
Abstract
There is biological and clinical evidence that mammalian tumors are highly heterogeneous at single-cell level, and that only a minority of the cancer cells have limitless replicative potential in vitro and in vivo. Here we review the different strategies currently used for the functional isolation of cancer cells with in vivo cancer initiation potential and self-renewal. These tools are currently used to better define their molecular, phenotypic, and drug-resistance characteristics in preclinical and clinical studies.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Cancer stem cells
- Cancer-initiating cells
- Methods
- FACS
- Side population
- Aldehyde dehydrogenase
- Tumorspheres
- Tumorigenicity
1 Introduction
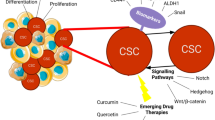
The majority of human cancers display a significant degree of intratumor heterogeneity in various phenotypic and functional features, such as differentiation grade, cellular morphology, gene expression, proliferative rate, and angiogenic and metastatic potential [1]. Growing evidence suggests that tumor cell heterogeneity might be attributed to a clonal evolutionary process , driven by genetic instability, and to differentiation of stem-like cells, the so-called cancer stem cells or cancer-initiating cells (CSCs/CICs) [2]. CSCs are described as a small population of undifferentiated cells characterized by indefinite self-renewal ability (through asymmetrical cell division), high tumorigenicity , and differentiation into non-CSC progeny that leads to the production of all cell types in the tumor, thus contributing to intratumor heterogeneity. According to the CSC hypothesis, tumorigenic stem-like cells within heterogeneous tumors are responsible for the tumor initiation and for the generation of distant metastasis [3, 4]. Moreover, since CSCs are highly resistant to chemotherapeutics, they are involved in local or metastatic tumor recurrence after therapy [5, 6].
Malignant stem cells were first identified in 1994 by John Dick and colleagues in acute myeloid leukemia (AML) [7]. The Dick laboratory isolated a phenotypically distinct subset of leukemic cells (CD34+CD38−) from the peripheral blood of AML patients, characterized by a rate of self-renewal higher than normal hematopoietic stem cells. Only these cells, with stem-like properties, were able to recapitulate the morphological features of the original malignancy upon transplantation into immunocompromised mice in limiting dilution assays. This pioneering work provided the paradigm for the general CSC model and laid the foundations for all CSC-related studies in solid tumors that followed.
Based on similar techniques previously applied to the hematopoietic model, Al-Hajj and colleagues have been the first to identify and characterize CSCs from breast cancer [8]. Since then, CSCs have been identified in various solid tumors , including melanoma [9], glioblastoma [10], lung carcinoma [11], prostate [12], and ovarian cancer [13], still using the same approach of combined flow cytometry and serial xenotransplantation in mice.
However, since the identification of rare malignant stem cells via flow cytometry is a critical step [14, 15], further methodologies for CSC isolation have been improved by using marker-independent strategies. Thus, the use of CSC-specific surface markers must be associated with other functional assays, namely (a) detection of side-population (SP) phenotypes by Hoechst 33342 dye exclusion [16, 17], (b) assessment of aldehyde dehydrogenase (ALDH) enzymatic activity [18, 19], and (c) evaluation of growth potential as floating spheres in serum-free medium [20, 21].
Since all the methods above mentioned have their limitations in identifying CSCs unequivocally, it might be more advantageous to use combinatorial markers and methodologies, especially when a whole tumor tissue is analyzed [22]. Indeed, prospective identification and isolation of CSCs from solid tumors, and a better understanding of the properties of these cells, will lead to the development of novel therapeutic strategies aimed to eradicate the engine of tumor progression. We describe here methods currently used for CSC identification and characterization, starting from primary solid tumors .
2 Materials
2.1 Laboratory Equipment
-
1.
Laminar flow biosafety cabinet (BSL2).
-
2.
Water bath.
-
3.
Hemocytometer.
-
4.
Inverted microscope.
-
5.
37 °C Humidified incubator with 5 % CO2.
-
6.
Falcon 40 μm cell strainers.
-
7.
0.20 μm Syringe filters.
2.2 Sample Acquisition and Dissociation
-
1.
Dissociation solution: Dulbecco’s modified Eagle’s medium:Ham’s nutrient mixture F-12 (1:1) (DMEM/F12) supplemented with 1× collagenase/hyaluronidase mixture (10× stock, Stem Cell Technologies) and 125 U/mL DNasi I (Life Technologies).
-
2.
RNAlater RNA Stabilization Reagent (QIAGEN).
-
3.
4 % Formaldehyde solution .
-
4.
Optimal cutting temperature (O.C.T.) solution (Bio Optica).
-
5.
Nalgene cryogenic vials.
-
6.
Nalgene Cryo 1 °C cell-freezing container.
-
7.
Dimethyl sulfoxide (DMSO).
-
8.
Liquid nitrogen transfer container.
-
9.
Dry ice.
-
10.
Forceps.
-
11.
Scalpels.
2.3 Cell Culture
-
1.
Complete growth medium: DMEM/F12 supplemented with 10 % fetal bovine serum (FBS) heat inactivated, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin.
-
2.
Trypsin-EDTA 1× in PBS without Phenol Red Ca2+ and Mg2+.
-
3.
PBS without Ca2+ and Mg2+.
-
4.
T-25 and T-75 tissue culture flasks.
-
5.
Various-sized multiwell plates .
2.4 Antigen Expression and Cell Sorting
-
1.
Navios Flow Cytometer (Beckman Coulter).
-
2.
Influx cell sorter (BD Biosciences).
-
3.
Ethylenediamine tetraacetic acid (EDTA).
-
4.
Bovine serum albumin (BSA).
-
5.
Red blood cell lysis buffer: 10 mM Potassium bicarbonate (KHCO3), 155 mM ammonium chloride (NH4Cl), 0.1 mM EDTA.
-
6.
7-Aminoactinomycin D (7-AAD) Viability Dye (Beckman Coulter).
-
7.
SYTO 16 Green Fluorescent Nucleic Acid Stain (Life Technologies).
-
8.
CD133/1 (AC133, Prominin1) (Miltenyi Biotec) and respective isotype control.
2.5 Marker-Independent Methods
-
1.
Verapamil (Sigma-Aldrich).
-
2.
Hoechst 33342 (Sigma-Aldrich).
-
3.
ALDEFLUOR™ kit (STEMCELL Technologies).
-
4.
Complete tumorsphere medium: DMEM/F12 supplemented with 5 μ/mL insulin, 20 ng/mL epidermal growth factor (EGF), 20 ng/mL basic fibroblast growth factor (bFGF), and 1× B27 supplement (50× stock, Life Technologies).
-
5.
Ultra-low-attachment plates.
2.6 In Vivo Tumorigenicity
-
1.
NOD/SCID interleukin-2 receptor γ (IL-2Rγ)–null (NSG) immunocompromised mice.
-
2.
BD Matrigel™ Basement Membrane Matrix (BD Biosciences).
-
3.
Round-bottomed 96-well microplates.
-
4.
Insulin syringes or Hamilton syringe (2biological Instruments).
-
5.
Avertin (2,2,2-Tribromoethanol) (Sigma-Aldrich).
-
6.
Hematoxylin and eosin.
3 Methods
3.1 Tumor Specimen Acquisition and Collection
Written informed consent and institutional review board (IRB) approval for research on human samples are basic prerequisites for all specimen acquisition. Fresh tissue sample from cancer resection should be collected in a sterile container as soon as possible after removal from the operating room . For nucleic acid isolation, it is crucial that the specimen must be frozen within 20 min in order to prevent degradation (RNA starts to degrade within minutes). Tumor fragments should be flash frozen and kept at −20 °C or −80 °C, or in liquid nitrogen for long-term storage. This provides excellent tissue integrity and a wide array of options for tissue analysis. Alternatively, immediately stabilize the sample in RNAlater RNA Stabilization Reagent (see Note 1 ). For xenotransplantation studies time is less critical, but the tissue should be kept refrigerated and in medium until transplantation. In our experience, patient-derived xenografts have been successfully established by implanting tumor samples after overnight storage at 4 °C. Additionally, save part of the specimen for further histological characterization by embedding a portion of the tumor in optimal cutting temperature (O.C.T.) solution, followed by flash freezing (see Note 2 ), or by fixing one part of the tumor in 4 % phosphate-buffered formalin and embedding in paraffin.
3.2 Sample Dissociation
Carry out all procedures in aseptic conditions under a laminar flow hood to minimize contaminations. Note that all fresh tissue should be handled in a biosafety cabinet using Biosafety Level 2 (BSL2) techniques.
-
1.
Prepare dissociation solution as described in Subheading 2.2 and place into a 50 mL conical tube.
-
2.
Transfer the tumor to a sterile petri dish using sterile forceps.
-
3.
Mince the sample with two scalpels in a cross-wise manner until tumor is reduce to a paste and the final tissue fragments are small enough to pass through the tip of a 5 mL pipette (see Note 3 ).
-
4.
Transfer the cut-up tumor to the tube containing the dissociation solution (prepared in step 1) and incubate into a 37 °C water bath (see Note 4 ).
-
5.
Mix up and down the tumor fragments every 15 min using a 5 mL pipette, and then a 1000 μL micropipette to further disintegrate cell clumps, until the tumor is well dissociated (see Note 5 ).
-
6.
Filter the cell suspension through a 40 μm nylon mesh cell strainer into a new 50 mL sterile tube and use the rubber end of the syringe plunger to break up clumps by gently squeezing cells against the cell strainer.
-
7.
Thoroughly rinse the strainer with PBS and recover any residual from underneath the cell strainer in order to minimize cell loss.
-
8.
Centrifuge for 5 min at 350 × g, discard the supernatant, and resuspend the cell pellet in a small volume of cold ammonium chloride lysis buffer (prepared as described in Subheading 2.4) to lyse red blood cells (see Note 6 ); incubate on ice for 5–10 min, wash with a 10× volume of PBS, and centrifuge again.
-
9.
Resuspend cells in DMEM/F12 and calculate the viable cell concentration with a hemocytometer and trypan blue.
3.3 In Vitro Expansion of Primary Tumor Cells
-
1.
After evaluation of viability, resuspend cells in complete growth medium described in Subheading 2.3.
-
2.
Plate the primary cell suspension in standard tissue culture flasks; use T25 flasks or multiwell plates depending on how many cells are available.
-
3.
Culture cells in a 37 °C humidified incubator with 5 % CO2; viable tumor cells should attach to the flask within 12–24 h.
-
4.
Inspect the culture regularly by inverted microscope to monitor cell growth and confluency; replace medium weekly, or when required as it looks depleted (orange/yellow color), by changing half volume with fresh complete culture medium (see Note 7 ).
-
5.
Split the culture when cells are 70–90 % confluent by trypsinization with an appropriate volume of 0.25 % trypsin and 0.02 % EDTA until all adherent cells detach (about 3–5 min).
-
6.
When all cells are detached, inactivate trypsin by adding an equal volume of complete DMEM/F12 and wash the flask with PBS to collect all detached cells into a 15 mL tube.
-
7.
Centrifuge at 350 × g for 5 min, discard supernatant, and resuspend the cell pellet in fresh complete culture medium.
-
8.
Perform a cell count using trypan blue exclusion assay on a hemacytometer (see Note 8 ).
-
9.
Replate cells at a concentration between ranges of 1 and 2 × 104 cells/cm2.
-
10.
Continue cell culture for up to five passages.
3.4 Methods to Characterize CSCs
The two main methods that are used to prospectively purify and characterize CSCs include marker-dependent and marker-independent strategies. Flow cytometry played a crucial role in identifying CSCs either by marker-dependent and by marker-independent methods, such as side-population analysis, aldefluor assay, and sphere formation assays, combined with limiting dilution xenotransplantation in immunodeficient mice. Here, we describe the principal in vitro and in vivo strategies used to identify CSCs. Importantly, the CSC-specific detection methods illustrated hereinafter have their limitations in identifying malignant stem cells unequivocally, especially when tumor tissue is analyzed. Therefore, the use of combinatorial markers and methodologies is imperative in order to isolate and characterize a putative CSC population.
3.4.1 Antigen Expression and Cell Sorting
CSCs are frequently identified and characterized by multiparametric flow cytometric analysis that represents a powerful technique to simultaneously interrogate the phenotype of single cells in suspension with high performance and reliability. Furthermore, it enables the separation of living cells by fluorescence-activated cell sorting (FACS), using antibodies directed at cell-surface markers in order to isolate rare CSCs from a large heterogeneous cell population.
-
1.
Harvest cells within 7 days of primary cultures, wash with PBS, and perform a cell count and viability analysis.
-
2.
Split cells in two aliquots for flow cytometric analysis and for cell sorting, respectively.
-
3.
Adjust cell number to a concentration of 1–5 × 106 cells/mL in FACS buffer (PBS with 2 mM EDTA and 0.5 % bovine serum albumin).
-
4.
Add the appropriate number of cells to be stained into FACS tubes.
-
5.
Add conjugated antibody (0.1–10 μg/mL, or a previously titrated amount) or respective isotype controls and vortex gently to mix. Incubate cells for at least 15 min at 4 °C in the dark (see Note 9 ).
-
6.
Remove any unbound antibody by washing cells in FACS buffer. Centrifuge at 350 × g for 5 min and decant the buffer.
-
7.
Resuspend stained cells in 200–400 μL of FACS buffer for flow cytometric analysis (see Note 10 ). Keep the cells at 4 °C until your scheduled time for analysis.
-
8.
Acquire cells (at least 1 × 106 total cells per sample) on a standard flow cytometer and perform analysis after selection of DNA+ (Syto16+) and viable (7-AAD−) cells. Set the analysis gates with the aid of “fluorescence minus one” isotype controls.
-
9.
The staining panel, both for analysis and cell sorting, includes various markers depending on the tumor type (an overview of markers used for the characterization of CSCs in different tumors is given in ref. 23). For instance, CD133 (prominin-1) has been recognized as a putative CSCs marker for glioblastoma, breast, colon, prostate, kidney, pancreatic, and lung carcinomas, liver and brain tumors , and melanoma [4] (see Note 11 ).
-
10.
For cell sorting, resuspend stained cells from step 7 in appropriate volume of FACS buffer so that the final cell concentration is 4–5 × 106 cells/mL.
-
11.
Sort CD133-positive and CD133-negative fractions using a three laser Influx high speed cell sorter (BD Biosciences) equipped with a Class I biosafety cabinet (see Note 12 ). The purity of CD133+ and CD133− sorted populations is routinely 90 %.
-
12.
Culture sorted cell populations in standard medium for further in vitro and in vivo experiments.
3.4.2 Side-Population Assay
The side-population (SP) discrimination assay is a flow cytometric strategy used to isolate CSCs by efflux of incorporated dyes like Hoechst 33342, via multi-drug resistance (MDR) and ATP-binding cassette (ABC) transporters expressed within the cell membrane. A peculiar characteristic of both normal and malignant stem cells is the overexpression of ABC transporters as efflux pumps protect stem cells from damage by xenobiotic substances [24]. Then, the flow cytometric profile of the cells which actively efflux the dye out has a distinctive feature and they form a distinct small cell population (<0.1 % of the total cell number), termed the “side population,” that shows little or no fluorescence with Hoechst 33342 [25]. Thus, the use of Hoechst 33342 to detect CSCs as an SP overcomes the lack of a tumor-specific phenotype and replaces it with a more direct and functional marker . Results obtained with this method must be rigorously validated anyway using other CSC-specific detection methods described in the previous and following sections.
-
1.
Count viable nucleated cells, resuspend in pre-warmed standard medium, and divide cells in two aliquots; adjust cell number to a concentration of 1 × 106 cells/mL in medium.
-
2.
Add to both cell suspensions the Hoechst 33342 stock solution to obtain a final concentration of 5 μg/mL.
-
3.
Add only to the aliquot control 50 μM verapamil (ABCB1 and ABCC1 transporter inhibitor, see Note 13 ) and leave the other aliquot untreated.
-
4.
Incubate the cell/dye suspensions for 90 min at 37 °C in the dark and gently invert the tubes every 15–20 min to avoid cell settling and clumping (see Note 14 ).
-
5.
After 90 min, spin the cells down at 4 °C (in a precooled rotor) and resuspend the cell pellets in an appropriate volume of cold FACS buffer (see Note 15 ).
-
6.
If the SP assay is combined with staining for cell surface phenotyping, the cells can now be processed for antibody staining as described in Subheading 3.4.1. Matching the SP assay with the classical cell-surface staining protocols can lead to a further characterization of putative CSC populations.
-
7.
Immediately prior to flow cytometry, add 7-AAD (to a final concentration of 2 μg/mL) and incubate cells for 15 min at 4 °C in the dark. Maintain cell suspensions at 4 °C and protected from light at all times.
-
8.
Acquire at least 500,000 total cells per sample on a suitable flow cytometer.
-
9.
The Hoechst 33342 dye is excited at 350 nm ultraviolet (UV). Therefore, an optimal SP resolution requires a flow cytometer equipped with a UV laser. To measure the resultant fluorescence at two wavelengths use a 450/20 band-pass filter and 675 long-pass filter for detection of Hoechst Blue and Red, respectively, and the 610 short-pass dichroic mirror to split the emission wavelengths.
-
10.
Create the following two-parameter plots (x-axis vs. y-axis) with all the parameters in linear scale:
-
(a)
Forward scatter (FSC) versus side scatter (SSC)
-
(b)
7-AAD versus SSC
-
(c)
Hoechst Red versus Hoechst Blue
-
(a)
-
11.
Exclude debris and dead cells in plots a and b, respectively.
-
12.
Identify the SP on the Hoechst Red versus Hoechst Blue plot (on the left side of the plot, showing low fluorescence intensity at both emission wavelengths).
-
13.
Optional: Sort SP-positive and SP-negative fractions and evaluate the purity. Culture sorted cell populations in standard medium for further in vitro and in vivo experiments.
3.4.3 Aldehyde Dehydrogenase (ALDH) Activity
The aldehyde dehydrogenase 1 (ALDH1) isoform catalyzes the conversion of retinol to retinoic acid in normal and malignant stem cells and its activity has been used as a functional stem cell marker to isolate CSCs in different tumor types [26]. Cells with ALDH1 enzymatic activity can be detected using a simple and rapid method that is now commercially available (STEMCELL Technologies) and it works with cultured cells as well as with cells isolated from solid tumors.
-
1.
Perform a viable cell count and resuspend cells in ALDEFLUOR™ assay buffer containing the ALDH substrate Bodipy™-aminoacetaldehyde (BAAA) and efflux inhibitor (see Notes 16 and 17 ).
-
2.
Transfer portion of cells from step 1 to negative control tube containing the ALDH enzyme inhibitor diethylamino-benzaldehyde (DEAB, 50 mmol/L).
-
3.
Incubate at 37 °C for 30–60 min.
-
4.
Spin the cells down at 4 °C (in a precooled rotor) and resuspend the cell pellets in an appropriate volume of cold FACS buffer (see Note 18 ).
-
5.
If the aldefluor assay is combined with staining for cell surface phenotyping, the cells can now be processed for antibody staining as described in Subheading 3.4.1.
-
6.
Measure the cellular fluorescence with the green fluorescence channel (520–540 nm) of a standard flow cytometer and perform analysis setting up proper gate according to instruction in ALDEFLUOR™ kit. Since only cells with an intact cellular membrane can retain BAA−, only viable ALDHbright cells are identified.
-
7.
Optional: Sort ALDH-positive and ALDH-negative fractions and evaluate the purity . Culture sorted cell populations in standard medium for further in vitro and in vivo experiments.
3.4.4 Sphere Formation Assay
In the sphere formation assay, cells from solid tumors are grown in serum-free, non-adherent conditions forming spherical structures (tumorspheres ). This assay can be used to estimate the percentage of CSCs present in a population of tumor cells since only malignant stem cells can survive and proliferate in this environment.
-
1.
Resuspend freshly isolated tumor cells or sorted cells into a single-cell suspension in complete tumorsphere medium described in Subheading 2.5. Importantly, B27 should be freshly added before use.
-
2.
Perform a viable cell count with trypan blue.
-
3.
Plate cell suspension onto ultra-low-attachment plates at appropriate density in triplicate. The optimum density is usually between 200 and 600 cells/cm2 depending on cell type.
-
4.
Maintain cells in a 5 % CO2-humidified incubator at 37 °C for 1 week without replenishing the medium so as not to disturb the formation of the tumorspheres .
-
5.
After 1-week incubation, analyze sphere formation under an inverted light microscope and count the number of tumorspheres (at 40× magnification) whose size can vary from less than 50 to 250 μm. They are easily distinguishable from aggregated cells as the cells seem to be fused together and single cells cannot be identified.
-
6.
Sphere-forming efficiency (%) is calculated according to the formula (number of spheres per wells/number of cells seeded per well) × 100.
3.4.5 In Vivo Tumorigenicity Evaluation
A crucial characteristic of CSCs is their ability to generate tumors as xenografts in immunodeficient mice. This unique feature must therefore be rigorously verified in animal models to prove the self-renewal and differentiation potential of the cellular subpopulation isolated with FACS and other marker-independent methods described above.
-
1.
Count viable cells, and serially dilute in PBS using a broad range of cell doses (from 102 to 106) to assess the optimum range of tumorigenicity.
-
2.
Thaw on ice an aliquot of Matrigel and mix equivalent volume of cell and Matrigel in a round-bottomed 96-well plate that is resting on ice (see Note 19 ).
-
3.
Prior to injection into mice, gently mix the cells/Matrigel suspensions and aspirate 20–100 μL per injection into cooled insulin syringe or a Hamilton syringe for the smallest volumes (see Note 20 ).
-
4.
Anesthetize mice with Avertin (375 mg/kg) intraperitoneally.
-
5.
Inject orthotopically at least five mice with each cell dose.
-
6.
Determine an approximate median of tumorigenic frequency and then use tighter dilutions to identify the minimum tumorigenic dose.
-
7.
Once xenografts are established, tumors should be compared histologically to the primary patient specimen (see Subheading 3.1) by hematoxylin and eosin staining.
4 Notes
-
1.
For RNAlater long-term storage at −20 °C, first incubate tumor fragments (placed in separate cryovials) overnight in the RNA stabilization reagent at 4 °C. Then transfer the tissue, in the reagent, to −20 °C for archival storage.
-
2.
Cover the cryomold with a thin layer of O.C.T. and, using sterile forceps, transfer the specimen to the cryomold. Fill it with more O.C.T. by paying attention to avoid air bubble formation, and freeze the mold in liquid nitrogen by holding the cryomold with a forceps over the opening of the liquid nitrogen transfer container (freezer gloves and a face shield should be used during this procedure).
-
3.
Many studies report that the implantation of tumor fragments in mice has a higher transplantation success rate than single-cell suspension injection . Then, in this step, it is important to keep aside small fragments (2–3 mm square) of tumor for both cryopreservation and direct implantation of the primary sample into immunodeficient mice. For cryopreservation: place no more than five fragments in a cryovial and fill it with a 90 % FBS + 10 % DMSO solution. Freeze the fragments by slowly decreasing the temperature 1 °C/min in a Nalgene Cryo 1 °C cell-freezing container filled with isopropanol. Place the container at −80 °C overnight before transferring the vials to liquid nitrogen cryotanks.
-
4.
Gentle agitation of tissue fragments may improve tumor dissociation. If use a shaker, or a tube rotator in a 37 °C incubator, set motion at low to moderate speed.
-
5.
The digestion progression can be monitored by gross observation and microscopic evaluation of the digestion medium (large undigested material may contain cell debris, fat, and extracellular matrix). Depending on the sample type, tumor dissociation should take from 30 min to 4 h. Since longer incubation times will reduce cell viability, the sample should not be left in the digestion medium for longer than necessary. Stop digestion by adding DMEM/F12 in equal amount of collagenase mix added.
-
6.
Adjust the pH to 7.2 and sterilize the solution using a 0.20 μm filter before use. Keep at 4 °C.
-
7.
At the first medium change, collect also culture supernatant containing unattached cells and plate it into a new flask to provide a backup culture.
-
8.
When possible, freeze earlier passages of the primary culture in 90 % FBS + 10 % DMSO and store in liquid nitrogen for further experiments.
-
9.
If you use primary unlabeled antibodies, after completing step 5, dilute the fluorochrome-labeled secondary antibody in FACS buffer (see individual antibody data sheet for the appropriate dilution) and then resuspend the cells in this solution. Incubate for at least 30 min at 4 °C in the dark. Wash the cells twice by centrifugation at 350 × g for 5 min and resuspend the cell pellet in 200–400 μL of FACS buffer for flow cytometric analysis.
-
10.
If you need to preserve cells for several days, after completing step 6 instead of resuspending cells in FACS buffer, add 100–200 μL 4 % paraformaldehyde and incubate for 15 min at room temperature. Centrifuge samples at 350 × g for 5 min and resuspend the cell pellet in 200–400 μL of FACS buffer. Fixation will stabilize the light scatter and inactivate most biohazardous agents. The fixation for different antigens will require optimization by the user.
-
11.
While CD133 has been efficiently used as a single marker for the isolation of CSCs in colorectal cancer and brain tumors , intratumoral heterogeneity makes it difficult to define universal stemness markers. Thereby, a set of markers are necessary to identify and characterize CSCs from other tumor types (see ref. 23).
-
12.
Continuously cool the samples to 4 °C during the procedure. Perform a forward scatter pulse height and side scatter analysis to exclude cell clusters and doublets. Perform a two-way cell sorting procedure with a 140 μm nozzle with a 5.5-PSI pressure, and with an event rate of 1000 events per second, using a sort pure mode (optimal conditions need to be optimized depending on the instrument and/or the cells sorted). Collect samples into sterile polypropylene tubes containing sterile FACS buffer, and use for further in vitro and in vivo studies.
-
13.
A significant number of ABC transporter inhibitors is available; based on both the tumor type and the type and content of ABC transporters, the use of a different inhibitor could be required (e.g., verapamil, cyclosporine A, and probenecid target the ABCB1 protein, while fumitremorgin C blocks the ABCG2 transporter).
-
14.
It is crucial that the temperature throughout the sample is maintained at 37 °C. To ensure a correct temperature control, the Hoechst incubation step is usually performed in a water bath. Otherwise, in order to avoid manual mixing of the tubes, dye incubation step can also be successfully carried out in a 37 °C incubator equipped with a gentle shaker.
-
15.
In order to inhibit further dye efflux, the cells must be kept at 4 °C for the rest of the experiment. Thus, steps following the Hoechst staining (e.g., washing, centrifugation, antibody staining, and data acquisition) should be carried out in the cold and in the dark, for minimizing metabolic activity and preserving the Hoechst profile.
-
16.
Perform the assay using different sample concentrations (range from 1 × 105 to 2 × 106 cells/mL) and select the cell concentration that better discerns between ALDHbright and ALDHlow cells minimizing background ratio.
-
17.
BAAA is a visible light excitable fluorochrome that diffuses freely across the plasma membrane of intact viable cells. Only cells with ALDH activity convert BAAA into the fluorescent product Bodipy™-aminoacetate (BAA−). The reaction product BAA− is retained inside cells since its exclusion through ABC transporters is blocked by efflux inhibitors contained into the assay buffer. To optimize the assay, it may be useful to add other efflux inhibitors (e.g., verapamil, probenecid, or 2-deoxy-d-glucose).
-
18.
In order to inhibit efflux of BAAA and of BAA− and to prevent loss of fluorescence intensity, the cells must be kept at 4 °C for the rest of the experiment.
-
19.
The final 1:1 cells/Matrigel suspension volume depends upon the injection site ranging from 20 μL (e.g., renal capsule and brain) to 100 μL (subcutaneous and mammary fat pad).
-
20.
Traditionally, CSC research has been carried out using severe combined immunodeficient (SCID) or nonobese diabetic SCID (NOD/SCID) mice. However, the use of more highly immunocompromised NOD/SCID interleukin-2 receptor γ (IL-2Rγ)–null (NSG) mice lacking T, B, and NK cells can increase the detection of tumorigenic cells.
References
Marusyk A, Polyak K (2010) Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 1805:105–117
Singh AK, Arya RK, Maheshwari S, Singh A, Meena S, Pandey P, Dormond O, Datta D (2015) Tumor heterogeneity and cancer stem cell paradigm: updates in concept, controversies and clinical relevance. Int J Cancer 136:1991–2000
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414:105–111
Ribatti D (2012) Cancer stem cells and tumor angiogenesis. Cancer Lett 321:13–17
Martin-Padura I, Marighetti P, Agliano A, Colombo F, Larzabal L, Redrado M, Bleau AM, Prior C, Bertolini F, Calvo A (2012) Residual dormant cancer stem-cell foci are responsible for tumor relapse after antiangiogenic metronomic therapy in hepatocellular carcinoma xenografts. Lab Invest 92:952–966
Dawood S, Austin L, Cristofanilli M (2014) Cancer stem cells: implications for cancer therapy. Oncology 28:1101–1107
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367:645–648
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983–3988
Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M (2005) A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 65:9328–9337
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828
Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15:504–514
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ (2005) Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 65:10946–10951
Bapat SA, Mali AM, Koppikar CB, Kurrey NK (2005) Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res 65:3025–3029
Zimmerlin L, Donnenberg VS, Donnenberg AD (2011) Rare event detection and analysis in flow cytometry: bone marrow mesenchymal stem cells, breast cancer stem/progenitor cells in malignant effusions, and pericytes in disaggregated adipose tissue. Methods Mol Biol 699:251–273
Tirino V, Desiderio V, d’Aquino R, De Francesco F, Pirozzi G, Graziano A, Galderisi U, Cavaliere C, De Rosa A, Papaccio G, Giordano A (2008) Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS One 3:e3469
Hadnagy A, Gaboury L, Beaulieu R, Balicki D (2006) SP analysis may be used to identify cancer stem cell populations. Exp Cell Res 312:3701–3710
Song J, Chang I, Chen Z, Kang M, Wang CY (2010) Characterization of side populations in HNSCC: highly invasive, chemoresistant and abnormal Wnt signaling. PLoS One 5:e11456
Ma I, Allan AL (2011) The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev 7:292–306
Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C (1999) Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci U S A 96:9118–9123
Liu JC, Deng T, Lehal RS, Kim J, Zacksenhaus E (2007) Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res 67:8671–8681
Zhong Y, Guan K, Guo S, Zhou C, Wang D, Ma W, Zhang Y, Li C, Zhang S (2010) Spheres derived from the human SK-RC-42 renal cell carcinoma cell line are enriched in cancer stem cells. Cancer Lett 299:150–160
Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, Laino L, De Francesco F, Papaccio G (2013) Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J 27:13–24
Greve B, Kelsch R, Spaniol K, Eich HT, Götte M (2012) Flow cytometry in cancer stem cell analysis and separation. Cytometry A 81:284–293
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275–284
Golebiewska A, Brons NH, Bjerkvig R, Niclou SP (2011) Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell 8:136–147
Sládek NE (2003) Human aldehyde dehydrogenases: potential pathological, pharmacological, and toxicological impact. J Biochem Mol Toxicol 17:7–23
Acknowledgments
Supported in part by AIRC (Associazione Italiana per la Ricerca sul Cancro), Fondazione Umberto Veronesi, and Ministero della Salute.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Orecchioni, S., Bertolini, F. (2016). Characterization of Cancer Stem Cells. In: Ribatti, D. (eds) Tumor Angiogenesis Assays. Methods in Molecular Biology, vol 1464. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3999-2_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3999-2_5
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3997-8
Online ISBN: 978-1-4939-3999-2
eBook Packages: Springer Protocols