
Abstract
The digital restriction enzyme analysis of methylation (DREAM) is a simple method for DNA methylation analysis at tens of thousands of CpG sites across the genome. The method creates specific signatures at unmethylated and methylated CpG sites by sequential digests of genomic DNA with restriction endonucleases SmaI and XmaI, respectively. Both enzymes have the same CCCGGG recognition site; however, they differ in their sensitivity to CpG methylation and their cutting pattern. SmaI cuts only unmethylated sites leaving blunt 5′-GGG ends. XmaI cuts remaining methylated CCmeCGG sites leaving 5′-CCGGG ends. Restriction fragments with distinct signatures at their ends are ligated to Illumina sequencing adaptors with sample-specific barcodes. High-throughput sequencing of pooled libraries follows. Sequencing reads are mapped to the restriction sites in the reference genome, and signatures corresponding to methylation status of individual DNA molecules are resolved. Methylation levels at target CpG sites are calculated as the proportion of sequencing reads with the methylated signature to the total number of reads mapping to the particular restriction site. Aligning the reads to the reference genome of any species is straightforward, since the method does not rely on bisulfite conversion of DNA. Sequencing of 25 million reads per human DNA library yields over 50,000 unique CpG sites with high coverage enabling accurate determination of DNA methylation levels. DREAM has a background less than 1 % making it suitable for accurate detection of low methylation levels. In summary, the method is simple, robust, highly reproducible, and cost-effective.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
DNA methylation at CpG sites is an important epigenetic mechanism determining chromatin configuration and accessibility of genes for expression in mammalian cells [1]. Aging is associated with erosion of the epigenomic integrity that can be detected as discrete methylation changes [2, 3]. The disruption of epigenome is frequently accentuated in cancer leading to vast disorganization of DNA methylation patterns [4]. Aberrant DNA methylation interacts with genetic mutations in cancer development and progression [5–7]. Cancer-specific DNA methylation changes are used as biomarkers [8–10]. Next-generation or high-throughput sequencing made possible to assess DNA methylation profiles genomewide. Affinity enrichment for methylcytosine , bisulfite conversion of unmethylated cytosines, and restriction enzymes distinguishing methylated and unmethylated CpG sites are three main approaches used in DNA methylation analyses [11, 12].
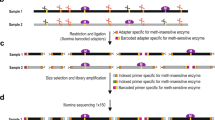
Here we present a simple method that can accurately measure DNA methylation levels at approximately 50,000–100,000 CpG sites across the genome [13]. The method is based on methylation-specific signatures created by restriction enzymes and deciphered by high-throughput sequencing. SmaI and XmaI are a unique pair of restriction endonucleases that target the same recognition site, CCCGGG, but differ in their sensitivity to CpG methylation and cutting pattern. SmaI cuts only unmethylated sites leaving blunt ends. The enzyme is completely blocked by CpG methylation. XmaI can cut both unmethylated and CpG-methylated sites (CCmeCGGG) leaving 5′CCGG overhangs. We utilize the enzymes sequentially. First, genomic DNA is exposed to SmaI that cuts all unmethylated sites, leaving all CCmeCGGG sites intact (Fig. 1a). Having digested all available unmethylated sites, we continue the cleavage by adding XmaI. The enzyme cuts the remaining sites that have been protected from SmaI by CpG methylation (Fig. 1b). The unmethylated sites thus have the GGG signature whereas the methylated sites have the CCGGG signature at the 5′ ends of the restriction fragments (Fig. 1c). Next, we stabilize the methylated signatures by filling the 3′ recesses using the exonuclease-deficient Klenow fragment DNA polymerase. This enzyme adds 3′-dA overhangs to all fragments for subsequent ligation of the sequencing adaptors. Standard procedures for making libraries and high-throughput sequencing follow. The sequencing reads are mapped to the SmaI/XmaI restriction sites in the reference genome. Methylation levels at individual CpG sites are calculated based on the counts of reads with methylated and unmethylated signatures mapping to the site. Paired end sequencing and 40 base read length is sufficient to map up to 200,000 unique CpG sites in the human genome. Using 25 million sequencing reads per sample typically yields more than 50,000 CpG sites covered with 20 and more reads. The assay is highly reproducible. Technical replicates show minimum differences and high correlation of methylation values at individual CpG sites (Figs. 2 and 3). Capture of consistent CpG sites with the high-sequencing depth makes the method suitable for analysis of large sample sets. Although the targets are restricted to accessible CCCGGG sites in the genome, the method has several advantages over bisulfite-based approaches. The background is below 1 %, since SmaI, the enzyme specific for unmethylated signature, is completely blocked by CpG methylation. Additionally, mapping the reads to the unconverted reference genome is computationally straightforward. The accuracy and reproducibility of DREAM is further increased by spiking the samples with standards of known methylation levels before enzymatic processing [13, 14]. These standards are used for fine adjustment of raw methylation levels and can be used for mitigating potential batch effects. We and others have successfully used DREAM as a robust, highly reproducible, and cost-effective method for global DNA methylation analysis of more than 1000 samples of human [3, 10, 13, 15–19], mouse [20], and zebrafish [21] cells and tissues.

Principle of the DREAM DNA methylation analysis. (a) SmaI restriction endonuclease cleaves CCCGGG sites in the middle. Only sites with unmethylated cytosines are cut, since CpG methylation completely blocks the enzyme from cutting. (b) XmaI restriction endonuclease added in the next step cleaves the remaining sites with methylated CpG cutting after the first cytosine. (c) Distinct GGG or CCGGG signatures are created at the 5′ ends of restriction fragments reflecting unmethylated or methylated CpG sites, respectively

Reproducibility of DREAM. Methylation differences at 66187 CpG sites covered with the minimum sequencing depth of 20 reads were evaluated in technical replicates. (a) Density plot shows bimodal distribution of methylation by the highest data density close to 0 % and 100 %. (b) Kernel density plot of methylation differences between technical replicates. Dotted lines: methylation differences smaller than 5 % were observed at 54740 CpG sites (83 % of total). Methylation differences 5–10 % were observed at 8066 CpG sites (12 % of total). Differences greater than 10 % were observed at 3381 CpG sites (5 % of total analyzed)

High correlation of methylation values in technical replicates. Methylation levels at CpG sites sequenced with 20 reads showed Pearson correlation r = 0.97 between technical replicates. This correlation was 0.997 for CpG sites sequenced with 100 reads
2 Materials
2.1 Making Methylation Standards
-
1.
E. coli genomic DNA, unsheared (Affymetrix, USB Cat# 14380).
-
2.
Lambda bacteriophage DNA (New England Biolabs).
-
3.
SmaI restriction endonuclease (New England Biolabs).
-
4.
HpaII CpG methyltransferase (New England Biolabs).
-
5.
HpaII restriction endonuclease (New England Biolabs).
-
6.
S-adenosylmethionine (SAM) (New England Biolabs).
-
7.
Oligonucleotide primers (Sigma, IDT or other suppliers).
-
8.
Taq DNA polymerase with ThermoPol® Buffer (New England Biolabs).
-
9.
TE buffer (TRIS 10 mM, EDTA 1 mM, pH 8.0).
-
10.
LTE buffer (TRIS 10 mM, EDTA 0.1 mM, pH 8.0).
-
11.
Sodium acetate 3 M, pH 5.0.
-
12.
Isopropanol .
-
13.
Ethanol.
-
14.
Bovine serum albumin (BSA).
-
15.
96-well PCR plates, not skirted.
-
16.
Adhesive seals for PCR plates.
-
17.
PCR thermal cycler.
-
18.
Agarose gel electrophoresis supplies.
-
19.
QIAquick PCR Purification Kit (Qiagen).
2.2 Construction of Sequencing Libraries
-
1.
SmaI restriction endonuclease (New England Biolabs).
-
2.
XmaI restriction endonuclease (New England Biolabs).
-
3.
Klenow fragment (3′ → 5′ exo-) (New England Biolabs).
-
4.
T4 DNA ligase (New England Biolabs).
-
5.
dNTP Set (100 mM each A,C,G,T) (GE Healthcare Life Sciences).
-
6.
NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1 and 2) (New England Biolabs).
-
7.
KAPA HiFi HotStart ReadyMix PCR Kit (Kapa Biosystems, Inc.).
-
8.
96-well PCR plates, not skirted.
-
9.
Adhesive seals for PCR plates.
-
10.
PCR thermal cycler.
2.3 Cleaning, Separation, and Quantitation of Sequencing Libraries
-
1.
Agencourt AMPure XP magnetic beads (Beckman Coulter).
-
2.
DynaMag-96 Side Magnetic Particle Concentrator (Invitrogen Cat. No. 123.31D).
-
3.
Ethanol.
-
4.
Molecular biology grade water.
-
5.
96-well PCR plates, not skirted.
-
6.
Qubit 2.0 fluorometer, dsDNA BR and HS Assay kits (Life Sciences).
-
7.
NanoDrop UV–vis Spectrophotometer (Thermo Scientific).
-
8.
Agilent 2100 Bioanalyzer (Agilent Technologies).
-
9.
Agilent High Sensitivity DNA Kit or DNA 1000 Kit (Agilent Technologies).
2.4 Next-Generation Sequencing
-
1.
Illumina HiSeq 2500 or a similar instrument for high-throughput sequencing.
-
2.
Linux server and/or bioinformatics support for processing of the sequencing data.
-
3.
Hard disk storage for the sequencing data.
3 Methods
3.1 Making Methylation Standard for Spiking in the Samples
For making an unmethylated standard LA168 as a PCR amplicon, use primers LA248F TCGAAAAAGAGCAGCACAGTGATGCCC and LA248R GTATGCCGCATTGCACTTT with lambda bacteriophage DNA (NEB #N3011) as the template:
-
1.
Mix the following reagents for PCR on ice: 800 ng lambda DNA in 340 μl of water, 40 μl of PCR buffer (final Mg2+ concentration 2 mM), 4 μL of 10 μM LA248F primer, 4 μl of 10 μM LA248R primer (final primer concentration 100 nM), 4 μl of 25 mM dNTP mix (final 250 μM), and 8 μl (40 U) of Taq polymerase. Aliquot the reaction mix to 16 wells in a PCR plate.
-
2.
Run the PCR program as follows: Initial denaturation at 94 °C for 3 min; 33 cycles consisting of denaturation at 94 °C for 15 s, annealing at 60 °C for 30 s, and elongation at 72 °C for 30 s; final extension at 72 °C for 5 min. Pool the PCR products from all aliquots into a single tube.
-
3.
Verify the presence of CCCGGG sites by SmaI digestion of 5 μl of the PCR product. Compare the digested and undigested PCR product using 2 % agarose gel electrophoresis . You should obtain a single band of 248 bp from the undigested PCR product and fragments of 168 bp, 53 and 27 bp after SmaI digestion. Purify the remaining PCR product using the PCR purification kit (Qiagen, QIAquick PCR Purification Kit #28104) following the manufacturer’s protocol. Measure DNA concentration of the unmethylated standard using NanoDrop spectrophotometer and/or Qubit fluorometer.
Making partially methylated (50 %) standard of E. coli genomic DNA
First, methylate completely E. coli genomic DNA with the HpaII methyltransferase at CpG sites in the CCGG recognition sequence. Check the completeness of methylation as the resistance to the HpaII restriction endonuclease . Next, make the 50 % methylated standard by mixing equal amounts of unmethylated and HpaII-methylated E. coli DNA (see Note 1 ).
-
4.
Methylate E. coli genomic DNA. Dissolve 10 mg of E. coli genomic DNA (Affymetrix, Part #14380) in 5 ml of TE buffer (10 mM Tris–HCl, EDTA 1 mM, pH 8.0). Set up methylation reaction M1 by pipetting 825 μl of nuclease-free water, 50 μl (100 μg) of E. coli gDNA, 100 μl of HpaII methylase buffer, and 2.5 μl of S-adenosylmethionine (SAM) 32 mM and 25 μl (100 units) of HpaII methyltransferase (NEB #M0214). SAM and the reaction buffer are provided with the enzyme. Set up a parallel tube M0 for unmethylated DNA: 825 μl of nuclease-free water, 50 μl (100 μg) of E. coli gDNA, 100 μl of HpaII methylase buffer, 2.5 μl of S-adenosylmethionine (SAM) 32 mM, and 25 μl of water instead of the HpaII methylase. Incubate both tubes overnight in a water bath at 37 °C. Incubate both tubes at 65 °C for 20 min to activate the enzyme.
-
5.
Check the completeness of methylation by HpaII restriction digest. Make the restriction buffer as follows: 360 μl of water, 40 μl of NEB Buffer 1 or NEB CutSmart® Buffer, and 4 μl of MgCl2 1 M. Mark four microcentrifuge tubes as E1, E2, E3, and E4. Add 40 μl of the restriction buffer to each tube. Add 10 μl of unmethylated E. coli DNA from tube M0 to tubes E1 and E2. Add 10 μl of HpaII-methylated E. coli reaction from tube M1 to tubes E3 and E4. Add 5 μl (50 units) of HpaII restriction endonuclease (NEB #R0171) in tubes E2 and E4. Incubate all tubes in a water bath at 37 °C for 1 h. Check the restriction digests by running electrophoresis of 20 μl of the reactions and a 100 bp DNA ladder (NEB #N3231) in 2 % agarose . Samples E1 and E3 are undigested controls. Sample E2 shows a smear of digested unmethylated DNA. If methylation of HpaII recognition sites (CCGG) is complete, sample E4 looks like E1 and E3 (Fig. 4).
Fig. 4 
Methylation of E. coli gDNA with HpaII methyltransferase. E. coli methylated at CCGG sites with HpaII methyltransferase is protected from cleavage with HpaII restriction endonuclease (lanes 7 and 8) while unmethylated DNA (lanes 3 and 4) is completely digested. Lanes 1, 2, 5, and 6 are controls without the restriction enzyme
-
6.
Clean the DNA in tubes M0 and M1 by alcohol precipitation and dissolve in TE. Add 111 μl of Na-acetate 3 M pH 5.0 to tubes M0 and M1 containing 1000 μl of DNA and mix by vortexing. Split the content of each tube in half by transferring 555 μl of the content to additional tubes M0 and M1. Add 389 μl (0.7 volume) of isopropanol to all tubes. Mix well by inverting and vortexing. Centrifuge the tubes at 12,000 g for 15 min. Pour out the supernatant while keeping the DNA pellet attached to the side of tubes. Wash the pellet with 1200 μl of 70 % ethanol two times. Spin briefly the empty tubes with DNA pellets after the second ethanol wash and carefully pipette out all traces of liquid using a fine tip. Add 100 μl of TE to each tube and dissolve the DNA pellets by incubation at 50 °C for 1 h followed by incubation at room temperature overnight. Pool the duplicate M0 and M1 tubes separately making M0 (unmethylated) and M1 (methylated) pools.
-
7.
Measure DNA concentration in M0 and M1 pools using spectrophotometer (NanoDrop) and/or fluorometer (Qubit). Make a stock solution of 50 % methylated E. coli standard by pooling of equal amounts of unmethylated (M0) and methylated (M1) DNA. Measure DNA concentration of the pool.
Making working solution of pooled methylation standards
-
8.
Make working solution of the methylation standards by diluting the stock of 50 % methylated E. coli solution to the concentration of 2 ng/μl in LTE buffer (10 mM Tris–HCl, 0.1 mM EDTA , pH 8.0) supplemented with bovine serum albumin (BSA) at 100 μg/ml. Add the unmethylated standard LA168, so that its final concentration is 2 pg/μl (i.e., 1000-fold lower than the concentration of E. coli DNA). Make 50 μl aliquots of the working solution of the methylation standards and keep them at −20 °C. Use 1 μl of the methylation standards to spike each sample before making DREAM libraries.

3.2 Preparing DNA Samples by Cleaning and Quantification
The DREAM assay requires unbroken genomic DNA free from traces of phenol, guanidinium salts or other potential contaminants that may interfere with enzymatic reactions. We recommend DNA purification using AMPure XP beads as the initial step before construction of sequencing libraries for all samples. The procedure removes small DNA fragments and traces of contaminants that may affect the activity of restriction enzymes and forming of methylation signatures:
-
1.
Measure DNA concentration in the samples using spectrophotometry (NanoDrop) and/or fluorometry (Qubit). Clean DNA should have OD ratio of 260/230 nm above 2.0 and 260/280 nm ratio in the range of 1.8–2.0. Check the integrity of gDNA by running electrophoresis of 200 ng of gDNA in 1 % agarose with 100 bp and 1 kb markers. Inspect the gel for the smear of fragmented DNA running below 500 bp and estimate the proportion of high molecular weight gDNA migrating above ~ 5 kb.
-
2.
Clean the genomic DNA sample and remove small fragments using AMPure XP beads at 0.5× beads to DNA ratio. Set up a 96-well PCR plate and mark wells for each DNA sample to be purified. We usually process 12 samples at a time. Ensure that AMPure XP beads have reached room temperature and resuspend them well before proceeding.
-
3.
Pipette aliquots of 2.5–10 μg of genomic DNA in the marked wells and adjust the volume to 70 μl with water (see Note 2 ). Add 35 μl of AMPure XP beads (0.4× ratio of beads to DNA) into the wells with DNA. Mix well by pipetting up/down. Incubate at room temperature for 15 min (see Note 3 ).
-
4.
Transfer the plate to magnetic stand and let it stand for 5 min to completely clear the solution of beads. With the plate on the magnetic stand, carefully remove 95 μl of clear binding buffer without drawing any beads. Save the supernatant containing low molecular weight DNA fragments in separate tubes for potential future use. High molecular weight DNA stays bound to the beads (see Note 4 ).
-
5.
Keep the plate on the magnetic stand and wash the beads with freshly prepared 80 % ethanol as follows: Add 200 μl of 80 % ethanol and allow to stand for 30 s. Remove and discard the supernatant. Repeat the wash with fresh 80 % ethanol one more time. Using a P20 pipette with a fine tip, carefully remove the remaining ethanol without disturbing the beads. Allow the beads air dry for 10 min or until fine cracks appear at the bead layer. Inspect each well carefully to ensure that all the ethanol has evaporated. It is critical that all residual ethanol be removed prior to continuing.
-
6.
Remove the plate from the magnet. Add 30 μl of room temperature nuclease-free water to the dried beads. Mix thoroughly to ensure all the beads are evenly suspended. Incubate at room temperature for 5 min (see Note 5 ).
-
7.
Place the plate on the magnetic stand. Incubate at room temperature for 2–5 min or longer until the supernatant gets completely clear. While keeping the plate on the magnet, carefully collect the eluted DNA, ensuring as few beads as possible are carried over and transfer the purified DNA to a fresh set of tubes.
-
8.
Measure DNA concentration in the samples using spectrophotometry (NanoDrop) and/or fluorometry (Qubit) and calculate the total amount of recovered DNA. One to two micrograms of purified genomic DNA are required for making high complexity DREAM libraries.
-
9.
Spike in 1 μl of methylation standards to each sample. It is important to add the standards in the samples before the restriction digests in the following steps.
3.3 Making DREAM Libraries for Illumina High-Throughput Sequencing
-
1.
Digest the DNA sample with spiked in standards with SmaI restriction endonuclease . The enzyme cuts all unmethylated CCCGGG sites creating blunt-ended fragments. Set up restriction digests in microcentrifuge tubes as follows: Add 21.5 μl of DNA sample (1–2 μg) spiked with 1 μl of methylation standards, 2.5 μl of CutSmart® Buffer, and 1 μl (20 units) of SmaI enzyme in the tube. Mix gently, spin briefly, and incubate in 25 °C water bath for 8 h.
-
2.
Continue the restriction digest by adding the second enzyme, XmaI restriction endonuclease. Add 20.5 μl of water, 2.5 μl of CutSmart® Buffer, and 2 μl (20 units) of the XmaI enzyme in each tube with the SmaI digest. Mix gently, spin briefly, and incubate in a 37 °C water bath overnight for 16 h.
-
3.
Fill the 3′ recesses left by the XmaI digest and add 3′ dA overhangs to all fragments. Add to each tube with SmaI/XmaI digests 2 μl of the CGA mix (dCTP, dGTP, and dATP, 10 mM each), 3 μl of Klenow fragment (3′ → 5′ exo–) DNA polymerase and mix. Spin briefly and incubate in a water bath at 37 °C for 30 min.
Purify the DNA fragments by AMPure XP beads (2.0× volume ratio of beads to DNA). Ensure that the beads have reached room temperature and resuspend them well before proceeding. Set up a 96-well PCR plate and mark wells for each sample to be purified.
-
4.
Add 110 μl of AMPure XP beads into the marked wells. Add the contents of the end-filling-A-tailing reactions (55 μl) in the wells with beads. Mix well by pipetting up and down. Incubate at room temperature for 15 min (see Note 3 ).
-
5.
Transfer the plate to the magnetic stand, and let it stand for 5 min until the solution becomes completely clear and the beads attached to the wall.
-
6.
Carefully remove 150 μl of the binding buffer and discard it. Leaving some of the volume behind minimizes bead loss at this step.
-
7.
Keep the plate on the magnetic stand and wash the beads with freshly prepared 80 % ethanol follows: Add 200 μl of 80 % ethanol and allow to stand for 30 s. Remove and discard the supernatant. Repeat the wash with fresh 80 % ethanol one more time. Using a P20 pipette with a fine tip, carefully remove the remaining ethanol without disturbing the beads. Allow the beads air dry for 10 min or until fine cracks appear at the bead layer. Inspect each well carefully to ensure that all the ethanol has evaporated. It is critical that all residual ethanol be removed prior to continuing.
-
8.
Remove the plate from the magnet. Add 30 μl of room temperature nuclease-free water to the dried beads. Mix thoroughly to ensure all the beads are evenly suspended. Incubate at room temperature for 5 min (see Note 5 ).
-
9.
Place the plate on the magnetic stand. Incubate at room temperature for 2–5 min or longer until the supernatant gets completely clear. While keeping the plate on the magnet, carefully collect the eluate with DNA, ensuring as few beads as possible are carried over and transfer the digested dA-tailed DNA to a fresh PCR plate labeled “Ligation.”
-
10.
Ligate sequencing adapters to the restriction fragments. Add to each well with DNA the following reagents: 3 μl of 10× ligation buffer (NEB #B0202S), 0.5 μl of NEBNext adaptor for Illumina 10 μM (from NEB kits #E7335 or #E7500), and 2 μl (800 U) of T4 DNA ligase (NEB #M0202). Mix by pipetting up and down. Cover the wells with adhesive seal. Spin briefly. Incubate the ligation reaction at 16 °C overnight.
-
11.
Cleave the hairpin loops in the NEBNext adaptors. Add 3 μl of USER enzyme to each reaction and pipette up and down to mix. Incubate at 37 °C for 15 min.
Clean up the ligated DNA and perform dual size selection using AMPure XP beads. First, remove large DNA fragments (>450 bp) by binding to beads used at the 0.6× ratio of AMPure beads to sample. At this step, collect the supernatant containing smaller DNA fragments (<450 bp). Next, transfer the supernatant into new wells and add additional beads. Addition of AMPure beads will increase the concentration of PEG and salt and result in binding of DNA fragments larger than 250 bp to the beads. The dual SPRI size selection will isolate target DNA fragments (250–450 bp) and leave behind unligated adaptors, self-ligated adaptor dimers, and large DNA fragments.
-
12.
Add 66.5 μl of water into the wells containing the 33.5 μl of the ligation reaction from the previous step to increase the DNA volume to 100 μl. Add 60 μl of AMPure XP beads (0.6× volume). Mix well by pipetting up and down. Incubate at room temperature for 15 min.
-
13.
Transfer the plate to the magnetic stand and let the beads completely separate from the supernatant for 5 min or more. Carefully collect 155 μl of the supernatant containing short DNA fragments (<450 bp) and transfer to a new set of wells. Note: The beads with large DNA fragments are left behind.
-
14.
Add 20 μl of AMPure XP beads to the 155 μl of clear supernatant in a new set of wells and mix thoroughly by pipetting up/down. Incubate at room temperature for 15 min.
-
15.
Transfer the plate to the magnetic stand and let the beads completely separate from the supernatant for 5 min or more.
-
16.
Carefully remove only 160 μl of the binding buffer and discard it. Leaving some of the volume behind minimizes bead loss at this step. DNA fragments larger than 250 bp are attached to the beads. Note: The beads should not disperse; instead, they will stay on the walls of the tubes. Significant loss of beads at this stage will impact the yield, so ensure beads are not removed with the binding buffer or the wash.
-
17.
With the plate still on the magnet, add 200 μl of freshly prepared 80 % ethanol and allow to stand for 1 min. Remove the ethanol wash using a pipette. Repeat the 80 % ethanol wash one more time, for a total of two washes. Using a fine tip, carefully remove all traces of ethanol from the bottom of each well. Note: It is critical to remove as much of the ethanol as possible after the final wash.
-
18.
Air dry the beads while keeping the plate on the magnet for a minimum of 10 min. Inspect each well carefully to ensure that all the ethanol has evaporated. It is critical that all residual ethanol be removed prior to continuing.
-
19.
Remove the plate from magnet. Add 25 μl of room temperature nuclease-free water to the dried beads. Mix thoroughly to ensure that beads in all wells are homogeneously resuspended. Incubate at room temperature for 5 min (see Note 5 ).
-
20.
Transfer the plate to the magnet and let it stand for 2 min or until the supernatant becomes clear.
-
21.
While keeping the plate on the magnet, carefully collect 22.5 μl of the eluate, ensuring as few beads as possible are carried over. Transfer the eluted DNA to a new plate labeled “PCR.”
Perform ligation-mediated PCR amplification. Use NEBNext oligos for Illumina (NEB #7335 with barcodes 1–12 and/or NEB #7500 with barcodes 13–27). Make sure to use a unique barcode for each sample.
-
22.
Transfer the PCR plate with DNA on ice. Add to each well 1.25 μl of NEBNext universal primer , 1.25 μl of the NEBNext Index i7 primer with a sample-specific barcode, and 25 μl of KAPA HiFi HotStart ReadyMix 2× (Kapa Biosystems, Inc. #KK2601). Seal the plate, spin briefly, and return on ice.
-
23.
Perform PCR amplification using the initial denaturation temperature of 98 °C for 45 s, followed by 11–12 cycles at 98 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. Add the final extension step at 72 °C for 1 min after the cycling (see Note 6 ).
Perform a post PCR cleanup with AMPure XP beads (1.2× volume beads to DNA ratio).
-
24.
Add 60 μl of AMPure beads into wells with PCR reactions. Mix well by pipetting up/down. Incubate at room temperature for 15 min.
-
25.
Transfer the plate to the magnet and let it stand for 5 min to completely clear the solution of beads.
-
26.
Carefully remove only 94 μl of the binding buffer and discard it. Leaving some of the volume behind minimizes bead loss at this step (see Note 4 ). With the plate still on the magnet, add 200 μl of freshly prepared 80 % ethanol and allow to stand for 1 min. Remove the ethanol wash using a pipette. Repeat the 80 % ethanol wash one more time, for a total of two washes. Using a fine tip, carefully remove all traces of ethanol from the bottom of each well (see Note 7 )
-
27.
Air dry the beads on the magnet for a minimum of 10 min. Inspect each well carefully to ensure that all the ethanol has evaporated (see Note 7 ).
-
28.
Remove the plate from the magnet. Add 30 μl room temperature nuclease-free water to the dried beads. Mix thoroughly to ensure all the beads are homogeneously resuspended. Incubate at room temperature for 5 min (see Note 5 ).
-
29.
Transfer tubes to magnet and let it stand for 2 min or until the supernatant clears out completely.
-
30.
While keeping the plate on the magnet, carefully collect the finished sequencing libraries in 25 μl of the eluate, ensuring as few beads as possible are carried over and transfer to a fresh set of tubes labeled with sample IDs.
3.4 Quality Control
-
1.
Measure DNA concentration in the samples using high sensitivity fluorometry (Qubit HS). Analyze the size distribution and the molarity of the library by Agilent 2100 Bioanalyzer electrophoresis using Agilent High Sensitivity DNA Kit (Fig. 5).
Fig. 5 
Size distribution of typical DREAM libraries. Agilent 2100 Bioanalyzer electrophoresis using standard sensitivity DNA 1000 kit. (a) DREAM library made from human blood gDNA. (b) DREAM library made from mouse liver gDNA
-
2.
Pool 12 samples with unique barcodes for Illumina sequencing using equal amount of femtomoles or nanograms of DNA for each sample (see Note 8 ).

3.5 High-Throughput Sequencing and Data Processing
-
1.
Sequence the pooled libraries on the Illumina high-throughput instrument using a paired end setup and 40–50 bases read length (see Note 9 ).
-
2.
Obtain the sequences in the fastq format. Align the sequences to the reference genome supplemented with the sequences of the methylation standards (see Note 10 ).
-
3.
Bowtie2 generates files in a generic Sequence Alignment/Map (SAM) format. Next, use SAMtools for conversion of SAM files into sorted indexed binary (BAM) files.
-
4.
Count methylation signatures and compute methylation levels. Use the sorted BAM files as input for the custom Python script which assigns the reads to individual CCCGGG sites positioned in the genome. The script counts the reads with methylated and unmethylated signatures and calculates raw methylation values as the percentage of reads with the methylated signature in the total number of reads. The Python script and annotation tables of target sites for several reference genomes are available at https://github.com/jmadzo/DREAM_project/ and https://github.com/jaroslavj/DREAM_tools.
-
5.
We recommend to normalize the raw methylation values using the observed and expected methylation values of the spiked in methylation standards [13, 14] (see Note 11 ).
4 Notes
-
1.
Complete methylation of CpG s in the CCGG recognition sequence is easily achievable by a single treatment of DNA with the HpaII methyltransferase (Fig. 4). All CCGG sites are inside the CCCGGG sites targeted by DREAM. In contrast, multiple enzymatic treatments are needed to achieve complete methylation of all genomic CpG sites using the M.SssI methyltransferase.
-
2.
The amount of DNA used for cleaning should be approximately 2.5–10 μg, depending on availability. The yield of the cleaning procedure depends on the purity and integrity of the DNA sample. One microgram of cleaned high molecular weight gDNA is sufficient for the DREAM assay. The minimum amount for making high complexity libraries is approximately 500 ng.
-
3.
Shaking a sealed plate on a microplate shaker at 1800 rpm may facilitate the binding of DNA to beads. Spin the plate briefly after removing from the shaker.
-
4.
The beads should not disperse; instead, they should stay attached to the side walls of the wells. Significant loss of beads will impact the yield, so ensure beads are not removed with the binding buffer or the wash.
-
5.
Shaking a sealed plate on a microplate shaker at 1800 rpm may facilitate the elution of DNA from the beads. Spin the plate briefly after removing from the shaker.
-
6.
Adjust the optimal number of PCR cycles so that the final concentration of DNA purified after the amplification is in the range of 10–20 ng/μl. Libraries with concentrations lower than 5 ng/μl could be re-amplified by additional 4–6 PCR cycles. Primers for Illumina termini P5 and P7 AATGATACGGCGACCACCGAGATCTACAC and CAAGCAGAAGACGGCATACGAGAT can be used for all libraries, since the sample-specific barcodes were set by the previous PCR (Subheading 3.3, steps 22–23).
-
7.
It is critical to remove as much of the ethanol as possible after the final wash.
-
8.
Pooling of 12 DREAM libraries works well for Illumina Rapid Run flow cell. The number of samples to be pooled should be adjusted to obtain approximately 25 million paired end reads per sample.
-
9.
Spike the sequencing pool with 10 % PhiX library to ensure sufficient diversity at first 5 bases. DREAM libraries have either C or G at bases number 1 and 2. Base 3 is invariably G. Spiking in a percentage of PhiX control library increases the nucleotide balance and makes clusters easier for the software to identify.
-
10.
We first build a bowtie2 index combining the reference genome, the E. coli genome, and the methylation standards added as extra chromosomes. We align the fastq reads using the Bowtie2 aligner with the sensitive option.
-
11.
We use the spiked in methylation standards to compensate for potential distortions of raw methylation values expressed as the percentage of methylated reads. We compare the expected and observed ratios of methylated to unmethylated reads mapped to the calibrators and calculate the correction coefficient. We first verify that the background methylation level of the completely unmethylated standard is less than 1 %. Next, we calculate the difference d of log ratios log (M/U) and log (m/u) for each standard, where M/U is the expected ratio of methylated and unmethylated reads and m and u are observed numbers of methylated and unmethylated reads (a). Correction factor c is expressed as an antilog of the average log difference (expected–observed) from all standards (b). Finally, we compute adjusted methylation values for each CpG site (c). We add 0.5 reads to the number of methylated and unmethylated reads to avoid division by zero.
-
(a)
d = log (M/U) – log (m/u)
-
(b)
c = exp(mean(d))
-
(c)
adjusted methylation = c*(m + 0.5)/[c*(m + 0.5) + u + 0.5]
-
(a)
References
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492
Issa JP (2014) Aging and epigenetic drift: a vicious cycle. J Clin Invest 124:24–29
Maegawa S, Gough SM, Watanabe-Okochi N, Lu Y, Zhang N, Castoro RJ, Estecio MR, Jelinek J, Liang S, Kitamura T, Aplan PD, Issa JP (2014) Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res 24:580–591
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128:683–692
Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jäger N, Jones DT, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJ, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FM, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, Van Meter T, Grajkowska W, Lach B, Kulozik AE, von Deimling A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A, Taylor MD (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506:445–450
Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, Goodell MA (2015) Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 125:629–638
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11:726–734
Issa JP (2012) DNA methylation as a clinical marker in oncology. J Clin Oncol 30:2566–2568
Chung W, Bondaruk J, Jelinek J, Lotan Y, Liang S, Czerniak B, Issa J-PJ (2011) Detection of bladder cancer using novel DNA methylation biomarkers in urine sediments. Cancer Epidemiol Biomarkers Prev 20:1483–1491
Foy JP, Pickering CR, Papadimitrakopoulou VA, Jelinek J, Lin SH, William WN, Frederick MJ, Wang J, Lang W, Feng L, Zhang L, Kim ES, Fan YH, Hong WK, El-Naggar AK, Lee JJ, Myers JN, Issa JP, Lippman SM, Mao L, Saintigny P (2015) New DNA methylation markers and global DNA hypomethylation are associated with oral cancer development. Cancer Prev Res (Phila) 8:1027–1035
Laird PW (2010) Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11:191–203
Plongthongkum N, Diep DH, Zhang K (2014) Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet 15:647–661
Jelinek J, Liang S, Lu Y, He R, Ramagli LS, Shpall EJ, Estecio MR, Issa JP (2012) Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics 7:1368–1378
Risso D, Ngai J, Speed TP, Dudoit S (2014) The role of spike-in standards in the normalization of RNA-seq. In: Datta S, Nettleton D (eds) Statistical analysis of next generation sequencing data. Frontiers in Probability and the Statistical Sciences. Springer International Publishing, Switzerland, pp 169–190
Malouf GG, Taube JH, Lu Y, Roysarkar T, Panjarian S, Estecio MR, Jelinek J, Yamazaki J, Raynal NJ, Long H, Tahara T, Tinnirello A, Ramachandran P, Zhang XY, Liang S, Mani SA, Issa JP (2013) Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol 14:R144
Jin C, Lu Y, Jelinek J, Liang S, Estecio MR, Barton MC, Issa JP (2014) TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res 42:6956–6971
Qin T, Si J, Raynal NJ, Wang X, Gharibyan V, Ahmed S, Hu X, Jin C, Lu Y, Shu J, Estecio MR, Jelinek J, Issa JP (2015) Epigenetic synergy between decitabine and platinum derivatives. Clin Epigenetics 7:97
Malouf GG, Tahara T, Paradis V, Fabre M, Guettier C, Yamazaki J, Long H, Lu Y, Raynal NJ, Jelinek J, Mouawad R, Khayat D, Brugières L, Raymond E, Issa JP (2015) Methylome sequencing for fibrolamellar hepatocellular carcinoma depicts distinctive features. Epigenetics 10:872–881
Yamazaki J, Jelinek J, Lu Y, Cesaroni M, Madzo J, Neumann F, He R, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Kantarjian HM, Liang S, Estecio MR, Godley LA Issa JP (2015) TET2 mutations affect non-CpG island DNA methylation at enhancers and transcription factor-binding sites in chronic myelomonocytic Leukemia. Cancer Res 75:2833–2843
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, Liang S, Lu Y, Darlington GJ, Meissner A, Issa JP, Godley LA, Li W, Goodell MA (2012) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44:23–31
van Esterik JC, Vitins AP, Hodemaekers HM, Kamstra JH, Legler J, Pennings JL, Steegenga WT, Lute C, Jelinek J, Issa JP, Dollé ME, van der Ven LT (2014) Liver DNA methylation analysis in adult female C57BL/6JxFVB mice following perinatal exposure to bisphenol A. Toxicol Lett 232:293–300
Acknowledgments
We thank Dr. Jean-Pierre Issa for the support and expert advice, Justin T. Lee and Bela Patel for the technical expertise in making of DREAM libraries, Dr. Yue-Sheng Li at Fox Chase Cancer Center Genomic Facility for the high-throughput sequencing, Dr. Matteo Cesaroni for the generous help with bioinformatics and data processing, and Dr. Amy B. Hart for the editorial help.
This work was supported by NIH grants R01-HD075203 from NICHD and P01-CA049639 from NCI.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Jelinek, J., Madzo, J. (2016). DREAM: A Simple Method for DNA Methylation Profiling by High-throughput Sequencing. In: Li, S., Zhang, H. (eds) Chronic Myeloid Leukemia. Methods in Molecular Biology, vol 1465. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-4011-0_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-4011-0_10
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-4009-7
Online ISBN: 978-1-4939-4011-0
eBook Packages: Springer Protocols