
Abstract
Osteoarthritis is a progressive disease of synovial joints. The ultimate consequence of the disease is the breakdown of cartilage and bone, resulting in joint pain, stiffness, and functional disability. Currently, there are no disease-modifying osteoarthritis drugs to treat the disease. Treatments for osteoarthritis focus on conservative measures to reduce pain and swelling, in combination with surgical stabilization of the injured joint if necessary. These interventions do not prevent the development of osteoarthritis. Based upon recent discoveries from our laboratory, we strongly believe that a cell membrane receptor tyrosine kinase, Discoidin Domain Receptor 2 (DDR2), may be an ideal therapeutic target for the development of disease-modifying osteoarthritis drugs. In this chapter, we discuss roles of DDR2 in a sequential chain of molecular events that lead to a normal joint becoming an osteoarthritic joint. If, as we suspect, DDR2 is one of the major contributors to progressive joint failure, then drugs that inhibit the kinase activity of DDR2 may be able to ameliorate osteoarthritis conditions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Feature of Osteoarthritis
Osteoarthritis (OA) is the most common form of arthritis [1] and has been recently defined as “a progressive disease of synovial joints that represents failed repair of joint damage that results from stresses that may be initiated by an abnormality in any of the synovial joint tissues.” This ultimately results in the breakdown of cartilage and bone, leading to symptoms of pain, stiffness, and functional disability [2]. Although the precise etiology of OA in most cases is unknown, it is the general consensus that OA is a multifactorial disorder involving both genetic and environmental components [1]. Genetic factors are mutations in genes that result in defects in articular cartilage matrix and chondrocyte metabolism. In the case of cartilage defects, age-dependent OA may occur as a result of even normal mechanical stresses on a defective joint. Environmental factors include obesity, overloading on joints, repetitive injury involving ligaments and menisci, loss of muscle strength, and joint malalignment. These conditions can result in abnormal mechanical stresses on a normal joint, which eventually leads to OA. It is obvious that the initiation of OA can involve multiple joint tissues with one, or a combination of, genetic and environmental factors. Thus, investigation of the detailed pathogenic mechanisms in all of these conditions remains a formidable challenge. However, regardless of the complexity of the etiology for OA, the ultimate consequence of articular cartilage breakdown in the development of OA follows a consistent pathological pattern [3–6].
Results from studies using animal models of OA and human OA tissues indicate that in the process of articular cartilage degeneration, the earliest indication of change is chondrocyte clustering and a general upregulation in synthetic activity of chondrocytes. For example, in our mouse models of OA, we found that the chondrocyte clusters and the overproduction of proteoglycans appeared in the uncalcified (superficial) layer of the articular cartilage prior to any overt pathologic sign of articular cartilage degeneration [7]. With regard to human OA cases, it is extremely difficult to obtain articular cartilage from joints at the earliest stage of articular cartilage degeneration. However, the chondrocyte clusters and the expansion of the pericellular matrix (enlarged lacunae) are seen in the remaining articular cartilage tissue of joints at late stages of OA [4]. Immediately following the upregulation in synthetic activity of chondrocytes, chondrocytes synthesize and release extracellular matrix (ECM )-degrading enzymes. The gradual loss of proteoglycans on the surface region of the articular cartilage follows; this is concurrent with type II collagen degradation. Cracks develop along the articular surface, producing the histological image termed fibrillation. At later stages of the degenerative process, fibrocartilage and osteophytes are formed. It is worth mentioning that two recent studies report an unexpected revelation that the disappearance of aggrecans in articular cartilage of knee joints in adult mice, due to the genetic inactivation of a transcription factor, sex-determining region Y box 9 (Sox9), or the conditional upregulated expression of bone morphogenetic protein 2 in the articular cartilage, does not initiate or accelerate the progression of cartilage degeneration [8, 9]. This completely opposes the prevailing notion that proteoglycans are one of the basic elements and are indispensible for the ability of articular cartilage to resist compressive pressure. One plausible explanation for this observation is that the loss of proteoglycans alone may not be sufficient to initiate or accelerate articular cartilage degeneration. Instead, the degradation of both proteoglycans and type II collagen may be required in the development of OA.
The similar pathological progressions of articular cartilage degeneration suggest that there may be multiple initiating pathways toward the common targets that contribute to articular cartilage degeneration. This eventually leads to OA. The similarities in the pathologic progression also indicate that studies of pathogenetic changes, even in cases of more rare forms of OA, are likely to provide significant information about disease mechanisms and therapeutic targets for treating common forms of the disease. Therefore, the identification of a sequence(s) of molecular events underlying the progression of articular cartilage degeneration may not only help us understand the disease better, but may also provide information for the design of new therapeutic strategies in the prevention and treatment of OA.
We also need to consider that pathogenetic mechanisms by which juvenile OA develops may be different from mechanisms that are responsible for development of OA in adults. It is conceivable that OA joints in juveniles result from immature (developing) joints. Under this condition, fully mature joints are never formed. However, OA joints in adults are the ultimate consequence of the degeneration of the mature articular cartilages of joints. In the remaining part of this chapter, we discuss the roles of DDR2 and other molecules in the development of OA in adults.
2 Matrix Metalloproteinase 13 in the Development of OA
Matrix metalloproteinase 13 (MMP-13 ) is one of the major matrix-degrading enzymes that are involved in the breakdown of articular cartilage, which eventually leads to OA. Proteoglycans and type II collagen are two major ECM components in articular cartilage. The degradation of these molecules inevitably leads to a normal joint becoming an OA joint; thus, it would be critical to determine which enzyme/enzymes are responsible for the degradation of proteoglycans and type II collagen. An ideal candidate would be an enzyme(s) that can degrade both components and should not be expressed in normal articular cartilage. Matrix metalloproteinase 13 (MMP-13) fits this profile exactly.
MMP-13, also known as collagenase-3 , was originally cloned from a cDNA library derived from a breast tumor in 1994 [10]. This enzyme can degrade both proteoglycans and type II collagen [11, 12]. Moreover, MMP13 degrades type II collagen more efficiently, in fact 10 times more, than MMP1. Expression of MMP13 is hardly detected in normal mature articular cartilage, but the activity and expression of this enzyme are increased in human OA cartilage and in joints of OA mouse models [13–16]. This is consistent with the observation that the constitutive expression of Mmp-13 in mouse cartilage results in OA-like changes in knee joints [17], and removal of this enzyme prevents articular cartilage erosion in a knee joint instability mouse model of OA [18]. The significant effects of MMP-13 in the development of OA are also highlighted by several other studies. A study by Verzijl et al. indicates that the half-life of type II collagen in humans is 117 years [19]. The long half-life of type II collagen indicates a slow turnover of the collagen in mature articular cartilages. This also suggests that it may not be needed for chondrocytes to continually produce type II collagen in mature articular cartilages. Thus, chondrocytes may possess limited potential for type II collagen replenishment once the collagen is degraded. Results from another study demonstrate that articular cartilage degradation is completely irreversible after induction of the MMP-mediated degradation of aggrecan and type II collagen, but is reversible in the presence of the aggrecanase-mediated aggrecan degradation [20]. If the nature of chondrocytes in mature articular cartilage limits their ability to produce type II collagen, and if the degradation of type II collagen is irreversible, then we must inhibit the activity and expression of MMP-13 in articular cartilage in order to intervene in the development of OA. In fact, numerous pharmacological companies have attempted to inhibit the activity of MMP-13 as a means of delaying the development of OA. However, the broad activity of MMP-13 precludes this enzyme to be a promising target for inhibitory drugs in the treatment of OA [21]. While this may be the case for inhibitors of enzymatic activity, this concern does not apply to molecules involved in the regulation of MMP-13 expression in articular chondrocytes .
Transcriptional analysis of MMP-13 shows that the human MMP-13 promoter contains a TATA box and binding sites for transcription factors such as AP-1 (e.g., c-Fos/c-Jun), PEA-3 (polyoma enhancer activator 3), Runx2 (Runt-related transcription factor 2), and C/EBPβ (CCAAT/enhancer binding protein) [22–26]. Mutations in these binding sites can partially or completely abolish the activity of the human MMP-13 promoter. Numerous transcriptional factors, such as E74-like factor 3, FosA family members, and HIF-2-alpha, have been shown to be involved in induction of MMP-13 in chondrocytes [27–29]. Questions still remain as to how these transcriptional factors are induced during the development of OA, and whether or not the removal of one or a combination of these transcriptional factors is sufficient to significantly attenuate or prevent the progression of OA.
Signaling pathways activated by inflammatory cytokines , such as TNF-α and IL-1β, have been shown to be involved in the transcriptional regulation of MMP-13 expression in chondrocytes [30–33]. It has also been shown that fibronectin fragments (FN-f) can stimulate chondrocytes to synthesize and release MMP-13 via integrin activation [34, 35]. Although these signaling pathways may play a significant role in transcriptional regulation of MMP-13 expression in chondrocytes, a certain degree of articular cartilage damage is required to stimulate the release of the inflammatory cytokines or generate the FN-f. One important question is whether or not a mechanism exists to induce MMP-13 in chondrocytes prior to any significant degradation of articular cartilage. Results from our recent investigations demonstrate that discoidin domain receptor 2 (DDR2) may be a molecule that induces expression of MMP-13 in chondrocytes prior to overt damage of articular cartilage .
3 DDR2 in the Pathogenesis of OA
What led us to investigate DDR2 in the pathogenesis of OA? The answer is the classical biochemistry event in living systems known as enzyme induction. One example is the inducible lactose-metabolizing enzyme by which the enzyme is induced by its own substrates, see Fig. 14.1.

Induction of β-galactosidase in bacteria E. coli by disaccharide lactose. In bacteria, β-galactosidase can convert disaccharide into monosaccharide, for example, from lactose to glucose and galactose. Then, bacteria can consume monosaccharide as energy resource
Bacterial Escherichia coli are not able to directly utilize disaccharide lactose for consumption. However, under the condition in which lactose is solely present, the enzyme β-galactosidase is induced in E. coli. The enzyme breaks down lactose into monosaccharides (galactose and glucose) that are then metabolized by E. coli. We believe that the induction of MMP-13 in chondrocytes may represent this similar situation. For example, MMP-13 is required for tissue turnover to remove ECM molecules such as collagens and proteoglycans, see Fig. 14.2.

Induction of the MMP-13 in chondrocytes by native type II collagen. Native type II collagen is too big to move into chondrocyte. Thus, the collagen needs to bind a cell surface receptor and send signal to induce MMP-13 in chondrocytes. MMP-13, in turn, degrades native type II collagen. This indicates that native type II collagen controls its own proteolysis
The native type II collagen cannot enter chondrocytes to physically induce MMP-13 by itself. Thus, the collagen has to interact with a cell surface molecule(s) and send signals into chondrocytes to induce and release MMP-13. However, if this occurs in relatively quiescent tissues in which there is little tissue turnover, such as mature (adult) articular cartilage, the induction of MMP-13 will result in articular cartilage degradation. If this is the case, the question becomes which cell surface molecule(s) transduce the signal into chondrocytes? Results from our recent investigations suggest that DDR2 may be such a molecule.
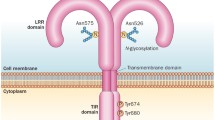
DDR2 was originally cloned as a cell surface receptor tyrosine kinase (RTKs) in 1993 [36–39]. The mRNA transcripts of these receptors are detected in several human and mouse tissues, mainly in cartilage, skeletal muscle, skin, and adipose. In 1997, two research groups reported native fibrillar collagens are ligands for DDR2 [40, 41]. Furthermore, two other independent research groups have identified specific amino acids on DDR2 and type II collagen, which are critical for the interaction of the receptor with type II collagen, see Fig. 14.3 [42–44]. A change in any of these amino acids will dramatically affect the affinity (interaction) of DDR2 with type II collagen. This suggests that the interaction of the receptor with type II collagen can be interrupted by the alteration of this specific amino acid peptide.

Critical amino acids for the interaction of DDR2 with type II collagen. An alteration of any of the amino acids in type II collagen and DDR2 shown in this figure will significantly affect the interaction of the two molecules
We examined the expression of DDR2 in human osteoarthritic tissues and mouse models of OA. We found that DDR2 protein was hardly detectable in normal articular cartilage. However, the expression of DDR2 was increased in human osteoarthritic tissues and mouse models of OA [45–49]. Moreover, DDR2 was colocalized with the increased activity and expression of MMP-13 in degenerative articular cartilage [50]. To understand whether or not the activation of DDR2 induced MMP-13 in chondrocytes, we performed a series of in vitro experiments. Results from these experiments showed that (1) the expression of MMP-13 was elevated in chondrocytes cultured on type II collagen. Surprisingly, we found that the expression of DDR2 was also increased in the chondrocytes. This indicated that chondrocytes exposed to native type II collagen were induced to express MMP-13 and the receptor itself. (2) When chondrocytes were cultured on denatured type II collagen (gelatin), the expression of MMP-13 and DDR2 were not induced, suggesting that the chondrocytes responded in a specific manner to triple-helical type II collagen. (3) The overexpression of full-length DDR2 cDNA resulted in increased expression of MMP-13, whereas the overexpression of a truncated DDR2 cDNA (lacking the protein tyrosine kinase) inhibited the increased expression of MMP-13. In addition, DDR2 lacking type II collagen-binding domain (discoidin domain) of the receptor had no effect on the expression of MMP-13 and the receptor itself. These results indicate that increased expression of MMP-13 in chondrocytes requires the interaction of DDR2 with type II collagen. (4) Ras/Raf/MEK/ERK and p38 signaling pathways were involved in the increased expression of MMP-13 in chondrocytes by type II collagen−DDR2 interaction. Based on the aforementioned finds, we carried out in vivo experiments to determine whether a reduction in expression of Ddr2 in heterozygous Ddr2 +/− condition could delay progression of articular cartilage degeneration, caused by either a deficiency in expression of collagen type XI or destabilization of the medial meniscus, in mice. We found that the decrease in the expression of Ddr2 indeed attenuated the progression of articular cartilage degeneration in mouse models of OA [51]. More importantly, three other independent research groups have confirmed the result from our experiments [52–54]. A question remains: how do chondrocytes silence DDR2 in normal mature articular cartilage?
Chondrocytes and their pericellular matrix are considered to be the primary structural and functional units, termed chondrons, of articular cartilage [55–61]. The concept of the chondron was first proposed by Benninghoff in 1925. Approximately 40 years later, Szirmai further evaluated the structure of the chondron by more systematic analysis. At the time, however, the chondron was not widely recognized as a functional unit. Some 20 years later, C.A. Poole’s research group completed additional experiments to physically isolate chondrons from cartilage and showed that chondrons are true anatomic and functional entities. Furthermore, Poole’s group demonstrated that the structure of the pericellular matrix of chondrocytes is disturbed in human OA cartilage. Chondrons consist of chondrocytes, the pericellular matrix, and a capsule surrounding the pericellular matrix. The pericellular matrix contains laminin, fibronectin, biglycan, decorin, fibromodulin, matrilin 3, and cartilage oligo matrix protein (COMP). The pericellular capsule is mostly composed of type VI and IX collagen, and proteoglycans. The capsule and the pericellular matrix separate chondrocytes from the adjacent interterritorial or territorial matrices containing type II collagen, see Fig. 14.4. Clearly, under normal conditions type II collagen is not exposed to chondrocytes. It is conceivable that the disruption or disappearance of the pericellular matrix enhances the exposure of chondrocytes to type II collagen. This, in turn, elicits the interaction of collagen type II with DDR2, resulting in induction of MMP-13, which leads to the destruction of articular cartilage.

A schematic illustration of articular cartilage . A chondrocyte is surrounded with the pericellular matrix to form a chondron. Matrix between two chondrons is termed as territorial matrix and matrix among more than three chondrons is termed as interterritorial matrix. Native type II collagens in the territorial and interterritorial matrices are separated from chondrocytes by the pericellular matrix. Thus, the type II collagen does not interact with its receptor, such as DDR2, in normal articular cartilage
Results from human and mouse genetic studies indicate the significant role of the pericellular matrix of chondrocytes in protecting articular cartilage against the development of OA. For example, the deficiency of one or a combination of two components of the pericellular matrix, such as type VI collagen, type IX collagen, matrilin 3, decorin, biglycan, and fibromodulin, result in early onset of OA in mice [46, 62–65]. In human genetic studies, mutations in type IX collagen and COMP are associated with OA [66–71].
We utilized a tetracycline-controlled gene expression system (Tet-Off system) in mice to investigate whether the pericellular matrix is indeed a critical structure to silence Ddr2. The system provides the ability to conditionally induce expression of Ddr2 in mature articular cartilage. This will prevent any potential abnormalities resulting from increased expression of the receptor during embryonic or early postnatal developmental stages. In the experiment, we induced the overexpression of Ddr2 in the articular cartilage of mouse knee joints at the age of 8 weeks old. It takes, in general, about 6 to 8 weeks for mice to become mature. Results from our experiments showed that the increased expression of Ddr2 did not induce expression of Mmp-13, and there were no OA-like pathologic changes observed in knee joints of the transgenic mice [50]. This indicates that a high-level expression of the inactive DDR2 does not cause the initiation/progression of OA. Our explanation is that although expression of Ddr2 was induced in articular cartilage of knee joints in the transgenic mice, the upregulated Ddr2 was surrounded by pericellular matrix. The pericellular matrix prevents Ddr2 from binding to type II collagen. Thus, Ddr2 was not activated (phosphorylated) and Mmp-13 would not be induced. Our data also suggested that the overexpressed Ddr2, without the interaction of the receptor with its ligand type II collagen, could not lead to autophosphorylation of the receptor itself.
In line with results from our experiments with the transgenic mice, another independent study by Vonk et al. demonstrate that DDR2 is silenced when chondrons are cultured on the collagen-coated plates [53]. It is clearly shown that DDR2 and MMP-13 are induced in chondrocytes in type II collagen-coated plates, whereas there is no induction of DDR2 and MMP-13 in chondrons when the chondrons are used in the type II collagen-coated plates. The results from these investigations support our hypothesis that the pericellular matrix of chondrocytes silences DDR2.
If disruption of the pericellular matrix is required for binding of DDR2 to type II collagen, then an enzyme/enzymes that are capable of degrading the pericellular molecules must play a critical role in development of OA. In fact, a study by Lee and Loeser suggests that proteases, not matrix metalloproteinases, may involve in the degradation of the pericellular matrix [72]. Numerous independent research groups have reported that a serine protease, high temperature requirement A1 (HTRA1), is highly expressed in human OA cartilage and in mouse models of OA. This suggests that HTRA1 may be involved in the development of OA.
4 HTRA1 in the Pathogenesis of OA
HTRA1 is one of four HTRA family members in human and mouse genomes [73, 74]. Mammalian HTRA1 was originally identified as a gene that was downregulated in SV40 transformed fibroblasts. Substrates of HTRA1 have been identified and include decorin, biglycan, fibromodulin, aggrecans, and fibronectin [75, 76]. All of these molecules are pericellular components of chondrocytes in articular cartilage. It is conceivable that upregulated expression and activity of this enzyme may cause degradation of the pericellular matrix of chondrocytes, which eventually leads to the destruction of a joint. In fact, HTRA1 has been implicated in rheumatoid arthritis (RA) and osteoarthritis (OA), and the level of HTRA1 is found to be higher in synovial fluids obtained from RA and OA patients [77–79]. HTRA1 is the most abundant protease in human OA cartilage. A study with a mouse model of RA demonstrates that increased expression of HtrA1 causes arthritic joints in the mice [80].
Results from our previous study also indicated that the protein level and activity of HtrA1 were increased in knee and temporomandibular joints of OA mouse models [81], including surgically induced models. This suggests that the altered mechanical stress in the surgical models can directly or indirectly induce HTRA1 in chondrocytes. More importantly, elevated expression of the enzyme was associated with disruption of the pericellular matrix of chondrocytes in the OA mouse models. We also found that the expression of this enzyme was increased prior to upregulation of Ddr2 and Mmp-13 in the mouse models. This indicates that HTRA1 may contribute to the development of OA through the degradation of the pericellular network, resulting in enhanced exposure of chondrocytes to type II collagen and activation of DDR2. Data from our experiments with the conditionally induced Ddr2 transgenic mice supports the pericellular matrix-degrading role of HTRA1 in the development of OA. We found that following a surgical destabilization of the medial meniscus (DMM), the progression of articular cartilage degeneration was accelerated in the upregulated Ddr2 transgenic mice, compared to that in wild-type littermates. Our explanation is that DMM surgery induces expression of HtrA1 that degrades the pericellular matrix of chondrocytes. Thus, in the Ddr2-induced transgenic mice, the disruption of the pericellular matrix coupled with the upregulated expression of Ddr2 accelerates the progression of articular cartilage degeneration. The data also indicates that DDR2 is able to accelerate the progression of the cartilage degeneration, but is not able to initiate the cartilage degeneration in the absence of biomechanical and/or biochemical disruption of the pericellular matrix. Therefore, maintaining the integrity of the pericellular matrix is one of the key factors against OA. Obviously, more in vivo experiments are needed to verify the role of HTRA1 in the pathogenesis of OA .
A critical question concerns which factor(s) cause induction of HTRA1 in chondrocytes. We recently investigated whether numerous factors, such as heating at 42 °C, hydrostatic pressure, tumor necrosis factor (TNFα), and transforming growth factor beta 1 (TGF-β1), were involved in the induction of HTRA1 in human primary articular chondrocytes [82]. We found that 42 °C, hydrostatic pressure, and TNFα treatment did not induce HTRA1 in the chondrocytes. In contrast, TGF-β1 induced HTRA1 in these cells.
5 TGF-β1 in the Pathogenesis of OA
Results from our experiments demonstrate that DDR2 may play a critical role in the development of OA. However, we consider that molecules that eventually lead to a condition resulting in activation of DDR2 are also crucial in the development of OA, such as HTRA1. In the following section, we also discuss about TGF-β1 as one of the molecules involved in the activation of DDR2.
TGF-β1 has been considered an anabolic factor to articular chondrocytes , based largely on results from in vitro and ex vivo experiments in which TGF-β1 can stimulate chondrocytes to synthesize and release extracellular matrix molecules, including proteoglycans and type II collagens [83–86]. In addition, results from two studies indicate that the genetic inactivation of Smad-3 or disruption of the interaction of Tgf-β1 with its receptor, Tgf-β type II receptor (Tgfbr2), in germline cells causes OA-like knee joints in mice [87, 88]. Moreover, a human genetic study reports that a two-nucleotide deletion, 741-742del AT (nonsense mutation), in SMAD-3 causes early-onset OA in a human family [89]. This is consistent with the results from the animal models, indicating that the lack of Tgf-β1 signaling in the germline cell results in OA. However, observations from other studies also suggest that the increased TGF-β1 signaling may initiate and accelerate articular cartilage degeneration in mature joints.
First, the enhanced production of extracellular matrix molecules , due to an increase in the synthetic activity of chondrocytes, is not necessarily beneficial or physiological in maintenance of the homeostasis of mature articular cartilage. For instance, one of the earliest pathological signs in articular cartilage degeneration is the overproduction of proteoglycans in mouse models of OA [5, 6]. A study by Van den Berg et al. reports that the constitutive overexpression of active TGF-β1 in adult mouse knee joints results in OA associated with increase in the production of proteoglycans in articular cartilage, hyperplasia of synovium and chondro-osteophyte formation [90]. Thus, the overproduction of proteoglycans in articular cartilage could be a pathologic response of the chondrocyte in the early stages of articular cartilage degeneration. This raises an interesting question: does TGF-β1 disrupt homeostasis of articular cartilage instead of repairing the damaged cartilage in mature joints? Second, the human genetic study reports that a nucleotide change, 859C > T or 782C > T in SMAD-3, increases the level of TGF-β1 and activity of the TGF-β1 signaling pathway in two human families associated with early-onset OA [89]. This is in agreement with the observation from two other studies indicating that the level of TGF-β1 is significantly higher in human osteoarthritic tissues than in healthy articular cartilages [91, 92]. Studies with animal models, by Itayem et al., suggest that intra-articular injections of TGF-β1 into adult rat knee joints may cause early onset of OA [93, 94]. Third, to confirm our in vitro observation that TGF-β1 induced HTRA1 in human chondrocytes, we investigated whether Tgf-β1 induced HtrA1 in articular chondrocytes of two mouse models of OA [7]. We found increases in the expression of Tgf-β1, p-Smad2/3, and HtrA1 in articular chondrocytes of knee joints in the mouse models and increased expressions of p-Smad2/3 and HtrA1 were colocalized in the chondrocyte. In addition, TGF-β1-induced expression of HTRA1 was inhibited by an ALK-5 inhibitor, SB431542, in human and mouse chondrocytes. This suggests that Tgf-β1 canonical signaling may be activated to induce HtrA1 in articular chondrocytes of the mouse models of OA. More importantly, results from another independent research group demonstrate that TGF-β1 induces HTRA1 in human primary chondrocytes [95].
Data from aforementioned investigations raise the question as to what the exact role of TGF-β1 is in the development of OA. Our explanations for this “conflicting” role of TGF-β1 in the pathogenesis of OA are (1) effective TGF-β1 signaling acts in a dose-dependent manner. In this scenario, an appropriate level of TGF-β1 is required for the development and maintenance of articular cartilages. Therefore, TGF-β1 below or above this level results in articular cartilage degeneration. (2) Effective TGF-β1 signaling acts in a developmental stage-dependent manner. In this scenario, TGF-β1 is required for the development of articular cartilage; however, once a joint is formed, TGF-β1 is no longer needed. Therefore, induction of TGF-β1 in an adult joint causes articular cartilage degeneration .
Recently, we started to investigate whether inhibition of Tgf-β1 signaling prevents degeneration of mature knee joints in mouse models of OA [96]. (1) By use of conditional knockout techniques in mice, we removed Tgfbr2 from articular cartilage of knee joints in mice at 8 weeks of age. We found neither the initiation and acceleration of articular cartilage degeneration in knee joints of mice at the age of 9 months old, nor hypertrophic chondrocytes in the articular cartilage of the mice. (2) We treated a spontaneous mouse model of OA, type XI collagen-haploinsufficiency (Col11a1 +/−) mice, with a neutralizing TGF-β1 antibody by intra-articular injection into the knee joints at 2 months of age. We found that the intra-articular injection of the neutralizing TGF-β1 antibody delayed articular cartilage degeneration approximately 3 months, compared to that in Col11a1 +/− mice without injection of the antibody. Approximately 15 months is required for Col11a1 +/− mice to develop OA knee joints (an average life span of the laboratory mice is about 30 months). (3) We treated surgically (DMM) induced OA mice with Losartan for 4 weeks immediately following surgery. Studies indicate that Losartan inhibits the activity of TGF-β1 signaling [96]. We found that treatment with Losartan delayed articular cartilage degeneration compared to that in control littermates. (4) We removed Tgfbr2 from knee joints of mice at 2 months of age. We then performed DMM surgery on knee joints of the mice. We found that the removal of the Tgfbr2 delayed articular cartilage degeneration at least 6 weeks, compared to that in wild-type littermates. Approximately 16 weeks is required for mice with DMM to develop OA knee joints. Based on the results from these experiments, we suggest that inhibition of TGF-β1 signaling attenuates articular cartilage degeneration in mature knee joints. Therefore, inhibition activity of TGF-β1, not application of TGF-β1, may be considered in treatment of OA in mature joints .
Data from our experiments demonstrated that the complete removal of Tgf-β1 signaling could, but not completely, delay the progression of articular cartilage degeneration in mice. This suggests that TGF-β1 may not be the only factor that induces HTRA1 in chondrocytes. In fact, data from several other studies indicate that other molecules, such as Wnt/β-catenin and LPS/TLR4 (toll-like receptor), can induce HTRA1 [80, 97].
6 A Molecular Pathway Underlying Articular Cartilage Degeneration
Based upon the data from our investigations and others’ studies, we propose a molecular pathway (see Fig. 14.5) underlying articular cartilage degeneration as follows: Excessive mechanical stresses induced by either a normal mechanical loading of defective joints or an overloading of normal joints can stimulate chondrocytes and other joint tissues to synthesize and release latent TGF-β1 into synovial fluid. This suggestion is supported by studies (1) a study by Lee et al. reports that mechanical injury of bovine cartilage explants causes a significant increase in TGF-β1 gene expression [98]. (2) Results from our study indicated that hydrostatic pressure on human articular chondrocytes in culture increased expression of TGF-β1 in the cells. (3) The latent TGF-β1 is then activated by mechanical shearing of synovial fluid [99, 100]. The active TGF-β1 binds to its cognate receptor, TGFβ receptor II, which induces expression of HTRA1 in chondrocytes. We notice that HTRA1 may be able to inhibit TGF-β1 signaling based on the structural and functional similarity of the Insulin-like growth factor-binding protein domain of HTRA1 with follistatin, a potent antagonist of activin (a TGF-β family protein) and the capability to cleave proTGF-β1 [73, 101]. This suggests that there may be a negative feedback loop between TGF-β1 and HTRA1, which regulates the activity and expression of these two molecules in chondrocytes. Consequences of induction of HTRA1 are degradation of the pericellular matrix and enhanced exposure of chondrocytes to type II collagen. Interaction of chondrocytes with type II collagen results in enhanced signaling through DDR2, which induces the expression of MMP-13 as well as expression of DDR2 itself. MMP-13 degradation of type II collagen and aggrecan results in type II collagen and aggrecan fragments, which in turn may activate signals that further increase the synthesis of MMP-13 [102]. The end result is a feedback amplification loop that causes irreversible articular cartilage degeneration. One legitimate question, from an evolutionary standpoint, is if this molecular pathway leads to irreversible articular cartilage degradation, why do we still retain it? One plausible explanation may be that this molecular pathway is needed for proper tissue turnover during development .

A schematic illustration of a molecular pathway underlying articular cartilage degeneration
References
Felson DT, Lawrence RC, Dieppe PA et al (2000) Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med 133:635–646
Lane NE, Brandt K, Hawker G et al (2011) OARSI-FDA initiative: defining the disease state of osteoarthritis. Osteoarthr Cartil 19(5):478–482. doi:10.1016/j.joca.2010.09.013
Hamerman D (1989) The biology of osteoarthritis. N Engl J Med 320(20):1322–1330
Lotz MK, Otsuki S, Grogan SP et al (2010) Cartilage cell clusters. Arthritis Rheum 62(8):2006–2018. doi:10.1002/art.27528
Xu L, Flahiff CM, Waldman BA et al (2003) Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of chondrodysplasia (cho) mice. Arthritis Rheum 48(9):2509–2518
Xu L, Polur I, Lim C et al (2009) Early-onset osteoarthritis of mouse temporomandibular joint induced by partial discectomy. Osteoarthr Cartil 17(7):917–922. doi:10.1016/j.joca.2009.01.002
Xu L, Golshirazian I, Asbury BJ et al (2014) Induction of high temperature requirement A1, a serine protease, by TGF-beta1 in articular chondrocytes of mouse models of OA. Histol Histopathol 29(5):609–618
Henry SP, Liang S, Akdemir KC et al (2012) The postnatal role of Sox9 in cartilage. J Bone Miner Res 27(12):2511–2525. doi:10.1002/jbmr.1696
Blaney Davidson EN, Vitters EL, Bennink MB et al (2015) Inducible chondrocyte-specific overexpression of BMP2 in young mice results in severe aggravation of osteophyte formation in experimental OA without altering cartilage damage. Ann Rheum Dis 74(6):1257–1264. doi:10.1136/annrheumdis-2013-204528
Freije JM, Díez-Itza I, Balbín M et al (1994) Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J Biol Chem 269(24):16766–16773
Mitchell PG, Magna HA, Reeves LM et al (1996) Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest 97(3):761–768
Fosang AJ, Last K, Knäuper V et al (1996) Degradation of cartilage aggrecan by collagenase-3 (MMP-13). FEBS Lett 380(1–2):17–20
Reboul P, Pelletier JP, Tardif G et al (1996) The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J Clin Invest 97(9):2011–2019
Shlopov BV, Lie WR, Mainardi CL et al (1997) Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum 40(11):2065–2074
Tetlow LC, Adlam DJ, Woolley DE (2001) Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum 44(3):585–594
Bau B, Gebhard PM, Haag J et al (2002) Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum 46(10):2648–2657
Neuhold LA, Killar L, Zhao W et al (2001) Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest 107(1):35–44
Little CB, Barai A, Burkhardt D et al (2009) Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 60(12):3723–3733. doi:10.1002/art.25002
Verzijl N, DeGroot J, Thorpe SR et al (2000) Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem 275(50):39027–39031
Karsdal MA, Madsen SH, Christiansen C et al (2008) Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis Res Ther 10(3):R63. doi:10.1186/ar2434
Hellio Le Graverand-Gastineau MP (2009) OA clinical trials: current targets and trials for OA. Choosing molecular targets: what have we learned and where we are headed? Osteoarthr Cartil 17(11):1393–1401. doi:10.1016/j.joca.2009.04.009
Pendas AM, Balbin M, Llano E et al (1997) Structural analysis and promoter characterization of the human collagenase-3 gene (MMP13). Genomics 40(2):222–233
Benbow U, Brinckerhoff CE (1997) The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol 15(8–9):519–526
Porte D, Tuckermann J, Becker M et al (1999) Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene 18(3):667–678
Hess J, Porte D, Munz C (2001) AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast specific element 2/AP-1 composite element. J Biol Chem 276(23):20029–20038
Hayashida M, Okazaki K, Fukushi J et al (2009) CCAAT/enhancer binding protein beta mediates expression of matrix metalloproteinase 13 in human articular chondrocytes in inflammatory arthritis. Arthritis Rheum 60(3):708–716. doi:10.1002/art.24332
Otero M, Plumb DA, Tsuchimochi K et al (2012) E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under pro-inflammatory stress. J Biol Chem 287(5):3559–3572. doi:10.1074/jbc.M111.265744
Ionescu A, Kozhemyakina E, Nicolae C et al (2012) FoxA family members are crucial regulators of the hypertrophic chondrocyte differentiation program. Dev Cell 22(5):927–939. doi:10.1016/j.devcel.2012.03.011
Saito T, Kawaguchi H (2010) HIF-2α as a possible therapeutic target of osteoarthritis. Osteoarthr Cartil 18(12):1552–1556. doi:10.1016/j.joca.2010.10.006
Mengshol JA, Vincenti MP, Coon CI et al (2000) Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum 43(4):801–811
Mengshol JA, Vincenti MP, Brinckerhoff CE (2001) IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res 29(21):4361–4372
Liacini A, Sylvester J, Li WQ et al (2002) Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor kappa B (NF-kappa B) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol 21(3):251–262
Vincenti MP, Brinckerhoff CE (2002) Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res 4(3):157–164
Forsyth CB, Pulai J, Loeser RF (2002) Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum 46(9):2368–2376
Loeser RF, Forsyth CB, Samarel AM et al (2003) Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem 278(27):24577–24585
Zerlin M, Julius MA, Goldfarb M (1993) NEP: a novel receptor-like tyrosine kinase expressed in proliferating neuroepithelia. Oncogene 8(10):2731–2739
Di Marco E, Cutuli N, Guerra L et al (1993) Molecular cloning of trkE, a novel trk-related putative tyrosine kinase receptor isolated from normal human keratinocytes and widely expressed by normal human tissues. J Biol Chem 268(32):24290–24295
Karn T, Holtrich U, Bräuninger A et al (1993) Structure, expression and chromosomal mapping of TKT from man and mouse: a new subclass of receptor tyrosine kinases with a factor VIII-like domain. Oncogene 8(12):3433–3440
Lai C, Gore M, Lemke G (1994) Structure, expression, and activity of Tyro 3, a neural adhesion-related receptor tyrosine kinase. Oncogene 9(3):877–883
Vogel W, Gish GD, Alves F et al (1997) The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell 1(1):13–23
Shrivastava A, Radziejewski C, Campbell E et al (1997) An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell 1(1):25–34
Leitinger B, Steplewski A, Fertala A (2004) The D2 period of collagen II contains a specific binding site for the human discoidin domain receptor, DDR2. J Mol Biol 344(4):993–1003
Konitsiotis AD, Raynal N, Bihan D et al (2008) Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. J Biol Chem 283(11):6861–6868. doi:10.1074/jbc.M709290200
Osamu I, Masanori O, Noritaka N et al (2007) Structural basis of the collagen-binding mode of discoidin domain receptor 2. EMBO J 26:4168–4176. doi:10.1038/sj.emboj.7601833
Xu L, Peng H, Wu D et al (2005) Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J Biol Chem 280(1):548–555
Hu K, Xu L, Cao L et al (2006) Pathogenesis of osteoarthritis-like changes in joints of type IX collagen-deficient mice. Arthritis Rheum 54(9):2891–2900
Xu L, Peng H, Glasson S et al (2007) Increased expression of a collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum 56(8):2663–2673
Lam NP, Li Y, Waldman AB et al (2007) Age-dependent increase of discoidin domain receptor 2 and matrix metalloproteinase 13 expression in temporomandibular joint cartilage of type IX and type XI collagen-deficient mice. Arch Oral Biol 52(6):579–584
Sunk IG, Bobacz K, Hofstaetter JG et al (2007) Increased expression of discoidin domain receptor 2 is linked to the degree of cartilage damage in human knee joints: a potential role in osteoarthritis pathogenesis. Arthritis Rheum 56(11):3685–3692
Xu L, Polur I, Servais JM et al (2011) Intact pericellular matrix of articular cartilage is required for un-activated discoidin domain receptor 2 in the mouse model. Am J Pathol 179(3):1338–1346. doi:10.1016/j.ajpath.2011.05.023
Xu L, Servais J, Polur I et al (2010) Attenuation of osteoarthritis progression by reduction of the discoidin domain receptor 2 in mice. Arthritis Rheum 62(9):2736–2744. doi:10.1002/art.27582
Klatt AR, Zech D, Kühn G et al (2009) Discoidin domain receptor 2 mediates the collagen II-dependent release of interleukin-6 in primary human chondrocytes. J Pathol 218(2):241–247. doi:10.1002/path.2529.]
Vonk LA, Doulabi BZ, Huang C et al (2011) Collagen-induced expression of collagenase-3 by primary chondrocytes is mediated by integrin α1 and discoidin domain receptor 2: a protein kinase C-dependent pathway. Rheumatology (Oxford) 50(3):463–472. doi:10.1093/rheumatology/keq305
Holt DW, Henderson ML, Stockdale CE et al (2012) Osteoarthritis-like changes in the heterozygous sedc mouse associated with the HtrA1-Ddr2-Mmp-13 degradative pathway: a new model of osteoarthritis. Osteoarthr Cartil 20(5):430–439. doi:10.1016/j.joca.2011.11.008
Poole CA, Flint MH, Beaumont BW (1988) Chondrons extracted from canine tibial cartilage: preliminary report on their isolation and structure. J Orthop Res 6(3):408–419
Poole CA, Ayad S, Schofield JR (1988) Chondrons from articular cartilage: I. Immunolocalization of type VI collagen in the pericellular capsule of isolated canine tibial chondrons. J Cell Sci 90(Pt 4):635–643
Poole CA, Wotton SF, Duance VC (1988) Localization of type IX collagen in chondrons isolated from porcine articular cartilage and rat chondrosarcoma. Histochem J 20(10):567–574
Poole CA, Honda T, Skinner SJ et al (1990) Chondrons from articular cartilage (II): analysis of the glycosaminoglycans in the cellular microenvironment of isolated canine chondrons. Connect Tissue Res 24(3–4):319–330
Poole CA, Matsuoka A, Schofield JR (1991) Chondrons from articular cartilage. III. Morphologic changes in the cellular microenvironment of chondrons isolated from osteoarthritic cartilage. Arthritis Rheum 34(1):22–35
Poole CA (1997) Articular cartilage chondrons: form, function and failure. J Anat 191(Pt1):1–13
Hunziker EB, Michel M, Studer D (1997) Ultrastructure of adult human articular cartilage matrix after cryotechnical processing. Microsc Res Tech 37(4):271–284
Alexopoulos LG, Youn I, Bonaldo P et al (2009) Developmental and osteoarthritic changes in Col6a1-knockout mice. Arthritis Rheum 60(3):771–779. doi:10.1002/art.24293
Wadhwa S, Embree M, Ameye L et al (2005) Mice deficient in biglycan and fibromodulin as a model for temporomandibular joint osteoarthritis. Cells Tissues Organs 181(3–4):136–143
Wadhwa S, Embree MC, Kilts T et al (2005) Accelerated osteoarthritis in the temporomandibular joint of biglycan/fibromodulin double-deficient mice. Osteoarthr Cartil 13(9):817–827
van der Weyden L, Wei L, Luo J et al (2006) Functional, knockout of the matrilin-3 gene causes premature chondrocyte maturation to hypertrophy and increases bone mineral density and osteoarthritis. Am J Pathol 169(2):515–527
Borochowitz ZU, Scheffer D, Adir V et al (2004) Spondylo-epi-metaphyseal dysplasia (SEMD) matrilin 3 type: homozygote matrilin 3 mutation in a novel form of SEMD. J Med Genet 41(5):366–372
Hecht JT, Nelson LD, Crowder E et al (1995) Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet 10(3):325–329
Muragaki Y, Mariman EC, van Beersum SE et al (1996) A mutation in the gene encoding the alpha 2 chain of the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nat Genet 12(1):103–105
Mustafa Z, Chapman K, Irven C et al (2000) Linkage analysis of candidate genes as susceptibility loci for osteoarthritis-suggestive linkage of COL9A1 to female hip osteoarthritis. Rheumatology (Oxford) 39(3):299–306
Bönnemann CG, Cox GF, Shapiro F et al (2000) A mutation in the alpha 3 chain of type IX collagen causes autosomal dominant multiple epiphyseal dysplasia with mild myopathy. PNAS 97(3):1212–1217
Czarny-Ratajczak M, Lohiniva J, Rogala P et al (2001) A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am J Hum Genet 69(5):969–980
Lee GM, Loeser RF (1998) Interactions of the chondrocyte with its pericellular matrix. Cell Mater 8:135–149
Zumbrunn J, Trueb B (1996) Primary structure of a putative serine protease specific for IGF-binding proteins. FEBS Lett 398(2–3):187–192
Clausen T, Southan C, Ehrmann M (2002) The HtrA family of proteases: implications for protein composition and cell fate. Mol Cell 10(3):443–455
Oka C, Tsujimoto R, Kajikawa M et al (2004) HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development 131(5):1041–1053
Tsuchiya A, Yano M, Tocharus J et al (2005) Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone 37(3):323–336
Hu SI, Carozza M, Klein M et al (1998) Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J Biol Chem 273(51):34406–34412
Grau S, Richards PJ, Kerr B et al (2006) The role of human HtrA1 in arthritic disease. J Biol Chem 281(10):6124–6129
Wu J, Liu W, Bemis A et al (2007) Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum 56(11):3675–3684
Hou Y, Lin H, Zhu L et al (2013) Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA-1 expression. Arthritis Rheum 65(11):2835–2846. doi:10.1002/art.38124
Polur I, Lee PL, Servais JM et al (2010) Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol Histopathol 25(5):599–608
Li YF, Li Y, Xu L et al (2011) Mechanical loading-induced TGF-ß1 mediates cartilage degradation caused by upregulation of HTRA1/DDR2. In: Abstracts of orthopaedic research society annual meeting, Long Beach, CA, January 13–16, 2011
Harvey AK, Hrubey PS, Chandrasekhar S (1991) Transforming growth factor-beta inhibition of interleukin-1 activity involves down-regulation of interleukin-1 receptors on chondrocytes. Exp Cell Res 195(2):376–385
Galéra P, Vivien D, Pronost S et al (1992) Transforming growth factor-beta 1 (TGF-beta 1) up-regulation of collagen type II in primary cultures of rabbit articular chondrocytes (RAC) involves increased mRNA levels without affecting mRNA stability and procollagen processing. J Cell Physiol 153(3):596–606
Rédini F, Mauviel A, Pronost S et al (1993) Transforming growth factor beta exerts opposite effects from interleukin-1 beta on cultured rabbit articular chondrocytes through reduction of interleukin-1 receptor expression. Arthritis Rheum 36(1):44–50
van Beuningen HM, van der Kraan PM, Arntz OJ et al (1994) Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab Invest 71(2):279–290
Serra R, Johnson M, Filvaroff EH et al (1997) Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol 139(2):541–552
Yang X, Chen L, Xu X et al (2001) TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol 153(1):35–46
van de Laar IM, Oldenburg RA, Pals G et al (2011) Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 43(2):121–126. doi:10.1038/ng.744
Bakker AC, van de Loo FA, van Beuningen HM et al (2001) Overexpression of active TGF-beta-1 in the murine knee joint: evidence for synovial-layer-dependent chondro-osteophyte formation. Osteoarthr Cartil 9(2):128–136
Schlaak JF, Pfers I, Meyer Zum Büschenfelde KH et al (1996) Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clin Exp Rheumatol 14(2):155–162
Kawamura I, Maeda S, Imamura K et al (2012) SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J Biol Chem 287(34):29101–29113. doi:10.1074/jbc.M112.349415
Itayem R, Mengarelli-Widholm S, Hulth A et al (1997) Ultrastructural studies on the effect of transforming growth factor-β1 on rat articular cartilage. APMIS 105(3):221–228
Itayem R, Mengarelli-WidholmI S, Reinholt F (1999) The lone-term effect of a short course of transforming growth factorβ1 on rat articular cartilage. APMIS 107(2):183–192
Urano T, Narusawa K, Kobayashi S et al (2010) Association of HTRA1 promoter polymorphism with spinal disc degeneration in Japanese women. J Bone Miner Metab 28(2):220–226. doi:10.1007/s00774-009-0124-0
Li Y, Chen R, Mian M et al (2014) Inhibition of Tgf-β1 attenuates articular cartilage degeneration in mature knee joints of mouse models of osteoarthritis. Paper presented at the world congress on osteoarthritis, Paris, France, April 24–27, 2014
Miclea RL, Siebelt M, Finos L et al (2011) Inhibition of Gsk3β in cartilage induces osteoarthritic features through activation of the canonical Wnt signaling pathway. Osteoarthr Cartil 19(11):1363–1372. doi:10.1016/j.joca.2011.07.014
Lee JH, Fitzgerald JB, Dimicco MA et al (2005) Mechanical injury of cartilage explants causes specific time-dependent changes in chondrocyte gene expression. Arthritis Rheum 52(8):2386–2395
Albro MB, Cigan AD, Nims RJ et al (2012) Shearing of synovial fluid activates latent TGF-β. Osteoarthr Cartil 20(11):1374–1382. doi:10.1016/j.joca.2012.07.006
Albro MB, Nims RJ, Cigan AD et al (2013) Accumulation of exogenous activated TGF-β in the superficial zone of articular cartilage. Biophys J 104(8):1794–1804. doi:10.1016/j.bpj.2013.02.052
Atsushi S, Hiroaki N, Akio Y et al (2011) Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-β1 via cleavage of proTGF-β1. Hum Mol Genet 20(9):1800–1810. doi:10.1093/hmg/ddr063
Fichter M, Körner U, Schömburg J et al (2006) Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J Orthop Res 24(1):63–70
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this chapter
Cite this chapter
Xu, L., Lee, P.L., Li, Y. (2016). Discoidin Domain Receptor 2 in Development of Osteoarthritis. In: Fridman, R., Huang, P. (eds) Discoidin Domain Receptors in Health and Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-6383-6_14
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6383-6_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-6381-2
Online ISBN: 978-1-4939-6383-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)