2012, 2013, 2015).
How do defects at the mitotic:meiotic switch interplay to promote tumorigenesis? The concurrent timing of aberrant proliferation, the mitotic:meiotic switch, and sex specification may provide clues to what signals are dysregulating the mitotic:meiotic switch and driving TGCT initiation in 129 mice. It has been previously shown that oogonia in both TGCT-resistant and susceptible mouse strains transiently express Ccnd1 from E12.5 to E15.5, just prior to initiating meiosis. Ccnd1 misexpression in TGCT-susceptible gonocytes occurs at the same developmental time-points that Ccnd1 is normally expressed in pre-meiotic oogonia (Heaney et al. 2012). These observations suggest that either a signal normally restricted to the developing ovary is aberrantly active or that activation of genes important to male gonocyte specification is delayed in the TGCT-susceptible testis. In the ovary, oogonia expression of Ccnd1 coincides with RA induction of Stra8 expression and the meiotic program from E13.5 to E15.5 (Koubova et al. 2006). In the embryonic testis RA is normally degraded by CYP26B1 expressed by Sertoli cells, prevents Stra8 induction and inhibits meiosis in gonocytes. However, recent evidence from Cyp26b1 and Stra8 double knockout mice, in which RA signaling is constitutively active in the embryonic testis but meiosis cannot be initiated, demonstrated that RA also has a Stra8-independent, pro-proliferative influence on gonocytes (Saba et al. 2014b). Importantly, this same study revealed that RA induced the expression of several genes normally restricted to pre-meiotic oogonia and adult spermatogonia, including Ccnd1, Ngn3, and Stra8. It has been previously demonstrated that these same genes are misexpressed by TGCT-susceptible gonocytes (Heaney et al. 2012). Furthermore, p15 INK4b, which is downregulated in TGCT-susceptible gonocytes, was found to be inhibited in the gonocytes of Cyp26b1 and Stra8 double knockout mice (Cook et al. 2011; Saba et al. 2014b). Thus, an aberrant RA signal could be altering the expression of positive and negative regulators of the G1-S transition and delay the mitotic arrest of gonocytes in 129/Sv testes.
Interestingly, Cyp26b1/Stra8 double knockout mice have rescued initiation of male differentiation, but still fail to enter mitotic arrest, similar to Cyp26b1 single knockout mice (Saba et al. 2014b). Therefore, the failure to enter mitotic arrest is not linked to an alternate pathway to initiate meiosis through the activation of Stra8. Intrinsic male gonocyte differentiation factors acting together with aberrant RA may be sufficient in the Cyp26b1/Stra8 double knockout mice to cause a failure of mitotic arrest. Convincingly, overexpression of NANOS2 in XX germ cells is sufficient to suppress meiosis and induce male specification (Suzuki and Saga 2008). In XY germ cells that are deficient for both Nanos2 and Stra8, normal male germ cell development is not rescued (Saba et al. 2014a), suggesting a possible role for Nanos2 in several aspects of sex specification in male gonocytes, in addition to the suppression of meiosis.
As alluded earlier, sex specification is intimately related with the developmental defects that contribute to the initiation of TGCTs in mice. The male or female specification pathways direct signaling from the microenvironment to control PGC specification, from E10.5, through the mitotic:meiotic switch starting at E13.5 to E15.5. The antagonism between signaling from female pathways for the induction of meiosis and normal male differentiation has been suggested for several decades (Wai-Sum and Baker 1976; McLaren 1984; Vigier et al. 1987; Yao et al. 2003; Kim et al. 2006). Additionally, studies in the mouse have postulated that decreased efficiency of intercellular somatic-germ cell signaling may lead to decreased activation of the male specification pathway and escape from induction of apoptosis. This hypothesis is exemplified in the reported differences in morphology of testis cords between TGCT-susceptible and resistant mouse strains; the 129 testes displayed large testis cords containing numerous germ cells, while C57Bl/6 testes had significantly smaller testis cords and fewer germ cells per cord (Western et al. 2011). Ideally, germ cells that are exposed to any aberrant signal are most likely removed by apoptosis. However, if the timing and the environment are just right, perhaps these mixed-up gonocytes initiate TGCTs.
As previously discussed, FGF9 signaling is also essential for XY germ cell survival and commitment to male specification (DiNapoli et al. 2006). Fgf9-null mice undergo sex reversal (Colvin et al. 2001; Schmahl et al. 2004; Bowles et al. 2010), but normal male differentiation can be rescued by deleting Wnt4 (Colvin et al. 2001; Kim et al. 2006). Conversely, loss of Wnt4 creates a partial sex reversal in XX gonads, and is not rescued by deleting Fgf9, indicating that FGF signaling is not necessary for the partial male characteristics developed in XX Wnt4-null gonads (Jameson et al. 2012). Thus, the pathways are not simply in opposition of each other, secondary components act in the male lineage to downregulate female pathways and promote male differentiation. This may provide insight as to how the “just right” condition comes about to promote tumor development in the face of preexisting developmental abnormalities .
Why has there been such an increase in TGCTs in the last 50 years? In this chapter, there has been a lengthy discussion of the genetic and developmental defects that give rise to TGCTs. But is there an environmental component that increases the prevalence of TGCTs? Studies have also found increased male reproductive health risks of undescended testis and hypospadias, and deteriorating semen quality in certain demographics and geographical regions (Skakkebaek et al. 2001). Xenoestrogens and other endocrine disrupting compounds may be to blame. Furthermore, the development of TGCTs has been linked to several disorders of gonadal development and sexual differentiation, which hints back to dysregulation of germ cell development. With a steady increase in incidence of Type II TGCTs in the last 40 years (Bernstein et al. 1999), in utero exposure to environmental factors may be interacting with genetic predispositions to disrupt normal germ cell development. (For more information on the discussion of the environment and hormone disruptors in spermatogonial development, refer to the chapter by Pat Hunt). Considering the large genetic component of developing TGCTs and the limited number of studies available to elucidate environmental risk, one could speculate that even a low risk of genetic susceptibility to developing TGCTs could interact with increased exposure to environmental triggers to increase rates of TGCTs.
How does epigenetics contribute to tumorigenesis? One of the areas that remains to be explored in the mouse model is the contribution of epigenetic changes to the genomic instability interacting with the developmental defects in the 129 background. There is evidence to reduced methylation contributing to increased malignancy of tumors in mice (Fraga et al. 2004), which may be the result of chromosomal instability. Reduced methylation has been shown to result in chromosomal instability in human glioblastomas (Cadieux et al. 2006) and colon cancers (Rodriguez et al. 2006). Hypomethylation of specific DNA sites is also associated with an erased (removed) pattern of genomic imprinting, found to be able to induce cancers in mice, related to the TRP53 and TGFβ pathway (Holm et al. 2005). Curiously, a few of the genes in susceptibility loci identified by GWAS, Dmrt1 and Prdm14, function to maintain epigenetic states in the developing PGC. DMRT1 functions as a DNA methylase (Krentz et al. 2009) and PRDM14 maintains a naïve pluripotent state by regulating DNA methylation (Leitch et al. 2013; Okashita et al. 2014). Considering the genome-wide demethylation that occurs during PGC specification, changes in methylation may be the initial instability that goes on to interact with the developmental defects, ultimately giving rise to TGCTs in 129 mice .
Germ cells that transform into ECCs , what switch is thrown? As PGCs begin sex specification around E11.5, these pluripotent cells have an increased capacity for teratoma formation. Seminal studies from Leroy Stevens demonstrated the high success rate (75–80%) in grafting genital ridges from E11.5 or E12.5129 mice into adult testes to form teratomas. As development progresses, however, the incidence of teratoma formation steeply declines as the age of the genital ridge increases. Grafted genital ridges from E13.5 embryos results in 16% teratoma incidence, and incidence falls to <10% from E14.5 or later (Stevens 1966). What signals can be received by nearly all E11.5 PGCs to transform these cells to ECCs, but can only transform a fraction of E13.5 gonocytes? From Steven’s studies, the window for exposing germ cells with developmental defects to the adult male signaling environment to induce tumor formation occurs prior to E13.5; starting at E13.5 gonocytes are refractory to ECCs transformation in an altered signaling environment. The timing of the mitotic:meiotic switch in this same developmental window has lead researchers to explore altered male and female germ cell specification as clues to inducing tumorigenesis .
7 Conclusion
The 129 inbred mouse model has provided a generous amount of insight to the pathology and etiology of TGCTs. There has been debate as to the applicability of this mouse model to the most common cancer in adolescent and young adult human males, considering the relatively young age of tumor presentation in mice compared to the activation of dormant GCNIS at puberty in humans. However, considering that the same genes in susceptibility loci induce tumors and that the first two loci were first identified as susceptibility genes on the 129/Sv background, the pathology of the disease may progress similarly between mouse and Type II TGCTs. The technology is available to understand the genetic susceptibility in humans and validate these findings in a suitable model. From human GWAS to the interplay between genetics and environmental exposures, future studies using mouse models are strategically poised to begin deconvoluting the tough questions at the intersection of cancer and developmental biology.
References
Abada PB, Howell SB (2014) Cisplatin induces resistance by triggering differentiation of testicular embryonal carcinoma cells. PLoS One 9(1):e87444
Adamah DJ, Gokhale PJ, Eastwood DJ, Rajpert De-Meyts E, Goepel J, Walsh JR, Moore HD, Andrews PW (2006) Dysfunction of the mitotic:meiotic switch as a potential cause of neoplastic conversion of primordial germ cells. Int J Androl 29(1):219–227
Adams IR, McLaren A (2002) Sexually dimorphic development of mouse primordial germ cells: switching from oogenesis to spermatogenesis. Development 129(5):1155–1164
Anderson PD, Nelson VR, Tesar PJ, Nadeau JH (2009) Genetic factors on mouse chromosome 18 affecting susceptibility to testicular germ cell tumors and permissiveness to embryonic stem cell derivation. Cancer Res 69(23):9112–9117
Andreassen KE, Kristiansen W, Karlsson R, Aschim EL, Dahl O, Fossa SD, Adami HO, Wiklund F, Haugen TB, Grotmol T (2013) Genetic variation in AKT1, PTEN and the 8q24 locus, and the risk of testicular germ cell tumor. Hum Reprod 28(7):1995–2002
Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31(6):785–799
Asada Y, Varnum DS, Frankel WN, Nadeau JH (1994) A mutation in the Ter gene causing increased susceptibility to testicular teratomas maps to mouse chromosome 18. Nat Genet 6(4):363–368
Atkin NB, Baker MC (1983) i(12p): specific chromosomal marker in seminoma and malignant teratoma of the testis? Cancer Genet Cytogenet 10(2):199–204
Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R (2003) Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17(1):126–140
Bahrami A, Ro JY, Ayala AG (2007) An overview of testicular germ cell tumors. Arch Pathol Lab Med 131(8):1267–1280
Barrios F, Filipponi D, Pellegrini M, Paronetto MP, Di Siena S, Geremia R, Rossi P, De Felici M, Jannini EA, Dolci S (2010) Opposing effects of retinoic acid and FGF9 on Nanos2 expression and meiotic entry of mouse germ cells. J Cell Sci 123(Pt 6):871–880
Bernstein L, Smith MA, Lihua L, Deapen D, Friedman DL (1999) Germ cell, trophoblastic and other gonadal neoplasms. In: Ries LAG, Smith MA, Gurney JC et al (eds) Cancer incidence and survival among childrem and adolescents: United States SEER program 1975–1995. National Cancer Institute, Bethesda, MD
Beumer TL, Roepers-Gajadien HL, Gademan IS, Kal HB, de Rooij DG (2000) Involvement of the D-type cyclins in germ cell proliferation and differentiation in the mouse. Biol Reprod 63(6):1893–1898
Bosl GJ, Motzer RJ (1997) Testicular germ-cell cancer. N Engl J Med 337(4):242–253
Bourc'his D, Bestor TH (2004) Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431(7004):96–99
Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294(5551):2536–2539
Bowles J, Feng CW, Spiller C, Davidson TL, Jackson A, Koopman P (2010) FGF9 suppresses meiosis and promotes male germ cell fate in mice. Dev Cell 19(3):440–449
Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, Yashiro K, Chawengsaksophak K, Wilson MJ, Rossant J, Hamada H, Koopman P (2006) Retinoid signaling determines germ cell fate in mice. Science 312(5773):596–600
Bowles J, Koopman P (2010) Sex determination in mammalian germ cells: extrinsic versus intrinsic factors. Reproduction 139(6):943–958
Brabrand S, Johannessen B, Axcrona U, Kraggerud SM, Berg KG, Bakken AC, Bruun J, Fossa SD, Lothe RA, Lehne G, Skotheim RI (2015) Exome sequencing of bilateral testicular germ cell tumors suggests independent development lineages. Neoplasia 17(2):167–174
Bromen K, Stang A, Baumgardt-Elms C, Stegmaier C, Ahrens W, Metz KA, Jockel KH (2004) Testicular, other genital, and breast cancers in first-degree relatives of testicular cancer patients and controls. Cancer Epidemiol Biomark Prev 13(8):1316–1324
Bujan L, Walschaerts M, Moinard N, Hennebicq S, Saias J, Brugnon F, Auger J, Berthaut I, Szerman E, Daudin M, Rives N (2013) Impact of chemotherapy and radiotherapy for testicular germ cell tumors on spermatogenesis and sperm DNA: a multicenter prospective study from the CECOS network. Fertil Steril 100(3):673–680
Bussey KJ, Lawce HJ, Himoe E, Shu XO, Heerema NA, Perlman EJ, Olson SB, Magenis RE (2001) SNRPN methylation patterns in germ cell tumors as a reflection of primordial germ cell development. Genes Chromosomes Cancer 32(4):342–352
Bussey KJ, Lawce HJ, Olson SB, Arthur DC, Kalousek DK, Krailo M, Giller R, Heifetz S, Womer R, Magenis RE (1999) Chromosome abnormalities of eighty-one pediatric germ cell tumors: sex-, age-, site-, and histopathology-related differences—a Children's Cancer Group study. Genes Chromosomes Cancer 25(2):134–146
Bustamante-Marin X, Garness JA, Capel B (2013) Testicular teratomas: an intersection of pluripotency, differentiation and cancer biology. Int J Dev Biol 57(2–4):201–210
Byskov AG (1978) The anatomy and ultrastructure of the rete system in the fetal mouse ovary. Biol Reprod 19(4):720–735
Byskov AG (1986) Differentiation of mammalian embryonic gonad. Physiol Rev 66(1):71–117
Cadieux B, Ching TT, VandenBerg SR, Costello JF (2006) Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res 66(17):8469–8476
Carouge D, Blanc V, Knoblaugh SE, Hunter RJ, Davidson NO, Nadeau JH (2016) Parent-of-origin effects of A1CF and AGO2 on testicular germ-cell tumors, testicular abnormalities, and fertilization bias. Proc Natl Acad Sci U S A 113(37):E5425–E5433
Carriere P, Baade P, Fritschi L (2007) Population based incidence and age distribution of spermatocytic seminoma. J Urol 178(1):125–128
Castedo SM, de Jong B, Oosterhuis JW, Seruca R, te Meerman GJ, Dam A, Schraffordt Koops H (1989) Cytogenetic analysis of ten human seminomas. Cancer Res 49(2):439–443
Chadalavada RS, Korkola JE, Houldsworth J, Olshen AB, Bosl GJ, Studer L, Chaganti RS (2007) Constitutive gene expression predisposes morphogen-mediated cell fate responses of NT2/D1 and 27X-1 human embryonal carcinoma cells. Stem Cells 25(3):771–778
Chanock S (2009) High marks for GWAS. Nat Genet 41(7):765–766
Chia NY, Chan YS, Feng B, Lu X, Orlov YL, Moreau D, Kumar P, Yang L, Jiang J, Lau MS, Huss M, Soh BS, Kraus P, Li P, Lufkin T, Lim B, Clarke ND, Bard F, Ng HH (2010) A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468(7321):316–320
Chia, V. M., Y. Li, L. R. Goldin, B. I. Graubard, M. H. Greene, L. Korde, M. V. Rubertone, R. L. Erickson and K. A. McGlynn (2009). "Risk of cancer in first- and second-degree relatives of testicular germ cell tumor cases and controls." Int J Cancer 124(4): 952–957
Childs AJ, Cowan G, Kinnell HL, Anderson RA, Saunders PT (2011) Retinoic Acid signalling and the control of meiotic entry in the human fetal gonad. PLoS One 6(6):e20249
Chin L, Andersen JN, Futreal PA (2011) Cancer genomics: from discovery science to personalized medicine. Nat Med 17(3):297–303
Chung CC, Kanetsky PA, Wang Z, Hildebrandt MA, Koster R, Skotheim RI, Kratz CP, Turnbull C, Cortessis VK, Bakken AC, Bishop DT, Cook MB, Erickson RL, Fossa SD, Jacobs KB, Korde LA, Kraggerud SM, Lothe RA, Loud JT, Rahman N, Skinner EC, Thomas DC, Wu X, Yeager M, Schumacher FR, Greene MH, Schwartz SM, McGlynn KA, Chanock SJ, Nathanson KL (2013) Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat Genet 45(6):680–685
Ciccodicola A, Dono R, Obici S, Simeone A, Zollo M, Persico MG (1989) Molecular characterization of a gene of the ‘EGF family’ expressed in undifferentiated human NTERA2 teratocarcinoma cells. EMBO J 8(7):1987–1991
Clark AT, Rodriguez RT, Bodnar MS, Abeyta MJ, Cedars MI, Turek PJ, Firpo MT, Reijo Pera RA (2004) Human STELLAR, NANOG, and GDF3 genes are expressed in pluripotent cells and map to chromosome 12p13, a hotspot for teratocarcinoma. Stem Cells 22(2):169–179
Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM (2001) Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 104(6):875–889
Cook MS, Coveney D, Batchvarov I, Nadeau JH, Capel B (2009) BAX-mediated cell death affects early germ cell loss and incidence of testicular teratomas in Dnd1(Ter/Ter) mice. Dev Biol 328(2):377–383
Cook MS, Munger SC, Nadeau JH, Capel B (2011) Regulation of male germ cell cycle arrest and differentiation by DND1 is modulated by genetic background. Development 138(1):23–32
Cools M, Wolffenbuttel KP, Drop SL, Oosterhuis JW, Looijenga LH (2011) Gonadal development and tumor formation at the crossroads of male and female sex determination. Sex Dev 5(4):167–180
Crockford GP, Linger R, Hockley S, Dudakia D, Johnson L, Huddart R, Tucker K, Friedlander M, Phillips KA, Hogg D, Jewett MA, Lohynska R, Daugaard G, Richard S, Chompret A, Bonaiti-Pellie C, Heidenreich A, Albers P, Olah E, Geczi L, Bodrogi I, Ormiston WJ, Daly PA, Guilford P, Fossa SD, Heimdal K, Tjulandin SA, Liubchenko L, Stoll H, Weber W, Forman D, Oliver T, Einhorn L, McMaster M, Kramer J, Greene MH, Weber BL, Nathanson KL, Cortessis V, Easton DF, Bishop DT, Stratton MR, Rapley EA (2006) Genome-wide linkage screen for testicular germ cell tumour susceptibility loci. Hum Mol Genet 15(3):443–451
Cutcutache I, Suzuki Y, Tan IB, Ramgopal S, Zhang S, Ramnarayanan K, Gan A, Lee HH, Tay ST, Ooi A, Ong CK, Bolthouse JT, Lane BR, Anema JG, Kahnoski RJ, Tan P, Teh BT, Rozen SG (2015) Exome-wide sequencing shows low mutation rates and identifies novel mutated genes in seminomas. Eur Urol 68(1):77–83
Czene K, Lichtenstein P, Hemminki K (2002) Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int J Cancer 99(2):260–266
Davies RJ, Miller R, Coleman N (2005) Colorectal cancer screening: prospects for molecular stool analysis. Nat Rev Cancer 5(3):199–209
Davis TL, Yang GJ, McCarrey JR, Bartolomei MS (2000) The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum Mol Genet 9(19):2885–2894
de Graaff WE, Oosterhuis JW, de JB, Dam A, van Putten WL, Castedo SM, Sleijfer DT, Schraffordt KH (1992) Ploidy of testicular carcinoma in situ. Lab Investig 66(2):166–168
de Haas EC, Altena R, Boezen HM, Zwart N, Smit AJ, Bakker SJ, van Roon AM, Postma A, Wolffenbuttel BH, Hoekstra HJ, van Leeuwen FE, Sleijfer DT, Gietema JA (2013) Early development of the metabolic syndrome after chemotherapy for testicular cancer. Ann Oncol 24(3):749–755
de Jong B, Oosterhuis JW, Castedo SM, Vos A, te Meerman GJ (1990) Pathogenesis of adult testicular germ cell tumors. A cytogenetic model. Cancer Genet Cytogenet 48(2):143–167
Deshpande A, Sicinski P, Hinds PW (2005) Cyclins and cdks in development and cancer: a perspective. Oncogene 24(17):2909–2915
DiNapoli L, Batchvarov J, Capel B (2006) FGF9 promotes survival of germ cells in the fetal testis. Development 133(8):1519–1527
Durcova-Hills G, Adams IR, Barton SC, Surani MA, McLaren A (2006) The role of exogenous fibroblast growth factor-2 on the reprogramming of primordial germ cells into pluripotent stem cells. Stem Cells 24(6):1441–1449
Feldman DR, Iyer G, Van Alstine L, Patil S, Al-Ahmadie H, Reuter VE, Bosl GJ, Chaganti RS, Solit DB (2014) Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin Cancer Res 20(14):3712–3720
Feng CW, Bowles J, Koopman P (2014) Control of mammalian germ cell entry into meiosis. Mol Cell Endocrinol 382(1):488–497
Filipczyk AA, Laslett AL, Mummery C, Pera MF (2007) Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Res 1(1):45–60
Forman D, Oliver RT, Brett AR, Marsh SG, Moses JH, Bodmer JG, Chilvers CE, Pike MC (1992) Familial testicular cancer: a report of the UK family register, estimation of risk and an HLA class 1 sib-pair analysis. Br J Cancer 65(2):255–262
Fraga MF, Herranz M, Espada J, Ballestar E, Paz MF, Ropero S, Erkek E, Bozdogan O, Peinado H, Niveleau A, Mao JH, Balmain A, Cano A, Esteller M (2004) A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res 64(16):5527–5534
Frigyesi A, Gisselsson D, Hansen GB, Soller M, Mitelman F, Hoglund M (2004) A model for karyotypic evolution in testicular germ cell tumors. Genes Chromosomes Cancer 40(3):172–178
Gidekel S, Pizov G, Bergman Y, Pikarsky E (2003) Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell 4(5):361–370
Gill ME, Hu YC, Lin Y, Page DC (2011) Licensing of gametogenesis, dependent on RNA binding protein DAZL, as a gateway to sexual differentiation of fetal germ cells. Proc Natl Acad Sci U S A 108(18):7443–7448
Gillis AJ, Verkerk AJ, Dekker MC, van Gurp RJ, Oosterhuis JW, Looijenga LH (1997) Methylation similarities of two CpG sites within exon 5 of human H19 between normal tissues and testicular germ cell tumours of adolescents and adults, without correlation with allelic and total level of expression. Br J Cancer 76(6):725–733
Ginsburg M, Snow MH, McLaren A (1990) Primordial germ cells in the mouse embryo during gastrulation. Development 110(2):521–528
Gori S, Porrozzi S, Roila F, Gatta G, De Giorgi U, Marangolo M (2005) Germ cell tumours of the testis. Crit Rev Oncol Hematol 53(2):141–164
Grabole N, Tischler J, Hackett JA, Kim S, Tang F, Leitch HG, Magnusdottir E, Surani MA (2013) Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep 14(7):629–637
Gutekunst M, Oren M, Weilbacher A, Dengler MA, Markwardt C, Thomale J, Aulitzky WE, van der Kuip H (2011) p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS One 6(4):e19198
Hacker A, Capel B, Goodfellow P, Lovell-Badge R (1995) Expression of Sry, the mouse sex determining gene. Development 121(6):1603–1614
Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA (2008) Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452(7189):877–881
Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA (2002) Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 117(1–2):15–23
Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA (2010) Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 329(5987):78–82
Harvey M, McArthur MJ, Montgomery CA Jr, Bradley A, Donehower LA (1993) Genetic background alters the spectrum of tumors that develop in p53-deficient mice. FASEB J 7(10):938–943
Hayashi K, Surani MA (2009) Self-renewing epiblast stem cells exhibit continual delineation of germ cells with epigenetic reprogramming in vitro. Development 136(21):3549–3556
Heaney JD, Anderson EL, Michelson MV, Zechel JL, Conrad PA, Page DC, Nadeau JH (2012) Germ cell pluripotency, premature differentiation and susceptibility to testicular teratomas in mice. Development 139(9):1577–1586
Heaney JD, Lam MY, Michelson MV, Nadeau JH (2008) Loss of the transmembrane but not the soluble kit ligand isoform increases testicular germ cell tumor susceptibility in mice. Cancer Res 68(13):5193–5197
Heaney JD, Michelson MV, Youngren KK, Lam MY, Nadeau JH (2009) Deletion of eIF2beta suppresses testicular cancer incidence and causes recessive lethality in agouti-yellow mice. Hum Mol Genet 18(8):1395–1404
Heaney JD, Nadeau JH (2008) Testicular germ cell tumors in mice: new ways to study a genetically complex trait. Methods Mol Biol 450:211–231
Heimdal K, Olsson H, Tretli S, Flodgren P, Borresen AL, Fossa SD (1996) Risk of cancer in relatives of testicular cancer patients. Br J Cancer 73(7):970–973
Heimdal K, Olsson H, Tretli S, Fossa SD, Borresen AL, Bishop DT (1997) A segregation analysis of testicular cancer based on Norwegian and Swedish families. Br J Cancer 75(7):1084–1087
Herrera RE, Chen F, Weinberg RA (1996) Increased histone H1 phosphorylation and relaxed chromatin structure in Rb-deficient fibroblasts. Proc Natl Acad Sci U S A 93(21):11510–11515
Hoei-Hansen CE, Holm M, Rajpert-De Meyts E, Skakkebaek NE (2003) Histological evidence of testicular dysgenesis in contralateral biopsies from 218 patients with testicular germ cell cancer. J Pathol 200(3):370–374
Hoff AM, Alagaratnam S, Zhao S, Bruun J, Andrews PW, Lothe RA, Skotheim RI (2016) Identification of novel fusion genes in testicular germ cell tumors. Cancer Res 76(1):108–116
Hoffner L, Deka R, Chakravarti A, Surti U (1994) Cytogenetics and origins of pediatric germ cell tumors. Cancer Genet Cytogenet 74(1):54–58
Holm TM, Jackson-Grusby L, Brambrink T, Yamada Y, Rideout WM 3rd, Jaenisch R (2005) Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell 8(4):275–285
Horwich A, Shipley J, Huddart R (2006) Testicular germ-cell cancer. Lancet 367(9512):754–765
Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, Korsay CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA (2012) SEER cancer statistics review, 1975–2009 (Vintage 2009 Populations). National Cancer Institute, Bethesda, MD
Hughes IA, Houk C, Ahmed SF, Lee PA, Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group (2006) Consensus statement on management of intersex disorders. J Pediatr Urol 2(3):148–162
Jameson SA, Lin YT, Capel B (2012) Testis development requires the repression of Wnt4 by Fgf signaling. Dev Biol 370(1):24–32
Jiang LI, Nadeau JH (2001) 129/Sv mice—a model system for studying germ cell biology and testicular cancer. Mamm Genome 12(2):89–94
Jonk LJ, de Jonge ME, Kruyt FA, Mummery CL, van der Saag PT, Kruijer W (1992) Aggregation and cell cycle dependent retinoic acid receptor mRNA expression in P19 embryonal carcinoma cells. Mech Dev 36(3):165–172
Jorgensen A, Lindhardt Johansen M, Juul A, Skakkebaek NE, Main KM, Rajpert-De Meyts E (2015) Pathogenesis of germ cell neoplasia in testicular dysgenesis and disorders of sex development. Semin Cell Dev Biol 5:124–137
Jorgensen A, Nielsen JE, Almstrup K, Toft BG, Petersen BL, Rajpert-De ME (2013) Dysregulation of the mitosis-meiosis switch in testicular carcinoma in situ. J Pathol 229(4):588–598
Jorgensen A, Nielsen JE, Blomberg Jensen M, Graem N, Rajpert-De Meyts E (2012) Analysis of meiosis regulators in human gonads: a sexually dimorphic spatio-temporal expression pattern suggests involvement of DMRT1 in meiotic entry. Mol Hum Reprod 18(11):523–534
Kadyrova LY, Habara Y, Lee TH, Wharton RP (2007) Translational control of maternal Cyclin B mRNA by Nanos in the Drosophila germline. Development 134(8):1519–1527
Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, Letrero R, Ciosek SL, Doody DR, Smith LM, Weaver J, Albano A, Chen C, Starr JR, Rader DJ, Godwin AK, Reilly MP, Hakonarson H, Schwartz SM, Nathanson KL (2009) Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet 41(7):811–815
Kanetsky PA, Mitra N, Vardhanabhuti S, Vaughn DJ, Li M, Ciosek SL, Letrero R, D'Andrea K, Vaddi M, Doody DR, Weaver J, Chen C, Starr JR, Hakonarson H, Rader DJ, Godwin AK, Reilly MP, Schwartz SM, Nathanson KL (2011) A second independent locus within DMRT1 is associated with testicular germ cell tumor susceptibility. Hum Mol Genet 20(15):3109–3117
Kaplan GW, Cromie WC, Kelalis PP, Silber I, Tank ES Jr (1988) Prepubertal yolk sac testicular tumors—report of the testicular tumor registry. J Urol 140(5 Pt 2):1109–1112
Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, Cecchini MJ, Spacek D, Batista LF, O'Brien M, Ng YH, Ang CE, Vaka D, Artandi SE, Dick FA, Brunet A, Sage J, Wernig M (2015) Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16(1):39–50
Karlsson R, Andreassen KE, Kristiansen W, Aschim EL, Bremnes RM, Dahl O, Fossa SD, Klepp O, Langberg CW, Solberg A, Tretli S, Magnusson PK, Adami HO, Haugen TB, Grotmol T, Wiklund F (2013) Investigation of six testicular germ cell tumor susceptibility genes suggests a parent-of-origin effect in SPRY4. Hum Mol Genet 22(16):3373–3380
Kee K, Angeles VT, Flores M, Nguyen HN, Reijo Pera RA (2009) Human DAZL, DAZ and BOULE genes modulate primordial germ-cell and haploid gamete formation. Nature 462(7270):222–225
Kehler J, Tolkunova E, Koschorz B, Pesce M, Gentile L, Boiani M, Lomeli H, Nagy A, McLaughlin KJ, Scholer HR, Tomilin A (2004) Oct4 is required for primordial germ cell survival. EMBO Rep 5(11):1078–1083
Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, Cummings OW, Wait C, Town A, Heinrich MC (2004) KIT mutations are common in testicular seminomas. Am J Pathol 164(1):305–313
Kim Y, Kobayashi A, Sekido R, DiNapoli L, Brennan J, Chaboissier MC, Poulat F, Behringer RR, Lovell-Badge R, Capel B (2006) Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol 4(6):e187
Kimura T, Suzuki A, Fujita Y, Yomogida K, Lomeli H, Asada N, Ikeuchi M, Nagy A, Mak TW, Nakano T (2003) Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development 130(8):1691–1700
Kleinsmith LJ, Pierce GB Jr (1964) Multipotentiality of single embryonal carcinoma cells. Cancer Res 24:1544–1551
Koide O, Iwai S, Baba K, Iri H (1987) Identification of testicular atypical germ cells by an immunohistochemical technique for placental alkaline phosphatase. Cancer 60(6):1325–1330
Kommoss F, Bibbo M, Talerman A (1990) Nuclear deoxyribonucleic acid content (ploidy) of endodermal sinus (yolk sac) tumor. Lab Investig 62(2):223–231
Koubova J, Hu YC, Bhattacharyya T, Soh YQ, Gill ME, Goodheart ML, Hogarth CA, Griswold MD, Page DC (2014) Retinoic acid activates two pathways required for meiosis in mice. PLoS Genet 10(8):e1004541
Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC (2006) Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc Natl Acad Sci U S A 103(8):2474–2479
Krag Jacobsen G, Barlebo H, Olsen J, Schultz HP, Starklint H, Sogaard H, Vaeth M (1984) Testicular germ cell tumours in Denmark 1976-1980. Pathology of 1058 consecutive cases. Acta Radiol Oncol 23(4):239–247
Kraggerud SM, Hoei-Hansen CE, Alagaratnam S, Skotheim RI, Abeler VM, Rajpert-de ME, Lothe RA (2013) Molecular characteristics of malignant ovarian germ cell tumors and comparison with testicular counterparts: implications for pathogenesis. Endocr Rev 34(3):339–376
Kraggerud SM, Skotheim RI, Szymanska J, Eknaes M, Fossa SD, Stenwig AE, Peltomaki P, Lothe RA (2002) Genome profiles of familial/bilateral and sporadic testicular germ cell tumors. Genes Chromosomes Cancer 34(2):168–174
Kratz CP, Han SS, Rosenberg PS, Berndt SI, Burdett L, Yeager M, Korde LA, Mai PL, Pfeiffer R, Greene MH (2011) Variants in or near KITLG, BAK1, DMRT1, and TERT-CLPTM1L predispose to familial testicular germ cell tumour. J Med Genet 48(7):473–476
Krentz AD, Murphy MW, Kim S, Cook MS, Capel B, Zhu R, Matin A, Sarver AL, Parker KL, Griswold MD, Looijenga LH, Bardwell VJ, Zarkower D (2009) The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc Natl Acad Sci U S A 106(52):22323–22328
Krentz AD, Murphy MW, Zhang T, Sarver AL, Jain S, Griswold MD, Bardwell VJ, Zarkower D (2013) Interaction between DMRT1 function and genetic background modulates signaling and pluripotency to control tumor susceptibility in the fetal germ line. Dev Biol 377(1):67–78
Kunwar PS, Siekhaus DE, Lehmann R (2006) In vivo migration: a germ cell perspective. Annu Rev Cell Dev Biol 22:237–265
Kurimoto K, Yamaji M, Seki Y, Saitou M (2008) Specification of the germ cell lineage in mice: a process orchestrated by the PR-domain proteins, Blimp1 and Prdm14. Cell Cycle 7(22):3514–3518
Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, Reik W (2003) Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 35(2):88–93
Lange UC, Adams DJ, Lee C, Barton S, Schneider R, Bradley A, Surani MA (2008) Normal germ line establishment in mice carrying a deletion of the Ifitm/Fragilis gene family cluster. Mol Cell Biol 28(15):4688–4696
Lanza DG, Dawson EP, Rao P, Heaney JD (2016) Misexpression of cyclin D1 in embryonic germ cells promotes testicular teratoma initiation. Cell Cycle 15(7):919–930
Lawson KA, Dunn NR, Roelen BA, Zeinstra LM, Davis AM, Wright CV, Korving JP, Hogan BL (1999) Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev 13(4):424–436
Lee J, Inoue K, Ono R, Ogonuki N, Kohda T, Kaneko-Ishino T, Ogura A, Ishino F (2002) Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 129(8):1807–1817
Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A, Surani MA, Hajkova P (2013) Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol 20(3):311–316
Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3(9):662–673
Lin Y, Page DC (2005) Dazl deficiency leads to embryonic arrest of germ cell development in XY C57BL/6 mice. Dev Biol 288(2):309–316
Lin YT, Capel B (2015) Cell fate commitment during mammalian sex determination. Curr Opin Genet Dev 32:144–152
Lindelof B, Eklund G (2001) Analysis of hereditary component of cancer by use of a familial index by site. Lancet 358(9294):1696–1698
Litchfield K, Holroyd A, Lloyd A, Broderick P, Nsengimana J, Eeles R, Easton DF, Dudakia D, Bishop DT, Reid A, Huddart RA, Grotmol T, Wiklund F, Shipley J, Houlston RS, Turnbull C (2015a) Identification of four new susceptibility loci for testicular germ cell tumour. Nat Commun 6:8690
Litchfield K, Levy M, Huddart RA, Shipley J, Turnbull C (2016) The genomic landscape of testicular germ cell tumours: from susceptibility to treatment. Nat Rev Urol 13(7):409–419
Litchfield K, Shipley J, Turnbull C (2015b) Common variants identified in genome-wide association studies of testicular germ cell tumour: an update, biological insights and clinical application. Andrology 3(1):34–46
Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, Renwick A, Seal S, Al-Saadi R, Broderick P, Turner NC, Houlston RS, Huddart R, Shipley J, Turnbull C (2015c) Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun 6:5973
Litchfield K, Thomsen H, Mitchell JS, Sundquist J, Houlston RS, Hemminki K, Turnbull C (2015d) Quantifying the heritability of testicular germ cell tumour using both population-based and genomic approaches. Sci Rep 5:13889
Looijenga LH, de LH, van OM, van Gurp RJ, Stoop H, Gillis AJ, de Gouveia Brazao CA, Weber RF, Kirkels WJ, van DT, von LM, Valk P, Lajos G, Olah E, Nesland JM, Fossa SD, Oosterhuis JW (2003a) Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res 63(22):7674–7678
Looijenga LH, Gillis AJ, van Gurp RJ, Verkerk AJ, Oosterhuis JW (1997) X inactivation in human testicular tumors. XIST expression and androgen receptor methylation status. Am J Pathol 151(2):581–590
Looijenga LH, Hersmus R, de Leeuw BH, Stoop H, Cools M, Oosterhuis JW, Drop SL, Wolffenbuttel KP (2010) Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab 24(2):291–310
Looijenga LH, Hersmus R, Gillis AJ, Pfundt R, Stoop HJ, van Gurp RJ, Veltman J, Beverloo HB, van Drunen E, van Kessel AG, Pera RR, Schneider DT, Summersgill B, Shipley J, McIntyre A, van der Spek P, Schoenmakers E, Oosterhuis JW (2006) Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res 66(1):290–302
Looijenga LH, Olie RA, van der Gaag I, van Sluijs FJ, Matoska J, Ploem-Zaaijer J, Knepfle C, Oosterhuis JW (1994) Seminomas of the canine testis. Counterpart of spermatocytic seminoma of men? Lab Investig 71(4):490–496
Looijenga LH, Oosterhuis JW (2002) Pathobiology of testicular germ cell tumors: views and news. Anal Quant Cytol Histol 24(5):263–279
Looijenga LH, Rosenberg C, van Gurp RJ, Geelen E, van Echten-Arends J, de Jong B, Mostert M, Wolter Oosterhuis J (2000) Comparative genomic hybridization of microdissected samples from different stages in the development of a seminoma and a non-seminoma. J Pathol 191(2):187–192
Looijenga LH, Stoop H, de Leeuw HP, de Gouveia Brazao CA, Gillis AJ, van Roozendaal KE, van Zoelen EJ, Weber RF, Wolffenbuttel KP, van DH, Honecker F, Bokemeyer C, Perlman EJ, Schneider DT, Kononen J, Sauter G, Oosterhuis JW (2003b) POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res 63(9):2244–2250
Looijenga LH, Stoop H, Hersmus R, Gillis AJ, Wolter Oosterhuis J (2007) Genomic and expression profiling of human spermatocytic seminomas: pathogenetic implications. Int J Androl 30(4):328–335; discussion 335–326
Looijenga LH, Verkerk AJ, Dekker MC, van Gurp RJ, Gillis AJ, Oosterhuis JW (1998) Genomic imprinting in testicular germ cell tumours. APMIS 106(1):187–195
Looijenga LH, Zafarana G, Grygalewicz B, Summersgill B, Debiec-Rychter M, Veltman J, Schoenmakers EF, Rodriguez S, Jafer O, Clark J, van Kessel AG, Shipley J, van Gurp RJ, Gillis AJ, Oosterhuis JW (2003c) Role of gain of 12p in germ cell tumour development. APMIS 111(1):161–171; discussion 172–163
MacLean G, Li H, Metzger D, Chambon P, Petkovich M (2007) Apoptotic extinction of germ cells in testes of Cyp26b1 knockout mice. Endocrinology 148(10):4560–4567
Magnusdottir E, Dietmann S, Murakami K, Gunesdogan U, Tang F, Bao S, Diamanti E, Lao K, Gottgens B, Azim Surani M (2013) A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat Cell Biol 15(8):905–915
Mahakali Zama A, Hudson FP 3rd, Bedell MA (2005) Analysis of hypomorphic KitlSl mutants suggests different requirements for KITL in proliferation and migration of mouse primordial germ cells. Biol Reprod 73(4):639–647
Mai PL, Friedlander M, Tucker K, Phillips KA, Hogg D, Jewett MA, Lohynska R, Daugaard G, Richard S, Bonaiti-Pellie C, Heidenreich A, Albers P, Bodrogi I, Geczi L, Olah E, Daly PA, Guilford P, Fossa SD, Heimdal K, Liubchenko L, Tjulandin SA, Stoll H, Weber W, Easton DF, Dudakia D, Huddart R, Stratton MR, Einhorn L, Korde L, Nathanson KL, Bishop DT, Rapley EA, Greene MH (2010) The International Testicular Cancer Linkage Consortium: a clinicopathologic descriptive analysis of 461 familial malignant testicular germ cell tumor kindred. Urol Oncol 28(5):492–499
Manivel JC, Reinberg Y, Niehans GA, Fraley EE (1989) Intratubular germ cell neoplasia in testicular teratomas and epidermoid cysts. Correlation with prognosis and possible biologic significance. Cancer 64(3):715–720
Manivel JC, Simonton S, Wold LE, Dehner LP (1988) Absence of intratubular germ cell neoplasia in testicular yolk sac tumors in children. A histochemical and immunohistochemical study. Arch Pathol Lab Med 112(6):641–645
Martin GR (1981) Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A 78(12):7634–7638
Matin A, Collin GB, Asada Y, Varnum D, Nadeau JH (1999) Susceptibility to testicular germ-cell tumours in a 129.MOLF-Chr 19 chromosome substitution strain. Nat Genet 23(2):237–240
Matin A, Collin GB, Varnum DS, Nadeau JH (1998) Testicular teratocarcinogenesis in mice—a review. APMIS 106(1):174–182
Matsui Y, Zsebo K, Hogan BL (1992) Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell 70(5):841–847
Matsui Y, Zsebo KM, Hogan BL (1990) Embryonic expression of a haematopoietic growth factor encoded by the Sl locus and the ligand for c-kit. Nature 347(6294):667–669
Mayer F, Honecker F, Looijenga LH, Bokemeyer C (2003) Towards an understanding of the biological basis of response to cisplatin-based chemotherapy in germ-cell tumors. Ann Oncol 14(6):825–832
McIntyre A, Summersgill B, Grygalewicz B, Gillis AJ, Stoop J, van Gurp RJ, Dennis N, Fisher C, Huddart R, Cooper C, Clark J, Oosterhuis JW, Looijenga LH, Shipley J (2005) Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res 65(18):8085–8089
McLaren A (1984) Meiosis and differentiation of mouse germ cells. Symp Soc Exp Biol 38:7–23
McLaren A (1999) Signaling for germ cells. Genes Dev 13(4):373–376
McLaren A (2000) Germ and somatic cell lineages in the developing gonad. Mol Cell Endocrinol 163(1–2):3–9
McLaren A (2003) Primordial germ cells in the mouse. Dev Biol 262(1):1–15
McLaren A, Southee D (1997) Entry of mouse embryonic germ cells into meiosis. Dev Biol 187(1):107–113
Medrano JV, Ramathal C, Nguyen HN, Simon C, Reijo Pera RA (2012) Divergent RNA-binding proteins, DAZL and VASA, induce meiotic progression in human germ cells derived in vitro. Stem Cells 30(3):441–451
Mendis SH, Meachem SJ, Sarraj MA, Loveland KL (2011) Activin A balances Sertoli and germ cell proliferation in the fetal mouse testis. Biol Reprod 84(2):379–391
Menke DB, Koubova J, Page DC (2003) Sexual differentiation of germ cells in XX mouse gonads occurs in an anterior-to-posterior wave. Dev Biol 262(2):303–312
Menke DB, Page DC (2002) Sexually dimorphic gene expression in the developing mouse gonad. Gene Expr Patterns 2(3–4):359–367
Mesnard D, Guzman-Ayala M, Constam DB (2006) Nodal specifies embryonic visceral endoderm and sustains pluripotent cells in the epiblast before overt axial patterning. Development 133(13):2497–2505
Miles DC, van den Bergen JA, Wakeling SI, Anderson RB, Sinclair AH, Western PS (2012) The proto-oncogene Ret is required for male foetal germ cell survival. Dev Biol 365(1):101–109
Miles DC, Wakeling SI, Stringer JM, van den Bergen JA, Wilhelm D, Sinclair AH, Western PS (2013) Signaling through the TGF beta-activin receptors ALK4/5/7 regulates testis formation and male germ cell development. PLoS One 8(1):e54606
Mintz B, Russell ES (1957) Gene-induced embryological modifications of primordial germ cells in the mouse. J Exp Zool 134(2):207–237
Moniot B, Ujjan S, Champagne J, Hirai H, Aritake K, Nagata K, Dubois E, Nidelet S, Nakamura M, Urade Y, Poulat F, Boizet-Bonhoure B (2014) Prostaglandin D2 acts through the Dp2 receptor to influence male germ cell differentiation in the foetal mouse testis. Development 141(18):3561–3571
Moreno SG, Attali M, Allemand I, Messiaen S, Fouchet P, Coffigny H, Romeo PH, Habert R (2010) TGFbeta signaling in male germ cells regulates gonocyte quiescence and fertility in mice. Dev Biol 342(1):74–84
Moshfegh C, Aires L, Kisielow M, Vogel V (2016) A gonogenic stimulated transition of mouse embryonic stem cells with enhanced control of diverse differentiation pathways. Sci Rep 6:25104
Mostert M, Rosenberg C, Stoop H, Schuyer M, Timmer A, Oosterhuis W, Looijenga L (2000) Comparative genomic and in situ hybridization of germ cell tumors of the infantile testis. Lab Investig 80(7):1055–1064
Mostofi FK, Sesterhenn IA, Davis CJ Jr (1988) Developments in histopathology of testicular germ cell tumors. Semin Urol 6(3):171–188
Muller AJ, Teresky AK, Levine AJ (2000) A male germ cell tumor-susceptibility-determining locus, pgct1, identified on murine chromosome 13. Proc Natl Acad Sci U S A 97(15):8421–8426
Muller J, Skakkebaek NE, Ritzen M, Ploen L, Petersen KE (1985) Carcinoma in situ of the testis in children with 45,X/46,XY gonadal dysgenesis. J Pediatr 106(3):431–436
Mummery CL, van Rooijen MA, van den Brink SE, de Laat SW (1987) Cell cycle analysis during retinoic acid induced differentiation of a human embryonal carcinoma-derived cell line. Cell Differ 20(2–3):153–160
Munsterberg A, Lovell-Badge R (1991) Expression of the mouse anti-mullerian hormone gene suggests a role in both male and female sexual differentiation. Development 113(2):613–624
Nathanson KL, Kanetsky PA, Hawes R, Vaughn DJ, Letrero R, Tucker K, Friedlander M, Phillips KA, Hogg D, Jewett MA, Lohynska R, Daugaard G, Richard S, Chompret A, Bonaiti-Pellie C, Heidenreich A, Olah E, Geczi L, Bodrogi I, Ormiston WJ, Daly PA, Oosterhuis JW, Gillis AJ, Looijenga LH, Guilford P, Fossa SD, Heimdal K, Tjulandin SA, Liubchenko L, Stoll H, Weber W, Rudd M, Huddart R, Crockford GP, Forman D, Oliver DT, Einhorn L, Weber BL, Kramer J, McMaster M, Greene MH, Pike M, Cortessis V, Chen C, Schwartz SM, Bishop DT, Easton DF, Stratton MR, Rapley EA (2005) The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am J Hum Genet 77(6):1034–1043
Nelson VR, Heaney JD, Tesar PJ, Davidson NO, Nadeau JH (2012) Transgenerational epigenetic effects of the Apobec1 cytidine deaminase deficiency on testicular germ cell tumor susceptibility and embryonic viability. Proc Natl Acad Sci U S A 109(41):E2766–E2773
Nicholls PK, Harrison CA, Gilchrist RB, Farnworth PG, Stanton PG (2009) Growth differentiation factor 9 is a germ cell regulator of Sertoli cell function. Endocrinology 150(5):2481–2490
Nitzsche B, Gloesenkamp C, Schrader M, Hoffmann B, Zengerling F, Balabanov S, Honecker F, Hopfner M (2012) Anti-tumour activity of two novel compounds in cisplatin-resistant testicular germ cell cancer. Br J Cancer 107(11):1853–1863
Noel EE, Yeste-Velasco M, Mao X, Perry J, Kudahetti SC, Li NF, Sharp S, Chaplin T, Xue L, McIntyre A, Shan L, Powles T, Oliver RT, Young BD, Shipley J, Berney DM, Joel SP, Lu YJ (2010) The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am J Pathol 176(6):2607–2615
Noguchi M, Watanabe C, Kobayashi T, Kuwashima M, Sakurai T, Katoh H, Moriwaki K (1996) The ter mutation responsible for germ cell deficiency nor ovarian teratocarcinogenesis in ter/ter congenic mice. Develop Growth Differ 38:59–69
Noguchi T, Stevens LC (1982) Primordial germ cell proliferation in fetal testes in mouse strains with high and low incidences of congenital testicular teratomas. J Natl Cancer Inst 69(4):907–913
Wai-Sum O, Baker TG (1976) Initiation and control of meiosis in hamster gonads in vitro. J Reprod Fertil 48(2):399–401
Ohinata Y, Ohta H, Shigeta M, Yamanaka K, Wakayama T, Saitou M (2009) A signaling principle for the specification of the germ cell lineage in mice. Cell 137(3):571–584
Ohinata Y, Payer B, O'Carroll D, Ancelin K, Ono Y, Sano M, Barton SC, Obukhanych T, Nussenzweig M, Tarakhovsky A, Saitou M, Surani MA (2005) Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 436(7048):207–213
Okashita N, Kumaki Y, Ebi K, Nishi M, Okamoto Y, Nakayama M, Hashimoto S, Nakamura T, Sugasawa K, Kojima N, Takada T, Okano M, Seki Y (2014) PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development 141(2):269–280
Oldenburg J, Kraggerud SM, Brydoy M, Cvancarova M, Lothe RA, Fossa SD (2007a) Association between long-term neuro-toxicities in testicular cancer survivors and polymorphisms in glutathione-s-transferase-P1 and -M1, a retrospective cross sectional study. J Transl Med 5:70
Oldenburg J, Kraggerud SM, Cvancarova M, Lothe RA, Fossa SD (2007b) Cisplatin-induced long-term hearing impairment is associated with specific glutathione s-transferase genotypes in testicular cancer survivors. J Clin Oncol 25(6):708–714
Oosterhuis JW, Castedo SM, de JB, Cornelisse CJ, Dam A, Sleijfer DT, Schraffordt KH (1989) Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab Investig 60(1):14–21
Oosterhuis JW, Looijenga LH (2005) Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer 5(3):210–222
Oosterhuis JW, Looijenga LH, van EJ, de JB (1997) Chromosomal constitution and developmental potential of human germ cell tumors and teratomas. Cancer Genet Cytogenet 95(1):96–102
Oshimori N, Fuchs E (2012) The harmonies played by TGF-beta in stem cell biology. Cell Stem Cell 11(6):751–764
Ottesen AM, Kirchhoff M, Rajpert De-Meyts E, Maahr J, Gerdes T, Rose H, Lundsteen C, Petersen PM, Philip J, Skakkebaek NE (1997) Detection of chromosomal aberrations in seminomatous germ cell tumours using comparative genomic hybridization. Genes Chromosomes Cancer 20(4):412–418
Ottesen AM, Skakkebaek NE, Lundsteen C, Leffers H, Larsen J, Rajpert-De Meyts E (2003) High-resolution comparative genomic hybridization detects extra chromosome arm 12p material in most cases of carcinoma in situ adjacent to overt germ cell tumors, but not before the invasive tumor development. Genes Chromosomes Cancer 38(2):117–125
Palumbo C, van RK, Gillis AJ, van Gurp RH, de MH, Oosterhuis JW, van Zoelen EJ, Looijenga LH (2002) Expression of the PDGF alpha-receptor 1.5 kb transcript, OCT-4, and c-KIT in human normal and malignant tissues. Implications for the early diagnosis of testicular germ cell tumours and for our understanding of regulatory mechanisms. J Pathol 196(4):467–477
Park SY, Jameson JL (2005) Minireview: transcriptional regulation of gonadal development and differentiation. Endocrinology 146(3):1035–1042
Pauklin S, Vallier L (2015) Activin/Nodal signalling in stem cells. Development 142(4):607–619
Payer B, Saitou M, Barton SC, Thresher R, Dixon JP, Zahn D, Colledge WH, Carlton MB, Nakano T, Surani MA (2003) Stella is a maternal effect gene required for normal early development in mice. Curr Biol 13(23):2110–2117
Pesce M, Scholer HR (2000) Oct-4: control of totipotency and germline determination. Mol Reprod Dev 55(4):452–457
Pesce M, Wang X, Wolgemuth DJ, Scholer H (1998) Differential expression of the Oct-4 transcription factor during mouse germ cell differentiation. Mech Dev 71(1–2):89–98
Peters U, Preisler-Adams S, Hebeisen A, Hahn M, Seifert E, Lanvers C, Heinecke A, Horst J, Jurgens H, Lamprecht-Dinnesen A (2000) Glutathione S-transferase genetic polymorphisms and individual sensitivity to the ototoxic effect of cisplatin. Anti-Cancer Drugs 11(8):639–643
Pierce GB, Stevens LC, Nakane PK (1967) Ultrastructural analysis of the early development of teratocarcinomas. J Natl Cancer Inst 39(4):755–773
Pleskacova J, Hersmus R, Oosterhuis JW, Setyawati BA, Faradz SM, Cools M, Wolffenbuttel KP, Lebl J, Drop SL, Looijenga LH (2010) Tumor risk in disorders of sex development. Sex Dev 4(4–5):259–269
Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W (2010) Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463(7284):1101–1105
Poynter JN, Hooten AJ, Frazier AL, Ross JA (2012) Associations between variants in KITLG, SPRY4, BAK1, and DMRT1 and pediatric germ cell tumors. Genes Chromosomes Cancer 51(3):266–271
Rajpert-De Meyts E (2006) Developmental model for the pathogenesis of testicular carcinoma in situ: genetic and environmental aspects. Hum Reprod Update 12(3):303–323
Rajpert-De Meyts E, Hanstein R, Jorgensen N, Graem N, Vogt PH, Skakkebaek NE (2004) Developmental expression of POU5F1 (OCT-3/4) in normal and dysgenetic human gonads. Hum Reprod 19(6):1338–1344
Rajpert-de Meyts E, Hoei-Hansen CE (2007) From gonocytes to testicular cancer: the role of impaired gonadal development. Ann N Y Acad Sci 1120:168–180
Rajpert-De Meyts E, Skakkebaek NE (1994) Expression of the c-kit protein product in carcinoma-in-situ and invasive testicular germ cell tumours. Int J Androl 17(2):85–92
Rapley EA, Crockford GP, Teare D, Biggs P, Seal S, Barfoot R, Edwards S, Hamoudi R, Heimdal K, Fossa SD, Tucker K, Donald J, Collins F, Friedlander M, Hogg D, Goss P, Heidenreich A, Ormiston W, Daly PA, Forman D, Oliver TD, Leahy M, Huddart R, Cooper CS, Bodmer JG, Easton DF, Stratton MR, Bishop DT (2000) Localization to Xq27 of a susceptibility gene for testicular germ-cell tumours. Nat Genet 24(2):197–200
Rapley EA, Nathanson KL (2010) Predisposition alleles for testicular germ cell tumour. Curr Opin Genet Dev 20(3):225–230
Rapley EA, Turnbull C, Al Olama AA, Dermitzakis ET, Linger R, Huddart RA, Renwick A, Hughes D, Hines S, Seal S, Morrison J, Nsengimana J, Deloukas P, Rahman N, Bishop DT, Easton DF, Stratton MR (2009) A genome-wide association study of testicular germ cell tumor. Nat Genet 41(7):807–810
Rodriguez E, Houldsworth J, Reuter VE, Meltzer P, Zhang J, Trent JM, Bosl GJ, Chaganti RS (1993) Molecular cytogenetic analysis of i(12p)-negative human male germ cell tumors. Genes Chromosomes Cancer 8(4):230–236
Rodriguez E, Mathew S, Reuter V, Ilson DH, Bosl GJ, Chaganti RS (1992) Cytogenetic analysis of 124 prospectively ascertained male germ cell tumors. Cancer Res 52(8):2285–2291
Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capella G, Ribas M, Peinado MA (2006) Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res 66(17):8462–9468
Ross JA, Schmidt PT, Perentesis JP, Davies SM (1999) Genomic imprinting of H19 and insulin-like growth factor-2 in pediatric germ cell tumors. Cancer 85(6):1389–1394
Rossant J, Papaioannou VE (1984) The relationship between embryonic, embryonal carcinoma and embryo-derived stem cells. Cell Differ 15(2–4):155–161
Ruark E, Seal S, McDonald H, Zhang F, Elliot A, Lau K, Perdeaux E, Rapley E, Eeles R, Peto J, Kote-Jarai Z, Muir K, Nsengimana J, Shipley J, Bishop DT, Stratton MR, Easton DF, Huddart RA, Rahman N, Turnbull C (2013) Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet 45(6):686–689
Saba R, Kato Y, Saga Y (2014a) NANOS2 promotes male germ cell development independent of meiosis suppression. Dev Biol 385(1):32–40
Saba R, Wu Q, Saga Y (2014b) CYP26B1 promotes male germ cell differentiation by suppressing STRA8-dependent meiotic and STRA8-independent mitotic pathways. Dev Biol 389(2):173–181
Saitou M (2009) Germ cell specification in mice. Curr Opin Genet Dev 19(4):386–395
Saitou M, Barton SC, Surani MA (2002) A molecular programme for the specification of germ cell fate in mice. Nature 418(6895):293–300
Saitou M, Kagiwada S, Kurimoto K (2012) Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development 139(1):15–31
Saitou M, Miyauchi H (2016) Gametogenesis from Pluripotent Stem Cells. Cell Stem Cell 18(6):721–735
Saitou M, Payer B, Lange UC, Erhardt S, Barton SC, Surani MA (2003) Specification of germ cell fate in mice. Philos Trans R Soc Lond Ser B Biol Sci 358(1436):1363–1370
Saitou M, Yabuta Y, Kurimoto K (2008) Single-cell cDNA high-density oligonucleotide microarray analysis: detection of individual cell types and properties in complex biological processes. Reprod Biomed Online 16(1):26–40
Saitou M, Yamaji M (2010) Germ cell specification in mice: signaling, transcription regulation, and epigenetic consequences. Reproduction 139(6):931–942
Sasaki H, Matsui Y (2008) Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet 9(2):129–140
Schemmer J, Arauzo-Bravo MJ, Haas N, Schafer S, Weber SN, Becker A, Eckert D, Zimmer A, Nettersheim D, Schorle H (2013) Transcription factor TFAP2C regulates major programs required for murine fetal germ cell maintenance and haploinsufficiency predisposes to teratomas in male mice. PLoS One 8(8):e71113
Schmahl J, Capel B (2003) Cell proliferation is necessary for the determination of male fate in the gonad. Dev Biol 258(2):264–276
Schmahl J, Eicher EM, Washburn LL, Capel B (2000) Sry induces cell proliferation in the mouse gonad. Development 127(1):65–73
Schmahl J, Kim Y, Colvin JS, Ornitz DM, Capel B (2004) Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 131(15):3627–3636
Schneider DT, Schuster AE, Fritsch MK, Hu J, Olson T, Lauer S, Gobel U, Perlman EJ (2001) Multipoint imprinting analysis indicates a common precursor cell for gonadal and nongonadal pediatric germ cell tumors. Cancer Res 61(19):7268–7276
Scholer HR (1991) Octamania: the POU factors in murine development. Trends Genet 7(10):323–329
Seki Y, Hayashi K, Itoh K, Mizugaki M, Saitou M, Matsui Y (2005) Extensive and orderly reprogramming of genome-wide chromatin modifications associated with specification and early development of germ cells in mice. Dev Biol 278(2):440–458
Seki Y, Yamaji M, Yabuta Y, Sano M, Shigeta M, Matsui Y, Saga Y, Tachibana M, Shinkai Y, Saitou M (2007) Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development 134(14):2627–2638
Sekido R, Lovell-Badge R (2008) Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453(7197):930–934
Shoji M, Tanaka T, Hosokawa M, Reuter M, Stark A, Kato Y, Kondoh G, Okawa K, Chujo T, Suzuki T, Hata K, Martin SL, Noce T, Kuramochi-Miyagawa S, Nakano T, Sasaki H, Pillai RS, Nakatsuji N, Chuma S (2009) The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev Cell 17(6):775–787
Shovlin TC, Bourc'his D, La Salle S, O'Doherty A, Trasler JM, Bestor TH, Walsh CP (2007) Sex-specific promoters regulate Dnmt3L expression in mouse germ cells. Hum Reprod 22(2):457–467
Silver SA, Wiley JM, Perlman EJ (1994) DNA ploidy analysis of pediatric germ cell tumors. Mod Pathol 7(9):951–956
Singh AM, Dalton S (2009) The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell 5(2):141–149
Singhera M, Lees K, Huddart R, Horwich A (2012) Minimizing toxicity in early-stage testicular cancer treatment. Expert Rev Anticancer Ther 12(2):185–193
Skakkebaek NE (1978) Carcinoma in situ of the testis: frequency and relationship to invasive germ cell tumours in infertile men. Histopathology 2(3):157–170
Skakkebaek NE, Rajpert-de ME, Main KM (2001) Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum Reprod 16(5):972–978
Skotheim RI, Monni O, Mousses S, Fossa SD, Kallioniemi OP, Lothe RA, Kallioniemi A (2002) New insights into testicular germ cell tumorigenesis from gene expression profiling. Cancer Res 62(8):2359–2364
Smiraglia DJ, Szymanska J, Kraggerud SM, Lothe RA, Peltomaki P, Plass C (2002) Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene 21(24):3909–3916
Soosay GN, Bobrow L, Happerfield L, Parkinson MC (1991) Morphology and immunohistochemistry of carcinoma in situ adjacent to testicular germ cell tumours in adults and children: implications for histogenesis. Histopathology 19(6):537–544
Souquet B, Tourpin S, Messiaen S, Moison D, Habert R, Livera G (2012) Nodal signaling regulates the entry into meiosis in fetal germ cells. Endocrinology 153(5):2466–2473
Spiller CM, Bowles J, Koopman P (2013) Nodal/Cripto signaling in fetal male germ cell development: implications for testicular germ cell tumors. Int J Dev Biol 57(2–4):211–219
Spiller CM, Feng CW, Jackson A, Gillis AJ, Rolland AD, Looijenga LH, Koopman P, Bowles J (2012) Endogenous Nodal signaling regulates germ cell potency during mammalian testis development. Development 139(22):4123–4132
Spiller CM, Wilhelm D, Koopman P (2010) Retinoblastoma 1 protein modulates XY germ cell entry into G1/G0 arrest during fetal development in mice. Biol Reprod 82(2):433–443
Stamp IM, Barlebo H, Rix M, Jacobsen GK (1993) Intratubular germ cell neoplasia in an infantile testis with immature teratoma. Histopathology 22(1):69–72
Stamp IM, Jacobsen GK (1995) Infant intratubular germ cell neoplasia. Am J Surg Pathol 19(4):489
Stevens L (1962) Testicular teratomas in fetal mice. J Natl Cancer Inst 28:247–267
Stevens L (1964) Experimental produciton of testicular teratomas in mice. Proc Natl Acad Sci U S A 52:654–661
Stevens L (1966) Development of resistance to teratocarcinogenesis by primordial germ cells in mice. J Natl Cancer Inst 37(6):859–867
Stevens L (1967a) The biology of teratomas. Adv Morphog 6:1–31
Stevens L (1967b) Origin of testicular teratomas from primordial germ cells in mice. J Natl Cancer Inst 38(4):549–552
Stevens L (1973) A new inbred subline of mice (129-terSv) with a high incidence of spontaneous congenital testicular teratomas. J Natl Cancer Inst 50(1):235–242
Stevens L, Bunker M (1964) Karyotpye and sex of primary testicualr teratomas in mice. J Natl Cancer Inst 33:65–78
Stevens L, Hummel K (1957) A description of spontaneous congenital testicular teratomas in strain 129 mice. J Natl Cancer Inst 18(5):719–747
Stevens L, Mackensen JA (1961) Genetic and environmental influences on teratocarcinogenesis in mice. J Natl Cancer Inst 27:443–453
Stevens LC (1958) Studies on transplantable testicular teratomas of strain 129 mice. J Natl Cancer Inst 20(6):1257–1275
Stevens LC (1981) Genetic influences on teratocarcinogenesis and parthenogenesis. Prog Clin Biol Res 45:93–104
Stock C, Ambros IM, Lion T, Haas OA, Zoubek A, Gadner H, Ambros PF (1994) Detection of numerical and structural chromosome abnormalities in pediatric germ cell tumors by means of interphase cytogenetics. Genes Chromosomes Cancer 11(1):40–50
Summersgill B, Goker H, Weber-Hall S, Huddart R, Horwich A, Shipley J (1998) Molecular cytogenetic analysis of adult testicular germ cell tumours and identification of regions of consensus copy number change. Br J Cancer 77(2):305–313
Summersgill B, Osin P, Lu YJ, Huddart R, Shipley J (2001) Chromosomal imbalances associated with carcinoma in situ and associated testicular germ cell tumours of adolescents and adults. Br J Cancer 85(2):213–220
Surani MA (2001) Reprogramming of genome function through epigenetic inheritance. Nature 414(6859):122–128
Surani MA, Hayashi K, Hajkova P (2007) Genetic and epigenetic regulators of pluripotency. Cell 128(4):747–762
Sutton KA (2000) Molecular mechanisms involved in the differentiation of spermatogenic stem cells. Rev Reprod 5(2):93–98
Suzuki A, Saga Y (2008) Nanos2 suppresses meiosis and promotes male germ cell differentiation. Genes Dev 22(4):430–435
Suzuki A, Tsuda M, Saga Y (2007) Functional redundancy among Nanos proteins and a distinct role of Nanos2 during male germ cell development. Development 134(1):77–83
Tam PP, Snow MH (1981) Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. J Embryol Exp Morphol 64:133–147
Taylor-Weiner A, Zack T, O'Donnell E, Guerriero JL, Bernard B, Reddy A, Han GC, AlDubayan S, Amin-Mansour A, Schumacher SE, Litchfield K, Turnbull C, Gabriel S, Beroukhim R, Getz G, Carter SL, Hirsch MS, Letai A, Sweeney C, Van Allen EM (2016) Genomic evolution and chemoresistance in germ-cell tumours. Nature 540(7631):114–118
Trautmann E, Guerquin MJ, Duquenne C, Lahaye JB, Habert R, Livera G (2008) Retinoic acid prevents germ cell mitotic arrest in mouse fetal testes. Cell Cycle 7(5):656–664
Tsuda M, Sasaoka Y, Kiso M, Abe K, Haraguchi S, Kobayashi S, Saga Y (2003) Conserved role of nanos proteins in germ cell development. Science 301(5637):1239–1241
Tsuneyoshi N, Sumi T, Onda H, Nojima H, Nakatsuji N, Suemori H (2008) PRDM14 suppresses expression of differentiation marker genes in human embryonic stem cells. Biochem Biophys Res Commun 367(4):899–905
Turnbull C, Rapley EA, Seal S, Pernet D, Renwick A, Hughes D, Ricketts M, Linger R, Nsengimana J, Deloukas P, Huddart RA, Bishop DT, Easton DF, Stratton MR, Rahman N (2010) Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat Genet 42(7):604–607
Ulbright TM, Amin MB, Balzer B, Berney DM, Epstein JI, Guo C, Idrees MT, Looijenga L, Paner G, Rajpert De-Meyts E, Skakkebaek NE, Tickoo SK, Yilmaz A, Oosterhuis JW (2016) Tumours of the testis and paratesticular tissue. In: Moch H, Humphrey PA, Ulbright TM, Reuter VE (eds) WHO classification of tumours of the urinary system and male genital organs, vol 8. International Agency for Research on Cancer, pp 185–227
Vallier L, Mendjan S, Brown S, Chng Z, Teo A, Smithers LE, Trotter MW, Cho CH, Martinez A, Rugg-Gunn P, Brons G, Pedersen RA (2009) Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 136(8):1339–1349
van Echten J (1995) Cytogenetic evidence that carcinoma in situ is the precursor lesion for invasive testicular germ cell tumors. Cancer Genet Cytogenet 85(2):133–137
van Gurp RJ, Oosterhuis JW, Kalscheuer V, Mariman EC, Looijenga LH (1994) Biallelic expression of the H19 and IGF2 genes in human testicular germ cell tumors. J Natl Cancer Inst 86(14):1070–1075
Vernet N, Dennefeld C, Rochette-Egly C, Oulad-Abdelghani M, Chambon P, Ghyselinck NB, Mark M (2006) Retinoic acid metabolism and signaling pathways in the adult and developing mouse testis. Endocrinology 147(1):96–110
Vigier B, Watrin F, Magre S, Tran D, Josso N (1987) Purified bovine AMH induces a characteristic freemartin effect in fetal rat prospective ovaries exposed to it in vitro. Development 100(1):43–55
Vincent SD, Dunn NR, Sciammas R, Shapiro-Shalef M, Davis MM, Calame K, Bikoff EK, Robertson EJ (2005) The zinc finger transcriptional repressor Blimp1/Prdm1 is dispensable for early axis formation but is required for specification of primordial germ cells in the mouse. Development 132(6):1315–1325
von Eyben FE (2004) Chromosomes, genes, and development of testicular germ cell tumors. Cancer Genet Cytogenet 151(2):93–138
Vos A, Oosterhuis JW, de Jong B, Buist J, Schraffordt KH (1990) Cytogenetics of carcinoma in situ of the testis. Cancer Genet Cytogenet 46(1):75–81
Walt H, Oosterhuis JW, STEVENS LC (1993) Experimental testicular germ cell tumorigenesis in mouse strains with and without spontaneous tumours differs from development of germ cell tumours of the adult human testis. Int J Androl 16(4):267–271
Weber S, Eckert D, Nettersheim D, Gillis AJ, Schafer S, Kuckenberg P, Ehlermann J, Werling U, Biermann K, Looijenga LH, Schorle H (2010) Critical function of AP-2 gamma/TCFAP2C in mouse embryonic germ cell maintenance. Biol Reprod 82(1):214–223
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42:D1001–D1006
Werling U, Schorle H (2002) Transcription factor gene AP-2 gamma essential for early murine development. Mol Cell Biol 22(9):3149–3156
Western P (2009) Foetal germ cells: striking the balance between pluripotency and differentiation. Int J Dev Biol 53(2–3):393–409
Western PS, Miles DC, van den Bergen JA, Burton M, Sinclair AH (2008) Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells 26(2):339–347
Western PS, Ralli RA, Wakeling SI, Lo C, van den Bergen JA, Miles DC, Sinclair AH (2011) Mitotic arrest in teratoma susceptible fetal male germ cells. PLoS One 6(6):e20736
Western PS, van den Bergen JA, Miles DC, Sinclair AH (2010) Male fetal germ cell differentiation involves complex repression of the regulatory network controlling pluripotency. FASEB J 24(8):3026–3035
Wu Q, Fukuda K, Weinstein M, Graff JM, Saga Y (2015) SMAD2 and p38 signaling pathways act in concert to determine XY primordial germ cell fate in mice. Development 142(3):575–586
Wu Q, Kanata K, Saba R, Deng CX, Hamada H, Saga Y (2013) Nodal/activin signaling promotes male germ cell fate and suppresses female programming in somatic cells. Development 140(2):291–300
Yamaguchi S, Kimura H, Tada M, Nakatsuji N, Tada T (2005) Nanog expression in mouse germ cell development. Gene Expr Patterns 5(5):639–646
Yamaji M, Seki Y, Kurimoto K, Yabuta Y, Yuasa M, Shigeta M, Yamanaka K, Ohinata Y, Saitou M (2008) Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet 40(8):1016–1022
Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, Nakato R, Yamada Y, Shirahige K, Saitou M (2013) PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell 12(3):368–382
Yan W, Samson M, Jegou B, Toppari J (2000) Bcl-w forms complexes with Bax and Bak, and elevated ratios of Bax/Bcl-w and Bak/Bcl-w correspond to spermatogonial and spermatocyte apoptosis in the testis. Mol Endocrinol 14(5):682–699
Yao HH, DiNapoli L, Capel B (2003) Meiotic germ cells antagonize mesonephric cell migration and testis cord formation in mouse gonads. Development 130(24):5895–5902
Yeom YI, Fuhrmann G, Ovitt CE, Brehm A, Ohbo K, Gross M, Hubner K, Scholer HR (1996) Germline regulatory element of Oct-4 specific for the totipotent cycle of embryonal cells. Development 122(3):881–894
Yi SE, LaPolt PS, Yoon BS, Chen JY, Lu JK, Lyons KM (2001) The type I BMP receptor BmprIB is essential for female reproductive function. Proc Natl Acad Sci U S A 98(14):7994–7999
Youngren KK, Coveney D, Peng X, Bhattacharya C, Schmidt LS, Nickerson ML, Lamb BT, Deng JM, Behringer RR, Capel B, Rubin EM, Nadeau JH, Matin A (2005) The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature 435(7040):360–364
Yu Z, Ji P, Cao J, Zhu S, Li Y, Zheng L, Chen X, Feng L (2009) Dazl promotes germ cell differentiation from embryonic stem cells. J Mol Cell Biol 1(2):93–103
Zhou X, Zhang Z, Yang X, Chen W, Zhang P (2009) Inhibition of cyclin D1 expression by cyclin D1 shRNAs in human oral squamous cell carcinoma cells is associated with increased cisplatin chemosensitivity. Int J Cancer 124(2):483–489
Zhu R, Heaney J, Nadeau JH, Ali S, Matin A (2010) Deficiency of splicing factor 1 suppresses the occurrence of testicular germ cell tumors. Cancer Res 70(18):7264–7272
Zhu R, Matin A (2014) Tumor loci and their interactions on mouse chromosome 19 that contribute to testicular germ cell tumors. BMC Genet 15:65
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this chapter

Cite this chapter
Lanza, D.G., Heaney, J.D. (2017). Testicular Germ Cell Tumors and Teratomas. In: Oatley, J., Griswold, M. (eds) The Biology of Mammalian Spermatogonia. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7505-1_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7505-1_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-7503-7
Online ISBN: 978-1-4939-7505-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)