
Abstract
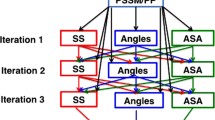
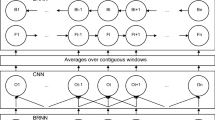
Predicting one-dimensional structure properties has played an important role to improve prediction of protein three-dimensional structures and functions. The most commonly predicted properties are secondary structure and accessible surface area (ASA) representing local and nonlocal structural characteristics, respectively. Secondary structure prediction is further complemented by prediction of continuous main-chain torsional angles. Here we describe a newly developed method SPIDER2 that utilizes three iterations of deep learning neural networks to improve the prediction accuracy of several structural properties simultaneously. For an independent test set of 1199 proteins SPIDER2 achieves 82 % accuracy for secondary structure prediction, 0.76 for the correlation coefficient between predicted and actual solvent accessible surface area, 19° and 30° for mean absolute errors of backbone φ and ψ angles, respectively, and 8° and 32° for mean absolute errors of Cα-based θ and τ angles, respectively. The method provides state-of-the-art, all-in-one accurate prediction of local structure and solvent accessible surface area. The method is implemented, as a webserver along with a standalone package that are available in our website: http://sparks-lab.org.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others

References
Lou W, Wang X, Chen F, Chen Y, Jiang B, Zhang H (2014) Sequence based prediction of DNA-binding proteins based on hybrid feature selection using random forest and Gaussian naïve Bayes. PLoS One 9(1)
Zhao H, Yang Y, Zhou Y (2011) Highly accurate and high-resolution function prediction of RNA binding proteins by fold recognition and binding affinity prediction. RNA Biol 8(6):988–996. doi:10.4161/rna.8.6.17813
Zhao H, Yang Y, von Itzstein M, Zhou Y (2014) Carbohydrate-binding protein identification by coupling structural similarity searching with binding affinity prediction. J Comput Chem 35(30):2177–2183
Zhao H, Wang J, Zhou Y, Yang Y (2014) Predicting DNA-binding proteins and binding residues by complex structure prediction and application to human proteome. PLoS One 9(5):e96694
Zhang T, Zhang H, Chen K, Ruan J, Shen S, Kurgan L (2010) Analysis and prediction of RNA-binding residues using sequence, evolutionary conservation, and predicted secondary structure and solvent accessibility. Curr Protein Peptide Sci 11(7):609–628
Zhang Z, Li Y, Lin B, Schroeder M, Huang B (2011) Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 27(15):2083–2088
Bradford JR, Westhead DR (2005) Improved prediction of protein–protein binding sites using a support vector machines approach. Bioinformatics 21(8):1487–1494
Folkman L, Yang Y, Li Z, Stantic B, Sattar A, Mort M, Cooper DN, Liu Y, Zhou Y (2015) DDIG-in: detecting disease-causing genetic variations due to frameshifting indels and nonsense mutations employing sequence and structural properties at nucleotide and protein levels. Bioinformatics 31(10):1599–1606
Zheng W, Zhang C, Hanlon M, Ruan J, Gao J (2014) An ensemble method for prediction of conformational B-cell epitopes from antigen sequences. Comput Biol Chem 49:51–58
Zhao H, Yang Y, Lin H, Zhang X, Mort M, Cooper DN, Liu Y, Zhou Y (2013) DDIG-in: discriminating between disease-associated and neutral non-frameshifting micro-indels, Genome Biology, 14, R43
Lyons J, Dehzangi A, Heffernan R, Yang Y, Zhou Y, Sharma A, Paliwal K (2015) Advancing the accuracy of protein fold recognition by utilizing profiles from Hidden Markov models, IEEE Transactions on NanoBioscience, 14, 761–772
Faraggi E, Yang Y, Zhang S, Zhou Y (2009) Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction. Structure 17(11):1515–1527. doi:10.1016/j.str.2009.09.006
Bradley P, Chivian D, Meiler J, Misura KM, Rohl CA, Schief WR, Wedemeyer WJ, Schueler-Furman O, Murphy P, Schonbrun J, Strauss CE, Baker D (2003) Rosetta predictions in CASP5: successes, failures, and prospects for complete automation. Proteins 53(Suppl 6):457–468. doi:10.1002/prot.10552
Handl J, Knowles J, Vernon R, Baker D, Lovell SC (2012) The dual role of fragments in fragment-assembly methods for de novo protein structure prediction. Proteins 80(2):490–504
Yang Y, Faraggi E, Zhao H, Zhou Y (2011) Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 27(15):2076–2082. doi:10.1093/bioinformatics/btr350
Zhang Y (2009) I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77(S9):100–113
Remmert M, Biegert A, Hauser A, Söding J (2011) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Meth 9(2):173–175
Cheng J, Wang Z, Tegge AN, Eickholt J (2009) Prediction of global and local quality of CASP8 models by MULTICOM series. Proteins 77(S9):181–184
Faraggi E, Zhang T, Yang Y, Kurgan L, Zhou Y (2011) SPINE X: improving protein secondary structure prediction by multi-step learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J Comput Chem 33:259–263
Yaseen A, Li YH (2014) Context-based features enhance protein secondary structure prediction accuracy. J Chem Inf Model 54(3):992–1002. doi:10.1021/Ci400647u
Wu S, Zhang Y (2008) ANGLOR: a composite machine-learning algorithm for protein backbone torsion angle prediction. PLoS One 3(10):e3400
Lyons J, Dehzangi A, Heffernan R, Sharma A, Paliwal K, Sattar A, Zhou Y, Yang Y (2014) Predicting backbone Calpha angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network. J Comput Chem 35(28):2040–2046. doi:10.1002/jcc.23718
Rost B, Sander C (1994) Conservation and prediction of solvent accessibility in protein families. Proteins 20(3):216–226
Gilis D, Rooman M (1997) Predicting protein stability changes upon mutation using database-derived potentials: solvent accessibility determines the importance of local versus non-local interactions along the sequence. J Mol Biol 272(2):276–290
Tuncbag N, Gursoy A, Keskin O (2009) Identification of computational hot spots in protein interfaces: combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics 25(12):1513–1520
Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55(3):379–400
Holbrook SR, Muskal SM, Kim SH (1990) Predicting surface exposure of amino acids from protein sequence. Protein Eng 3(8):659–665
Pollastri G, Baldi P, Fariselli P, Casadio R (2002) Prediction of coordination number and relative solvent accessibility in proteins. Proteins 47(2):142–153
Dor O, Zhou Y (2007) Real-SPINE: an integrated system of neural networks for real-value prediction of protein structural properties. Proteins 68(1):76–81
Garg A, Kaur H, Raghava GP (2005) Real value prediction of solvent accessibility in proteins using multiple sequence alignment and secondary structure. Proteins 61(2):318–324. doi:10.1002/prot.20630
Yuan Z, Huang B (2004) Prediction of protein accessible surface areas by support vector regression. Proteins 57(3):558–564. doi:10.1002/prot.20234
Ahmad S, Gromiha MM, Sarai A (2003) Real value prediction of solvent accessibility from amino acid sequence. Proteins 50(4):629–635. doi:10.1002/prot.10328
Adamczak R, Porollo A, Meller J (2004) Accurate prediction of solvent accessibility using neural networks-based regression. Proteins 56(4):753–767. doi:10.1002/prot.20176
Heffernan R, Paliwal K, Lyons J, Dehzangi A, Sharma A, Wang J, Sattar A, Yang Y, Zhou Y (2015) Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci Rep 5:11476
Bengio Y, Lamblin P, Popovici D, Larochelle H (2007) Greedy layer-wise training of deep networks. Adv Neural Inform Process Syst 19:153
Hinton GE (2007) Learning multiple a layers of representation. Trends Cogn Sci 11(10):428–434. doi:10.1016/J.Tics.2007.09.004
Bengio Y (2009) Learning deep architectures for AI. Found Trends Mach Learn 2(1):1–127
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Acknowledgements
This work was supported in part by National Health and Medical Research Council (1059775) of Australia and Australian Research Council’s Linkage Infrastructure, Equipment and Facilities funding scheme (project number LE150100161), the Taishan Scholars Program of Shandong province of China, National Natural Science Foundation of China (61540025) to Y.Z. and National Natural Science Foundation of China (61271378) to Y.Y. and J.W. We also gratefully acknowledge the support of the Griffith University eResearch Services Team and the use of the High Performance Computing Cluster “Gowonda” to complete this research. This research/project has also been undertaken with the aid of the research cloud resources provided by the Queensland Cyber Infrastructure Foundation (QCIF).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media New York
About this protocol
Cite this protocol
Yang, Y. et al. (2017). SPIDER2: A Package to Predict Secondary Structure, Accessible Surface Area, and Main-Chain Torsional Angles by Deep Neural Networks. In: Zhou, Y., Kloczkowski, A., Faraggi, E., Yang, Y. (eds) Prediction of Protein Secondary Structure. Methods in Molecular Biology, vol 1484. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6406-2_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6406-2_6
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6404-8
Online ISBN: 978-1-4939-6406-2
eBook Packages: Springer Protocols