
Abstract
Metabolically competent, inexpensive, and robust in vitro cell models are needed for studying liver drug-metabolizing enzymes and hepatotoxicity. Human hepatoma HuH-7 cells develop into a differentiated in vitro model resembling primary human hepatocytes after a 2-week dimethyl sulfoxide (DMSO) treatment. DMSO-treated HuH-7 cells express elevated cytochrome P450 3A4 (CYP3A4) enzyme gene expression and activity compared to untreated HuH-7 cells. This cell model could be used to study CYP3A4 inhibition by reversible and time-dependent inhibitors, including drugs, food-related substances, and environmental chemicals. The DMSO-treated HuH-7 model is also a suitable tool for investigating hepatotoxicity. This chapter describes a detailed methodology for developing DMSO-treated HuH-7 cells, which are subsequently used for CYP3A4 inhibition and hepatotoxicity studies.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
Metabolism is a key function of liver , which is carried out by drug-metabolizing enzymes, such as cytochromes P450. Over the years, several in vitro cell models have been developed to mimic liver function, but many lack drug-metabolizing enzyme activities, which is true, for example, with HepG2 cells [1]. Other models, such as primary human hepatocytes and HepaRG cells, express drug-metabolizing enzyme activities, but are expensive, scarce, or require higher levels of maintenance [2]. Therefore, a cost-effective, metabolically competent, and robust in vitro cell model is needed for medium to high-throughput screening purposes.
Human hepatoma HuH-7 cells were derived from a Japanese male with well differentiated hepatocellular carcinoma in 1982 [3]. Studies have shown that HuH-7 cells could be induced by DMSO and differentiate into primary human hepatocyte-like cells [4, 5]. The DMSO-treated HuH-7 cells resemble primary human hepatocyte characteristics, such as, polygonal shape and binucleated cells (Fig. 1). Furthermore, DMSO-treated HuH-7 cells express the functional CYP 3A4 enzyme, the most abundant liver enzyme which mediates the metabolism of more than 50 % of all marketed drugs [5]. Inhibition of CYP3A4 activity could cause drug–drug or food–drug interactions, which may lead to adverse effects [6].

Phase contrast photomicrographs of DMSO -treated HuH-7 cells with arrows pointing to binucleated cells . Bar represents 100 μm
This chapter describes a protocol to establish DMSO-treated HuH-7 cells for measuring CYP3A4 inhibition and hepatotoxicity . A general timeline for developing DMSO-treated HuH-7 cell model is illustrated in Fig. 2. The cells are passaged for two or three times after thawing, then seeded in a flask for confluence and DMSO treatment. Finally, the cells are assayed for both reversible and time-dependent CYP3A4 inhibition, as well as tested for hepatotoxicity using control compounds.

Development of DMSO -treated HuH-7 cells. Timeline begins two to three passages after initial thawing of cells
2 Materials
2.1 Equipment
-
1.
BMG FLUOstar Omega multimode microplate reader.
-
2.
New Brunswick Galaxy 48R CO2 incubator.
-
3.
Inverted microscope.
-
4.
Countess, automated cell counter; counting slides; Trypan blue stain 0.4 %.
-
5.
96-well, black, clear flat bottom, tissue culture-treated polystyrene microplates, with lids, sterile.
-
6.
96-well, white, opaque flat bottom, untreated polystyrene microplates, nonsterile.
-
7.
Vacuum filter/storage bottle system, 0.22 μm pore, cellulose acetate membrane filter, sterile.
-
8.
8-channel pipette and tips; multichannel reservoir.
2.2 Cells and Culture Medium
-
1.
Human hepatoma HuH-7 cells, Health Science Research Resources Bank, Japan Health Sciences Foundation.
-
2.
Dulbecco’s Minimal Essential Medium (DMEM), low glucose, GlutaMAX Supplement, pyruvate.
-
3.
100× MEM nonessential amino acids and 1 M HEPES.
-
4.
Fetal bovine serum, Atlanta Biologicals Premium Select.
2.3 Reagents and Solutions
-
1.
Rat tail collagen type I.
-
2.
Glacial acetic acid.
-
3.
Dulbecco’s phosphate-buffered saline (DPBS, no calcium, no magnesium) and Versene (0.02 % EDTA).
-
4.
Trypsin–EDTA (0.5 %), without phenol red.
-
5.
Promega P450-Glo CYP3A4 (luciferin-IPA substrate) and CellTiter-Glo assays; beetle luciferin, potassium salt.
-
6.
DMSO, ketoconazole , troleandomycin , salicylamide , nitrofurantoin , and nefazodone hydrochloride .
3 Methods
3.1 Cell Culture
-
1.
Coat tissue culture surface using rat tail collagen type I (Table 1). Dissolve collagen in 0.02 N acetic acid to make a 100 μg/mL solution. Coat tissue culture surfaces at 10 μg/cm2. Incubate coated vessels at room temperature for at least 2 h and rinse with DPBS. Use fresh or air dry and store at 2–8 °C for up to 1 month.
Table 1 Collagen coating -
2.
Prepare basal medium (Table 2) and 1 % DMSO supplemented basal medium. Measure and mix all components under sterile conditions, followed by filter sterilization using a 0.22 μM filter. Medium is stable for 1 month if stored at 2–8 °C. 1 % DMSO supplemented basal medium is prepared fresh for medium change after cells reach confluence.
Table 2 Basal medium -
3.
Thaw and recover HuH-7 cells into T25 or T75 flasks using basal medium. Passage cells two or three times when reach 85–90 % confluence.
-
4.
When desired cell amount is reached, remove the basal medium from flask using aspiration and rinse twice with Versene solution.
-
5.
Add 0.025 % trypsin–EDTA reagent (prepared using 0.5 % trypsin–EDTA and DPBS) and rinse the flask quickly. Remove extra trypsin reagent and leave 1 mL in the flask. Incubate the flask (37 °C) until cell detachment (3–5 min).
-
6.
Use a microscope to observe. When cells are completely detached, add 3–4 mL prewarmed (37 °C) basal medium to stop trypsinization. Failure to add medium promptly can result in over-trypsinization and significant cell death.
-
7.
Determine the viable cell concentration (cells/mL) using a cell counter. Adjust cell concentration using basal medium as needed.
-
8.
Seed HuH-7 cells at 6.3 × 104 cells/cm2 onto collagen coated T25 or T75 flasks using basal medium. Wait 10 min at room temperature before returning to the incubator. Replenish medium twice per week until 100 % confluence, which takes about 3–4 days after seeding.
-
9.
Upon cell confluence, switch basal medium to 1 % DMSO supplemented basal medium (fresh prepared each time). Replenish medium twice per week for 2 weeks using 1 % DMSO supplemented basal medium.
-
10.
After the 2-week DMSO treatment, seed DMSO-treated HuH-7 cells at 3.1 × 105 cells/cm2 onto a collagen coated clear bottom 96-well plate using basal medium. Use plated cells for assays within 48 h after seeding (see Note 1 ).
3.2 CYP3A4 Reversible Inhibition Assay
-
1.
Reagent preparation.
-
Serum-free medium, prepare according to Table 2, leave out fetal bovine serum, and compensate volume with DMEM.
-
Luciferin-IPA substrate, thaw 3 mM stock solution, protect from light.
-
Salicylamide , Phase II conjugation inhibitor, prepare 3 M in DMSO fresh each time.
-
Beetle luciferin, prepare 2 mM stock in H2O, serial dilute using serum-free medium (0.2–80 nM, see Note 2 ).
-
Luciferin detection reagent, equilibrate to room temperature.
-
Ketoconazole , prepare 2 mM stock in DMSO and conduct serial dilution using DMSO (0.01–2 mM).
-
Dissolve luciferin-IPA substrate (3 μM), salicylamine (3 mM) and various concentrations of ketoconazole (0.01–2 μM) using serum-free medium (see Note 3 ) and prewarm to 37 °C. Preparation without ketoconazole serves as the control.
-
-
2.
CYP3A4 reversible inhibition.
Wash DMSO-treated HuH-7 cells on 96-well plates with serum-free medium and incubate with ketoconazole (0.01–2 μM) simultaneously with CYP3A4 substrate luciferin-IPA (3 μM, Km value) at 37 °C (100 μL/well) for 30 min. Include beetle luciferin standards (100 μL) in blank wells.
-
3.
Assay detection.
Terminate reaction by transferring 50 μL of incubation medium from each well to a separate white flat bottom plate with wells containing 50 μL of luciferin detection reagent at room temperature. Mix well and incubate the plate for 20 min, then measure luminescence using a plate reader (Fig. 3a).
Fig. 3 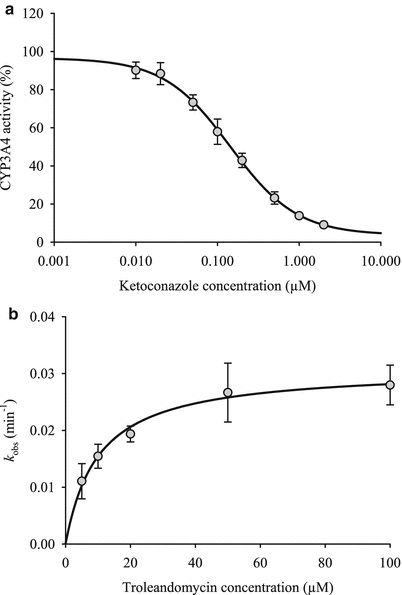
(a) CYP3A4 r eversible inhibition by ketoconazole and (b) time-dependent inhibition by troleandomycin in DMSO-t reated HuH-7 cells. Figure reproduced from reference [5]
-
4.
Calculate IC 50, the half-maximal inhibitory concentration of ketoconazole determined using a nonlinear regression Hill model.

3.3 CYP3A4 Time-Dependent Inhibition Assay
-
1.
Reagent preparation.
-
Serum-free medium, prepare according to Table 2, leave out fetal bovine serum, and compensate volume with DMEM.
-
Luciferin-IPA substrate, thaw 3 mM stock solution, protect from light.
-
Salicylamide , Phase II conjugation inhibitor, prepare 3 M in DMSO fresh each time.
-
Beetle luciferin, prepare 2 mM stock in H2O, serial dilute using serum-free medium (0.2–80 nM, see Note 2 ).
-
Luciferin detection reagent, equilibrate to room temperature.
-
Troleandomycin , prepare 10 mM in DMSO and conduct serial dilution using DMSO (0.2–10 mM). Dissolve each concentration using serum-free medium (5–100 μM) and prewarm to 37 °C. Incubation without troleandomycin serves as the control.
-
Dissolve luciferin-IPA substrate (3 μM) and salicylamide (3 mM) using serum-free medium and prewarm to 37 °C.
-
-
2.
CYP3A4 time-dependent inhibition: preincubation.
Wash DMSO-treated HuH-7 cells on 96-well plates with serum-free medium and incubate with troleandomycin (5–100 μM, 100 μL/well) for 0, 15, 30, and 60 min at 37 °C.
-
3.
CYP3A4 time-dependent inhibition: substrate incubation.
At different time points, wash cells with serum-free medium and incubate with CYP3A4 substrate luciferin-IPA (3 μM, Km value) at 37 °C (100 μL/well) for an additional 30 min. Include beetle luciferin standards (100 μL) in blank wells.
-
4.
Assay detection.
Terminate reaction by transferring 50 μL of incubation medium from each well to a separate white flat bottom plate with wells containing 50 μL of luciferin detection reagent at room temperature. Mix well and incubate the plate for 20 min, then measure luminescence using a plate reader (Fig. 3b).
-
5.
Calculate Parameters.
Obtain inactivation rate constant k obs by plotting the natural logarithm of the remaining CYP3A4 activity (%) against preincubation time with troleandomycin [I].
Calculate kinetic parameters k inact and K I by fitting data to the following equation using a nonlinear regression,
$$ {k}_{\mathrm{obs}}=\frac{k_{\mathrm{inact}}\left[\mathrm{I}\right]}{K_{\mathrm{I}}+\left[\mathrm{I}\right]} $$
3.4 Hepatotoxicity Assay
-
1.
Reagent preparation.
Nitrofurantoin and nefazodone, prepare 100 mM in DMSO, protect from light. Conduct serial dilution using basal medium (1–100 μM) on treatment day.
CellTiter-Glo assay, reconstitute and equilibrate at room temperature on assay day (24 h after treatment day).
-
2.
Treatment.
Incubate DMSO-treated HuH-7 cells with nitrofurantoin or nefazodone (100 μL/well) for 24 h at 37 °C. Incubation without chemicals serves as the control.
-
3.
Toxicity assay.
After treatment, equilibrate HuH-7 cells at room temperature for 30 min. Add 100 μL of CellTiter-Glo reagent to each well and mix well. Incubate the plate for additional 10 min, then measure luminescence using a plate reader (Fig. 4).
Fig. 4 
Cytotoxicity of nitrofurantoin and nefazodone (24 h) in DMSO-t reated HuH-7 cells. Figure reproduced from reference [5]
-
4.
Calculate EC 50, the half-maximal cytotoxic concentration of hepatotoxicants determined using a nonlinear regression Hill model.

4 Notes
-
1.
DMSO-treated HuH-7 cells maintain CYP3A4 activity in basal medium for at least 48 h after plating.
-
2.
Add 100 μL/well of beetle luciferin standards (0.2–80 nM) to blank wells on the 96-well plate, proceed together with samples for inhibition studies and luminescence detection. When calculate enzyme activity, use beetle luciferin standard curve range 0.1–40 nM.
-
3.
Prepare luciferin-IPA and salicylamide in serum-free medium, then dissolve various concentrations of ketoconazole using a multichannel reservoir.
References
Lin J, Schyschka L, Muhl-Benninghaus R, Neumann J, Hao L, Nussler N, Dooley S, Liu L, Stockle U, Nussler AK, Ehnert S (2012) Comparative analysis of phase I and II enzyme activities in 5 hepatic cell lines identifies Huh-7 and HCC-T cells with the highest potential to study drug metabolism. Arch Toxicol 86(1):87–95. doi:10.1007/s00204-011-0733-y
Donato MT, Jover R, Gomez-Lechon MJ (2013) Hepatic cell lines for drug hepatotoxicity testing: limitations and strategies to upgrade their metabolic competence by gene engineering. Curr Drug Metab 14(9):946–968
Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J (1982) Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42(9):3858–3863
Choi S, Sainz B Jr, Corcoran P, Uprichard S, Jeong H (2009) Characterization of increased drug metabolism activity in dimethyl sulfoxide (DMSO)-treated Huh7 hepatoma cells. Xenobiotica 39(3):205–217. doi:10.1080/00498250802613620
Liu Y, Flynn TJ, Xia M, Wiesenfeld PL, Ferguson MS (2015) Evaluation of CYP3A4 inhibition and hepatotoxicity using DMSO-treated human hepatoma HuH-7 cells. Cell Biol Toxicol 31(4-5):221–230. doi:10.1007/s10565-015-9306-9
Hisaka A, Ohno Y, Yamamoto T, Suzuki H (2010) Prediction of pharmacokinetic drug-drug interaction caused by changes in cytochrome P450 activity using in vivo information. Pharmacol Ther 125(2):230–248. doi:10.1016/j.pharmthera.2009.10.011
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Liu, Y. (2016). Study Liver Cytochrome P450 3A4 Inhibition and Hepatotoxicity Using DMSO-Differentiated HuH-7 Cells. In: Zhu, H., Xia, M. (eds) High-Throughput Screening Assays in Toxicology. Methods in Molecular Biology, vol 1473. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6346-1_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6346-1_7
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6344-7
Online ISBN: 978-1-4939-6346-1
eBook Packages: Springer Protocols